 Amanda K. Huber
Amanda K. Huber Patrick C. Duncker
Patrick C. Duncker David N. Irani
David N. Irani- Department of Neurology, University of Michigan Medical School, Ann Arbor, MI, USA
The central nervous system (CNS), once viewed as an immune-privileged site protected by the blood–brain barrier (BBB), is now known to be a dynamic immunological environment through which immune cells migrate to prevent and respond to events such as localized infection. During these responses, endogenous glial cells, including astrocytes and microglia, become highly reactive and may secrete inflammatory mediators that regulate BBB permeability and recruit additional circulating immune cells. Here, we discuss the various roles played by astrocytes, microglia, and infiltrating immune cells during host immunity to non-tumor antigens in the CNS, focusing first on bacterial and viral infections, and then turning to responses directed against self-antigens in the setting of CNS autoimmunity.
Introduction
The central nervous system (CNS) was previously viewed as an immune-privileged area, fully isolated from the immune system by the blood–brain barrier (BBB). In early studies, Ehrlich reported that while various organs were strongly stained following intravenous, intra-arterial, or subcutaneous injection of intravital dyes, the brain was only weakly stained or not at all (1). Other studies found that tissue grafts were not rejected when implanted into the brains of test animals (2), leading to the idea that the CNS was fully “immune-privileged.” This viewpoint had to be altered, however, after it was discovered that a graft within the CNS could be rejected if a second graft was placed subcutaneously into the same animal (3). This finding clearly demonstrated that foreign antigens are recognized in the CNS if peripheral priming occurs (3). It is now accepted that the BBB is a dynamic, interactive, and regulatory tissue interface that allows bi-directional communication between the CNS and the immune system (4, 5).
The BBB, formed by complex interactions between capillary endothelial cells (ECs), astrocyte end-feet, pericytes, and microglia (6, 7), is the largest and most stringent barrier that impedes the paracellular movement of ions, solutes, proteins, water, and leukocytes into the CNS (8). However, the BBB can also be influenced by peripheral immune events, creating what has now come to be known as the neuro-immune axis (4, 9, 10). The neuro-immune axis is not only responsible for establishing the blood–CNS barrier at baseline, but it also regulates communication between the CNS and the immune system during pathological conditions such as viral or bacterial infections, ischemia, or inflammatory autoimmune disorders such as multiple sclerosis (MS) (11). It achieves this state by responding to secreted factors from both immune and CNS cells, as well as by regulating the exchange of chemokines, cytokines, and immune cells between the blood and the CNS (4, 9, 10). Therefore, the original concept of the BBB being a purely anatomical barrier has now shifted to one where the BBB is considered a highly reactive interface controlled by signals from ECs, glial cells, pericytes, and neurons in the CNS, as well as from immune responses in the periphery (12–21).
Structural Characteristics of the BBB
The BBB is composed of capillary ECs ensheathed by astrocyte end-feet, pericytes, and microglia (6, 7). Astrocyte end-feet completely surround the abluminal surface of brain capillaries forming a layer known as the glial limitans, but direct contact with EC is inhibited by a dense basement membrane (22). While astrocytes are necessary to maintain BBB integrity by secreting factors that alter barrier permeability (6, 23), they are not actually required to form the BBB, which develops even before these astrocytic processes are present (24–26). Astrocytes control blood flow to the CNS by regulating vascular tone through fluctuating calcium currents (27). Pericytes are essential to barrier formation, as the BBB is compromised in pericyte-deficient mice (28, 29). These cells regulate gene expression in EC and induce the polarization of astrocyte end-feet (28). Microglia play a role at the BBB by regulating substrate transport across EC and by linking the brain to systemic immune activity (30).
Blood–brain barrier EC forms a highly sophisticated barrier via a network of tight junctions (TJ) and adherens junctions (AJ) (8, 31, 32). The EC of the CNS are unique in that the TJ restrict the paracellular passage of solutes, have no pinocytic activity, and have few if any fenestrations (33–39). This causes the BBB to have high endothelial electrical resistance (40, 41), some 50–100 times higher than peripheral microvessels (42–44). The TJ are composed of a parallel network of intramembranous protein strands, composed of claudins, occludin, and zonula occludin (ZO) proteins (37). Claudins, specifically claudin-3, -4, and -12, compose the TJ backbone (45–47). Occludin is not required for TJ formation (48); instead, it plays a role in “sealing” the junction thereby increasing electrical resistance (49, 50). CNS microvessel TJ are also abundant in ZO-2, and to a lesser extent, ZO-1, that are cytoplasmic accessory proteins that serve to anchor the transmembrane proteins of the TJ to the actin cytoskeleton of the ECs (51, 52).
The choroid plexus (CP) is a villous structure located on the roof of the four cerebral ventricles where cerebrospinal fluid (CSF) is actively secreted. The CP is highly vascular and contains the blood–CSF barrier (BCSFB) (51). Unlike the BBB, however, the BCSFB arises from cuboidal choroid plexus epithelial cells (CPE) with a very different TJ structure. The CPE express ZO-1 and ZO-2 in different amounts (51), and have a different claudin signature, expressing claudin-1, -2, -3, and -11 (51, 53, 54). Furthermore, capillaries within the CP villi are fenestrated (51, 55, 56), reflected by a much lower endothelial electrical resistance than the BBB (57). For these reasons, the BBB is considered more of an absolute barrier, while the BCSFB may be where most normal immune surveillance of the CNS occurs (58).
Immune Surveillance and Infiltration of the CNS
It is now accepted there is a constant need for immune surveillance of the normal CNS as part of host defense (11, 59, 60), with mechanisms present that simultaneously keep excessive inflammation in check (61). To assist in maintaining this control, the healthy CNS is relatively devoid of antigen-presenting cells (APC), lacks constitutive human leukocyte antigen (HLA) class I and II protein expression on parenchymal cells, and does not maintain typical lymphatic vessels (11). CD4+ T cells, having first encountered antigens in deep cervical lymph nodes (62), carry out routine surveillance of the CNS by searching for their cognate antigens presented by macrophages in the CSF (11, 63). Resting lymphocytes fail to enter the CNS (64), while activated T cells of all specificities can traverse the BBB and/or BCSFB (65). Those cells that do not encounter their cognate antigen within a few hours then circulate out of the CNS (66, 67).
The first steps of pathogenic neuroinflammation involve changes at the BBB, including increased production of chemokines and up-regulation of adhesion molecules by the EC resulting in leukocytes traversing the BBB and accumulating in the perivascular space of post-capillary venules (11, 68). Even during these early events, however, cellular recruitment remains tightly controlled as parenchymal lymphocytes express a unique adhesion molecule profile, different from peripheral T cells (69–71). Once in the perivascular space, T cells encounter the glial limitans as well as astrocytes that express and release factors that induce apoptosis (72), inhibit proliferation (72), induce differentiation into a regulatory (Treg) phenotype (73). Microglia and neurons also assist in controlling neuroinflammation. Microglia do so by expressing a homolog of the co-stimulatory molecule B7, programed death protein (PD)-1, which negatively regulates T cell activation and cytokine production (74). Neurons secrete transforming growth factor (TGF)-β, exert cell contact-dependent effects that support the conversion of CD4 T cells to Tregs, and can be induced to express the PD-1 ligand, PD-L1 (75). Thus, while the BBB is not the impenetrable barrier it was once thought to be, CD4+ T cell surveillance of the CNS is still a tightly controlled process.
Host Immune Responses to Bacterial Infections of the CNS
Bacterial infections of the CNS are rare, but often life threatening, events (76). Excluding direct inoculation following CNS trauma, bacteria typically gain CNS entry following hematogenous dissemination from distant sites (lungs and heart valves) or by direct extension from parameningeal foci of infection (inner ear and sinuses). Penetration of the BBB may occur via three potential mechanisms: (1) direct destruction of capillary ECs (77, 78), (2) disruption of intercellular TJ and migration in between ECs (79), and (3) transcytosis via intracellular vesicles directly through ECs (80). Once inside, numerous innate immune receptors and pathways are activated (Figure 1).
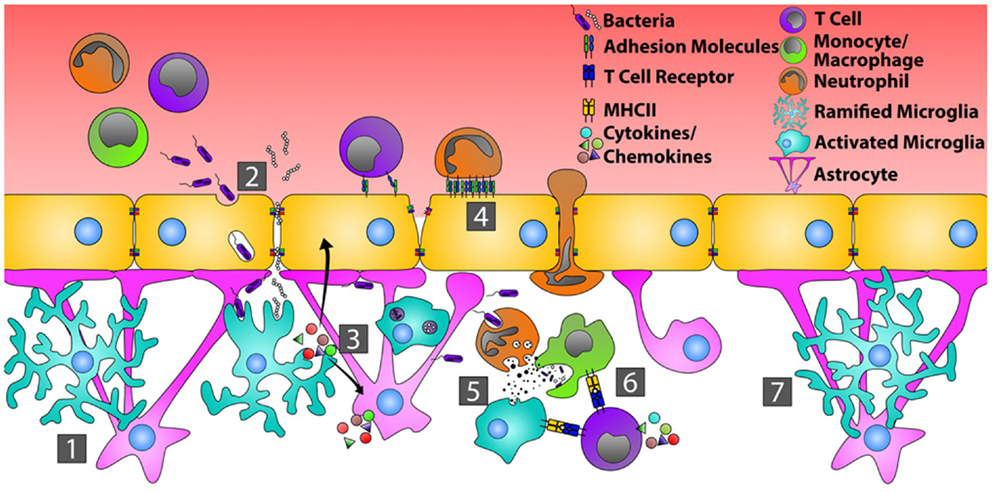
Figure 1. Orchestration of the immune response during bacterial infection of the CNS. In the quiescent CNS (1), bacteria typically gain entry by transcytosis across the endothelial cells of the BBB, or by passing in between endothelial cells where tight junctions have been disrupted (2). Common bacterial motifs (PAMPs) are recognized by pattern recognition receptors (PRRs) on microglia and astrocytes resulting in their activation. This causes changes in glial cell morphology, enhanced production of inflammatory mediators that recruit neutrophils, monocytes, and T cells, and increased endothelial cell expression of adhesion molecules, including ICAM-1 and VCAM-1 (3). Circulating neutrophils, monocytes, and T cells then bind and extravasate into the infected CNS (4). Neutrophils directly phagocytize and kill bacteria through the release of defensins, lytic enzymes, and anti-microbial peptides (5). Neutrophils also produce MMPs, IL-6, IL-8, CXCL9, and CXCL10 that further open the BBB and shift the chemotactic profile toward the recruitment of T cells. Bacterial antigens are presented to T cells by microglia and/or infiltrating monocytes, transitioning from innate immunity toward an adaptive immune response (6). Resolution of bacterial infection returns tight junctions to normal and microglia and astrocytes to a resting state (7).
