 Marcin Teodorczyk
Marcin Teodorczyk Mirko H. H. Schmidt
Mirko H. H. Schmidt- Molecular Signal Transduction Laboratories, Institute for Microscopic Anatomy and Neurobiology, Focus Program Translational Neuroscience (FTN), Rhine Main Neuroscience Network (rmn2), Johannes Gutenberg University of Mainz School of Medicine, Mainz, Germany
Notch receptors play an essential role in the regulation of central cellular processes during embryonic and postnatal development. The mammalian genome encodes for four Notch paralogs (Notch 1–4), which are activated by three Delta-like (Dll1/3/4) and two Serrate-like (Jagged1/2) ligands. Further, non-canonical Notch ligands such as epidermal growth factor like protein 7 (EGFL7) have been identified and serve mostly as antagonists of Notch signaling. The Notch pathway prevents neuronal differentiation in the central nervous system by driving neural stem cell maintenance and commitment of neural progenitor cells into the glial lineage. Notch is therefore often implicated in the development of brain tumors, as tumor cells share various characteristics with neural stem and progenitor cells. Notch receptors are overexpressed in gliomas and their oncogenicity has been confirmed by gain- and loss-of-function studies in vitro and in vivo. To this end, special attention is paid to the impact of Notch signaling on stem-like brain tumor-propagating cells as these cells contribute to growth, survival, invasion, and recurrence of brain tumors. Based on the outcome of ongoing studies in vivo, Notch-directed therapies such as γ-secretase inhibitors and blocking antibodies have entered and completed various clinical trials. This review summarizes the current knowledge on Notch signaling in brain tumor formation and therapy.
Introduction
Malignant gliomas represent the most futile type of brain tumor in adults with an annual incidence of 5 per 100,000 individuals (1, 2). According to the World Health Organization (WHO) guidelines, they can be characterized as astrocytomas, oligodendrogliomas, ependymomas, or oligo-astrocytomas (mixed gliomas). Another WHO classification is based on the malignancy of the neoplasms and ranges from grade I, corresponding to low-proliferative non-invasive tumors, up to grade IV, assigned to cytologically malignant, highly infiltrative, mitotically active, and necrosis-prone glioblastoma multiforme (GBM, malignant glioma). Most of the diagnosed GBMs (90–95%) are primary tumors, although secondary glioblastomas might also arise from low-grade tumors (3, 4). Medulloblastomas, the most frequent neoplasms in children, are of a cerebellar origin and are therefore not included in this glioma classification system (5). WHO grades reflect patients’ prognosis: grade I tumors are generally curable by surgical resection alone (1), while the current standard care for GBM involves maximal surgical resection followed by temozolomide (TMZ) chemotherapy and radiation. Unfortunately, this treatment regimen has severe side-effects and barely extends the median survival from 12.1 to 14.6 months (6). Currently, the genetics and molecular biology of brain tumors are the focus of extensive studies. It is well established that glioma-driving mutations affect pathways regulating cellular parameters such as cell growth, apoptosis, migration, and angiogenesis (7–9). Master regulators of these biological processes frequently mutated in glioma are TP53, PTEN, PDGFR, NF1 or epidermal growth factor receptor (EGFR) and there is growing evidence that developmental signaling cues such as Notch are deregulated in malignant brain tumors as well.
Mammalian Notch Receptors and Ligands
Notch receptors are single-pass transmembrane proteins formed by two non-covalently associated polypeptide chains. The Drosophila melanogaster genome encodes only a single Notch gene, but four receptors (Notch 1–4) are found in mammals. After the synthesis of a single-chain precursor, the receptor undergoes a so-called S1 cleavage mediated by furin-like proteases in the trans-Golgi network. S1 generates an N-terminal extracellular domain (NECD) and a C-terminal fragment corresponding to the transmembrane domain (NTM) extending into the cytoplasm (intracellular Notch domain, NICD). The resulting heterodimer, held together by non-covalent bonds, is inserted into the plasma membrane (10). The NECD consists of multiple EGF-like repeats, which partially bind calcium ions and are required for ligand interaction (11). The Notch1 receptor contains 36 EGF repeats in the intracellular domain (12), while Notch2 contains 35 repeats (13), Notch3 34 repeats (14), and Notch4 29 repeats (15). The NECD negative regulatory region (NRR) is composed of three cysteine-rich Lin12/Notch repeats (LNR) (16) and a juxtamembrane heterodimerization domain. As the name suggests, NRR is responsible for the auto-inhibition of the Notch receptor (17, 18) and binds to a short extracellular region of NTM (19). The intracellular domain NICD of the Notch receptor is involved in cellular signaling and includes the recombination signal-binding protein Jκ (RBP-Jκ) associated module (RAM) (20), seven ankyrin (ANK) repeats (21), two nuclear-localization signals (NLS) (22), a transactivation domain (TAD) (23), and a C-terminal PEST sequence (rich in proline, glutamic acid, serine, and threonine) (24).
