 Grégoire B. Morand
Grégoire B. Morand Sabrina Daniela da Silva
Sabrina Daniela da Silva Michael P. Hier1
Michael P. Hier1 Moulay A. Alaoui-Jamali
Moulay A. Alaoui-Jamali- 1Department of Otolaryngology-Head and Neck Surgery, Sir Mortimer B. Davis-Jewish General Hospital, McGill University, Montreal, QC, Canada
- 2Departments of Medicine and Oncology, Segal Cancer Centre and Lady Davis Institute for Medical Research, Sir Mortimer B. Davis-Jewish General Hospital, McGill University, Montreal, QC, Canada
Vitamin D is a key regulator of calcium metabolism and has been implicated as a cancer preventive agent. However, clinical studies have revealed conflicting results on its cancer preventive properties, attributed in part to multiple metabolic and regulatory factors susceptible to affect individual responses to exogenous vitamin D. Vitamin D is obtained from dietary sources and sun exposure, which depends on numerous parameters such as skin type, latitude, and lifestyle factors. Focusing on thyroid cancer (TC), we document that genetic and epigenetic determinants can greatly impact individual response to vitamin D and may outweigh the classical clinical correlative studies that focus on sun exposure/dietary intake factors. In particular, genetic determinants innate to host intrinsic metabolic pathways such as highly polymorphic cytochromes P450s responsible for the metabolic activation of vitamin D are expressed in many organs, including the thyroid gland and can impact vitamin D interaction with its nuclear receptor (VDR) in thyroid tissue. Moreover, downstream regulatory pathways in vitamin D signaling as well as VDR are also subject to wide genetic variability among human populations as shown by genome-wide studies. These genetic variations in multiple components of vitamin D pathways are critical determinants for the revaluation of the potential preventive and anticancer properties of vitamin D in TC.
Introduction
Thyroid cancer (TC) is the most common endocrine cancer malignancy worldwide (1) with a rising incidence in particular among young patients and women (2–4). Overdiagnosis of subclinical disease, previously proposed as a contributor for the rising incidence, cannot explain the full extent of the increase (5, 6). Risk factors such as exposure to ionizing radiation (7–10), chemical genotoxins (11–13), and obesity (14–17), as well as a lack of protective factors, such as vitamin D deficiency have been implicated in TC increased incidence (18–21).
Vitamin D, an active ingredient of cod-liver oil, was first identified as a cure for rickets in the nineteenth century and has emerged as a principal regulator of calcium homeostasis (22). Cutaneous exposure to sun and dietary intake are the two main natural sources of vitamin D. Vitamin D activity depends on metabolic activation through hydroxylation of the 25 followed by the 1 position of the molecule by cytochromes P450s, which generate the biologically active metabolite 1,25(OH)2D3. The action of vitamin D occurs mainly through its binding to the nuclear vitamin D receptor (VDR), which acts as a hormone-regulated transcription factor (23). Upon activation, the VDR forms a heterodimer with related retinoid-X receptors and binds to vitamin D response elements (VDREs) on chromatin regions resulting in the regulation of the expression of several target genes (24–26). VDRE binding by the VDR provides the principle mechanism by which the receptor can activate gene transcription. However, the hormone-bound receptor can also repress gene transcription by a variety of mechanisms (27). Downstream targets of the receptor are involved in mineral metabolism, but VDR also regulates a variety of other metabolic pathways, many of which are components of immune response and cancer signaling (28, 29).
Independent studies support that circulating levels of vitamin D are inversely correlated to several malignancies, including colorectal cancer (30, 31), prostate cancer (32), breast cancer (33, 34), and head and neck squamous cell carcinoma (35, 36). As well, a more recent meta-analysis reported a correlation between vitamin D deficiency and poorer prognosis in several tumor types (37). In TC, several studies point toward a role for impaired 1,25(OH)2D3-VDR signaling in the occurrence and progression of the disease (38). This review addresses new insights into genetic and epigenetic determinants of vitamin D response in relation to cancer risk focusing on TC. We provide a systematic review and analysis of experimental and clinical data and the impact of genome-wide analyses on individual susceptibility to TC.
Materials and Methods
Genomic Database
The UCSC Cancer Genomics Browser (39), a set of web-based tools to display, was used to investigate and analyze cancer genomics data and its clinical information associated with VDR. The browser provides whole-genome to base-pair level views of several different types of genomic data, including next-generation sequencing platforms. Biological pathways, collections of genes, genomic or clinical information were used to sort, aggregate, and zoom into a group of samples. The current release (2013) displays an expanding set of data from various sources, including 201 datasets from 22 The Cancer Genome Atlas (TCGA) cancers as well as data from Cancer Cell Line Encyclopedia and Stand Up To Cancer (39).
Database of Somatic Mutations
To collect data on TC related to VDR mutation, the web-software BioMart Central Portal and the Catalog of Somatic Mutations in Cancer (COSMIC) database (40) were used. BioMart offers a one-stop shop solution to access a wide array of biological databases, such as the major biomolecular sequence, pathway, and annotation databases such as Ensembl, Uniprot, Reactome, HGNC, Wormbase, and PRIDE (41). The Cancer BioMart web-interface with the following criteria was used: (1) Primary site = “thyroid”; (2) Mutation ID is not empty. The first criterion ensures that the mutation occurs in thyroid tissues, and the second criterion helps to exclude the samples without mutation in a specific gene. Thereby, we obtained the list of mutations in TC.
Catalog of Somatic Mutations in Cancer (40) stores and displays somatic mutation information and related details on human cancers. COSMIC was developed, and is currently maintained, at the Welcome Trust Sanger Institute. It is designed to gather, curate, and organize information on somatic mutations in cancer and to make it freely available on-line. It combines cancer mutation data, manually curate from the scientific literature, with the output from the Cancer Genome Project (CGP). Genes are selected for full literature curation using the Cancer Gene Census. The current release (v64) describes over 913,166 coding mutations of 24,394 genes from almost 847,698 tumor samples. All genes selected for the COSMIC database came from studies in the literature and are somatically mutated in human cancer (42). Based on this authority resource, a dataset of TC mutation was constructed.
Data Extraction
Information was carefully extracted from all eligible publications including clinical and experimental studies assessing any relation between vitamin D and non-medullary TC. A search for studies in the electronic databases Ovid Medline, Ovid Embase, Web of Science, AMED, and the Cochrane Library was run using an elaborated search strategy (Supplemental Material). In order not to miss any appropriate study, no time or language limits were applied for the search. Review articles were included only temporarily to provide a manual search tool.
The selection of studies involved an initial screening of the title and the abstract. In doubtful cases, the full text was obtained. Articles were entered in the data management software and the duplicates were eliminated (Endnote 6®, Thomson Reuters Inc.). For clinical studies, detailed information about participants (number of patients, study location(s), and demographics variables), exposure (sun irradiation, dietary intake, and vitamin D serum level), comparison group, and outcome was assessed.
The search retrieved 471 references published until July 4th, 2013, 12 from the Cochrane Library, 176 from Ovid Medline, 188 from Ovid Embase and AMED, and 95 from Web of Sciences. Crosschecking the references of the reviews led to the inclusion of four supplementary articles (43–46). No clinical trial was available. The flow chart of study selection is shown in Figure 1.
