 Alona Zer
Alona Zer Natasha Leighl
Natasha Leighl- Division of Medical Oncology, Princess Margaret Cancer Centre, University of Toronto, Toronto, ON, Canada
Lung adenocarcinoma is the most common subtype of lung cancer today. With the discovery of epidermal growth factor receptor (EGFR) mutations, anaplastic lymphoma kinase (ALK) rearrangements, and effective targeted therapy, personalized medicine has become a reality for patients with lung adenocarcinoma. Here, we review potential additional targets and novel therapies of interest in lung adenocarcinoma including targets within the cell surface (receptor tyrosine kinases EGFR, human epidermal growth factor receptor 2, RET, ROS1, mesenchymal–epidermal transition, TRK), targets in intracellular signal transduction (ALK, RAS–RAF–MEK, PI3K–AKT–PTEN, WNT), nuclear targets such as poly-ADP ribose polymerase, heat shock protein 90, and histone deacetylase, and selected pathways in the tumor environment. With the evolving ability to identify specific molecular aberrations in patient tumors in routine practice, our ability to further personalize therapy in lung adenocarcinoma is rapidly expanding.
Introduction
In recent years, we have witnessed a transformation of the treatment paradigm for advanced non-small cell lung cancer (NSCLC). Previously, patients were offered platinum-based chemotherapy, followed by second-line chemotherapy with docetaxel or pemetrexed, and erlotinib after chemotherapy failure, yielding modest benefits in an unselected population (1). Using molecular selection, clinical trials of targeted therapy have demonstrated major improvements in response, quality of life, and progression-free survival compared to chemotherapy, using epidermal growth factor receptor (EGFR) TKI in EGFR mutant NSCLC and crizotinib in anaplastic lymphoma kinase (ALK) rearranged NSCLC (2, 3). Survival is similar in many of these trials, given the high rate of crossover from chemotherapy to the more active agent upon progression.
It is now standard of care to test non-squamous lung carcinoma for the presence of EGFR mutation and ALK rearrangement upon diagnosis of advanced disease (4), in order to select patients for initial EGFR TKI and ALK inhibitor therapy. The remarkable activity of these agents in molecularly selected lung cancer patients has led to a rapid increase in studies evaluating new targets and novel targeted agents. These targets include oncogenic driver mutations (genomic alterations that initiate malignant transformation of the normal cell), signal transduction proteins, tumor angiogenesis, and factors in the tumor environment supporting cancer cell proliferation (for example, immune-modulating proteins) (Figure 1; Table 1). In this review, we discuss selected new and promising targets as well as targeted therapies currently under investigation in non-squamous NSCLC, specifically adenocarcinoma.
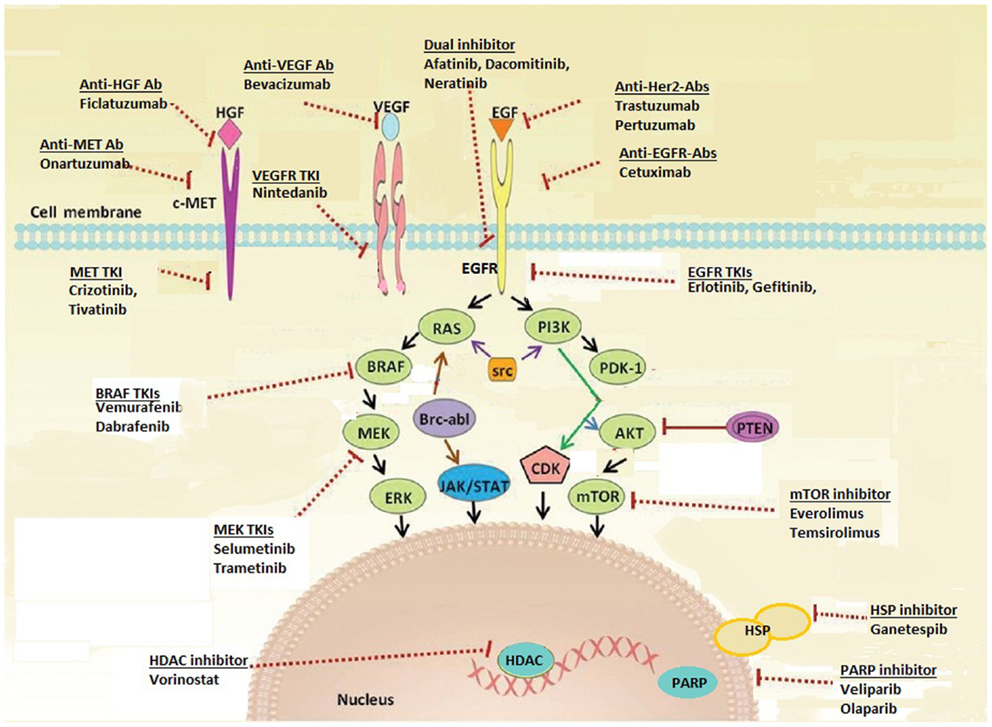
Figure 1. Targetable pathways in the non-squamous NSCLC cell.