The canonical Notch ligands belong to the so-called Delta-Serrate-Lag2 (DSL) family and include the five mammalian type I transmembrane proteins Delta-like 1 (Dll1) (25), Dll3 (26), Dll4 (27), Jagged1 (28), and Jagged2 (29). The N-terminal region, the DSL domain and the first two EGF-like repeats are necessary for the interaction with EGF-like repeats of Notch receptors (30, 31). In addition, several transmembrane and soluble proteins have been described as non-canonical ligands, e.g., F3/contactin (32), Delta-like 1 (Dlk1), Dlk2, Delta and Notch-like EGF-related receptor (DNER), or the EGF-like protein 7 (EGFL7) (33–35). Common structural features of this group are the presence of EGF-like repeats and the absence of DSL domain. Dlk1, Dlk2, and DNER are transmembrane proteins (although Dlk1 and Dlk2 also exist in soluble forms), while EGFL7 is a bona fide secreted factor. Interestingly, DNER stimulates Notch signaling while current evidence indicates an inhibitory function of Dlk1/2 and EGFL7 (36).
Notch Signaling Pathway
Both Notch receptors and canonical ligands are transmembrane proteins, thus requiring close proximity of the plasma membranes in which they are embedded for interaction. The interaction between neighboring cells is referred to as in-trans interaction and switches Notch signaling on (Figure 1). This type of association relies on the EGF-like repeats 11 + 12 of Notch1/2/4 and repeats 10 + 11 of Notch3, respectively (11, 36). In-cis interaction between receptors and ligands expressed on the same cell inhibit the Notch pathway (37–39) and involves the EGF-like repeats 24–29 of Notch1 receptor (40). In-trans activation triggers the ubiquitination and internalization of the respective ligand and disrupts the hydrophobic interactions between NECD and NTM in the Notch receptor. This in turn exposes NTM to the extracellular S2 cleavage by “a disintegrin and metalloprotease” 10 (ADAM10) or ADAM17 (41). The phenotype of ADAM10 knock-out mice resembles Notch deficiencies (42, 43); however, cell culture-based experiments indicate that ADAM10 and 17 may share substrates including Notch receptors in vitro (44, 45). Both proteases create an intermediate membrane-tethered Notch extracellular truncation (NEXT), which is subsequently processed by the γ-secretase–presenilin complex (19). This so-called S3 cleavage releases the intracellular Notch domain NICD, which translocates into the nucleus (46) and binds to a protein complex containing DNA-binding proteins of the CSL family (RBP-Jκ/CBF-1/KBF2 in mammals) and mediates its conversion from a repressor to an activator of transcription followed by the recruitment of the co-activator mastermind-like 1 (MAML1) (47). In turn, the NICD–RBP-Jκ–MAML1 ternary complex recruits further components of the RNA polymerase II holoenzyme such as the histone acetyltransferases CBP/p300 (48) or PCAF/GCN5 (49). Ultimately, these events lead to the transcriptional de-repression of several genes that are often themselves transcriptional repressors such as Hairy/Enhancer of Split (Hes) and Hey (subfamily of Hes, related with YRPW motif) proteins (50–52). Hes-1, Hes-5, and Hey-1 are well-described direct Notch targets (53, 54), and growing evidence suggests Hes-7, Hey-2, and Hey-L as direct target genes (55). The list of genes regulated by Notch is still expanding and includes transcription factors such as NFκB (56, 57), PPAR (58), c-Myc (59–61), Sox2 (62), Pax6 (63), as well as cell cycle regulators such as cyclin D1 (64), and p21/Waf1 (65) among many others.