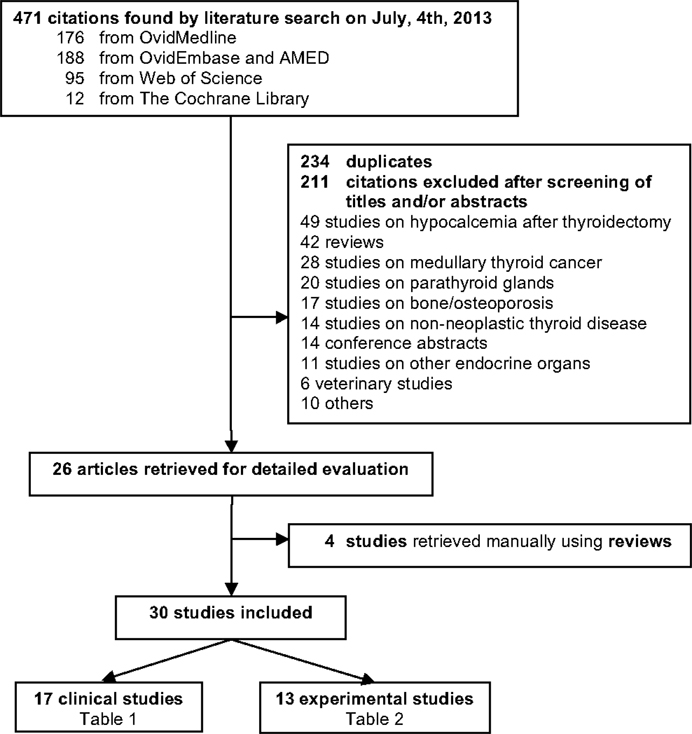
Figure 1. Flow chart of study selection for systematic review.
Overall 30 articles were included, of which 17 were clinical studies (Table 1) and 13 experimental studies (Table 2). These studies were published in English language from 1987 to 2013. Of the 17 clinical studies, 8 (47.0%) showed protective effect of vitamin D (44, 45, 47–52), 6 (35.3%) no significant relationship (43, 46, 53–57), and 2 (11.7%) revealed an increased TC risk with high vitamin D intake (58, 59). No comparison could be drawn from the remaining study (5.8%) (60). TC incidence was assessed in all of these studies, mortality in two (45, 47); and one report assessed both (45). Except for three studies involving Arab populations (51, 56, 60), all studies included Europeans’ descendants and/or Hispanic whites.
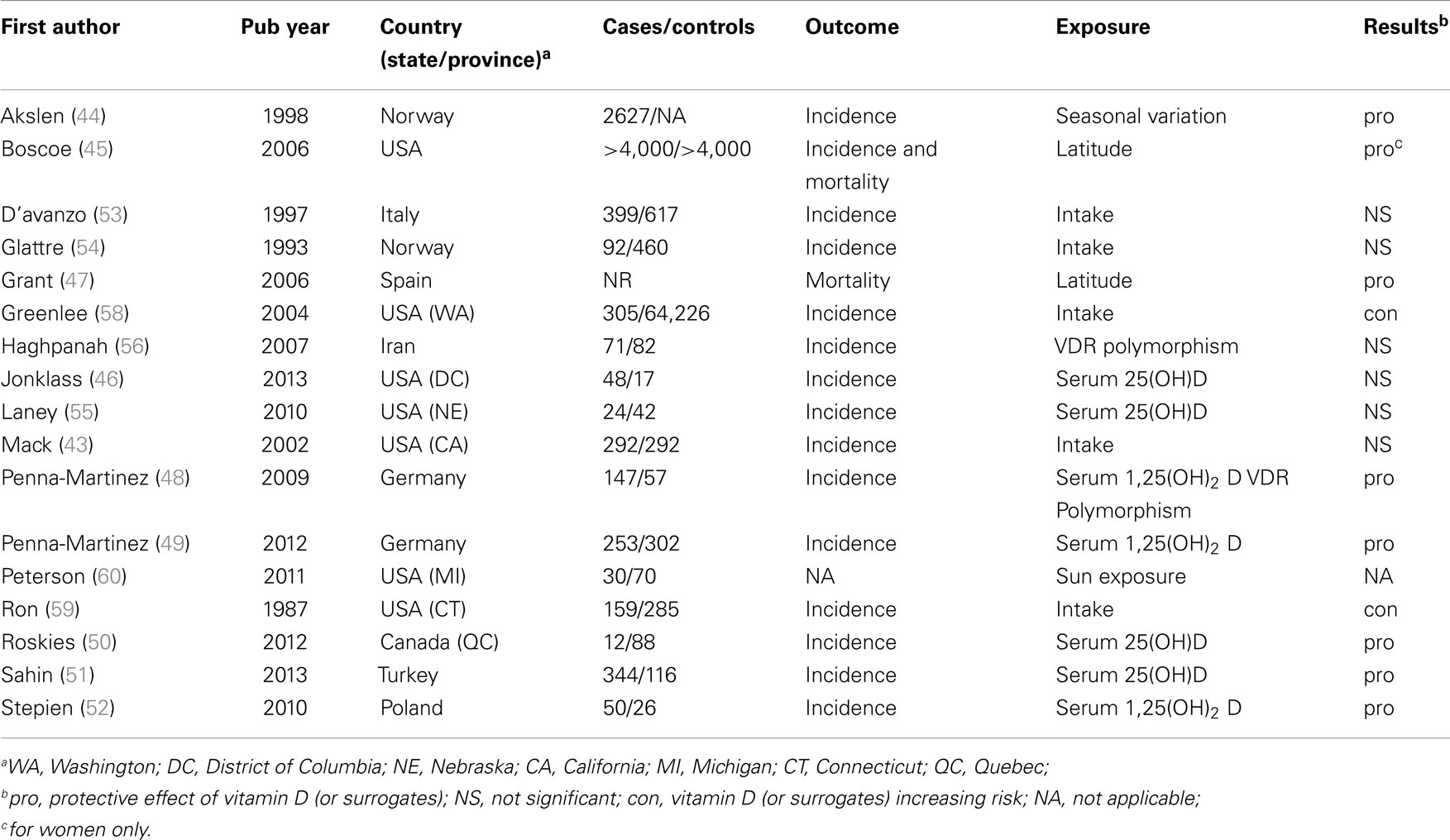
Table 1. Summary of clinical studies reporting an association between thyroid cancer and vitamin D.
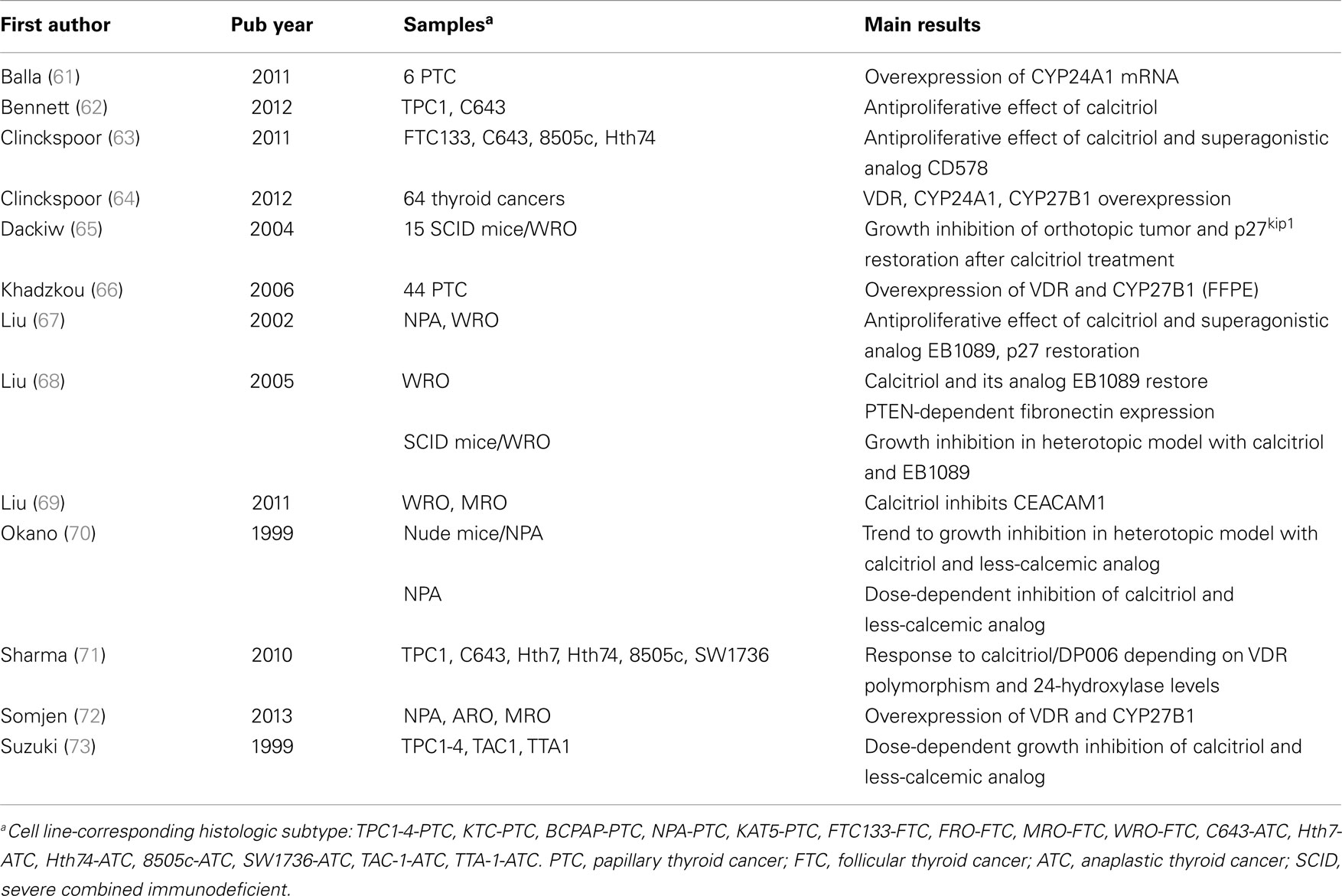
Table 2. Experimental studies using cell lines or preclinical models to assess vitamin D effect on thyroid cancer.