Microglia
Analogous to peripheral tissues, resident CNS immune cells known as microglia bear a wide range of innate immune receptors. Common bacterial motifs, referred to as pathogen associated molecular patterns (PAMP), are recognized by cognate pattern recognition receptors (PRR), including Toll-like receptors (TLR), on the surface and in the cytoplasm of microglia, and to a lesser extent, on astrocytes (81–83). Microglial activation, triggered either by intact bacteria or bacterial cell wall antigens (84, 85), results in rapid changes in cellular morphology in vivo (86). Similar to tissue resident macrophages found in the periphery, microglia can phagocytize bacteria and present bacterial antigens via HLA to infiltrating CD4 T cells in vivo (84, 87, 88). These cells also rapidly produce pro-inflammatory cytokines and chemokines that recruit peripheral leukocytes to the area of infection and activate astrocytes. For example, during both experimental Streptococcus pneumoniae and Staphylococcus aureus infections of the CNS, microglia produce tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-12, C-X-C motif ligand (CXCL)1, CXCL2, C-C motif ligand (CCL)2, CCL3, and CCL5 ex vivo, mediators that recruit neutrophils (CXCL1 and CXCL2), monocytes (CCL2 and CCL3), and T cells (CCL5) (84, 85, 89–91). These activated microglia also secrete matrix metalloproteinases (MMP) that enhance BBB breakdown and facilitate additional leukocyte extravasation into the CNS (92). Finally, microglia can have direct bactericidal activity, being capable of producing reactive oxygen species (ROS), reactive nitrogen intermediates, and other proteases that kill bacteria in vivo (93–96).
Astrocytes
Microglia partner with astrocytes to eliminate infection as quickly as possible in order to minimize neuronal damage (86, 97, 98). In the normal CNS, astrocytes contribute to gap junction stability of the BBB (99). Their release of pro-inflammatory mediators such as IL-1β (100, 101), nitric oxide (102), TGF-β (103), and MMPs (92) in vitro suggest these cells may compromise BBB integrity in the setting of bacterial infection. Astrocytes are activated by bacterial PAMP or mediators produced by microglia; this changes their morphology and further triggers their release of innate inflammatory mediators both in vitro and in vivo. These mediators can include complement proteins, IL-1β, IL-6, and the chemokines, CCL2, CCL3, CXCL1, and CXCL10 (104–111), which further help recruit neutrophils, monocytes, and T cells. In response to interferon (IFN)-γ, TNF-α, and/or IL-1β, astrocytes also up-regulate the cell surface adhesion molecules, intercellular adhesion molecule (ICAM)-1, and vascular cell adhesion molecule (VCAM)-1 in vitro (112–116), which would enhance the infiltration of monocytes and T cells into the CNS in vivo.
Neutrophils
As in the periphery (117, 118), neutrophils are one of the primary lines of host defense during CNS bacterial infections (112, 119, 120). Studies in knockout mice show that the main chemokines driving neutrophil recruitment to the CNS are the C-X-C motif receptor (CXCR)-2 ligands, CXCL1 and CXCL2 (121). Furthermore, CSF samples from patients with bacterial meningitis show elevated levels of neutrophil attracting chemokines compared to controls (122, 123). Neutrophils, like microglia, respond to PAMP through various TLR, and are activated by cytokines such as TNF-α and IFN-γ in vitro (124). Neutrophils activated in the periphery up-regulate adhesion molecules that enhance their migration into tissues (125), while BBB EC express E-selectin and P-selectin during CNS bacterial infection (126), suggesting a mechanism that allows for the migration of neutrophils during these infections. Once neutrophils recognize a bacterial pathogen, they can directly phagocytize these organisms (127), as well as release MMP, defensins, lytic enzymes, and anti-microbial peptides that aid in clearing the infection (128). The inflammatory cytokine, TNF-α, induces neutrophils to produce IL-6, IL-8, CXCL9, and CXCL10 in vivo (129, 130), thereby shifting the chemotactic profile toward the recruitment of T cells and driving the adaptive immune response.
T Cells
Adaptive immune responses are important in fighting CNS bacterial infections (131). During bacterial meningitis, T cell production of IFN-γ leads to the generation of chemokines that preferentially recruit monocytes and more T cells (132), supporting the transition from an innate to an adaptive immune response. Furthermore, IFN-γ, potentially made locally by T cells, increases the antigen-presenting capacity of microglial cells in vitro via up-regulation of HLA class I and II molecules, the co-stimulatory molecules, B7-1 and B7-2, and CD40 (133, 134). Bacterial antigen presentation by microglia activates T cells (135), driving further T cell proliferation and greater production of IFN-γ.
Host Immune Responses to Viral infections of the CNS
Viruses use a variety of mechanisms to gain entry into the CNS. In the case of alphaherpesviruses (i.e., herpes simplex virus and varicella-zoster virus) and rabies virus, infection of peripheral nerves allows viral particles to travel by anterograde axonal transport into the CNS. Human immunodeficiency virus and human T cell leukemia virus-I enter the CNS parenchyma by infecting host immune cells in the periphery, and using them as “Trojan horses” to carry viral particles across the BBB. Finally, Epstein–Barr virus and West Nile virus directly infect the ECs of the BBB, resulting in barrier disruption and enhanced migration of immune cells into the parenchyma (136).
Because viruses can infect microglia, astrocytes, oligodendrocytes, as well as terminally differentiated and non-renewable cells such as neurons, the ensuing immune response within the CNS must avoid extensive cytolytic damage of virus-infected target cells (137). In general, innate anti-viral immunity such as the generation of type-I IFN occurs very rapidly, while the adaptive immune response is slower because it must first develop in the periphery (138). Important components of adaptive anti-viral immunity involve IFN-γ production by T cells as well as the expansion and migration of virus-specific antibody secreting cells (ASC) (138, 139) (Figure 2).
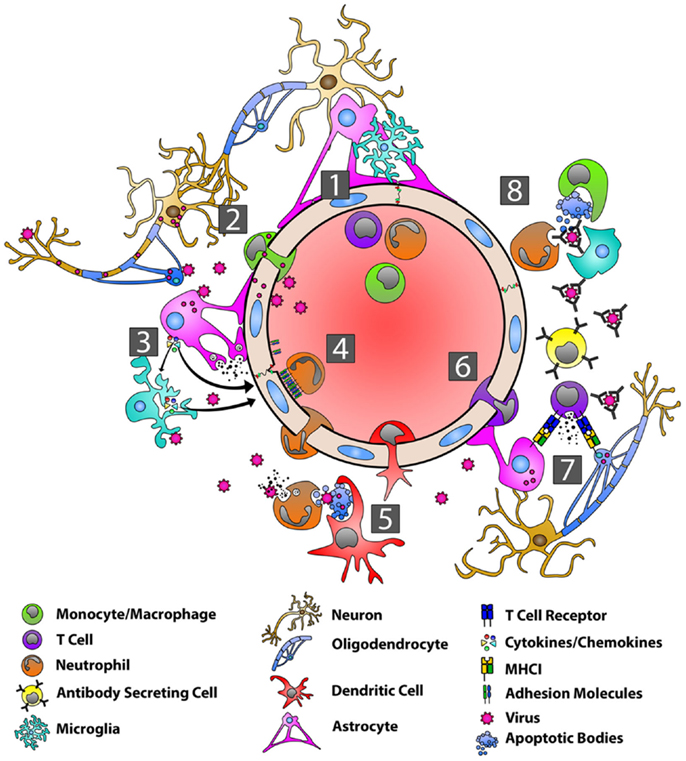
Figure 2. Orchestration of the immune response during viral infection of the CNS. With the BBB in a resting state (1), viruses can gain entry into the CNS by infecting peripheral nerves and traveling by anterograde axonal transport into the CNS, by infecting host immune cells in the periphery and using these cells as “Trojan horses” to carry them across the BBB, or by directly infecting BBB endothelial cells (2). Viral PAMPs then activate microglia, astrocytes, and oligodendrocytes (3). Microglia and astrocytes produce a range of anti-viral/pro-inflammatory cytokines, including type-I IFNs, IL-6, TNF-α, IL-12, IL-1α, and IL-1β (3). Astrocytes also produce MMP-3 and MMP-12 resulting in the up-regulation of adhesion molecules on endothelial cells (3). Interactions between adhesion molecules and neutrophils contribute to BBB breakdown via the production of MMP-9 and the disassembly of the tight junctions (4). DCs are seen in the CNS within several days and migrate to draining lymph nodes where they activate and expand virus-specific T cells (5). Chemokines produced by astrocytes are responsible for recruiting virus-specific CD4+ and CD8+ T cells as well as ASCs to the CNS (6). CD8+ T cells produce IFN-γ and lytic molecules, including granzyme B and perforin, to eliminate virus from astrocytes, while IFN-γ controls viral replication in oligodendrocytes (7). Virus-specific antibodies control virus replication in cells such as neurons via complement–independent, non-cytolytic mechanisms. These antibodies inhibit virus budding and replication, viral RNA transcription, and cell-to-cell virus spread.