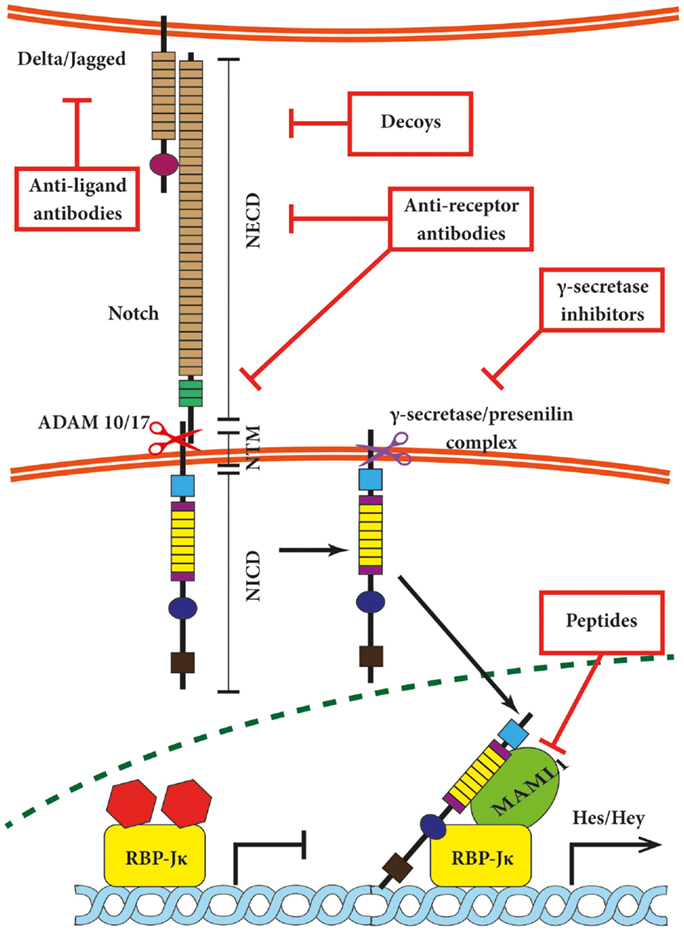
Figure 1. Canonical Notch signaling with points of intervention of current therapies. The interaction between Delta/Jagged-type ligands and Notch receptors leads to S2 cleavage on the extracellular site by “a disintegrin and metalloprotease” 10 (ADAM10) or ADAM17, which is followed by S3 cleavage by the γ-secretase–presenilin complex. The S3 cleavage gives rise to an intracellular Notch fragment (NICD) that translocates into the nucleus, where NICD binds to a protein complex containing recombination signal-binding protein Jκ (RBP-Jκ). This mediates the conversion of RBP-Jκ from a repressor to a transcriptional activator and is followed by the recruitment of the co-activator mastermind-like 1 (MAML1). These events lead to the de-repression of transcription of hairy/enhancer of split (Hes) and Hey. Several stages of the Notch signaling pathway are prone to pharmacological intervention and are labeled in the figure. Gamma-secretase inhibitors and blocking antibodies are already in clinical trials and decoys have been tested in animal models. Peptide inhibitors represent potential future treatment modalities. NECD, Notch extracellular domain; NTM, Notch transmembrane domain.
Role of Notch Signaling in the Healthy Developing Brain
Notch signaling is an evolutionary conserved pathway that prevents equipotent cells from acquiring identical cell fates. This can be accomplished through the so-called lateral inhibition; a process in which a cell that stochastically acquires enhanced ligand expression stimulates neighboring cells. The in-cis inhibition of Notch on the ligand-expressing cells renders this interaction unilateral. In neural development, the signal-sending cell will differentiate into a neuronal precursor while the signal receiving cell will remain as an uncommitted progenitor. This correlates with a decreased expression of Hes-1, Hes-5, and proneural genes as well as Notch ligands in uncommitted progenitors (66, 67).
Notch signaling plays a pivotal role in biological processes including apoptosis, cell proliferation, differentiation, and cell lineage decision in stem cells. Therefore, Notch governs embryonic development and is highly active in undifferentiated cells of the embryonic central nervous system, while its expression is reduced and spatially restricted in the adult brain (66, 68, 69). The relevance of Notch is in part due to its impact on the maintenance of neural stem (NSCs) and progenitor cells (NPCs) as well as the stimulation of their glial differentiation at the expense of their neuronal fate (70–72). Notch1 knock-out mice die before E11.5, approximately the time of neuronal maturation, among other reasons due to a loss of neuroblasts and premature neuronal differentiation (73, 74).