Results and Discussion
Determinants of Vitamin D Levels and Impact in TC
Solar UVB irradiation is the primary source of vitamin D and can be estimated by latitude of the living area. In TC, large epidemiological studies support an inverse relation between TC incidence and latitude (45, 47) (Table 1). These studies performed a multivariate analysis to adjust for confounding factors. However, vitamin D levels were not measured. Consequently, it is unclear if the multivariate analysis resulted in accurate vitamin D estimates. Indeed, vitamin D deficiency is highly prevalent among latitudes that benefit from high solar irradiation such as Africa, the Middle East, and Southern Asia. This may be due to skin pigmentation, traditional clothing, and sun avoidance seen in southern heat-exposed populations (60, 74). In contrast, fair-skinned northern populations usually seek sun exposure and may also benefit from high intake of vitamin D rich diet such as fatty fish and cod-liver oil (74). Further, a mutation in the cutaneous structural protein filaggrin, which occurs in up to 10% of Europeans was shown to lead to higher circulating vitamin D levels (75). Nonetheless, North American and European studies have shown seasonal variations of vitamin D levels due to insufficient sun irradiation during winter (76). In TC, one study from Norway reported higher proliferation values for tumors resected during winter compared to other seasons (44). These results comply with above-mentioned studies showing an inverse relation between TC incidence and latitude (45, 47). For studies estimating vitamin D consumption and TC risk, however, no convincing associations have been shown (Table 1) (43, 53, 54, 58, 59). This may be due to the general poor correlation between vitamin D deficiency and estimates of vitamin D consumption (57).
A more accurate way to assess vitamin D is biological monitoring. Association studies investigating the relationship between levels of serum vitamin D and TC risk mostly point toward a protective effect of vitamin D (48–52, 55, 77) (Table 1). Pooling the data among these studies is not possible due to different cut-off levels for different vitamin D derivatives and control groups used in each of these studies. This would greatly limit the validity of a meta-analysis. The lack of consensus in cut-off levels may reflect the fact that those are differently defined depending on targeted clinical endpoints (78, 79). Classical vitamin D targets, i.e., those implicated in calcium and bone homeostasis, do not allow conclusions on optimal level of vitamin D having anticancer properties. While doses up to 4,000 IU of daily vitamin D supplementation have been considered safe, studies have reported hypercalcemia, nephrolithiasis, vascular, and soft tissue calcification with high doses of vitamin D and also U-shaped relationship between vitamin D levels above 75 nmol/l and certain cancer subtypes (80, 81). One additional issue of most of these association studies is that vitamin D levels were measured only once, which does not permit distinction between outcome and exposure. Indeed, some studies have reported low serum vitamin D as a result of malignancy (82).
Above-mentioned skin types, alimentary, and social habits yet do not fully explain vitamin D variability among populations (83). One major determinant of individual susceptibility to vitamin D is the activity of vitamin D metabolizing enzymes. Three major cytochrome P-450 (CYP) hydroxylases are responsible for vitamin D activation through 25- followed by 1α-hydroxylation of the molecule, and deactivation through 24-hydroxylation. Multiple enzymes have been reported as vitamin D 25-hydroxylases, a step occurring constitutively and primarily in the liver. Unlike 25-hydroxylation, 1α-hydroxylation of 25(OH)D3 by the CYP27B1 is a tightly regulated and rate-limiting step. It is regulated by calcium, 1α,25(OH)2D3 itself, PTH, calcitonin, and phosphate levels. Recently, fibroblast growth factor 23 (FGF23) was identified as a novel antagonist of PTH and is thought to play an important role in vitamin D regulation pathway (84). Although CYP27B1 and CYP24A1 are primarily expressed in the kidney, recent studies showed that they are expressed in many other tissues, including the thyroid (61, 62). In TC, there is evidence that polymorphisms leading to impaired CYP27B1 function and/or increased CYP24A1 activity are associated with increased TC risk (49). Transcriptional profiling studies show that both enzymes are overexpressed in early TC (61), but their expression tends to decrease along with tumor progression (64, 66).
Determinants of Predicted Response to Vitamin D
The action of vitamin D mainly occurs through binding to the VDR (23), whose levels are subject to genetic variations. Using the UCSC genomic database, we analyzed 552 thyroid samples that underwent genomic profiling using RNA Seq. The expression of VDR was down regulated in benign thyroid samples and up regulated in most TC cases (Figure 2). These results are confirmed by a few in vitro studies using TC cell lines (72) and independent clinical samples (64, 66). However, VDR levels alone may translate poorly with response to vitamin D stimulation if polymorphisms of VDR are not taken into account (71, 85, 86). The analysis of the genomic organization of the VDR locus at chromosome 12q13.1 revealed the large VDR gene (about 100 Kb) with an extensive promoter region capable of generating multiple tissue-specific transcripts (87). In view of the observed genome-wide frequency of single nucleotide polymorphisms (88), one can predict >100 functional polymorphisms to be present in the VDR region alone, including the promoter region (Figure 3). Point mutations in the VDR gene have been identified in various regions, including the VDR DNA binding domain (DBD) and the ligand-binding domain (LBD) (89). Such mutations can disrupt ligand-binding affinity to the receptor (90), heterodimerization of VDR with RXR (91), or interactions of the VDR receptor with partners such as coactivators (92). Other mutations such as in the initiation codon can create a premature termination (93) or alternative translation start sites to result in alternative splicing and formation of truncated proteins (94, 95). The analysis of the COSMIC database showed a high proportion of missense mutations that were re-identified (67.44%), while complex mutations were not detected (Table 3). The distribution of the mutations observed in the VDR gene in TC is shown in Figure 4. Only two studies investigated the association between VDR polymorphisms and TC risk, one showed an increased TC risk for patients with particular VDR polymorphism (48), while another could not point out any significant difference (56).