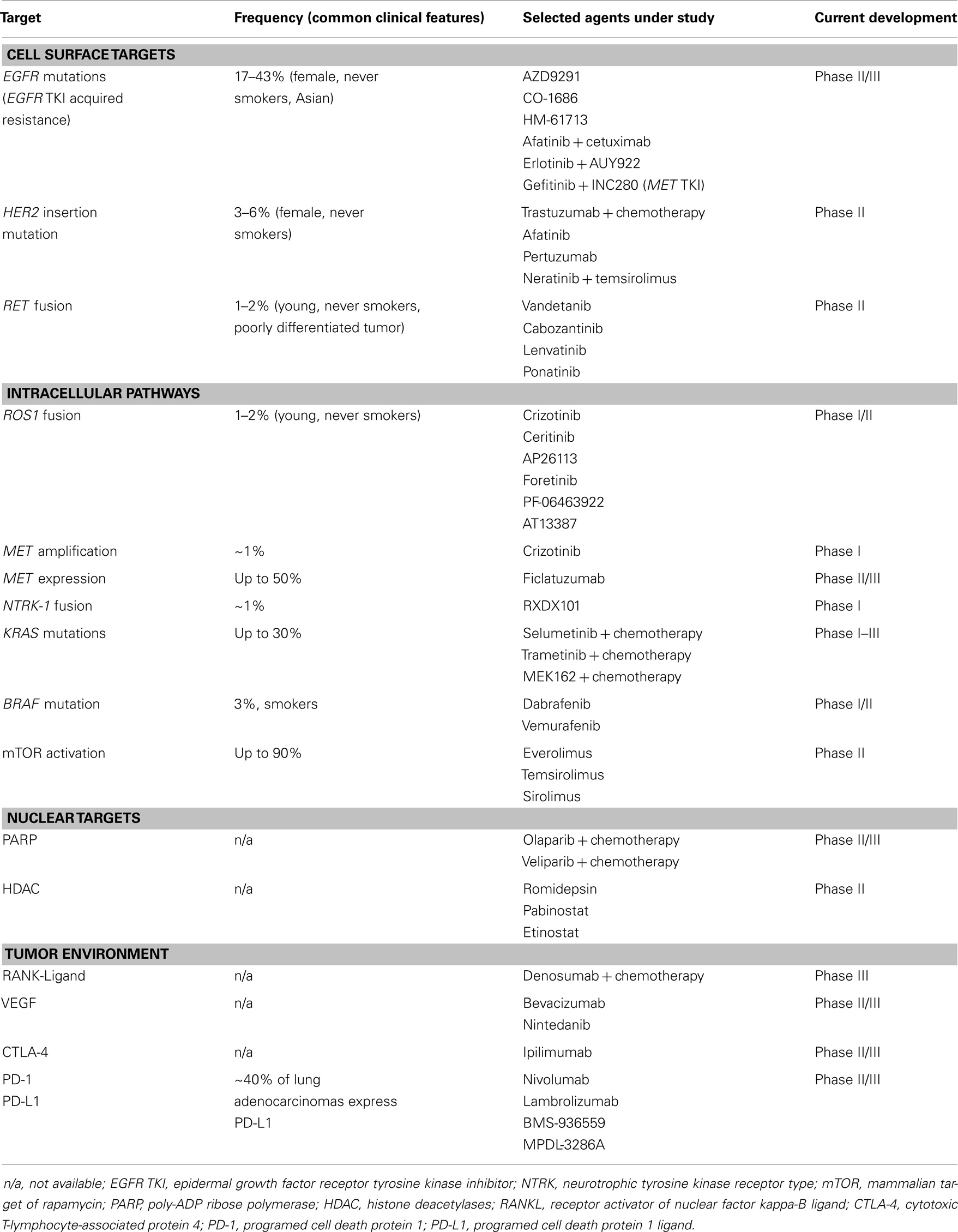
Table 1. Selected targets and selected targeted agents in lung adenocarcinoma.
Targets within the Cell Surface
Epidermal Growth Factor Receptor
Targeting EGFR has led to a breakthrough in understanding of lung cancer biology, and the NSCLC treatment paradigm. Mutations in EGFR, resulting in greater affinity for ATP binding by the EGFR tyrosine kinase domain and constitutive activation, are found in ~15% of lung cancers in Caucasians and 40% in Asians (5, 6). Activating mutations are significantly associated with response to EGFR TKIs, with erlotinib, gefitinib, and afatinib established as initial standard therapy. However, resistance mutations have been identified, such as T790M in exon 20. There are multiple agents in development with enhanced affinity for T790M mutant lung cancer that may spare wild type EGFR, potentially avoiding toxicities like rash and diarrhea. AZD9291 and CO-1686 are examples of such agents, and have reported responses in 58–64% of patients with acquired EGFR TKI resistance and documented T790M mutation (7, 8). There are other strategies in development, targeting acquired EGFR TKI resistance including chemotherapy with intercalated EGFR TKI, combinations with mesenchymal–epidermal transition (MET), dual EGFR and heat shock protein 90 (HSP90) inhibitors, and more. For example, combination of afatinib and cetuximab has demonstrated activity in patients with acquired EGFR TKI resistance and T790M positive and negative tumors (9), and the addition of AUY922 to erlotinib has restored sensitivity in 22% of patients with acquired resistance to erlotinib (10).