Role of Notch in Brain Neoplasms
Due to the central role of Notch in differentiation, its deregulation leads to multiple malignancies. The first evidence of the tumorigenic potential of Notch came from the translocation t(7; 9) in T cell acute lymphoblastic leukemia (T-ALL), which leads to a fusion of genes encoding the β chain of T cell receptors and the TAN1/NOTCH1 gene. The product of this gene acts like as a constitutively active form of NICD (75). Subsequently, the components of the Notch pathway have been described to be deregulated in numerous hematological malignancies and brain tumors including gliomas and medulloblastomas (76). The frequency and intensity of Notch2 expression in medulloblastoma is higher than that of Notch1 (77, 78). Moreover, Notch2 was shown to promote tumorigenesis of medulloblastoma, whereas, Notch1 inhibited tumor growth (77). In glioma, however, the correlation between tumor grade and expression of Notch isoforms has not been fully clarified, yet. The tumor-suppressive role of Notch1 is supported by the fact that it has been detected in all gliomas but a subset of grade IV tumors (79). Along the line, Notch1 expression has been shown to be higher in grade II and III malignancies than in glioblastomas (80). Furthermore, nuclear Notch1 staining has been correlated with a better outcome in high-grade glioma subtypes (81). These data suggest a favorable prognosis for patients carrying Notch1-positive tumors. However, according to other reports, the expression of Notch1 exhibits a positive correlation with glioma progression (82, 83), and high expression of Notch1 protein has been reported to be an independent predictor of poor survival in glioma (82). The oncogenic potential of Notch2 is indicated by the fact that the loss of Notch2 positively correlated with a favorable prognosis in small groups of patients diagnosed with oligodendroglioma and GBM (84). However, this conclusion has not yet been fully supported by experimental data.
Various other components of the Notch pathway have been used as markers for different stages of glioma. Transcription of Dll1 has been described to be regulated by the neurogenic transcription factor Hash-1 (85). Both Hash-1 and Dll1 has been found to be upregulated in progressive astrocytoma (grade II and III) as well as in secondary GBM, accompanied by a decrease in the Notch target gene Hes-1. These data indicate that enhanced Dll1 expression inhibits Notch signaling in these subgroups of gliomas. In primary GBM, on the other hand, there is the opposite pattern of Hash-1 and Hes-1 expression (86), implying that enhanced Dll1 expression inhibits Notch signaling while active Notch characterizes primary GBM. Interestingly, Phillips et al. proposed a different classification of gliomas from that of the WHO (81), in which the tumor is assigned into one of three subtypes based on genomic data: proneural, proliferative, or mesenchymal. In terms of prognosis, the proneural group correlates with longer survival as compared to the other two groups. This group contains most of grade III and secondary grade IV tumors (87) and displays the expression of neuroblast and developing neurons markers including Hash-1 and Dll1. These data are in accordance with other publications that show that Notch is active in primary GBM, while low-grade astrocytomas express the ligands Dll1 and Jagged1 (88, 89).
Notch receptors and ligands are post-translationally regulated by ubiquitylation and endocytosis. Two proteins regulating these processes have attracted attention in the cancer field, namely Neuralized1 (Neurl1) and Numb. The former is known to bind and to monoubiquinate Jagged1 in mammalian cells (90). The chromosomal fragment 10q25.1 encoding Neurl1 has been found to be frequently lost in grade II astrocytomas and GBM. Moreover, expression of human Neurl1 was nearly absent in high-grade astrocytoma and the majority of investigated glioma cell lines in contrast to the normal brain tissue (91). Analysis of Neurl1 expression in medulloblastoma led to similar results as it was downregulated compared to normal cerebral tissue (92). Numb, another antagonist of Notch signaling, is not an ubiquitin ligase itself but rather serves as an adapter protein for the E3 ubiquitin ligase Itch (also known as AIP4) (Figure 2) (93). Overexpression of Numb led to the proteasomal degradation of NICD (94) but the degradation of Numb by ligand of Notch protein X (LNX) overcame Notch downregulation (95). Expression of LNX is reduced in gliomas of different grades (96), offering another putative mechanism for enhanced Notch signaling in brain tumors.