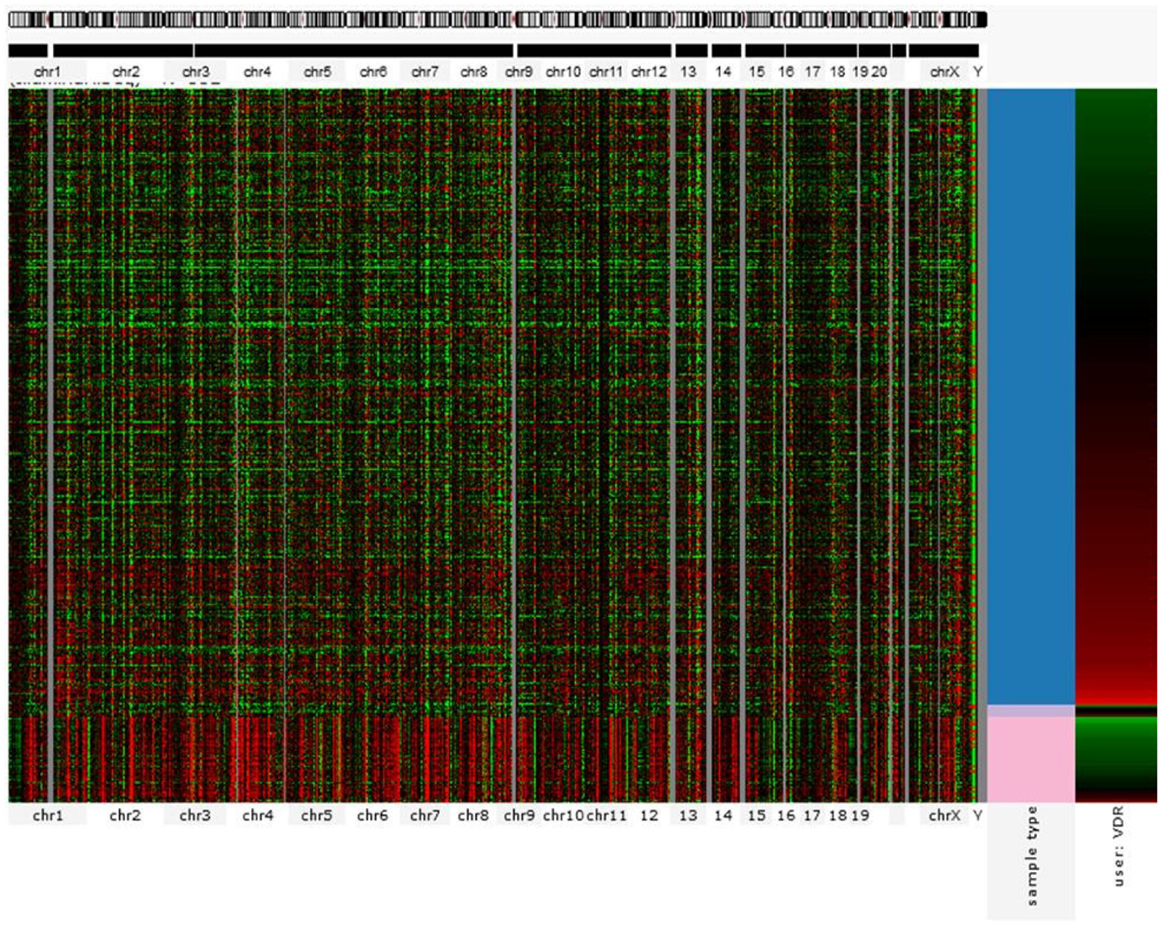
Figure 2. Gene expression profile showing VDR signature for 552 thyroid cancer cases (RNA Seq). Each row corresponds to sample from a single case. Columns from the left correspond to genomic heatmap according to chromosomal location. The last two columns represent VDR expression profile (represented by red for overexpression and green for downregulation) in normal (pink) versus cancer (red) tissues. VDR is mostly overexpressed in malignant samples but almost absent in benign tissues. Source: UC Santa Cruz – Cancer Genomics Browser.
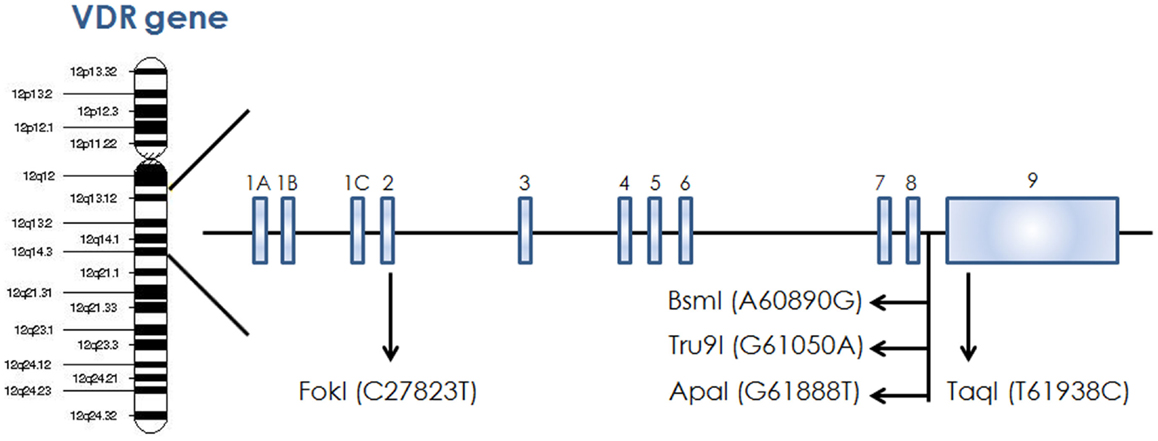
Figure 3. Schematic diagram of VDR gene showing different restriction site on chromosome 12.
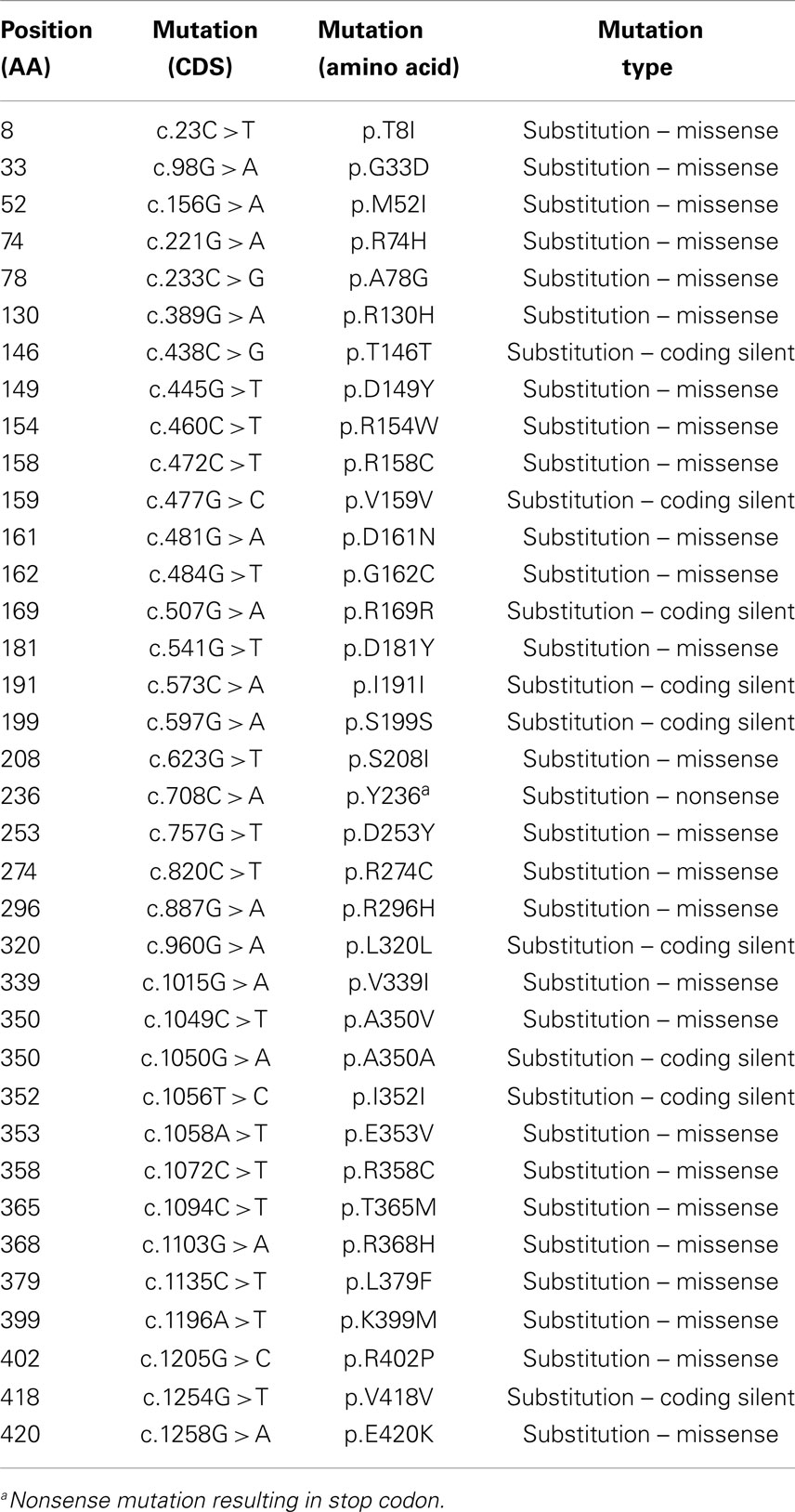
Table 3. Mutations identified in VDR.