Human Epidermal Growth Factor Receptor 2
Human epidermal growth factor receptor 2 is a cell surface receptor, and member of the erbB receptor tyrosine kinase family. It is activated by heterodimerization with other ligand-bound members of the erbB family, or by homodimerization. HER2 is a key oncogene in breast cancer, and is associated with improved outcomes with trastuzumab (anti-HER2 monoclonal antibody) (11, 12). In NSCLC, HER2 protein overexpression is found in 6–35% of patients and HER2 gene amplification is found in 10–20% (13). Trastuzumab has shown minimal activity in lung cancer, both as a single agent and in combination with chemotherapy, particularly in patients with FISH positive or IHC 3+ tumors (14, 15).
HER2 mutations are seen in 2.8–6% of lung adenocarcinomas (16, 17), more commonly in women and non-smokers. These mutations are commonly exon 20 in-frame insertions. Activity has been seen with trastuzumab-based therapy and afatinib (13, 18). A phase I trial of neratinib (an irreversible pan-HER inhibitor) and temsirolimus (mTOR inhibitor) suggested benefit in five patients with HER2-mutant NSCLC (19). A phase II trial assessing this combination is underway. Other trials include studies of HER2-directed antibodies (trastuzumab, pertuzumab), TKIs (neratinib, dacomitinib, and afatinib), and a peptide vaccine (www.clinicaltrials.gov).
RET
RET (rearranged during transfection), is a known oncogene in thyroid cancer, with both activating mutations and gene rearrangements observed (20). Approximately 1.5% of NSCLC cases have RET translocations, typically in younger, non-smoking adenocarcinoma patients (21). Fusion variants include KIF5B-RET in adenocarcinoma, CCDC6, NCO4, and TRIM33 also found in thyroid cancer (22, 23).
Vandetanib, sunitinib, sorafenib, lenvatinib, ponatinib, and cabozantinib are all multi-targeted kinase inhibitors that target RET. Activity has been seen in RET-positive lung cancer patients with cabozantinib and vandetinib, and multiple trials are ongoing in this population with a recent halt in a ponatinib study for safety concerns (24, 25).
ROS1
ROS1 encodes a receptor tyrosine kinase of the insulin receptor super family, with no known ligand and little known about its normal function. ROS1 fusion genes, with oncogenic transformation potential, have been described in multiple tumor cell lines, including lung cancer. The prevalence of ROS1 rearrangement in NSCLC is estimated at 1–2%, and can be detected using FISH or IHC. Patients, similar to those with ALK-rearranged lung cancer, tend to be younger, never smokers, and have adenocarcinoma histology, although cases in squamous carcinoma have been reported (26). A response rate of 60% has been reported with crizotinib in 35 patients with ROS-1-rearranged lung cancer, including two patients with complete response, and median PFS was not reached (27). Multiple other agents are under development including AP26113, foretinib, PF-06463922, ceritinib, and HSP90 inhibitors such as AT13387 (NCT01712217).
Mesenchymal–Epidermal Transition Receptor
Mesenchymal–epidermal transition is a receptor tyrosine kinase, which undergoes homodimerization by binding its ligand, hepatocyte growth factor (HGF), to trigger intracellular signaling cascades, including PI3K–AKT–mTOR and RAS–RAF–MAPK pathways. In lung cancer, MET mutations are rare, but amplification is seen in up to 21%, resulting in constitutive MET activation and is believed to be a potential mechanism of acquired EGFR TKI resistance (28, 29). MET expression is seen in at least one-third of lung cancers, including adenocarcinoma and squamous histology (30).
Targeting MET protein-expressing lung cancer has not been successful to date, with negative phase III trials of onartuzumab (anti-MET monoclonal antibody), and TKIs including tivantinib (31, 32). Crizotinib activity has been reported in MET-amplified tumors (33), with ongoing studies in EGFR TKI-resistant lung cancer of MET and HGF-targeted agents, such as ficlatuzumab (anti-HGF monoclonal antibody, NCT02034981).
NTRK1 Fusions
These have recently been described in never smokers with adenocarcinoma that is ALK and EGFR wild type. NTRK-1 fusions have been identified in 3 of 91 lung adenocarcinoma samples that were EGFR/KRAS/ALK-1/ROS-1 negative (34). RXDX101 has demonstrated activity in TRK-fusion positive lung cancer in a recent phase I trial (35).