Microglia, Astrocytes, and Oligodendrocytes
During CNS viral infections, virus-specific PAMP activate individual TLR present on microglia, astrocytes, and oligodendrocytes. The former two cell populations, in particular, respond by producing anti-viral and pro-inflammatory mediators. During experimental mouse hepatitis virus (MHV) infection, astrocytes and microglia produce both type-I IFN (IFN-α and IFN-β), as well as IL-6, TNF-α, IL-12, IL-1α, and IL-1β in vivo (140–142). Furthermore, MHV infection triggers MMP-3 and MMP-12 release from astrocytes and oligodendrocytes (142), which along with IL-6 and the up-regulation of adhesion molecules on cerebrovascular endothelium, enhance cellular migration across the BBB (143). Astrocytes produce CXCL10, CXCL11, and CCL5 in vivo that recruit virus-specific CD4+ and CD8+ T cells (144–146), as well as ASC (147, 148), to the CNS to promote viral clearance. CXCL9 production from microglia is dependent on IFN-γ, while CXCL10 and CXCL11 are up-regulated by type-I IFN and TNF-α (149–152).
Myeloid Lineage Cells
Neutrophils and macrophages are recruited to the CNS following viral infection (153, 154). Thus far, macrophages appear to have more limited anti-viral activity in the CNS (155), but neutrophils contribute to the breakdown of the BBB by interacting with EC via adhesion molecules to promote the disassembly of tight junction complexes (156). Neutrophils also secrete MMP-9 that degrades the extracellular matrix and basal lamina of the BBB and further opens the BBB (157). This has been most clearly demonstrated in the MHV model, where depletion of MMP-9 inhibited lymphocyte infiltration into the CNS (157, 158). Dendritic cells (DC) are seen in the CNS within a few days after CNS viral infection. These cells rely on the chemokine CCL3 to migrate to cervical lymph nodes draining the CNS, where they prime virus-specific T cells (159).
T Cells
In the MHV model, virus-specific CD8+ T cells are detected in local lymph nodes prior to CNS infiltration and then accumulate in the CNS (160). Both CD4+ and CD8+ T cells are in part recruited to the CNS by the chemokines, CXCL9 and CXCL10, acting through their cognate receptor, CXCR3 (161–163). T cell expression of CCR2 and CCR5 likely contribute to CNS recruitment as well (164, 165). The role of CD4+ T cells in this setting is to support CD8+ T cell function via the production of IFN-γ (166). CD8+ T cells are the main anti-viral effector cells in the CNS during infection and are essential for clearing virus from glial cells (142, 167, 168). CD8+ T cells produce IFN-γ and lytic molecules, including granzyme B and perforin (169). These lytic molecules eliminate virally infected astrocytes (170), while IFN-γ serves to control viral replication in oligodendrocytes (171, 172). In both the MHV and Sindbis virus (SINV) encephalitis models, T cells promote B cell proliferation and differentiation (173, 174), in part by secreting the cytokines, IL-10 and IL-21 (175–177).
B Cells and Anti-Viral Antibodies
Virus-specific ASC help control viruses in the CNS through potent complement-independent, non-cytolytic mechanisms (141, 178–183). These ASC arise either from ectopic lymphoid follicle-like structures within the CNS (152) or migrate from cervical lymph nodes where they have expanded and up-regulated CXCR3 and CXCR4 on their surface prior to entering the CNS (184). ASC recruitment to the CNS has been most extensively studied in the SINV encephalitis model. The initial ASC entering the CNS have an HLA class II positive, plasmablast-like phenotype, but these cells gradually lose HLA class II expression and acquire a more plasma cell-like phenotype (139, 141). Virus-specific antibodies function to neutralize both extracellular virus as well as virus particles budding from infected cell membranes. During SINV infection, antibodies that bind the E2 viral envelope glycoprotein inhibit virus replication (185) and prevent viral budding from infected neurons without actually killing target cells (182, 186). Similarly, during rabies virus infection, antibodies against the RV glycoprotein inhibit viral RNA transcription and prevent cell-to-cell viral spread (187). Antibodies can also trigger natural killer (NK) cells and macrophages to induce antibody dependent cell-mediated cytolysis of virally infected cells (152). Finally, in exchange for non-cytolytic viral clearance in the acute setting, virus-specific ASC must persist in the CNS long term to prevent viral reactivation at a later date since viral RNA is never fully eradicated from target tissues (139).
Host Immune Responses to Self (Myelin) Antigens in the CNS
Multiple Sclerosis
Multiple sclerosis, an autoimmune disease characterized by infiltrating immune cells targeting myelin antigens in the CNS, is the most common cause of neurologic disability in persons younger than 40 years of age (188). Pathologically, MS lesions are characterized by focal inflammation, demyelination, and axonal damage (189). MS is a complex disease whose occurrence and progression are influenced by both genetic (190–192) and environmental (193, 194) risk factors. Evidence derived from both human genetic studies and a related mouse model, experimental autoimmune encephalomyelitis (EAE), suggest that encephalitogenic CD4+ T cells are primary initiators of disease. Genome-wide association studies show that MS risk alleles are confined to immune related genes governing antigen presentation as well as the proliferation and survival of T cells, including HLA class II (HLA-DRB1*1501), IL-2R, and IL-7R (190–192). Moreover, EAE in mice is induced by immunizing animals with various myelin peptides (195), or via the adoptive transfer of myelin-specific CD4+ T cells, resulting in a disease having some clinical and pathological similarities to human MS (196, 197). In MS patients, CD4+ T cells localize within CNS lesions present in the brain (198) and spinal cord (199), and elevated frequencies of myelin-reactive CD4+ T cells can be found in circulating the blood (200, 201). Although not described in detail here due to space constraints, many MS lesions also contain abundant CD8+ T cells whose specificity and role in disease pathogenesis remain poorly understood. Likewise, therapies targeted specifically at B cells have proven highly effective in MS patients, highlighting an emerging role for this cell type in both relapsing and progressive forms of disease.
Role of CD4+ T Cells in Autoimmune Inflammation of the CNS
During both MS and EAE, self-reactive T cells are likely activated in the periphery (189), where they undergo initial differentiation and expansion (124). Upon entry into the CNS, these cells are reactivated by myelin epitopes presented by an as of yet unidentified local DC (202, 203). Production of cytokines such as IFN-γ and TNF-α from activated CD4+ T cells results in local activation of resting microglia, leading to the up-regulation of HLA class I and II as well as co-stimulatory molecules (B7-1, B7-2, and CD40) on the surface of these cells (133, 134, 204, 205). These activated microglia are capable of serving as APC for infiltrating myelin-specific CD4+ T cells in vivo thus sustaining this pathogenic local T cell response (97). Production of cytokines, chemokines, and MMPs by microglia (206) facilitate local inflammation by causing BBB breakdown and recruiting more immune cells into the CNS. These include circulating monocytes capable of differentiating into inflammatory DC and macrophages upon tissue entry (207), culminating in demyelination (124). Furthermore, microglial production of IL-23 and IL-1β promotes granulocyte macrophage colony-stimulating factor (GM-CSF) secretion by CD4+ T cells (208). GM-CSF has been shown in EAE to promote CNS inflammation by mobilizing Ly6Chi monocytes from the bone marrow into the periphery, thereby increasing the number of circulating monocytes available for recruitment to the CNS (207). GM-CSF can also increase HLA class II expression and pro-inflammatory cytokine production by microglia, macrophages, and DC in vitro (209, 210). IL-17 producing T cells have been detected within CNS lesions during both EAE and MS (211, 212). IL-17 promotes brain inflammation, inducing the production of pro-inflammatory cytokines, TNF-α, IL-6, and IL-1β most probably from astrocytes, microglia, or macrophages. It also stimulates the release of chemokines responsible for recruiting neutrophils to the CNS, particularly CXCL1 and CXCL2 (213, 214). Finally, IL-17 can disrupt TJ in the BBB, allowing further migration of CD4+ T cells to the CNS (212, 215).
Role of Glial Cells in Autoimmune Inflammation of the CNS
Microglia
Microglia play important roles in augmenting CNS inflammation, demyelination, and neuronal damage in both EAE and MS (67, 216–218). Activation of microglia occurs rapidly following the induction of EAE and results in the release of cytokines, chemokines, ROS, and tissue-degrading MMP (206). One mediator, TNF-like weak inducer of apoptosis (TWEAK), triggers proliferation, angiogenesis, inflammation, is associated with extensive myelin loss, and induces astrocyte cell death during MS (219). IL-17 produced by microglia (220) worsens brain inflammation by stimulating GM-CSF production, as well as increasing IL-6, inflammatory proteins, nitric oxide, and adhesion molecule expression by macrophages. Moreover, expression of myeloperoxidase (MPO) and ROS by microglia results in direct myelin degradation and neuronal damage (216, 218). Paradoxically, microglia also can play a neuroprotective role during CNS autoimmunity. These cells can promote remyelination, protect neurons, and suppress the adaptive immune response within the CNS (221, 222). Within MS lesions, microglia and macrophages express the neurotrophic factors, nerve growth factor (NGF), and brain-derived neurotrophic factor (BDNF), supporting neuronal survival (220, 223, 224). Furthermore, microglia secrete the anti-inflammatory cytokines, IL-10 and TGF-β, and express the inhibitory receptor, PD-L1, responsible for inhibiting T cell proliferation and cytokine production (74, 225).