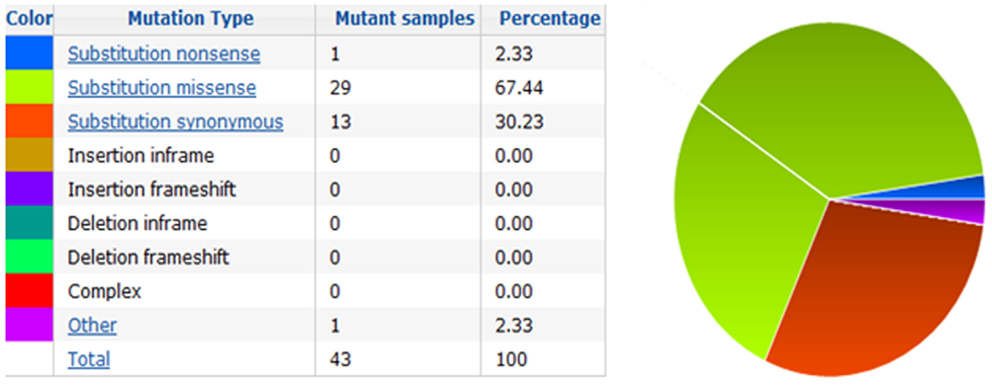
Figure 4. Pie-chart showing the percentage of the mutation type in VDR in thyroid cancer according to COSMIC database.
Downstream Impact of VDR Activation
Upon activation by vitamin D, VDR binds as a heterodimer with retinoid-X receptors to specific VDREs (84). VDREs usually bear a consensus sequence known as DR3 element located in the promoter region of the target genes. In addition to this classic mechanism, recent chromatin-immunoprecipitation (ChIP-seq) studies allowed to gain genome-wide insights of the binding sites of VDR (96). These studies showed that the ligand-bound heterodimer can bind to ~2000–8000 sites in the genome. Interestingly, the majority of the binding sites do not bear the classical DR3-type sequence (84). A significant enrichment was seen in regions associated with active chromatin and histone modifications thus supporting a broad genetic and epigenetic regulatory role of vitamin D. Further enrichment of VDR binding was also found in proximity of genes involved in autoimmune diseases (e.g., multiple sclerosis, type-I diabetes, and Crohn’s disease) and colorectal or breast cancer (97). For TC, only data relying on classical in vitro experiments is available.
In agreement with experimental studies in other cancer types, exposure of a variety of TC cells to vitamin D in vitro leads to antiproliferative and pro-differentiation properties (62, 63, 67, 70, 71, 73) (Table 2). These results have been confirmed by in vivo studies (65, 68). Most studies are testing vitamin D itself and synthetic vitamin D analogs, as patient’s exposure to pharmacologically high doses of vitamin D can be limited by the side-effects, mainly hypercalcemia (63, 67, 70, 71, 73).
Mechanistically, vitamin D was shown to inhibit proliferation through c-mac mRNA inhibition, which is a well-known proto-oncogene (70). Further, it can induce a growth arrest effect in part through stimulating accumulation of the cyclin-dependent kinase inhibitor p27kip1 in the nucleus (67). Treatment with vitamin D is thought to prevent p27kip1 phosphorylation, which was shown to increase its ubiquitin-dependent proteasome degradation (67). Further, vitamin D was shown to enhance cell–cell adhesion through PTEN-dependent fibronectin upregulation (68). Those results could be confirmed in vivo. Interestingly, the antiproliferative effect of vitamin D was abolished when knocking down fibronectin (68) and was shown to be independent of CEACAM1 expression, a tumor-suppressive adhesion molecule (69).
Conclusion and Perspectives
The pleiotropic roles of vitamin D in cancer have been recognized through seminal preclinical studies although the preventive and therapeutic potential of vitamin D or its analogs remain debated due in part to the complex mode of action of this vitamin. Recent progress in high-throughput technologies to interrogate human genomic and epigenomic events has provided additional levels of regulatory loops and individual genetic variations that can impact on individual susceptibility to vitamin D. This knowledge opens up new tools to address confounding factors that contribute to discrepant results seen in previous association studies, in particular in relation to cancer prevention. As well, this knowledge impels an exciting avenue in the discovery of novel vitamin D analogs with enhanced preventive or therapeutic efficiency and limited side-effects.
Author Contributions
Gregoire B. Morand performed the literature search, the retrieval of the studies, the data extraction, and wrote the main part of the manuscript under Sabrina Daniela da Silva and Moulay A. Alaoui-Jamali’s supervision. All the authors participated substantially to the final manuscript and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Gregoire B. Morand is supported by the Swiss Cancer League (BIL KFS-3002-08-2012). The authors would like to thank Dr. John White (Department of Physiology, McGill University) for his precious review of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fonc.2014.00309/abstract
References
1. Curado M, Edwards B, Shin H, Storm H, Ferlay J, Heanue M, et al. Cancer Incidence in Five Continents Vol. IX. Lyon: IARC Scientific Publication No 160 (2007).