Targets within Intracellular Pathways
Anaplastic Lymphoma Kinase
Anaplastic lymphoma kinase fusion genes, resulting in ALK fusion proteins, are present in 3–5% of lung adenocarcinomas, commonly in never smokers and younger patients. The presence of ALK fusion strongly predicts response to ALK TKIs, such as crizotinib, ceritinib, and others. This topic is discussed in length in a separate review article in this issue.
RAS–RAF–MEK pathway
The RAS family of oncogenes includes H-RAS, K-RAS, and N-RAS. RAS proteins encode a membrane-bound GTP-ase that mediates signal transduction from various tyrosine kinase receptors (e.g., EGFR, HER2) to the RAF/MEK/ERK pathway and others, regulating cell growth, proliferation, and apoptosis (36). KRAS mutations are seen in ~30% of Western adenocarcinoma cases, fewer in Asian populations, most commonly in codons 12 or 13. NRAS and HRAS mutations are less common in lung cancer, <1% (37).
K-RAS
The role of mutant KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) as a prognostic or predictive marker in NSCLC remains controversial. An analysis of LACE-BIO suggests that it is not prognostic in early stage lung cancer, nor does it predict for adjuvant chemotherapy benefit (38). Several studies suggest that it is a potential negative predictor of benefit from EGFR TKI (39). While KRAS mutations have been identified in patients with and without smoking histories, never smokers are more likely to have transition mutations. Transversion mutations are found almost exclusively in smokers (40).
The most promising agents in development for KRAS mutant lung cancer have been MEK inhibitors combined with chemotherapy. Selumetinib, a MEK1/2 inhibitor, significantly improved PFS and response when added to docetaxel versus docetaxel plus placebo (HR = 0.58, p = 0.014, RR 37 vs. 0%, p < 0.0001), with a trend toward greater survival (41); a phase III trial is ongoing. Trametinib, another MEK inhibitor, showed activity in combination with docetaxel as well as with pemetrexed (42, 43). The response rate with single agent trametinib is 12%, with similar activity to docetaxel in pre-treated KRAS mutant lung cancer patients (44).
BRAF
BRAF, a serine-threonine kinase, lies downstream of KRAS and directly activates MEK by phosphorylation, which in turn activates ERK. BRAF (v-Raf murine sarcoma viral oncogene homolog B) mutations and BRAF inhibitors first gained attention in melanoma where 40–60% of tumors harbor activating V600E BRAF mutations. Three percent of lung adenocarcinomas harbor BRAF mutations, half of the V600E subtype, inducing constitutive kinase activity. These mutations occur more frequently in smokers. Dabrafenib, a BRAF kinase inhibitor, demonstrated a 54% RR in 17 BRAF V600E-mutated NSCLC patients (45). Vemurafenib is another BRAF kinase inhibitor that shown activity in this population. There are ongoing clinical trials assessing BRAF, MEK, and AKT inhibitors in this population.
PI3K–AKT–PTEN pathway
The phosphatidylinositol 3-kinase (PI3K)–AKT–mTOR (mammalian target of rapamycin) signaling pathway is one of the most dysregulated pathways in human cancers, including NSCLC. PI3K can be activated by transmembrane receptor tyrosine kinases like EGFR or RAS, through phosphorylation of AKT. This inhibits pro-apoptotic proteins and promotes cell survival. Activated mTOR complexes (mTORC1), downstream of PI3K–AKT, result in increased ribosomal protein synthesis and further AKT activation (mTORC2). PI3K-dependent signal transduction can be terminated by PTEN, a tumor suppressor intracellular protein (46).
PIK3CA
PIK3CA encodes the catalytic subunit of PI3K, and mutations and amplification are seen in 2 and 12–17% of NSCLC cases (47, 48). These mutations can co-exist with other known driver mutations in lung adenocarcinoma, including EGFR and KRAS and in the setting of acquired EGFR TKI resistance (49, 50), suggesting that this may not be a driver mutation in itself. Trials of PI3K specific kinase inhibitors are ongoing.
PTEN, AKT, mTOR
Loss of PTEN protein expression, with subsequent AKT overexpression, occurs in a third of NSCLC cases, and is associated with poor prognosis in lung cancer (51). This may be related to epigenetic silencing, as PTEN mutations are rare in NSCLC (52). AKT activation and mTOR phosphorylation is found in 51% of NSCLC cases, although AKT mutations are rare (<1%). Given the high level of activation and “crosstalk” with the RAS–RAF–MEK pathway, studies of mTOR and AKT inhibitors are of major interest in lung cancer. Everolimus (RAD001), temsirolimus, and other mTOR inhibitors are being investigated in combination with other targeted agents, including EGFR TKIs, although toxicity of these agents remains challenging, with high rates of fatigue and stomatitis (53, 54).