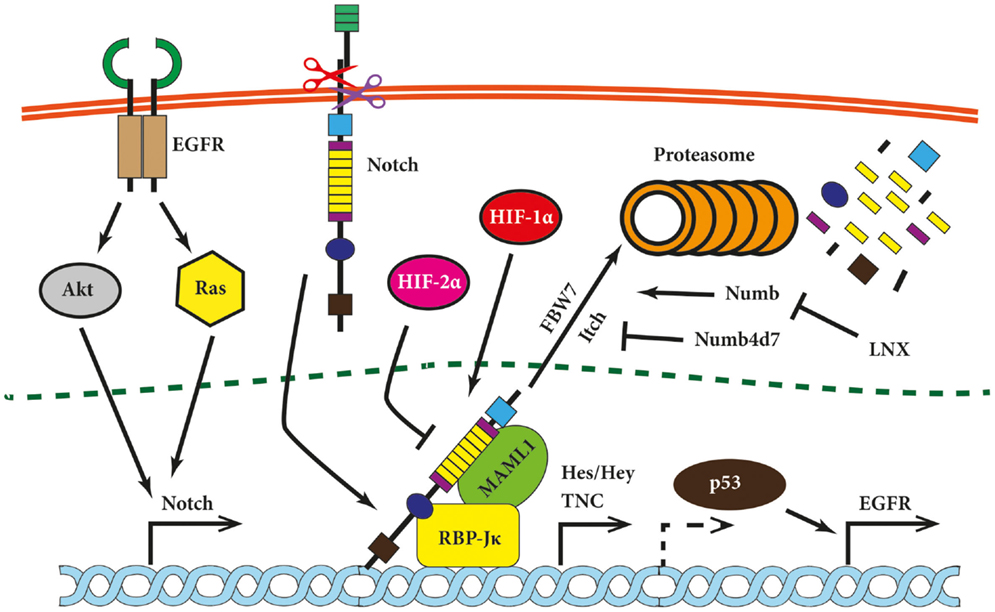
Figure 2. Notch signaling modules relevant for brain tumors. Notch signaling has been shown to be modulated on multiple levels in glioma cells and is linked upstream and downstream to other tumorigenic pathways. Its expression is induced by Ras and Akt, while Notch itself induces expression of epidermal growth factor (EGFR) via p53 (indirectly; hence dashed arrow) and pro-migratory glycoprotein tenascin C (TNC). Hypoxia-inducible factor 1α (HIF-1α) and HIF-2α compete for NICD binding. The Notch inhibitor HIF-2α is displaced by HIF-1α under hypoxic conditions. Further, several proteins modulate the Notch pathway at the level of NICD. Examples are Numb, which promotes Notch degradation via ubiquitin ligases such as FBW7 or Itch, or Numb4d7 and ligand of Notch protein X (LNX), which stimulate Notch signaling.
Notch activity is often measured by the expression levels of its direct target genes. Consequently, there are reports linking the expression of Hes/Hey transcriptional repressors to cancer prognosis. Expression of Hey-1 correlated with a twofold shorter disease-free survival compared to patients carrying Hey-1-negative tumors. In addition, Hey-1 is more frequently expressed in GBM as compared to low-grade astrocytomas while no expression was found in normal brain tissue or in neuroblastoma (97). Further, these observations have been confirmed by clinical data from 62 GBM patients, where the expression of Hey-1 correlated with a shorter overall survival. In Hey-1-negative cases, on the other hand, survival was significantly longer and accompanied by a high number of long-term survivors (98).
Non-canonical Notch ligands represent the least explored group of glioma-related markers. Dlk1 has been found to be upregulated in a subset of GBM as compared to the healthy brain. The protein has been suggested as a Notch inhibitor, which is supported by the finding that Hes-1 was downregulated in glioma cell lines stably expressing Dlk1 or treated with Dlk1-conditioned medium. Moreover, GBM cell lines transfected with this ligand exhibited an augmented proliferation (99).
Notch in Glioma Cells
The response of brain tumor cells to the modulation of Notch signaling has been investigated by Purow and colleagues (80), who demonstrated that the siRNA-mediated knock-down of Notch1 in glioma cell lines led to an increased cell death, decreased proliferation as well as cell cycle arrest. Further, the treatment affected cell morphology and triggered the outgrowth of neurite-like extensions (80). Another group reported an upregulation of the astrocyte marker glial fibrillary acidic protein (GFAP) and a downregulation of the mesenchymal marker vimentin as well as reduced proliferation of glioma cell lines upon Notch inhibition (88). These morphologic features are indicative of differentiation, suggesting that remaining in an undifferentiated state enhances the oncogenic potential of glioma cells. These in vitro data suggest that Notch1 acts as an oncogene in glioma, which has been confirmed in an intracranial xenograft model, where mice injected with control U251MG cells died sooner as compared to mice that received cells in which Notch1 or Dll1 were downregulated by siRNA. Knock-down of Jagged1 did not improve survival and caused a milder inhibition of proliferation in vitro as compared to the Dll1 knock-down (80). Furthermore, a recent report compared the role of the Notch paralogs Notch1 and Notch2, which displayed opposing effects on the propagation of glioma cells. Equally, knocking-down Notch1 or overexpressing Notch2 suppressed cell growth and invasion in addition to enhancing apoptosis of subcutaneously engrafted U251 and A172 glioma cells (100).