2. Liu S, Semenciw R, Ugnat AM, Mao Y. Increasing thyroid cancer incidence in Canada, 1970-1996: time trends and age-period-cohort effects. Br J Cancer (2001) 85(9):1335–9. doi: 10.1054/bjoc.2001.2061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973–2002. JAMA (2006) 295(18):2164–7. doi:10.1001/jama.295.18.2164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Veiga LH, Neta G, Aschebrook-Kilfoy B, Ron E, Devesa SS. Thyroid cancer incidence patterns in Sao Paulo, Brazil and the U.S. SEER program, 1997-2008. Thyroid (2013). 23(6):748–57. doi:10.1089/thy.2012.0532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Morris LG, Sikora AG, Tosteson TD, Davies L. The increasing incidence of thyroid cancer: the influence of access to care. Thyroid (2013) 23(7):885–91. doi:10.1089/thy.2013.0045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Morris LGT, Myssiorek D. Improved detection does not fully explain the rising incidence of well-differentiated thyroid cancer: a population-based analysis. Am J Surg (2010) 200(4):454–61. doi:10.1016/j.amjsurg.2009.11.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Hamatani K, Mukai M, Takahashi K, Hayashi Y, Nakachi K, Kusunoki Y. Rearranged anaplastic lymphoma kinase (ALK) gene in adult-onset papillary thyroid cancer amongst atomic bomb survivors. Thyroid (2012) 22(11):1153–9. doi:10.1089/thy.2011.0511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Goyal N, Camacho F, Mangano J, Goldenberg D. Thyroid cancer characteristics in the population surrounding three mile island. Laryngoscope (2012) 122(6):1415–21. doi:10.1002/lary.23314
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Fridman MV, Savva NN, Krasko OV, Zborovskaya AA, Mankovskaya SV, Kurt Werner S, et al. Clinical and pathologic features of “sporadic” papillary thyroid carcinoma registered in the years 2005 to 2008 in children and adolescents of Belarus. Thyroid (2012) 22(10):1016–24. doi:10.1089/thy.2011.0005
10. Dom G, Tarabichi M, Unger K, Thomas G, Oczko-Wojciechowska M, Bogdanova T, et al. A gene expression signature distinguishes normal tissues of sporadic and radiation-induced papillary thyroid carcinomas. Br J Cancer (2012) 107(6):994–1000. doi:10.1038/bjc.2012.302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Biondi BM, Arpaia D, Montuori P, Ciancia G, Ippolito S, Pettinato G, et al. Under the shadow of Vesuvius: a risk for thyroid cancer? Thyroid (2012) 22(12):1296–7. doi:10.1089/thy.2012-0002
12. Pellegriti G, De Vathaire F, Scollo C, Attard M, Giordano C, Arena S, et al. Papillary thyroid cancer incidence in the volcanic area of Sicily. J Natl Cancer Inst (2009) 101(22):1575–83. doi:10.1093/jnci/djp354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Lise M, Franceschi S, Buzzoni C, Zambon P, Falcini F, Crocetti E, et al. Changes in the incidence of thyroid cancer between 1991 and 2005 in Italy: a geographical analysis. Thyroid (2012) 22(1):27–34. doi:10.1089/thy.2011.0038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Zhao ZG, Guo XG, Ba CX, Wang W, Yang YY, Wang J, et al. Overweight, obesity and thyroid cancer risk: a meta-analysis of cohort studies. J Int Med Res (2012) 40(6):2041–50. doi:10.1177/030006051204000601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Cheng SP, Liu CL, Hsu YC, Chang YC, Huang SY, Lee JJ. Expression and biologic significance of adiponectin receptors in papillary thyroid carcinoma. Cell Biochem Biophys (2013) 65(2):203–10. doi:10.1007/s12013-012-9419-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Cheng SP, Liu CL, Hsu YC, Chang YC, Huang SY, Lee JJ. Regulation of leptin receptor expression in human papillary thyroid cancer cells. Biomed Pharmacother (2012) 66(6):469–73. doi:10.1016/j.biopha.2012.03.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Brindel P, Doyon F, Rachedi F, Boissin JL, Sebbag J, Shan L, et al. Anthropometric factors in differentiated thyroid cancer in French Polynesia: a case-control study. Cancer Causes Control (2009) 20(5):581–90. doi:10.1007/s10552-008-9266-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Michikawa T, Inoue M, Shimazu T, Sasazuki S, Iwasaki M, Sawada N, et al. Green tea and coffee consumption and its association with thyroid cancer risk: a population-based cohort study in Japan. Cancer Causes Control (2011) 22(7):985–93. doi:10.1007/s10552-011-9771-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Jung SK, Kim K, Tae K, Kong G, Kim MK. The effect of raw vegetable and fruit intake on thyroid cancer risk among women: a case-control study in South Korea. Br J Nutr (2013) 109(1):118–28. doi:10.1017/S0007114512000591
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Clero E, Doyon F, Chungue V, Rachedi F, Boissin JL, Sebbag J, et al. Dietary patterns, goitrogenic food, and thyroid cancer: a case-control study in French Polynesia. Nutr Cancer (2012) 64(7):929–36. doi:10.1080/01635581.2012.713538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Kohrle J, Gartner R. Selenium and thyroid. Best Pract Res Clin Endocrinol Metab (2009) 23(6):815–27. doi:10.1016/j.beem.2009.08.002
22. Rajakumar K. Vitamin D, cod-liver oil, sunlight, and rickets: a historical perspective. Pediatrics (2003) 112(2):e132–5. doi:10.1542/peds.112.2.e132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Brown AJ. Vitamin D analogues. Am J Kidney Dis (1998) 32(2 Suppl 2):S25–39. doi:10.1053/ajkd.1998.v32.pm9808141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature (1992) 355(6359):446–9. doi:10.1038/355446a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Lin R, White JH. The pleiotropic actions of vitamin D. Bioessays (2004) 26(1):21–8. doi:10.1002/bies.10368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science (2001) 294(5548):1866–70. doi:10.1126/science.294.5548.1866
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Dimitrov V, Salehi-Tabar R, An BS, White JH. Non-classical mechanisms of transcriptional regulation by the vitamin D receptor: insights into calcium homeostasis, immune system regulation and cancer chemoprevention. J Steroid Biochem Mol Biol (2014) 144(Pt A):74–80. doi:10.1016/j.jsbmb.2013.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. White JH. Vitamin D signaling, infectious diseases, and regulation of innate immunity. Infect Immun (2008) 76(9):3837–43. doi:10.1128/IAI.00353-08
29. Liu PT, Stenger S, Li H, Wenzel L, Tan BH, Krutzik SR, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science (2006) 311(5768):1770–3. doi:10.1126/science.1123933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Pilz S, Kienreich K, Tomaschitz A, Ritz E, Lerchbaum E, Obermayer-Pietsch B, et al. Vitamin d and cancer mortality: systematic review of prospective epidemiological studies. Anticancer Agents Med Chem (2013) 13(1):107–17. doi:10.2174/187152013804487407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Ma Y, Zhang P, Wang F, Yang J, Liu Z, Qin H. Association between vitamin D and risk of colorectal cancer: a systematic review of prospective studies. J Clin Oncol (2011) 29(28):3775–82. doi:10.1200/JCO.2011.35.7566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Gandini S, Boniol M, Haukka J, Byrnes G, Cox B, Sneyd MJ, et al. Meta-analysis of observational studies of serum 25-hydroxyvitamin D levels and colorectal, breast and prostate cancer and colorectal adenoma. Int J Cancer (2011) 128(6):1414–24. doi:10.1002/ijc.25439
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Bauer SR, Hankinson SE, Bertone-Johnson ER, Ding EL. Plasma vitamin D levels, menopause, and risk of breast cancer: dose-response meta-analysis of prospective studies. Medicine (2013) 92(3):123–31. doi:10.1097/MD.0b013e3182943bc2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Chen P, Hu P, Xie D, Qin Y, Wang F, Wang H. Meta-analysis of vitamin D, calcium and the prevention of breast cancer. Breast Cancer Res Treat (2010) 121(2):469–77. doi:10.1007/s10549-009-0593-9
35. Prudencio J, Akutsu N, Benlimame N, Wang T, Bastien Y, Lin R, et al. Action of low calcemic 1alpha,25-dihydroxyvitamin D3 analogue EB1089 in head and neck squamous cell carcinoma. J Natl Cancer Inst (2001) 93(10):745–53. doi:10.1093/jnci/93.10.745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Satake K, Takagi E, Ishii A, Kato Y, Imagawa Y, Kimura Y, et al. Anti-tumor effect of vitamin A and D on head and neck squamous cell carcinoma. Auris Nasus Larynx (2003) 30(4):403–12. doi:10.1016/S0385-8146(03)00091-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Li M, Chen P, Li J, Chu R, Xie D, Wang H. Review: the impacts of circulating 25-hydroxyvitamin D levels on cancer patient outcomes: a systematic review and meta-analysis. J Clin Endocrinol Metab (2014) 99(7):2327–36. doi:10.1210/jc.2013-4320
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Clinckspoor I, Verlinden L, Mathieu C, Bouillon R, Verstuyf A, Decallonne B. Vitamin D in thyroid tumorigenesis and development. Prog Histochem Cytochem (2013) 48(2):65–98. doi:10.1016/j.proghi.2013.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Goldman M, Craft B, Swatloski T, Ellrott K, Cline M, Diekhans M, et al. The UCSC cancer genomics browser: update 2013. Nucleic Acids Res (2013) 41(Database issue):D949–54. doi:10.1093/nar/gks1008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, et al. COSMIC (the catalogue of somatic mutations in cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res (2010) 38(Database issue):D652–7. doi:10.1093/nar/gkp995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Haider S, Ballester B, Smedley D, Zhang J, Rice P, Kasprzyk A. BioMart central portal – unified access to biological data. Nucleic Acids Res (2009) 37(Web Server issue):W23–7. doi:10.1093/nar/gkp265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, et al. The COSMIC (catalogue of somatic mutations in cancer) database and website. Br J Cancer (2004) 91(2):355–8. doi:10.1038/sj.bjc.6601894
43. Mack WJ, Preston-Martin S, Bernstein L, Qian D. Lifestyle and other risk factors for thyroid cancer in Los Angeles county females. Ann Epidemiol (2002) 12(6):395–401. doi:10.1016/S1047-2797(01)00281-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Akslen LA, Sothern RB. Seasonal variations in the presentation and growth of thyroid cancer. Br J Cancer (1998) 77(7):1174–9. doi:10.1038/bjc.1998.195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Boscoe FP, Schymura MJ. Solar ultraviolet-B exposure and cancer incidence and mortality in the United States, 1993-2002. BMC Cancer (2006) 6:264. doi:10.1186/1471-2407-6-264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Jonklaas J, Danielsen M, Wang H. A pilot study of serum selenium, vitamin D, and thyrotropin concentrations in patients with thyroid cancer. Thyroid (2013) 23(9):1079–86. doi:10.1089/thy.2012.0548
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Grant WB. An ecologic study of cancer mortality rates in Spain with respect to indices of solar UVB irradiance and smoking. Int J Cancer (2007) 120(5):1123–8. doi:10.1002/ijc.22386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Penna-Martinez M, Ramos-Lopez E, Stern J, Hinsch N, Hansmann ML, Selkinski I, et al. Vitamin D receptor polymorphisms in differentiated thyroid carcinoma. Thyroid (2009) 19(6):623–8. doi:10.1089/thy.2008.0388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Penna-Martinez M, Ramos-Lopez E, Stern J, Kahles H, Hinsch N, Hansmann ML, et al. Impaired vitamin D activation and association with CYP24A1 haplotypes in differentiated thyroid carcinoma. Thyroid (2012) 22(7):709–16. doi:10.1089/thy.2011.0330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Roskies M, Dolev Y, Caglar D, Hier MP, Mlynarek A, Majdan A, et al. Vitamin D deficiency as a potentially modifiable risk factor for thyroid cancer. J Otolaryngol Head Neck Surg (2012) 41(3):160–3.