Wnt-beta-catenin pathway
The Wnt signaling pathway is highly active in lung cancer and correlates with metastasis and proliferation, and is believed to maintain cancer stem cells. Activated Wnt signaling inhibits the proteolysis of beta-catenin. Accumulated beta-catenin in cytoplasm moves to the nucleus where it initiates transcription factors promoting cell growth and chemo- and radio-resistance. Down-regulation of Wnt inhibitors is common in NSCLC samples and associated with poor prognosis (55). WNT mutations are rare in lung cancer and mutations in Beta-catenin are detected in 2% of lung adenocarcinoma (56). Several targeted therapies against the Wnt pathway are being investigated in early phase trials, including PRI-724, a small molecule beta-catenin inhibitor.
Nuclear Targets
Poly-ADP Ribose Polymerase
BRCA1, BRCA2, and PALB2 are proteins responsible for repair of DNA double-strand breaks through the homologous repair pathway; breaks that are not repaired lead to apoptosis. This repair pathway can be disrupted by mutations in BRCA1, BRCA2, or ATM (ataxia telangiectasia mutated), found in 7% of lung adenocarcinomas. High levels of BRCA1 protein expression in lung cancer correlate with poor survival, while decreased expression predicts response to platinum-based chemotherapy (57, 58). The poly-ADP ribose polymerase (PARP) enzyme is key in repairing single-strand DNA breaks, which may lead to double-strand breaks. BRCA deficient or mutated cells are sensitive to PARP inhibition, which may also sensitize cancer cells to alkylator or platinum damage via DNA single- or double-strand breaks. Despite a negative study with iniparib and chemotherapy, veliparib, and olaparib are being evaluated in combination with platinum-based therapy or EGFR TKI in NSCLC.
Heat shock protein 90
Heat shock protein 90 is a chaperone protein that assists posttranslational folding of several proteins to stabilize and protect them from cellular stresses like heat or hypoxia, including critical proteins in lung cancer such as EGFR, HER2, MET, ALK, and others. HSP90 inhibitors have shown activity in EGFR mutant lung cancer after the development of resistance, in ALK-rearranged tumors and more recently in EGFR wild type adenocarcinoma when combined with chemotherapy (59). A phase III clinical trial of docetaxel plus or minus ganetespib in chemo-naïve adenocarcinoma is ongoing. Other HSP90 inhibitors under active investigation in lung cancer include retaspimycin (IPI-504), AUY992, and AT13387.
Histone deacetylase
Histones are a family of proteins bound to DNA strands that maintain the helical structure of DNA. DNA expression is regulated by acetylation and deacetylation of histones. Deacetylation results in condensed DNA and reduced transcription. But histone deacetylase (HDAC), highly expressed in most cancers, may also alter activity of various proteins involved in carcinogenesis including HSP90, STAT3, and p53. HDAC inhibitors have multiple effects on DNA transcription, including induction of HSP90 acetylation (see above), disrupting its function, and resulting in tumor apoptosis. Vorinostat, FDA approved for treatment of cutaneous T-cell lymphoma, showed initial promise when added to chemotherapy in advanced NSCLC, although the subsequent phase III trial was negative (60). Other HDAC inhibitors being studied include etinostat, romidepsin, pabinostat, pivanex, and CI-994.
Targets in the Tumor Environment
Angiogenesis
Vascular endothelial growth factor (VEGF) is a pro-angiogenic factor, with a key role in tumor angiogenesis. Its high expression in a variety of tumors, including NSCLC, is associated with poor prognosis (61). Although multiple agents targeting VEGF and VEGF receptors have been studied in lung cancer, only bevacizumab and more recently nintedanib have improved survival in advanced non-squamous NSCLC. Bevacizumab, combined with paclitaxel and carboplatin, improved response, PFS, and survival in the practice-changing ECOG4599 trial (62), although subsequent bevacizumab trials have not improved survival compared to chemotherapy alone. Nintedanib, a multi-targeted VEGF- and FGFR-1 receptor TKI demonstrated greater OS in a subgroup of adenocarcinoma patients when added to docetaxel versus chemotherapy alone (63). Trials of multiple other agents have not demonstrated positive results, although trials with VEGF/R inhibitors, including in different molecular subtype of adenocarcinoma, are ongoing.
Vascular disrupting agents, such as vadimezan, target vasculature directly, not through VEGF/VEGFR. To date, trials of these and multiple other anti-angiogenic agents have not yet yielded benefit.
Immune Modulation
The immune system plays an active role in eradication of malignant cells. However, the evolution of cancer includes developing mechanisms to escape the immune system. Several approaches are now being investigated to boost anti-cancer immune response, either by inhibiting immune checkpoints (as CTLA-4, PD-1, and PD-L1) or by developing vaccines of cancer antigens. This topic is discussed in length in a separate review article, with the PD-1 checkpoint inhibitors as the most promising current target in immune therapy of lung cancer, with demonstrated single agent activity in both adenocarcinoma and squamous carcinoma (64).
There are multiple other potential targets in lung adenocarcinoma that are not reviewed here, such as the cell surface receptor insulin-like growth factor 1 receptor, apoptotic receptors, and proteins including TRAIL, BCL-1, IAP proteins including survivin, and the proteasome. Additional targets in the tumor environment include adhesion molecules such as integrins, and even osteoclasts, all potentially important targets in lung cancer with ongoing trials of targeted agents.