Astrocytes
Astrocytes are a major source of CCL2 and CXCL10 in the CNS, regulating the migration of monocytes into the brain (CCL2) and microglia into the lesion site (CXCL10) (111, 226–228). One study suggested these cells play a more prominent role in regulating the recruitment of peripheral monocytes into the CNS (229). CXCL12, a chemokine that induces the expression of CXCL8 and CCL2, is also expressed by astrocytes in MS lesions (230). CXCL12 can be cleaved by MMP-2, also expressed by astrocytes in MS and EAE, into a neurotoxic peptide that causes further neuronal damage (231). Similar to microglia, astrocytes also play a protective role during MS and EAE. Homeostatic astrocyte functions include buffering potassium, removing extracellular glutamate that can accumulate to toxic levels, adjusting water balance, and controlling synaptic activity and blood flow in the CNS (8). These cells are also able to produce neurotrophins and the anti-inflammatory cytokine, IL-10 (232).
Conclusion
The vast complexity of cellular interconnections within the CNS, and the non-renewable nature of many neural cells, mandate that some local immune responses be tightly controlled while others (i.e., cytolytic ones) be excluded to the fullest extent possible. The BBB is a dynamic and highly regulated tissue interface that helps make the CNS a unique immunological environment. It responds to signals from both neurons and glial cells on one side while simultaneously being able to sample immunological events passing through intravascular compartments. Immune cells perform normal surveillance of the CNS by searching for antigens previously encountered in extraneural sites such as the deep cervical lymph nodes. Pathological conditions such as infections caused by viruses or bacteria elicit changes at the BBB, including the up-regulation of a unique subset of adhesion molecules as well as heightened release of chemokines by ECs. Mediators produced by astrocytes and microglia further increase BBB permeability and recruit additional circulating leukocytes into the CNS. The ensuing immune response must then be tightly controlled in order to avoid collateral tissue damage. As such, astrocytes and microglia maintain mechanisms to dampen inflammatory responses. In some settings, immune cells such as ASC persist long term within the CNS to prevent viral reactivation. When normal control mechanisms fail, neuroinflammatory diseases such as MS can result. For this reason alone, it is imperative that the complexity of immune reactions within the CNS be better understood.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ehrlich P, Bolduan C. Collected Studies on Immunity. 1st ed. New York, NY: John Wiley & Sons (1906). 586 p.
2. Murphy JB, Sturm E. Conditions determining the transplantability of tissues in the brain. J Exp Med (1923) 38(2):183–97. doi: 10.1084/jem.38.2.183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol (1948) 29(1):58–69.
4. Banks WA. The blood-brain barrier in neuroimmunology: tales of separation and assimilation. Brain Behav Immun (2014). doi:10.1016/j.bbi.2014.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Abbott NJ, Mendonca LL, Dolman DE. The blood-brain barrier in systemic lupus erythematosus. Lupus (2003) 12(12):908–15. doi:10.1191/0961203303lu501oa
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci (2006) 7(1):41–53. doi:10.1038/nrn1824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Brown RC, Morris AP, O’Neil RG. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res (2007) 1130(1):17–30. doi:10.1016/j.brainres.2006.10.083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. De Bock M, Vandenbroucke RE, Decrock E, Culot M, Cecchelli R, Leybaert L. A new angle on blood-CNS interfaces: a role for connexins? FEBS Lett (2014) 588(8):1259–70. doi:10.1016/j.febslet.2014.02.060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Engelhardt B. The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr Pharm Des (2008) 14(16):1555–65. doi:10.2174/138161208784705432
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B. Review: leucocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol Appl Neurobiol (2011) 37(1):24–39. doi:10.1111/j.1365-2990.2010.01140.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Engelhardt B, Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol (2005) 26(9):485–95. doi:10.1016/j.it.2005.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis (2010) 37(1):13–25. doi:10.1016/j.nbd.2009.07.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Lok J, Gupta P, Guo S, Kim WJ, Whalen MJ, van Leyen K, et al. Cell-cell signaling in the neurovascular unit. Neurochem Res (2007) 32(12):2032–45. doi:10.1007/s11064-007-9342-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Amar J, Burcelin R, Ruidavets JB, Cani PD, Fauvel J, Alessi MC, et al. Energy intake is associated with endotoxemia in apparently healthy men. Am J Clin Nutr (2008) 87(5):1219–23.
15. Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One (2008) 3(6):e2516. doi:10.1371/journal.pone.0002516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Brenchley JM, Price DA, Schacker TW, Asher TE, Silvestri G, Rao S, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med (2006) 12(12):1365–71. doi:10.1038/nm1511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res (2009) 50(1):90–7. doi:10.1194/jlr.M800156-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Maes M, Kubera M, Leunis JC. The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol Lett (2008) 29(1):117–24.
19. Ng QY, Lee KW, Byrne C, Ho TF, Lim CL. Plasma endotoxin and immune responses during a 21-km road race under a warm and humid environment. Ann Acad Med Singapore (2008) 37(4):307–14.
20. Sikkeland LI, Skogstad M, Ovstebø R, Brusletto B, Haug KB, Kongerud J, et al. Circulating lipopolysaccharides in the blood from “bioprotein” production workers. Occup Environ Med (2008) 65(3):211–4. doi:10.1136/oem.2007.032938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, et al. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol (2009) 206(1–2):121–4. doi:10.1016/j.jneuroim.2008.09.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M. Signaling at the gliovascular interface. J Neurosci (2003) 23(27):9254–62.
23. Gimsa U, Mitchison NA, Brunner-Weinzier MC. Immune privilege as an intrinsic CNS property: astrocytes protect the CNS against T-cell-mediated neuroinflammation. Mediators Inflamm (2013) 2013:320519. doi:10.1155/2013/320519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol (2007) 28(1):5–11. doi:10.1016/j.it.2006.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Saunders NR, Habgood MD, Dziegielewska KM. Barrier mechanisms in the brain, II. Immature brain. Clin Exp Pharmacol Physiol (1999) 26(2):85–91. doi:10.1046/j.1440-1681.1999.02987.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Saunders NR, Ek CJ, Habgood MD, Dziegielewska KM. Barriers in the brain: a renaissance? Trends Neurosci (2008) 31(6):279–86. doi:10.1016/j.tins.2008.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature (2004) 431(7005):195–9. doi:10.1038/nature02827
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood-brain barrier. Nature (2010) 468(7323):557–61. doi:10.1038/nature09522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature (2010) 468(7323):562–6. doi:10.1038/nature09513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sa-Pereira I, Brites D, Brito MA. Neurovascular unit: a focus on pericytes. Mol Neurobiol (2012) 45(2):327–47. doi:10.1007/s12035-012-8244-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Engelhardt S, Patkar S, Ogunshola OO. Cell-specific blood-brain barrier regulation in health and disease: a focus on hypoxia. Br J Pharmacol (2014) 171(5):1210–30. doi:10.1111/bph.12489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Muoio V, Persson PB, Sendeski MM. The neurovascular unit – concept review. Acta Physiol (Oxf) (2014) 210(4):790–8. doi:10.1111/apha.12250
33. Antonelli-Orlidge A, Saunders KB, Smith SR, D’Amore PA. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci U S A (1989) 86(12):4544–8. doi:10.1073/pnas.86.12.4544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Frigerio S, Gelati M, Ciusani E, Corsini E, Dufour A, Massa G, et al. Immunocompetence of human microvascular brain endothelial cells: cytokine regulation of IL-1beta, MCP-1, IL-10, sICAM-1 and sVCAM-1. J Neurol (1998) 245(11):727–30. doi:10.1007/s004150050275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol (1969) 40(3):648–77. doi:10.1083/jcb.40.3.648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol (1967) 34(1):207–17. doi:10.1083/jcb.34.1.207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Ge S, Song L, Pachter JS. Where is the blood-brain barrier really? J Neurosci Res (2005) 79(4):421–7. doi:10.1002/jnr.20313
38. Ek CJ, Dziegielewska KM, Stolp H, Saunders NR. Functional effectiveness of the blood-brain barrier to small water-soluble molecules in developing and adult opossum (Monodelphis domestica). J Comp Neurol (2006) 496(1):13–26. doi:10.1002/cne.20885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Ribatti D, Nico B, Crivellato E, Artico M. Development of the blood-brain barrier: a historical point of view. Anat Rec B New Anat (2006) 289(1):3–8. doi:10.1002/ar.b.20087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Crone C, Olesen SP. Electrical resistance of brain microvascular endothelium. Brain Res (1982) 241(1):49–55. doi:10.1016/0006-8993(82)91227-6
42. Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol (2004) 5(4):261–70. doi:10.1038/nrm1357
43. Nagasawa K, Chiba H, Fujita H, Kojima T, Saito T, Endo T, et al. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J Cell Physiol (2006) 208(1):123–32. doi:10.1002/jcp.20647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Prat A, Biernacki K, Wosik K, Antel JP. Glial cell influence on the human blood-brain barrier. Glia (2001) 36(2):145–55. doi:10.1002/glia.1104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol (2002) 38(6):323–37. doi:10.1016/S1537-1891(02)00200-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol (2003) 161(3):653–60. doi:10.1083/jcb.200302070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lippoldt A, Liebner S, Andbjer B, Kalbacher H, Wolburg H, Haller H, et al. Organization of choroid plexus epithelial and endothelial cell tight junctions and regulation of claudin-1, -2 and -5 expression by protein kinase C. Neuroreport (2000) 11(7):1427–31. doi:10.1097/00001756-200005150-00015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, et al. Complex phenotype of mice lacking occludin, a component of tight junction strands. Mol Biol Cell (2000) 11(12):4131–42. doi:10.1091/mbc.11.12.4131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. McCarthy M, Wood C, Fedoseyeva L, Whittemore SR. Media components influence viral gene expression assays in human fetal astrocyte cultures. J Neurovirol (1995) 1(3–4):275–85. doi:10.3109/13550289509114024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Davies DC. Blood-brain barrier breakdown in septic encephalopathy and brain tumours. J Anat (2002) 200(6):639–46. doi:10.1046/j.1469-7580.2002.00065.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Kratzer I, Vasiljevic A, Rey C, Fevre-Montange M, Saunders N, Strazielle N, et al. Complexity and developmental changes in the expression pattern of claudins at the blood-CSF barrier. Histochem Cell Biol (2012) 138(6):861–79. doi:10.1007/s00418-012-1001-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev (2005) 57(2):173–85. doi:10.1124/pr.57.2.4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Johansson PA, Dziegielewska KM, Liddelow SA, Saunders NR. The blood-CSF barrier explained: when development is not immaturity. Bioessays (2008) 30(3):237–48. doi:10.1002/bies.20718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Redzic Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: similarities and differences. Fluids Barriers CNS (2011) 8(1):3. doi:10.1186/2045-8118-8-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Del MR. Bigio, ependymal cells: biology and pathology. Acta Neuropathol (2010) 119(1):55–73. doi:10.1007/s00401-009-0624-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Dermietzel R. Junctions in the central nervous system of the cat. IV. Interendothelial junctions of cerebral blood vessels from selected areas of the brain. Cell Tissue Res (1975) 164(1):45–62.