51. Sahin M, Ucan B, Ginis Z, Topaloglu O, Gungunes A, Bozkurt NC, et al. Vitamin D3 levels and insulin resistance in papillary thyroid cancer patients. Med Oncol (2013) 30(2):589. doi:10.1007/s12032-013-0589-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Stepien T, Krupinski R, Sopinski J, Kuzdak K, Komorowski J, Lawnicka H, et al. Decreased 1-25 dihydroxyvitamin D3 concentration in peripheral blood serum of patients with thyroid cancer. Arch Med Res (2010) 41(3):190–4. doi:10.1016/j.arcmed.2010.04.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. D’Avanzo B, Ron E, La Vecchia C, Francaschi S, Negri E, Zleglar R. Selected micronutrient intake and thyroid carcinoma risk. Cancer (1997) 79(11):2186–92. doi:10.1002/(SICI)1097-0142(19970601)79:11<2186::AID-CNCR17>3.3.CO;2-K
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Glattre E, Haldorsen T, Berg JP, Stensvold I, Solvoll K. Norwegian case-control study testing the hypothesis that seafood increases the risk of thyroid cancer. Cancer Causes Control (1993) 4(1):11–6. doi:10.1007/BF00051708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Laney N, Meza J, Lyden E, Erickson J, Treude K, Goldner W. The prevalence of vitamin D deficiency is similar between thyroid nodule and thyroid cancer patients. Int J Endocrinol (2010) 2010:805716. doi:10.1155/2010/805716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Haghpanah V, Ghaffari SH, Rahimpour P, Abbasi A, Saeedi M, Pak H, et al. Vitamin D receptor gene polymorphisms in patients with thyroid cancer. Gene Ther Mol Biol (2007) 11B:299–304.
57. Millen AE, Bodnar LM. Vitamin D assessment in population-based studies: a review of the issues. Am J Clin Nutr (2008) 87(4):1102S–5S.
58. Greenlee H, White E, Patterson RE, Kristal AR. Supplement use among cancer survivors in the vitamins and lifestyle (VITAL) study cohort. J Altern Complement Med (2004) 10(4):660–6. doi:10.1089/acm.2004.10.660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Ron E, Kleinerman RA, Boice JDJ, Livolsi VA, Flannery JT, Fraumeni JFJ. A population-based case-control study of thyroid cancer. J Natl Cancer Inst (1987) 79(1):1–12.PMID
60. Peterson L, Soliman A, Ruterbusch JJ, Smith N, Schwartz K. Comparison of exposures among Arab American and non-Hispanic white female thyroid cancer cases in metropolitan detroit. J Immigr Minor Health (2011) 13(6):1033–40. doi:10.1007/s10903-011-9485-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Balla B, Kosa JP, Tobias B, Halaszlaki C, Takacs I, Horvath H, et al. Marked increase in CYP24A1 gene expression in human papillary thyroid cancer. Thyroid (2011) 21(4):459–60. doi:10.1089/thy.2010.0420
62. Bennett RG, Wakeley SE, Hamel FG, High RR, Korch C, Goldner WS. Gene expression of vitamin D metabolic enzymes at baseline and in response to vitamin D treatment in thyroid cancer cell lines. Oncology (2012) 83(5):264–72. doi:10.1159/000342093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Clinckspoor I, Verlinden L, Overbergh L, Korch C, Bouillon R, Mathieu C, et al. 1,25-dihydroxyvitamin D3 and a superagonistic analog in combination with paclitaxel or suberoylanilide hydroxamic acid have potent antiproliferative effects on anaplastic thyroid cancer. J Steroid Biochem Mol Biol (2011) 124(1–2):1–9. doi:10.1016/j.jsbmb.2010.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Clinckspoor I, Hauben E, Verlinden L, Van den Bruel A, Vanwalleghem L, Poorten VV, et al. Altered Expression of key players in vitamin D metabolism and signaling in malignant and benign thyroid tumors. J Histochem Cytochem (2012) 60(7):502–11. doi:10.1369/0022155412447296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Dackiw AP, Ezzat S, Huang P, Liu W, Asa SL. Vitamin D3 administration induces nuclear p27 accumulation, restores differentiation, and reduces tumor burden in a mouse model of metastatic follicular thyroid cancer. Endocrinology (2004) 145(12):5840–6. doi:10.1210/en.2004-0785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Khadzkou K, Buchwald P, Westin G, Dralle H, Akerstrom G, Hellman P. 25-hydroxyvitamin D3 1alpha-hydroxylase and vitamin D receptor expression in papillary thyroid carcinoma. J Histochem Cytochem (2006) 54(3):355–61. doi:10.1369/jhc.5A6734.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Liu W, Asa SL, Fantus IG, Walfish PG, Ezzat S. Vitamin D arrests thyroid carcinoma cell growth and induces p27 dephosphorylation and accumulation through PTEN/akt-dependent and -independent pathways. Am J Pathol (2002) 160(2):511–9. doi:10.1016/S0002-9440(10)64870-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Liu W, Asa SL, Ezzat S. 1alpha,25-dihydroxyvitamin D3 targets PTEN-dependent fibronectin expression to restore thyroid cancer cell adhesiveness. Mol Endocrinol (2005) 19(9):2349–57. doi:10.1210/me.2005-0117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Liu W, Guo M, Ezzat S, Asa SL. Vitamin D inhibits CEACAM1 to promote insulin/IGF-I receptor signaling without compromising anti-proliferative action. Lab Invest (2011) 91(1):147–56. doi:10.1038/labinvest.2010.144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Okano K, Ohtsuru A, Tsukazaki T, Miyazaki Y, Yonekura A, Namba H, et al. Effect of 22-oxa-1,25-dihydroxyvitamin D-3 on human thyroid cancer cell growth. Endocr J (1999) 46(2):243–52. doi:10.1507/endocrj.46.243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Sharma V, Fretwell D, Crees Z, Kerege A, Klopper JP. Thyroid cancer resistance to vitamin D receptor activation is associated with 24-hydroxylase levels but not the ff FokI polymorphism. Thyroid (2010) 20(10):1103–11. doi:10.1089/thy.2010.0096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Somjen D, Grafi-Cohen M, Posner GH, Sharon O, Kraiem Z, Stern N. Vitamin D less-calcemic analog modulates the expression of estrogen receptors, vitamin D receptor and 1-hydroxylase 25-hydroxy vitamin D in human thyroid cancer cell lines. J Steroid Biochem Mol Biol (2013) 136:80–2. doi:10.1016/j.jsbmb.2012.09.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Suzuki S, Takenoshita S, Furukawa H, Tsuchiya A. Antineoplastic activity of 1,25(OH)2D3 and its analogue 22-oxacalcitriol against human anaplastic thyroid carcinoma cell lines in vitro. Int J Mol Med (1999) 4(6):611–4.