Much less is known about molecules exerting a Notch-driven malignant phenotype in glioma. One report suggests that Notch expression is linked to enhanced cell migration mediated by tenascin C (TNC), whose promoter is activated by the Notch-induced transcription factor RBP-Jκ (Figure 2) (78). In brief, TNC is a matrix glycoprotein that induced proliferation and migration of neuronal precursors and its expression increases in GBM as compared to grade III astrocytomas. Moreover, patients lacking TSC in the extracellular matrix (ECM) survived significantly longer than patients with TNC-positive lesions (101).
Notch in Stem-Like Brain Tumor-Propagating Cells
Glioblastoma, like other cancers, result from the accumulation of genetic and epigenetic mismatches (102). Until recently, it was presumed to originate solely from glial cells, i.e., astrocytes or oligodendrocytes, residing within the brain parenchyma. However, the discovery of proliferating cells in the adult brain led to a modification of this hypothesis. According to studies performed in genetically modified mouse models, gliomas may arise from NSCs or NPCs (103–105). More differentiated cells were also investigated: experiments with mosaic inactivation of Tp53 and Nf1 in NSCs showed that the most rapid phase of tumor growth occurs when the cells migrate out of the stem cell niche and become Olig2-positive oligodendroglial progenitor cells (106). In a different genetic model, non-stem cell progenitor cells were shown to generate astrocytomas and oligodendrogliomas upon the transgenic induction of v-erbB combined with Tp53 deletion (107). Another strain utilized to investigate the tumorigenic potential of differentiated cells is Ink4A/Arf knock-out mice. These animals are prone to develop brain tumors as compared to wild-type mice. The Ink4A/Arf locus encodes two tumor suppressors: p16INK4a, which prevents Rb phosphorylation by binding CDK4, and p14/19ARF, which prevents p53 degradation via MDM2 inhibition. Both processes are crucial for cell cycle regulation. Astrocytes derived from these animals were shown to undergo de-differentiation and induce glioma upon the expression of the EGFRvIII oncogene or stimulation with EGF or PDGF in vivo (103, 108).
The term “cancer stem cell” has been coined in order to describe tumor-propagating cells (109). These cells differ from normal tissue stem cells as they lack a well-defined and conserved hierarchy (110). Further confusion is caused by referring to them as tumor-initiating cells, which is supposed to reflect that they are the origin of tumors. In our opinion, the term stem-like brain tumor-propagating cell (BTPC) is more appropriate to “cancer stem cell” or “tumor-initiating cell” in the context of glioma formation as it refers to the most solid biological feature of this cell type, namely, the propagation of glioma in vivo. Relying on the expression of cell markers (or the lack thereof) to name this cell type may be misleading, as the transcriptome of BTPCs is highly variable (111) and this cell type expresses all types of cellular markers (e.g., nestin, GFAP, or beta III-tubulin) in parallel (own observations). Initially, BTPCs were isolated from gliomas based on CD133 expression, propagated in serum-free medium and shown to recapitulate glioma growth in vivo (112). However, CD133 cannot be considered a universal BTPC marker because CD133-negative cells also give rise to tumors, and some tumors are fully devoid of CD133-positive cells (113). It should be pointed out that several groups have reported that CD133 is also expressed by endothelial cells (114–116) and might thus be better suited as a marker of enhanced angiogenesis. In support of this hypothesis, vascular CD133, but not tumor-expressed CD133, was found to correlate with glioma grade (117). Several other extracellular molecules have been proposed as BTPC markers including CD44, CD15, and integrin α6. However, they have not yet been verified by the glioma community as bona fide BTPCs markers (118). Furthermore, considerable attention has been paid to stem cell-specific intracellular proteins like the transcription factors Sox2, Oct-4, Bmi-1, and Id4, RNA-binding protein Musashi-1 or the intermediate filament nestin, a stem cell marker and transcriptional target of Notch (119, 120). There is evidence that the expression levels of Musashi-1 and nestin positively correlate with high glioma grade and poor survival (121).