Conclusion
Striking therapeutic advances in metastatic NSCLC have been observed with targeted agents using molecular selection, notable for patients with EGFR mutant or ALK-rearranged lung cancer. Testing for these oncogenic drivers is now standard of care in advanced lung adenocarcinoma, but they are found in only ~20% of lung adenocarcinomas in Western populations, while remaining patients are eligible only for standard chemotherapy. However, this “success story,” as well as improved understanding of molecular pathways of lung carcinogenesis, had led to rapid progress in the identification of novel targets in adenocarcinoma and potential therapies. Despite this enthusiasm, there are still barriers to overcome, including how to approach tumors without single oncogene addiction, i.e., targeting multiple pathways, and also how to overcome primary and secondary resistance to targeted therapies. Finally, the development of accurate, rapid, tissue-, and cost-conserving assays to identify multiple targets simultaneously, including targets beyond genomic sequencing, is urgently needed. In the meantime, drug development and discovery of novel targets in lung adenocarcinoma remain one of the fastest growing areas of research and development in oncology today.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Leighl NB. Treatment paradigms for patients with metastatic non-small-cell lung cancer: first-, second- and third-line. Curr Oncol (2012) 19(Suppl 1):S52–8. doi: 10.3747/co.19.1114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi:10.1056/NEJMoa0810699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med (2013) 368(25):2385–94. doi:10.1056/NEJMoa1214886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol (2013) 8(7):823–59. doi:10.1097/JTO.0b013e318290868f
5. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med (2009) 361(10):958–67. doi:10.1056/NEJMoa0904554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Shi Y, Au JS, Thongprasert S, Srinivasan S, Tsai CM, Khoa MT, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol (2014) 9(2):154–62. doi:10.1097/JTO.0000000000000033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Finlay MR, Anderton M, Ashton S, Ballard P, Bethel PA, Box MR, et al. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients (pts) with EGFR inhibitor-resistant non-small cell lung cancer (NSCLC). J Clin Oncol (2014) 32(15_suppl: abstr. 8009):5s. doi:10.1021/jm500973a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Sequist LV, Soria J-C, Gadgeel SM, Wakelee HA, Camidge DR, Varga A, et al. First-in-human evaluation of CO-1686, an irreversible, highly selective tyrosine kinase inhibitor of mutations of EGFR (activating and T790M). J Clin Oncol (2014) 32(15_suppl: abstr. 8010):5s.
9. Janjigian YY, Azzoli CG, Krug LM, Pereira LK, Rizvi NA, Pietanza MC, et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin Cancer Res (2011) 17(8):2521–7. doi:10.1158/1078-0432.CCR-10-2662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Johnson ML, Hart EM, Rademaker A, Weitner BB, Urman A, Simm HD, et al. A phase II study of HSP90 inhibitor AUY922 and erlotinib for patients with EGFR-mutant lung cancer and acquired resistance to EGFR tyrosine kinase inhibitors. J Clin Oncol (2013) 31(Suppl):abstr 8036.
11. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med (2001) 344(11):783–92. doi:10.1056/NEJM200103153441101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med (2005) 353:1673–84. doi:10.1056/NEJMoa052122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol (2013) 31(16):1997–2003. doi:10.1200/JCO.2012.45.6095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Clamon G, Herndon J, Kern J, Govindan R, Garst J, Watson D, et al. Lack of trastuzumab activity in nonsmall cell lung carcinoma with overexpression of erb-B2: 39810: a phase II trial of cancer and leukemia group B. Cancer (2005) 103(8):1670–5. doi:10.1002/cncr.20950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Gatzemeier U. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann Oncol (2004) 15(1):19–27. doi:10.1093/annonc/mdh031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Shigematsu H, Takahashi T, Nomura M, Majmudar K, Suzuki M, Lee H, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res (2005) 65(5):1642–6. doi:10.1158/0008-5472.CAN-04-4235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Arcila ME, Chaft JE, Nafa K, Roy-Chowdhuri S, Lau C, Zaidinski M, et al. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res (2012) 18(18):4910–8. doi:10.1158/1078-0432.CCR-12-0912
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Falchook GS, Janku F, Tsao AS, Bastida CC, Stewart DJ, Kurzrock R. Non-small-cell lung cancer with HER2 exon 20 mutation: regression with dual HER2 inhibition and anti-VEGF combination treatment. J Thorac Oncol (2013) 8(2):e19–20. doi:10.1097/JTO.0b013e31827ce38e
19. Gandhi L, Bahleda R, Tolaney SM, Kwak EL, Cleary JM, Pandya SS, et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol (2014) 32(2):68–75. doi:10.1200/JCO.2012.47.2787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Wells SA Jr, Santoro M. Targeting the RET pathway in thyroid cancer. Clin Cancer Res (2009) 15(23):7119–23. doi:10.1158/1078-0432.CCR-08-2742
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol (2012) 30(35):4352–9. doi:10.1200/JCO.2012.44.1477
22. Kohno T, Tsuta K, Tsuchihara K, Nakaoku T, Yoh K, Goto K. RET fusion gene: translation to personalized lung cancer therapy. Cancer Sci (2013) 104(11):1396–400. doi:10.1111/cas.12275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Borrello MG, Ardini E, Locati LD, Greco A, Licitra L, Pierotti MA. RET inhibition: implications in cancer therapy. Expert Opin Ther Targets (2013) 17(4):403–19. doi:10.1517/14728222.2013.758715
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov (2013) 3(6):630–5. doi:10.1158/2159-8290.CD-13-0035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Gautschi O, Zander T, Keller FA, Strobel K, Hirschmann A, Aebi S, et al. A patient with lung adenocarcinoma and RET fusion treated with vandetanib. J Thorac Oncol (2013) 8(5):e43–4. doi:10.1097/JTO.0b013e31828a4d07
26. Davies KD, Le AT, Theodoro MF, Skokan MC, Aisner DL, Berge EM, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res (2012) 18(17):4570–9. doi:10.1158/1078-0432.CCR-12-0550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Efficacy and safety of crizotinib in patients with advanced ROS1-rearranged non-small cell lung cancer. J Clin Oncol (2013) 31(Suppl):abstr 8032.
28. Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol (2008) 3(4):331–9. doi:10.1097/JTO.0b013e318168d9d4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316(5827):1039–43. doi:10.1126/science.1141478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Tsao MS, Liu N, Chen JR, Pappas J, Ho J, To C, et al. Differential expression of Met/hepatocyte growth factor receptor in subtypes of non-small cell lung cancers. Lung Cancer (1998) 20:1–16. doi:10.1016/S0169-5002(98)00007-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Spigel DR, Edelman MJ, O’Byrne K, Paz-Ares L, Shames DS, Yu W, et al. Onartuzumab plus erlotinib versus erlotinib in previously treated stage IIIB or IV NSCLC: results from the pivotal phase III randomized multicenter placebo-controlled METLung (OAM4971g) global trial. J Clin Oncol (2014) 32(Suppl; abstr 8000):5s.
32. Sequist LV, von Pawel J, Garmey EG, Akerley WL, Brugger W, Ferrari D, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol (2011) 29(24):3307–15. doi:10.1200/JCO.2010.34.0570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Camidge DR, Shapiro G, Otterson GA, Villaruz LC, Villalona-Calero MA, Iafrate AJ, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer. J Clin Oncol (2014) 32(Suppl; abstr 8001):5s.
34. Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med (2013) 19:1469–72. doi:10.1038/nm.3352
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. De Braud FG, Pilla L, Niger M, Damian S, Bardazza B, Martinetti A, et al. Phase I open label, dose escalation study of RXDX101, an oral pan-trk, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol (2014) 32(Suppl; abstr 2502):5s.
36. Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy. Cancer Lett (2009) 283(2):125–34. doi:10.1016/j.canlet.2009.01.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II, et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA (2014) 311:1998–2006. doi:10.1001/jama.2014.3741
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Shepherd FA, Domerg C, Hainaut P, Jänne PA, Pignon JP, Graziano S, et al. Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy. J Clin Oncol (2013) 31(17):2173–81. doi:10.1200/JCO.2012.48.1390
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada clinical trials group study BR.21. J Clin Oncol (2008) 26(26):4268–75. doi:10.1200/JCO.2007.14.8924
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res (2008) 14(18):5731–4. doi:10.1158/1078-0432.CCR-08-0646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Jänne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol (2013) 14(1):38–47. doi:10.1016/S1470-2045(12)70489-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Gandara DR, Hiret S, Blumenschein GR, Barlesi F, Madelaine J, Infante JR, et al. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with docetaxel in KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. J Clin Oncol (2013) 31(Suppl):abstr 8028.
43. Kelly K, Mazieres U, Leighl NB, Barlesi F, Zalcman G, Gordon MS, et al. Oral MEK1/MEK2 inhibitor trametinib (GSK1120212) in combination with pemetrexed in KRAS-mutant and wild-type (WT) advanced non-small cell lung cancer (NSCLC): a phase I/Ib trial. J Clin Oncol (2013) 31(Suppl):abstr 8027.
44. Blumenschein GR, Smit EF, Planchard D, Kim D-W, Cadranel J, De Pas T, et al. MEK114653: a randomized, multicenter, phase II study to assess efficacy and safety of trametinib (T) compared with docetaxel (D) in KRAS-mutant advanced non–small cell lung cancer (NSCLC). J Clin Oncol (2013) 31(Suppl):abstr 8027.