57. Saito Y, Wright EM. Regulation of bicarbonate transport across the brush border membrane of the bull-frog choroid plexus. J Physiol (1984) 350:327–42.
58. Shechter R, London A, Schwartz M. Orchestrated leukocyte recruitment to immune-privileged sites: absolute barriers versus educational gates. Nat Rev Immunol (2013) 13(3):206–18. doi:10.1038/nri3391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol (2007) 28(1):12–8. doi:10.1016/j.it.2006.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi:10.1038/nri3265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Liblau RS, Gonzalez-Dunia D, Wiendl H, Zipp F. Neurons as targets for T cells in the nervous system. Trends Neurosci (2013) 36(6):315–24. doi:10.1016/j.tins.2013.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Weller RO, Engelhardt B, Phillips MJ. Lymphocyte targeting of the central nervous system: a review of afferent and efferent CNS-immune pathways. Brain Pathol (1996) 6(3):275–88. doi:10.1111/j.1750-3639.1996.tb00855.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res (1991) 28(2):254–60. doi:10.1002/jnr.490280213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci (1986) 9(6):271–7. doi:10.1016/0166-2236(86)90077-9
65. Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol (1991) 1(2):97–105. doi:10.1111/j.1750-3639.1991.tb00646.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. McMenamin PG. Distribution and phenotype of dendritic cells and resident tissue macrophages in the dura mater, leptomeninges, and choroid plexus of the rat brain as demonstrated in wholemount preparations. J Comp Neurol (1999) 405(4):553–62. doi:10.1002/(SICI)1096-9861(19990322)405:4<553::AID-CNE8>3.0.CO;2-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol (1998) 161(7):3767–75.
68. Raine CS, Cannella B, Duijvestijn AM, Cross AH. Homing to central nervous system vasculature by antigen-specific lymphocytes. II. Lymphocyte/endothelial cell adhesion during the initial stages of autoimmune demyelination. Lab Invest (1990) 63(4):476–89.
69. Zeine R, Owens T. Direct demonstration of the infiltration of murine central-nervous-system by Pgp-1/Cd44high Cd45rblow Cd4+ T-cells that induce experimental allergic encephalomyelitis. J Neuroimmunol (1992) 40(1):57–70. doi:10.1016/0165-5728(92)90213-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Engelhardt B, Martin-Simonet MT, Rott LS, Butcher EC, Michie SA. Adhesion molecule phenotype of T lymphocytes in inflamed CNS. J Neuroimmunol (1998) 84(1):92–104. doi:10.1016/S0165-5728(97)00237-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Engelhardt B, Conley FK, Kilshaw PJ, Butcher EC. Lymphocytes infiltrating the CNS during inflammation display a distinctive phenotype and bind to VCAM-1 but not to MAdCAM-1. Int Immunol (1995) 7(3):481–91. doi:10.1093/intimm/7.3.481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Bechmann I, Mor G, Nilsen J, Eliza M, Nitsch R, Naftolin F. FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: evidence for the existence of an immunological brain barrier. Glia (1999) 27(1):62–74. doi:10.1002/(SICI)1098-1136(199907)27:1<62::AID-GLIA7>3.0.CO;2-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Meinl E, Aloisi F, Ertl B, Weber F, de Waal Malefyt R, Wekerle H, et al. Multiple sclerosis. Immunomodulatory effects of human astrocytes on T cells. Brain (1994) 117(Pt 6):1323–32. doi:10.1093/brain/117.6.1323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Magnus T, Schreiner B, Korn T, Jack C, Guo H, Antel J, et al. Microglial expression of the B7 family member B7 homolog 1 confers strong immune inhibition: implications for immune responses and autoimmunity in the CNS. J Neurosci (2005) 25(10):2537–46. doi:10.1523/JNEUROSCI.4794-04.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Liu Y, Teige I, Birnir B, Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med (2006) 12(5):518–25. doi:10.1038/nm1402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Sarrazin JL, Bonneville F, Martin-Blondel G. Brain infections. Diagn Interv Imaging (2012) 93(6):473–90. doi:10.1016/j.diii.2012.04.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Coureuil M, Mikaty G, Miller F, Lécuyer H, Bernard C, Bourdoulous S, et al. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science (2009) 325(5936):83–7. doi:10.1126/science.1173196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Marriott HM, Mitchell TJ, Dockrell DH. Pneumolysin: a double-edged sword during the host-pathogen interaction. Curr Mol Med (2008) 8(6):497–509. doi:10.2174/156652408785747924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Attali C, Durmort C, Vernet T, Di Guilmi AM. The interaction of Streptococcus pneumoniae with plasmin mediates transmigration across endothelial and epithelial monolayers by intercellular junction cleavage. Infect Immun (2008) 76(11):5350–6. doi:10.1128/IAI.00184-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Ring A, Weiser JN, Tuomanen EI. Pneumococcal trafficking across the blood-brain barrier. Molecular analysis of a novel bidirectional pathway. J Clin Invest (1998) 102(2):347–60. doi:10.1172/JCI2406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Koedel U. Toll-like receptors in bacterial meningitis. Curr Top Microbiol Immunol (2009) 336:15–40. doi:10.1007/978-3-642-00549-7_2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. J Neuropathol Exp Neurol (2002) 61(11):1013–21.
83. Deng GM, Liu ZQ, Tarkowski A. Intracisternally localized bacterial DNA containing CpG motifs induces meningitis. J Immunol (2001) 167(8):4616–26. doi:10.4049/jimmunol.167.8.4616
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Hanisch UK, Prinz M, Angstwurm K, Häusler KG, Kann O, Kettenmann H, et al. The protein tyrosine kinase inhibitor AG126 prevents the massive microglial cytokine induction by pneumococcal cell walls. Eur J Immunol (2001) 31(7):2104–15. doi:10.1002/1521-4141(200107)31:7<2104::AID-IMMU2104>3.0.CO;2-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev (2004) 17(4):942–64. doi:10.1128/CMR.17.4.942-964.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kreutzberg GW. Microglia, the first line of defence in brain pathologies. Arzneimittelforschung (1995) 45(3A):357–60.
87. Peterson PK, Gekker G, Hu S, Sheng WS, Anderson WR, Ulevitch RJ, et al. CD14 receptor-mediated uptake of nonopsonized Mycobacterium tuberculosis by human microglia. Infect Immun (1995) 63(4):1598–602.
88. Rock RB, Hu S, Gekker G, Sheng WS, May B, Kapur V, et al. Mycobacterium tuberculosis-induced cytokine and chemokine expression by human microglia and astrocytes: effects of dexamethasone. J Infect Dis (2005) 192(12):2054–8. doi:10.1086/498165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Kielian T, Mayes P, Kielian M. Characterization of microglial responses to Staphylococcus aureus: effects on cytokine, costimulatory molecule, and toll-like receptor expression. J Neuroimmunol (2002) 130(1–2):86–99. doi:10.1016/S0165-5728(02)00216-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kielian T, Esen N, Bearden ED. Toll-like receptor 2 (TLR2) is pivotal for recognition of S. aureus peptidoglycan but not intact bacteria by microglia. Glia (2005) 49(4):567–76. doi:10.1002/glia.20144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kielian T, Phulwani NK, Esen N, Syed MM, Haney AC, McCastlain K, et al. MyD88-dependent signals are essential for the host immune response in experimental brain abscess. J Immunol (2007) 178(7):4528–37. doi:10.4049/jimmunol.178.7.4528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Leppert D, Lindberg RL, Kappos L, Leib SL. Matrix metalloproteinases: multifunctional effectors of inflammation in multiple sclerosis and bacterial meningitis. Brain Res Brain Res Rev (2001) 36(2–3):249–57. doi:10.1016/S0165-0173(01)00101-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Banati RB, Gehrmann J, Schubert P, Kreutzberg GW. Cytotoxicity of microglia. Glia (1993) 7(1):111–8. doi:10.1002/glia.440070117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Banati RB, Rothe G, Valet G, Kreutzberg GW. Detection of lysosomal cysteine proteinases in microglia: flow cytometric measurement and histochemical localization of cathepsin B and L. Glia (1993) 7(2):183–91. doi:10.1002/glia.440070208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Colton CA, Gilbert DL. Microglia, an in vivo source of reactive oxygen species in the brain. Adv Neurol (1993) 59:321–6.