Notch has been suggested to play an important role in the maintenance of BTPCs as it regulates the maintenance and differentiation of NSCs (122–124). First results hinted that glioma cells cultured under stem cell conditions express Notch1, Notch4, Dll1, and Dll3 (125). In another publication, nine glioma-derived cell lines were divided into two groups based on sphere-forming capacity, CD133 expression and high invasiveness. Two of the transcripts belonging to the Notch cascade were overexpressed in the tumorigenic group while their expression was not increased in any cell line from the less stem-like/invasive group (126). Overexpressing NICD in the human glioma cell line SHG-44 led to enhanced proliferation as well as higher colony- and sphere-formation potentials. Moreover, the sphere-forming cells displayed BTPC characteristics such as the expression of the NSC marker nestin and the ability to differentiate into all three neural lineages based on immunofluorescence staining for GFAP, MAP2, and GalC (89). When NICD-overexpressing BTPCs were intracranially implanted into nude mice, they formed highly vascularized tumors containing large vessels with a central lumen. The cells were, however, hardly disseminating in contrast to control cells, which infiltrated both hemispheres (127). Taken together, these results suggest that due to the activation of multiple cell responses, Notch may play different roles in BTPCs depending on the cellular and environmental context. Furthermore, intracellular modifiers of Notch signaling, such as Numb proteins, are involved in the regulation of BTPCs. Numb4 promotes Notch degradation via FBW7 ubiquitin ligase assembly, while its truncated form, Numb4 delta 7 (Numb4d7), generated by alternative splicing, increases Notch signaling (Figure 2). Although Numb4d7 opposes growth-inhibitory effect of Numb4, both isoforms promote expression of stem cell markers in BTPCs. Thus, it appears Numb4 can affect BTPC differentiation independent from Notch inhibition itself (128).
Lastly, microvascular proliferation is an important feature of glioblastoma, underlining the essential role of angiogenesis in brain tumor development (1, 129). Formation of blood vessels is a response to hypoxia that results in the stabilization of the hypoxia-inducible factors (HIF)-1α and -2α and the subsequent upregulation of pro-angiogenic factors like VEGF. Notch signaling has been shown to be activated by hypoxia in normal and neoplastic cells as evidenced by the increased expression of Notch1, Hes-1, Hey-1, Dll1, and Dll4 (130, 131). Notch plays a central role in maintaining NSCs undifferentiated under hypoxic conditions (132), and therefore, its impact on BPTCs has been investigated. Treatment of medulloblastoma-derived BTPCs with immobilized recombinant Dll4 under hypoxic conditions (2% oxygen) led to the expansion of CD133-positive and nestin-positive cells. Inhibition of the Notch pathway resulted in the opposite effect as tumor cells underwent neuronal differentiation when treated with the γ-secretase inhibitor (GSI) N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) to inhibit Notch signaling (133). Further, the maintenance of glioma BTPCs can be hindered by the depletion of HIF-1α or inactivation of Notch signaling, partly because of the interaction between HIF-1α and NICD (134). This interaction has been confirmed by another group, which also described competition between HIF-1α and HIF-2α for NICD binding. HIF-2α inhibited Notch and was displaced by HIF-1α under hypoxic conditions (135).
Cross-Talk of Notch with the EGFR Pathway
Overexpression and increased activity of EGFR is one of the hallmarks of primary GBM (136, 137). EGFR is a receptor tyrosine kinase that initiates multiple cellular pathways such as mitogen-activated protein kinase (MAPK) cascade or phosphoinositide 3-kinase (PI3K) pathway. Aberrant enhancement of EGFR signaling, which can be caused by overexpression, increase in gene copy number or ligand-independent mutated receptors, leads to cancer-driving processes such as augmented proliferation, angiogenesis, migration/invasion, and impaired apoptosis (138). Such activities, albeit strictly regulated, are especially crucial during ontogeny, including brain development and neurogenesis. Therefore, EGFR signaling has been investigated in the context of other neurogenic pathways such as Notch. Aguirre et al. presented evidence that EGFR and Notch have opposite effects on cells derived from subventricular zone (SVZ). Enhanced EGFR signaling caused an expansion of NPCs at the expense of self-renewing NSCs and downregulated Notch signaling via Numb (139). This antagonism ceased in the later stages of cell differentiation, as both EGFR and Notch have been shown to direct cells toward a glial fate (140, 141). Along this line, a synergy between the EGFR and Notch pathways in glioma was indicated by Purow et al. who showed that EGFR is under the transcriptional control of Notch signaling (Figure 2). Experiments involving silencing or overexpression of Notch and p53 indicate that this regulation was mediated by p53. Moreover, Notch1 and EGFR expression correlated in high-grade astrocytomas that were negative for the amplification of the EGFR gene (142).