45. Planchard D, Mazieres J, Riely GJ, Rudin CM, Barlesi F, Quoix EA, et al. Interim results of phase II study BRF113928 of dabrafenib in BRAF V600E mutation–positive non-small cell lung cancer (NSCLC) patients. J Clin Oncol (2013) 31(Suppl):abstr 8009.
46. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol (2010) 28(6):1075–83. doi:10.1200/JCO.2009.25.3641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kawano O, Sasaki H, Endo K, Suzuki E, Haneda H, Yukiue H, et al. PIK3CA mutation status in Japanese lung cancer patients. Lung Cancer (2006) 54(2):209–15. doi:10.1016/j.lungcan.2006.07.006
48. Kawano O, Sasaki H, Okuda K, Yukiue H, Yokoyama T, Yano M, et al. PIK3CA gene amplification in Japanese non-small cell lung cancer. Lung Cancer (2007) 58(1):159–60. doi:10.1016/j.lungcan.2007.06.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Pietanza MC, et al. Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma-rationale for comprehensive mutation profiling. Mol Cancer Ther (2012) 11(2):485–91. doi:10.1158/1535-7163.MCT-11-0692
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med (2011) 3(75):75ra26. doi:10.1126/scitranslmed.3002003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Tang JM, He QY, Guo RX, Chang XJ. Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer (2006) 51(2):181–91. doi:10.1016/j.lungcan.2005.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, et al. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol (2005) 36(7):768–76. doi:10.1016/j.humpath.2005.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Besse B, Leighl N, Bennouna J, Papadimitrakopoulou VA, Blais N, Traynor AM, et al. Phase II study of everolimus-erlotinib in previously treated patients with advanced non-small-cell lung cancer. Ann Oncol (2014) 25(2):409–15. doi:10.1093/annonc/mdt536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Ekman S, Wynes MW, Hirsch FR. The mTOR pathway in lung cancer and implications for therapy and biomarker analysis. J Thorac Oncol (2012) 7(6):947–53. doi:10.1097/JTO.0b013e31825581bd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Stewart DJ. Wnt signaling pathway in non-small cell lung cancer. J Natl Cancer Inst (2014) 106(1):djt356. doi:10.1093/jnci/djt356
56. Sequist LV, Heist RS, Shaw AT, Fidias P, Rosovsky R, Temel JS, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol (2011) 22(12):2616–24. doi:10.1093/annonc/mdr489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Rosell R, Skrzypski M, Jassem E, Taron M, Bartolucci R, Sanchez JJ, et al. BRCA1: a novel prognostic factor in resected non-small-cell lung cancer. PLoS One (2007) 2(11):e1129. doi:10.1371/journal.pone.0001129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Reguart N, Cardona AF, Carrasco E, Gomez P, Taron M, Rosell R. BRCA1: a new genomic marker for non-small-cell lung cancer. Clin Lung Cancer (2008) 9(6):331–9. doi:10.3816/CLC.2008.n.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Ramalingam SS, Goss GD, Andric ZG, Bondarenko I, Zaric B, Ceric T, et al. A randomized study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel versus docetaxel alone for second-line therapy of lung adenocarcinoma (GALAXY-1). J Clin Oncol (2013) 31(Suppl):abstr CRA8007.
60. Ramalingam SS, Maitland ML, Frankel P, Argiris AE, Koczywas M, Gitlitz B, et al. Carboplatin and paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol (2010) 28(1):56–62. doi:10.1200/JCO.2009.24.9094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Otrock ZK, Hatoum HA, Musallam KM, Awada AH, Shamseddine AI. Is VEGF a predictive biomarker to anti-angiogenic therapy? Crit Rev Oncol Hematol (2011) 79(2):103–11. doi:10.1016/j.critrevonc.2010.07.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med (2006) 355(24):2542–50. doi:10.1056/NEJMoa061884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, et al. Nintedanib plus docetaxel in NSCLC patients progressing after first-line chemotherapy: LUME Lung 1, a randomized, double-blind phase III trial. J Clin Oncol (2013) 31(Suppl):abstr LBA8011. doi:10.1016/S1470-2045(13)70586-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: NSCLC, nuclear targets, intracellular pathways, EGFR, ALK, novel targets, non-squamous
Citation: Zer A and Leighl N (2014) Promising targets and current clinical trials in metastatic non-squamous NSCLC. Front. Oncol. 4:329. doi: 10.3389/fonc.2014.00329
Received: 17 July 2014; Accepted: 31 October 2014;
Published online: 25 November 2014.
Edited by:
Barbara Melosky, British Columbia Cancer Agency, CanadaReviewed by:
Marina Chiara Garassino, Istituto Nazionale dei Tumori, ItalyMeng Xu Welliver, The Ohio State University James Cancer Center, USA
Copyright: © 2014 Zer and Leighl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natasha Leighl, Division of Medical Oncology, Princess Margaret Cancer Centre, University of Toronto, 5-105 610 University Avenue, Toronto, ON M5G 2M9, Canada e-mail:bmF0YXNoYS5sZWlnaGxAdWhuLmNh