74. Mithal A, Wahl DA, Bonjour JP, Burckhardt P, Dawson-Hughes B, Eisman JA, et al. Global vitamin D status and determinants of hypovitaminosis D. Osteoporos Int (2009) 20(11):1807–20. doi:10.1007/s00198-009-0954-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Thyssen JP, Thuesen B, Huth C, Standl M, Carson CG, Heinrich J, et al. Skin barrier abnormality caused by filaggrin (FLG) mutations is associated with increased serum 25-hydroxyvitamin D concentrations. J Allergy Clin Immunol (2012) 130(5):1204–7e2. doi:10.1016/j.jaci.2012.06.046
76. Kasahara AK, Singh RJ, Noymer A. Vitamin D (25OHD) serum seasonality in the United States. PLoS One (2013) 8(6):e65785. doi:10.1371/journal.pone.0065785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Atlas of Cancer Mortality and Other Causes of Death in Spain 1978-1992. Fundacion Cientifica de la Asociacion Espanola Contra el Cancer Madrid (1996). Available from: http://www2.uca.es/hospital/atlas92/www/Atlas92.html
78. Dawson-Hughes B, Heaney RP, Holick MF, Lips P, Meunier PJ, Vieth R. Estimates of optimal vitamin D status. Osteoporosis international (2005) 16(7):713–6.
79. Souberbielle JC, Cavalier E. Supplementation, optimal status, and analytical determination of vitamin D: where are we standing in 2012? Anticancer Agents Med Chem (2013) 13(1):36–44. doi:10.2174/1871520611307010036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the institute of medicine: what clinicians need to know. J Clin Endocrinol Metab (2011) 96(1):53–8. doi:10.1210/jc.2010-2704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. White JH. Vitamin D and human health: more than just bone. Nat Rev Endocrinol (2013) 9(10):623. doi:10.1038/nrendo.2013.75-c1
82. Dev R, Del Fabbro E, Schwartz GG, Hui D, Palla SL, Gutierrez N, et al. Preliminary report: vitamin D deficiency in advanced cancer patients with symptoms of fatigue or anorexia. Oncologist (2011) 16(11):1637–41. doi:10.1634/theoncologist.2011-0151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Wang TJ, Zhang F, Richards JB, Kestenbaum B, van Meurs JB, Berry D, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet (2010) 376(9736):180–8. doi:10.1016/S0140-6736(10)60588-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Goltzman D, Hendy GN, White JH. Vitamin D and its receptor during late development. Biochim Biophys Acta (2014). doi:10.1016/j.bbagrm.2014.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Levin GP, Robinson-Cohen C, de Boer IH, Houston DK, Lohman K, Liu Y, et al. Genetic variants and associations of 25-hydroxyvitamin D concentrations with major clinical outcomes. JAMA (2012) 308(18):1898–905. doi:10.1001/jama.2012.17304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Serrano JC, De Lorenzo D, Cassanye A, Martin-Gari M, Espinel A, Delgado MA, et al. Vitamin D receptor BsmI polymorphism modulates soy intake and 25-hydroxyvitamin D supplementation benefits in cardiovascular disease risk factors profile. Genes Nutr (2013) 8(6):561–9. doi:10.1007/s12263-013-0356-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Crofts LA, Hancock MS, Morrison NA, Eisman JA. Multiple promoters direct the tissue-specific expression of novel N-terminal variant human vitamin D receptor gene transcripts. Proc Natl Acad Sci U S A (1998) 95(18):10529–34. doi:10.1073/pnas.95.18.10529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Wang DG, Fan JB, Siao CJ, Berno A, Young P, Sapolsky R, et al. Large-scale identification, mapping, and genotyping of single-nucleotide polymorphisms in the human genome. Science (1998) 280(5366):1077–82. doi:10.1126/science.280.5366.1077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr Rev (1999) 20(2):156–88. doi:10.1210/er.20.2.156
90. Whitfield GK, Selznick SH, Haussler CA, Hsieh JC, Galligan MA, Jurutka PW, et al. Vitamin D receptors from patients with resistance to 1,25-dihydroxyvitamin D3: point mutations confer reduced transactivation in response to ligand and impaired interaction with the retinoid X receptor heterodimeric partner. Mol Endocrinol (1996) 10(12):1617–31. doi:10.1210/mend.10.12.8961271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell (2000) 5(1):173–9. doi:10.1016/S1097-2765(00)80413-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Malloy PJ, Xu R, Peng L, Clark PA, Feldman D. A novel mutation in helix 12 of the vitamin D receptor impairs coactivator interaction and causes hereditary 1,25-dihydroxyvitamin D-resistant rickets without alopecia. Mol Endocrinol (2002) 16(11):2538–46. doi:10.1210/me.2002-0152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Malloy PJ, Hochberg Z, Tiosano D, Pike JW, Hughes MR, Feldman D. The molecular basis of hereditary 1,25-dihydroxyvitamin D3 resistant rickets in seven related families. J Clin Invest (1990) 86(6):2071–9. doi:10.1172/JCI114944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Ma NS, Malloy PJ, Pitukcheewanont P, Dreimane D, Geffner ME, Feldman D. Hereditary vitamin D resistant rickets: identification of a novel splice site mutation in the vitamin D receptor gene and successful treatment with oral calcium therapy. Bone (2009) 45(4):743–6. doi:10.1016/j.bone.2009.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Hawa NS, Cockerill FJ, Vadher S, Hewison M, Rut AR, Pike JW, et al. Identification of a novel mutation in hereditary vitamin D resistant rickets causing exon skipping. Clin Endocrinol (1996) 45(1):85–92. doi:10.1111/j.1365-2265.1996.tb02064.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Tuoresmaki P, Vaisanen S, Neme A, Heikkinen S, Carlberg C. Patterns of genome-wide VDR locations. PLoS One (2014) 9(4):e96105. doi:10.1371/journal.pone.0096105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Ramagopalan SV, Heger A, Berlanga AJ, Maugeri NJ, Lincoln MR, Burrell A, et al. A ChIP-seq defined genome-wide map of vitamin D receptor binding: associations with disease and evolution. Genome Res (2010) 20(10):1352–60. doi:10.1101/gr.107920.110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: thyroid cancer, vitamin D, VDR, genome-wide studies, cancer susceptibility
Citation: Morand GB, da Silva SD, Hier MP and Alaoui-Jamali MA (2014) Insights into genetic and epigenetic determinants with impact on vitamin D signaling and cancer association studies: the case of thyroid cancer. Front. Oncol. 4:309. doi: 10.3389/fonc.2014.00309
Received: 19 August 2014; Paper pending published: 12 September 2014;
Accepted: 17 October 2014; Published online: 04 November 2014.
Edited by:
Thian-Sze Wong, The University of Hong Kong, ChinaReviewed by:
Wen-Hung Wang, Taipei and Fu-Jen Catholic University School of Medicine, TaiwanWei Gao, The University of Hong Kong, China
Copyright: © 2014 Morand, da Silva, Hier and Alaoui-Jamali. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Moulay A. Alaoui-Jamali, Departments of Medicine and Oncology, Segal Cancer Centre and Lady Davis Institute for Medical Research, Sir Mortimer B. Davis-Jewish General Hospital, McGill University, 3755 Côte Ste-Catherine Road, Montreal, QC H3T 1E2, Canada e-mail:bW91bGF5LmFsYW91aS1qYW1hbGlAbWNnaWxsLmNh