96. Colton CA, Keri JE, Chen WT, Monsky WL. Protease production by cultured microglia: substrate gel analysis and immobilized matrix degradation. J Neurosci Res (1993) 35(3):297–304. doi:10.1002/jnr.490350309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol (2010) 119(1):7–35. doi:10.1007/s00401-009-0619-8
99. Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia (2013) 61(12):1939–58. doi:10.1002/glia.22575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. John GR, Scemes E, Suadicani SO, Liu JS, Charles PC, Lee SC, et al. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc Natl Acad Sci U S A (1999) 96(20):11613–8. doi:10.1073/pnas.96.20.11613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Duffy HS, John GR, Lee SC, Brosnan CF, Spray DC. Reciprocal regulation of the junctional proteins claudin-1 and connexin43 by interleukin-1beta in primary human fetal astrocytes. J Neurosci (2000) 20(23):RC114.
102. Bolanos JP, Medina JM. Induction of nitric oxide synthase inhibits gap junction permeability in cultured rat astrocytes. J Neurochem (1996) 66(5):2091–9. doi:10.1046/j.1471-4159.1996.66052091.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Reuss B, Unsicker K. Regulation of gap junction communication by growth factors from non-neural cells to astroglia: a brief review. Glia (1998) 24(1):32–8. doi:10.1002/(SICI)1098-1136(199809)24:1<32::AID-GLIA4>3.0.CO;2-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Allaman I, Belanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci (2011) 34(2):76–87. doi:10.1016/j.tins.2010.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. McKimmie CS, Graham GJ. Astrocytes modulate the chemokine network in a pathogen-specific manner. Biochem Biophys Res Commun (2010) 394(4):1006–11. doi:10.1016/j.bbrc.2010.03.111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci (2007) 10(11):1355–60. doi:10.1038/nn1988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Vesce S, Rossi D, Brambilla L, Volterra A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int Rev Neurobiol (2007) 82:57–71. doi:10.1016/S0074-7742(07)82003-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol (2007) 44(5):999–1010. doi:10.1016/j.molimm.2006.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Panenka W, Jijon H, Herx LM, Armstrong JN, Feighan D, Wei T, et al. P2X7-like receptor activation in astrocytes increases chemokine monocyte chemoattractant protein-1 expression via mitogen-activated protein kinase. J Neurosci (2001) 21(18):7135–42.
110. Liu MT, Chen BP, Oertel P, Buchmeier MJ, Armstrong D, Hamilton TA, et al. The T cell chemoattractant IFN-inducible protein 10 is essential in host defense against viral-induced neurologic disease. J Immunol (2000) 165(5):2327–30. doi:10.4049/jimmunol.165.5.2327
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Ransohoff RM, Hamilton TA, Tani M, Stoler MH, Shick HE, Major JA, et al. Astrocyte expression of mRNA encoding cytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J (1993) 7(6):592–600.
112. Carpentier PA, Begolka WS, Olson JK, Elhofy A, Karpus WJ, Miller SD. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia (2005) 49(3):360–74. doi:10.1002/glia.20117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Girvin AM, Gordon KB, Welsh CJ, Clipstone NA, Miller SD. Differential abilities of central nervous system resident endothelial cells and astrocytes to serve as inducible antigen-presenting cells. Blood (2002) 99(10):3692–701. doi:10.1182/blood-2001-12-0229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Lee SJ, Park JY, Hou J, Benveniste EN. Transcriptional regulation of the intercellular adhesion molecule-1 gene by proinflammatory cytokines in human astrocytes. Glia (1999) 25(1):21–32. doi:10.1002/(SICI)1098-1136(19990101)25:1<21::AID-GLIA3>3.0.CO;2-R
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Rosenman SJ, Shrikant P, Dubb L, Benveniste EN, Ransohoff RM. Cytokine-induced expression of vascular cell adhesion molecule-1 (VCAM-1) by astrocytes and astrocytoma cell lines. J Immunol (1995) 154(4):1888–99.
116. Shrikant P, Chung IY, Ballestas ME, Benveniste EN. Regulation of intercellular adhesion molecule-1 gene expression by tumor necrosis factor-alpha, interleukin-1 beta, and interferon-gamma in astrocytes. J Neuroimmunol (1994) 51(2):209–20. doi:10.1016/0165-5728(94)90083-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Borregaard N. Neutrophils, from marrow to microbes. Immunity (2010) 33(5):657–70. doi:10.1016/j.immuni.2010.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol (2006) 6(3):173–82. doi:10.1038/nri1785
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Lacroix S, Feinstein D, Rivest S. The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol (1998) 8(4):625–40. doi:10.1111/j.1750-3639.1998.tb00189.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediators Inflamm (2013) 2013:480739. doi:10.1155/2013/480739
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol (2001) 166(7):4634–43. doi:10.4049/jimmunol.166.7.4634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Kastenbauer S, Angele B, Sporer B, Pfister HW, Koedel U. Patterns of protein expression in infectious meningitis: a cerebrospinal fluid protein array analysis. J Neuroimmunol (2005) 164(1–2):134–9. doi:10.1016/j.jneuroim.2005.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Spanaus KS, Nadal D, Pfister HW, Seebach J, Widmer U, Frei K, et al. C-X-C and C-C chemokines are expressed in the cerebrospinal fluid in bacterial meningitis and mediate chemotactic activity on peripheral blood-derived polymorphonuclear and mononuclear cells in vitro. J Immunol (1997) 158(4):1956–64.
124. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest (2012) 122(4):1164–71. doi:10.1172/JCI58644
125. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol (2011) 11(8):519–31. doi:10.1038/nri3024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Bernardes-Silva M, Anthony DC, Issekutz AC, Perry VH. Recruitment of neutrophils across the blood-brain barrier: the role of E- and P-selectins. J Cereb Blood Flow Metab (2001) 21(9):1115–24. doi:10.1097/00004647-200109000-00009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Neal JW, Gasque P. How does the brain limit the severity of inflammation and tissue injury during bacterial meningitis? J Neuropathol Exp Neurol (2013) 72(5):370–85. doi:10.1097/NEN.0b013e3182909f2f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Cederlund A, Gudmundsson GH, Agerberth B. Antimicrobial peptides important in innate immunity. FEBS J (2011) 278(20):3942–51. doi:10.1111/j.1742-4658.2011.08302.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Cardona AE, Li M, Liu L, Savarin C, Ransohoff RM. Chemokines in and out of the central nervous system: much more than chemotaxis and inflammation. J Leukoc Biol (2008) 84(3):587–94. doi:10.1189/jlb.1107763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Müller M, Carter S, Hofer MJ, Campbell IL. Review: the chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity – a tale of conflict and conundrum. Neuropathol Appl Neurobiol (2010) 36(5):368–87. doi:10.1111/j.1365-2990.2010.01089.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Kwok LY, Miletic H, Lütjen S, Soltek S, Deckert M, Schlüter D. Protective immunosurveillance of the central nervous system by Listeria-specific CD4 and CD8 T cells in systemic listeriosis in the absence of intracerebral Listeria. J Immunol (2002) 169(4):2010–9. doi:10.4049/jimmunol.169.4.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Häusler KG, Prinz M, Nolte C, Weber JR, Schumann RR, Kettenmann H, et al. Interferon-gamma differentially modulates the release of cytokines and chemokines in lipopolysaccharide- and pneumococcal cell wall-stimulated mouse microglia and macrophages. Eur J Neurosci (2002) 16(11):2113–22. doi:10.1046/j.1460-9568.2002.02287.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Aloisi F, Ria F, Penna G, Adorini L. Microglia are more efficient than astrocytes in antigen processing and in Th1 but not Th2 cell activation. J Immunol (1998) 160(10):4671–80.
134. Frei K, Siepl C, Groscurth P, Bodmer S, Schwerdel C, Fontana A. Antigen presentation and tumor cytotoxicity by interferon-gamma-treated microglial cells. Eur J Immunol (1987) 17(9):1271–8. doi:10.1002/eji.1830170909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Ford AL, Foulcher E, Lemckert FA, Sedgwick JD. Microglia induce CD4 T lymphocyte final effector function and death. J Exp Med (1996) 184(5):1737–45. doi:10.1084/jem.184.5.1737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe (2013) 13(4):379–93. doi:10.1016/j.chom.2013.03.010
137. Wraith DC, Nicholson LB. The adaptive immune system in diseases of the central nervous system. J Clin Invest (2012) 122(4):1172–9. doi:10.1172/JCI58648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Griffin DE, Metcalf T. Clearance of virus infection from the CNS. Curr Opin Virol (2011) 1(3):216–21. doi:10.1016/j.coviro.2011.05.021
139. Metcalf TU, Griffin DE. Alphavirus-induced encephalomyelitis: antibody-secreting cells and viral clearance from the nervous system. J Virol (2011) 85(21):11490–501. doi:10.1128/JVI.05379-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Savarin C, Bergmann CC. Neuroimmunology of central nervous system viral infections: the cells, molecules and mechanisms involved. Curr Opin Pharmacol (2008) 8(4):472–9. doi:10.1016/j.coph.2008.05.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Tschen SI, Stohlman SA, Ramakrishna C, Hinton DR, Atkinson RD, Bergmann CC. CNS viral infection diverts homing of antibody-secreting cells from lymphoid organs to the CNS. Eur J Immunol (2006) 36(3):603–12. doi:10.1002/eji.200535123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Bergmann CC, Lane TE, Stohlman SA. Coronavirus infection of the central nervous system: host-virus stand-off. Nat Rev Microbiol (2006) 4(2):121–32. doi:10.1038/nrmicro1343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Ishihara K, Hirano T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev (2002) 13(4–5):357–68. doi:10.1016/S1359-6101(02)00027-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Lane TE, Hardison JL, Walsh KB. Functional diversity of chemokines and chemokine receptors in response to viral infection of the central nervous system. Curr Top Microbiol Immunol (2006) 303:1–27.
145. Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, et al. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol (2005) 79(17):11457–66. doi:10.1128/JVI.79.17.11457-11466.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med (2005) 202(8):1087–98. doi:10.1084/jem.20042530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Lane TE, Asensio VC, Yu N, Paoletti AD, Campbell IL, Buchmeier MJ. Dynamic regulation of alpha- and beta-chemokine expression in the central nervous system during mouse hepatitis virus-induced demyelinating disease. J Immunol (1998) 160(2):970–8.
148. Phares TW, Stohlman SA, Hinton DR, Bergmann CC. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J Virol (2013) 87(6):3382–92. doi:10.1128/JVI.03307-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Oh JW, Schwiebert LM, Benveniste EN. Cytokine regulation of CC and CXC chemokine expression by human astrocytes. J Neurovirol (1999) 5(1):82–94. doi:10.3109/13550289909029749
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Cole KE, Strick CA, Paradis TJ, Ogborne KT, Loetscher M, Gladue RP, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med (1998) 187(12):2009–21. doi:10.1084/jem.187.12.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J Exp Med (1987) 166(4):1084–97. doi:10.1084/jem.166.4.1084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Phares TW, Stohlman SA, Bergmann CC. Intrathecal humoral immunity to encephalitic RNA viruses. Viruses (2013) 5(2):732–52. doi:10.3390/v5020732
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Templeton SP, Kim TS, O’Malley K, Perlman S. Maturation and localization of macrophages and microglia during infection with a neurotropic murine coronavirus. Brain Pathol (2008) 18(1):40–51. doi:10.1111/j.1750-3639.2007.00098.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Zuo J, Stohlman SA, Hoskin JB, Hinton DR, Atkinson R, Bergmann CC. Mouse hepatitis virus pathogenesis in the central nervous system is independent of IL-15 and natural killer cells. Virology (2006) 350(1):206–15. doi:10.1016/j.virol.2006.01.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Xue S, Sun N, Van Rooijen N, Perlman S. Depletion of blood-borne macrophages does not reduce demyelination in mice infected with a neurotropic coronavirus. J Virol (1999) 73(8):6327–34.
156. Kjeldsen L, Sengeløv H, Lollike K, Nielsen MH, Borregaard N. Isolation and characterization of gelatinase granules from human neutrophils. Blood (1994) 83(6):1640–9.
157. Zhou J, Stohlman SA, Hinton DR, Marten NW. Neutrophils promote mononuclear cell infiltration during viral-induced encephalitis. J Immunol (2003) 170(6):3331–6. doi:10.4049/jimmunol.170.6.3331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Zhou J, Stohlman SA, Atkinson R, Hinton DR, Marten NW. Matrix metalloproteinase expression correlates with virulence following neurotropic mouse hepatitis virus infection. J Virol (2002) 76(15):7374–84. doi:10.1128/JVI.76.15.7374-7384.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Trifilo MJ, Lane TE. The CC chemokine ligand 3 regulates CD11c+ CD11b+CD8alpha- dendritic cell maturation and activation following viral infection of the central nervous system: implications for a role in T cell activation. Virology (2004) 327(1):8–15. doi:10.1016/j.virol.2004.06.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Marten NW, Stohlman SA, Zhou J, Bergmann CC. Kinetics of virus-specific CD8+ -T-cell expansion and trafficking following central nervous system infection. J Virol (2003) 77(4):2775–8. doi:10.1128/JVI.77.4.2775-2778.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Stiles LN, Hosking MP, Edwards RA, Strieter RM, Lane TE. Differential roles for CXCR3 in CD4+ and CD8+ T cell trafficking following viral infection of the CNS. Eur J Immunol (2006) 36(3):613–22. doi:10.1002/eji.200535509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Stiles LN, Liu MT, Kane JA, Lane TE. CXCL10 and trafficking of virus-specific T cells during coronavirus-induced demyelination. Autoimmunity (2009) 42(6):484–91. doi:10.1080/08916930902810708
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Walsh KB, Edwards RA, Romero KM, Kotlajich MV, Stohlman SA, Lane TE. Expression of CXC chemokine ligand 10 from the mouse hepatitis virus genome results in protection from viral-induced neurological and liver disease. J Immunol (2007) 179(2):1155–65. doi:10.4049/jimmunol.179.2.1155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Chen BP, Kuziel WA, Lane TE. Lack of CCR2 results in increased mortality and impaired leukocyte activation and trafficking following infection of the central nervous system with a neurotropic coronavirus. J Immunol (2001) 167(8):4585–92. doi:10.4049/jimmunol.167.8.4585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Glass WG, Lane TE. Functional expression of chemokine receptor CCR5 on CD4+ T cells during virus-induced central nervous system disease. J Virol (2003) 77(1):191–8. doi:10.1128/JVI.77.1.191-198.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Weinger JG, Marro BS, Hosking MP, Lane TE. The chemokine receptor CXCR2 and coronavirus-induced neurologic disease. Virology (2013) 435(1):110–7. doi:10.1016/j.virol.2012.08.049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Bergmann CC, Altman JD, Hinton D, Stohlman SA. Inverted immunodominance and impaired cytolytic function of CD8+ T cells during viral persistence in the central nervous system. J Immunol (1999) 163(6):3379–87.
168. Bergmann CC, Parra B, Hinton DR, Chandran R, Morrison M, Stohlman SA. Perforin-mediated effector function within the central nervous system requires IFN-gamma-mediated MHC up-regulation. J Immunol (2003) 170(6):3204–13. doi:10.4049/jimmunol.170.6.3204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Ramakrishna C, Stohlman SA, Atkinson RA, Hinton DR, Bergmann CC. Differential regulation of primary and secondary CD8+ T cells in the central nervous system. J Immunol (2004) 173(10):6265–73. doi:10.4049/jimmunol.173.10.6265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Lin MT, Stohlman SA, Hinton DR. Mouse hepatitis virus is cleared from the central nervous systems of mice lacking perforin-mediated cytolysis. J Virol (1997) 71(1):383–91.
171. González JM, Bergmann CC, Ramakrishna C, Hinton DR, Atkinson R, Hoskin J, et al. Inhibition of interferon-gamma signaling in oligodendroglia delays coronavirus clearance without altering demyelination. Am J Pathol (2006) 168(3):796–804. doi:10.2353/ajpath.2006.050496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Parra B, Hinton DR, Marten NW, Bergmann CC, Lin MT, Yang CS, et al. IFN-gamma is required for viral clearance from central nervous system oligodendroglia. J Immunol (1999) 162(3):1641–7.
173. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, et al. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med (2010) 207(2):353–63. doi:10.1084/jem.20091738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, et al. Biology of interleukin-10. Cytokine Growth Factor Rev (2010) 21(5):331–44. doi:10.1016/j.cytogfr.2010.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Phares TW, Marques CP, Stohlman SA, Hinton DR, Bergmann CC. Factors supporting intrathecal humoral responses following viral encephalomyelitis. J Virol (2011) 85(6):2589–98. doi:10.1128/JVI.02260-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Metcalf TU, Baxter VK, Nilaratanakul V, Griffin DE. Recruitment and retention of B cells in the central nervous system in response to alphavirus encephalomyelitis. J Virol (2013) 87(5):2420–9. doi:10.1128/JVI.01769-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Puntambekar SS, Bergmann CC, Savarin C, Karp CL, Phares TW, Parra GI, et al. Shifting hierarchies of interleukin-10-producing T cell populations in the central nervous system during acute and persistent viral encephalomyelitis. J Virol (2011) 85(13):6702–13. doi:10.1128/JVI.00200-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Griffin D, Levine B, Tyor W, Ubol S, Desprès P. The role of antibody in recovery from alphavirus encephalitis. Immunol Rev (1997) 159:155–61. doi:10.1111/j.1600-065X.1997.tb01013.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Ramakrishna C, Bergmann CC, Atkinson R, Stohlman SA. Control of central nervous system viral persistence by neutralizing antibody. J Virol (2003) 77(8):4670–8. doi:10.1128/JVI.77.8.4670-4678.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Hooper DC, Phares TW, Fabis MJ, Roy A. The production of antibody by invading B cells is required for the clearance of rabies virus from the central nervous system. PLoS Negl Trop Dis (2009) 3(10):e535. doi:10.1371/journal.pntd.0000535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Fragkoudis R, Ballany CM, Boyd A, Fazakerley JK. In Semliki Forest virus encephalitis, antibody rapidly clears infectious virus and is required to eliminate viral material from the brain, but is not required to generate lesions of demyelination. J Gen Virol (2008) 89(Pt 10):2565–8. doi:10.1099/vir.0.2008/002238-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Levine B, Hardwick JM, Trapp BD, Crawford TO, Bollinger RC, Griffin DE. Antibody-mediated clearance of alphavirus infection from neurons. Science (1991) 254(5033):856–60. doi:10.1126/science.1658936
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Lee H, Sunden Y, Ochiai K, Umemura T. Experimental intracerebral vaccination protects mouse from a neurotropic virus by attracting antibody secreting cells to the CNS. Immunol Lett (2011) 139(1–2):102–9. doi:10.1016/j.imlet.2011.05.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Marques CP, Kapil P, Hinton DR, Hindinger C, Nutt SL, Ransohoff RM, et al. CXCR3-dependent plasma blast migration to the central nervous system during viral encephalomyelitis. J Virol (2011) 85(13):6136–47. doi:10.1128/JVI.00202-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Griffin DE. Recovery from viral encephalomyelitis: immune-mediated noncytolytic virus clearance from neurons. Immunol Res (2010) 47(1–3):123–33. doi:10.1007/s12026-009-8143-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Ubol S, Levine B, Lee SH, Greenspan NS, Griffin DE. Roles of immunoglobulin valency and the heavy-chain constant domain in antibody-mediated downregulation of Sindbis virus replication in persistently infected neurons. J Virol (1995) 69(3):1990–3.
187. Dietzschold B, Kao M, Zheng YM, Chen ZY, Maul G, Fu ZF, et al. Delineation of putative mechanisms involved in antibody-mediated clearance of rabies virus from the central nervous system. Proc Natl Acad Sci U S A (1992) 89(15):7252–6. doi:10.1073/pnas.89.15.7252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Niedziela N, damczyk-Sowa MA, Pierzchala K. Epidemiology and clinical record of multiple sclerosis in selected countries: a systematic review. Int J Neurosci (2014) 124(5):322–30. doi:10.3109/00207454.2013.840618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Pierson E, Simmons SB, Castelli L, Goverman JM. Mechanisms regulating regional localization of inflammation during CNS autoimmunity. Immunol Rev (2012) 248(1):205–15. doi:10.1111/j.1600-065X.2012.01126.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Islam T, Gauderman WJ, Cozen W, Hamilton AS, Burnett ME, Mack TM. Differential twin concordance for multiple sclerosis by latitude of birthplace. Ann Neurol (2006) 60(1):56–64. doi:10.1002/ana.20871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Patsopoulos NA, Barcellos LF, Hintzen RQ, Schaefer C, van Duijn CM, Noble JA, et al. Fine-mapping the genetic association of the major histocompatibility complex in multiple sclerosis: HLA and non-HLA effects. PLoS Genet (2013) 9(11):e1003926. doi:10.1371/journal.pgen.1003926
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. International Multiple Sclerosis Genetics Consortium (IMSGC), Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet (2013) 45(11):1353–60. doi:10.1038/ng.2770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. van der Mei IA, Ponsonby AL, Dwyer T, Blizzard L, Simmons R, Taylor BV, et al. Past exposure to sun, skin phenotype, and risk of multiple sclerosis: case-control study. BMJ (2003) 327(7410):316. doi:10.1136/bmj.327.7410.316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Lassmann H, Niedobitek G, Aloisi F, Middeldorp JM, NeuroproMiSe EBV Working Group. Epstein-Barr virus in the multiple sclerosis brain: a controversial issue – report on a focused workshop held in the Centre for Brain Research of the Medical University of Vienna, Austria. Brain (2011) 134(Pt 9):2772–86. doi:10.1093/brain/awr197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Stromnes IM, Goverman JM. Active induction of experimental allergic encephalomyelitis. Nat Protoc (2006) 1(4):1810–9. doi:10.1038/nprot.2006.285
196. Stromnes IM, Goverman JM. Passive induction of experimental allergic encephalomyelitis. Nat Protoc (2006) 1(4):1952–60. doi:10.1038/nprot.2006.284
197. Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM. IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med (2008) 205(7):1535–41. doi:10.1084/jem.20080159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Oksenberg JR, Panzara MA, Begovich AB, Mitchell D, Erlich HA, Murray RS, et al. Selection for T-cell receptor V beta-D beta-J beta gene rearrangements with specificity for a myelin basic protein peptide in brain lesions of multiple sclerosis. Nature (1993) 362(6415):68–70. doi:10.1038/362068a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Vogt J, Paul F, Aktas O, Müller-Wielsch K, Dörr J, Dörr S, et al. Lower motor neuron loss in multiple sclerosis and experimental autoimmune encephalomyelitis. Ann Neurol (2009) 66(3):310–22. doi:10.1002/ana.21719
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature (1990) 346(6280):183–7. doi:10.1038/346183a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Allegretta M, Nicklas JA, Sriram S, Albertini RJ. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science (1990) 247(4943):718–21. doi:10.1126/science.1689076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Leech MD, Barr TA, Turner DG, Brown S, O’Connor RA, Gray D, et al. Cutting edge: IL-6-dependent autoimmune disease: dendritic cells as a sufficient, but transient, source. J Immunol (2013) 190(3):881–5. doi:10.4049/jimmunol.1202925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med (2005) 11(3):328–34. doi:10.1038/nm1197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol (2004) 173(6):3916–24. doi:10.4049/jimmunol.173.6.3916
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Olson JK, Girvin AM, Miller SD. Direct activation of innate and antigen-presenting functions of microglia following infection with Theiler’s virus. J Virol (2001) 75(20):9780–9. doi:10.1128/JVI.75.20.9780-9789.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Mayo L, Quintana FJ, Weiner HL. The innate immune system in demyelinating disease. Immunol Rev (2012) 248(1):170–87. doi:10.1111/j.1600-065X.2012.01135.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood (2009) 113(14):3190–7. doi:10.1182/blood-2008-07-168575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Li J, Gran B, Zhang GX, Ventura ES, Siglienti I, Rostami A, et al. Differential expression and regulation of IL-23 and IL-12 subunits and receptors in adult mouse microglia. J Neurol Sci (2003) 215(1–2):95–103. doi:10.1016/S0022-510X(03)00203-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol (2011) 12(6):568–75. doi:10.1038/ni.2031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol (2011) 12(6):560–7. doi:10.1038/ni.2027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
211. Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, et al. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol (2006) 177(1):566–73. doi:10.4049/jimmunol.177.1.566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, et al. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med (2007) 13(10):1173–5. doi:10.1038/nm1651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med (2001) 194(4):519–27. doi:10.1084/jem.194.4.519
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Kimizuka Y, Kimura S, Saga T, Ishii M, Hasegawa N, Betsuyaku T, et al. Roles of interleukin-17 in an experimental Legionella pneumophila pneumonia model. Infect Immun (2012) 80(3):1121–7. doi:10.1128/IAI.05544-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J (2010) 24(4):1023–34. doi:10.1096/fj.09-141978
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Bauer J, Sminia T, Wouterlood FG, Dijkstra CD. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J Neurosci Res (1994) 38(4):365–75. doi:10.1002/jnr.490380402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Benveniste EN. Cytokines: influence on glial cell gene expression and function. Chem Immunol (1997) 69:31–75. doi:10.1159/000058653
218. Huizinga R, van der Star BJ, Kipp M, Jong R, Gerritsen W, Clarner T, et al. Phagocytosis of neuronal debris by microglia is associated with neuronal damage in multiple sclerosis. Glia (2012) 60(3):422–31. doi:10.1002/glia.22276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
219. Serafini B, Magliozzi R, Rosicarelli B, Reynolds R, Zheng TS, Aloisi F. Expression of TWEAK and its receptor Fn14 in the multiple sclerosis brain: implications for inflammatory tissue injury. J Neuropathol Exp Neurol (2008) 67(12):1137–48. doi:10.1097/NEN.0b013e31818dab90
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H, et al. Production and functions of IL-17 in microglia. J Neuroimmunol (2008) 194(1–2):54–61. doi:10.1016/j.jneuroim.2007.11.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Doring A, Yong VW. The good, the bad and the ugly. Macrophages/microglia with a focus on myelin repair. Front Biosci (Schol Ed) (2011) 3:846–56. doi:10.2741/191
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev (2011) 91(2):461–553. doi:10.1152/physrev.00011.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
223. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol (2009) 27:119–45. doi:10.1146/annurev.immunol.021908.132528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, Lassmann H. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain (2002) 125(Pt 1):75–85. doi:10.1093/brain/awf015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Duncan DS, Miller SD. CNS expression of B7-H1 regulates pro-inflammatory cytokine production and alters severity of Theiler’s virus-induced demyelinating disease. PLoS One (2011) 6(4):e18548. doi:10.1371/journal.pone.0018548
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Van Der Voorn P, Tekstra J, Beelen RH, Tensen CP, Van Der Valk P, De Groot CJ. Expression of MCP-1 by reactive astrocytes in demyelinating multiple sclerosis lesions. Am J Pathol (1999) 154(1):45–51. doi:10.1016/S0002-9440(10)65249-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Majumder S, Zhou LZ, Ransohoff RM. Transcriptional regulation of chemokine gene expression in astrocytes. J Neurosci Res (1996) 45(6):758–69. doi:10.1002/(SICI)1097-4547(19960915)45:6<758::AID-JNR12>3.0.CO;2-S
228. Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, et al. Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol (1996) 156(11):4363–8.
229. Toft-Hansen H, Fuchtbauer L, Owens T. Inhibition of reactive astrocytosis in established experimental autoimmune encephalomyelitis favors infiltration by myeloid cells over T cells and enhances severity of disease. Glia (2011) 59(1):166–76. doi:10.1002/glia.21088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
230. Calderon TM, Eugenin EA, Lopez L, Kumar SS, Hesselgesser J, Raine CS, et al. A role for CXCL12 (SDF-1alpha) in the pathogenesis of multiple sclerosis: regulation of CXCL12 expression in astrocytes by soluble myelin basic protein. J Neuroimmunol (2006) 177(1–2):27–39. doi:10.1016/j.jneuroim.2006.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
231. Zhang K, McQuibban GA, Silva C, Butler GS, Johnston JB, Holden J, et al. HIV-induced metalloproteinase processing of the chemokine stromal cell derived factor-1 causes neurodegeneration. Nat Neurosci (2003) 6(10):1064–71. doi:10.1038/nn1127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Nash B, Ioannidou K, Barnett SC. Astrocyte phenotypes and their relationship to myelination. J Anat (2011) 219(1):44–52. doi:10.1111/j.1469-7580.2010.01330.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: neuroimmunology, non-tumor antigens, glial cells, CNS autoimmunity, blood–brain barrier, CNS infections
Citation: Huber AK, Duncker PC and Irani DN (2014) Immune responses to non-tumor antigens in the central nervous system. Front. Oncol. 4:328. doi: 10.3389/fonc.2014.00328
Received: 02 October 2014; Accepted: 30 October 2014;
Published online: 13 November 2014.
Edited by:
Lois A. Lampson, Harvard Medical School, USACopyright: © 2014 Huber, Duncker and Irani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David N. Irani, Department of Neurology, University of Michigan Medical School, 109 Zina Pitcher Place, 4007 Biomedical Sciences Research Building, Ann Arbor, MI 48109-2200, USA e-mail:ZGF2aWRpcmFAbWVkLnVtaWNoLmVkdQ==