Epidermal growth factor receptor stimulation leads to the activation of multiple signal mediators including the Ras family of small GTPases. Ras-transformed astrocytes have been shown to express higher levels of Notch1 as compared to their non-transformed counterparts and inhibition of Notch signaling reduced their aggressive phenotype. However, introduction of NICD did not lead to a transformation of the immortalized astrocytes on its own and did not enhance cell growth of Ras-transformed cells (88). The tumorigenic cooperation between Ras and Notch might occur at an earlier stage of cell development as the overexpression of NICD and K-Ras in glial progenitors induced periventricular lesions with stem-like characteristics in the SVZ (based on retention of proliferation and nestin expression). K-Ras and Notch acted in a synergistic manner in this model as the individual overexpression of each did not cause a comparable phenotype (119). Further evidence showed that both Ras and Akt induce Notch1 expression in a mouse glioma model (143).
Notch-Based Brain Tumor Therapies in Clinical Trials
γ-Secretase Inhibitors
Brain tumor-propagating cells have been shown to exhibit a higher resistance to chemo- and radiotherapy than bulk tumor cells (144, 145). However, they seem equally sensitive to treatments targeting stem cell pathways such as Notch (146, 147). Cleavage of Notch receptors and the formation of the NICD fragments is inhibited by GSIs in order to shut off Notch signaling (Figure 1). Several of them have been successfully applied in animals. The first GSI tested in a brain tumor model was DAPT. Treatment of mice harboring D283 medulloblastoma xenografts with DAPT resulted in decreased proliferation and increased apoptosis of tumor cells (148). The promising results of DAPT treatments prompted the development and testing of other inhibitors such as GSI-18 (149). This drug has been tested in two different subcutaneous brain tumor models. In the first setting, pretreatment of DAOY medulloblastoma cells hindered the growth of subcutaneous xenografts (147). The same group applied tumorspheres isolated from primary GBMs for subcutaneous implantation studies. Spheres pre-treated with GSI-18 did not form tumors anymore. These observations were confirmed in intracranial implantation studies in a relatively small cohort of animals. Mice treated with GSI-18 survived significantly longer post-engraftment as compared to the control group (146).
Chemotherapy is often based on a combination of drugs and thus GSIs have been tested together with already established anti-tumor agents. Ex vivo treatment with TMZ and DAPT lowered the tumorigenicity of U87NS and U373NS cells in subcutaneous mouse xenograft models. Moreover, administration of GSI (LY411, 575) and TMZ in vivo blocked tumor progression in four out of eight mice carrying U87NS xenografts (150). Administration of TMZ plus RO4929097 (Roche) was partially effective in mice intracranially injected with Hs683 cells. The authors identified the ECM protein EFEMP1 as a mediator of TMZ resistance and a target of the GSI. Further, the analysis of publicly available glioblastoma databases revealed that EFEMP1 expression correlated with the resistance to TMZ treatment and such could serve as a marker for the application of GSIs (151). GSIs (DAPT and L685, 458) were also shown to dramatically increase cell death of irradiated glioma BTPCs in vitro. This phenotype was reversed by the overexpression of NICD of Notch1 or Notch2, confirming that Notch is essentially involved in the survival of glioma BTPCs post irradiation. Furthermore, siRNA-mediated knock-down of Notch1 or Notch2 impaired tumor formation in xenografts but unfortunately, GSIs were not tested in this setting (152).
Several GSIs have been tested in clinical studies in various tumor types (Table 1). RO4929097 has been shown to extend the median survival rate in an intracranial mouse glioma model (153). This GSI has completed several phase I studies: applied alone (154), in combination with the VEGF tyrosine kinase inhibitor ceridanib (155) or in combination with gemcitabine (156). Additionally, a phase Ib trial tested the combination of RO4929097 and the mTOR inhibitor temsirolimus (157). All four studies showed that the drug is tolerable, and few patients showed a partial response. Although patients bearing a broad range of solid tumors were recruited, the drug was not tested on brain tumor patients. Currently, there are three ongoing trials directed against malignant glioma: a phase I study that combines the GSI RO4929097 with TMZ and radiation therapy (NCT01119599), a phase I/II study in which the drug is tested in combination with bevacizumab, a monoclonal anti-VEGF antibody (NCT01189240) and applied alone in a phase II trial (NCT01122901). The outcome of these treatments still awaits publication but the results of two different phase II cancer studies have been published. Unfortunately, none of 33 treated patients with colorectal metastatic cancer showed an objective radiographic response and the median progression-free survival was only 1.8 months in total (158). The outcome of a study on patients previously treated for metastatic pancreatic carcinoma did not meet its goals either. Consequently, the development of RO4929097 was discontinued by the sponsor (159).
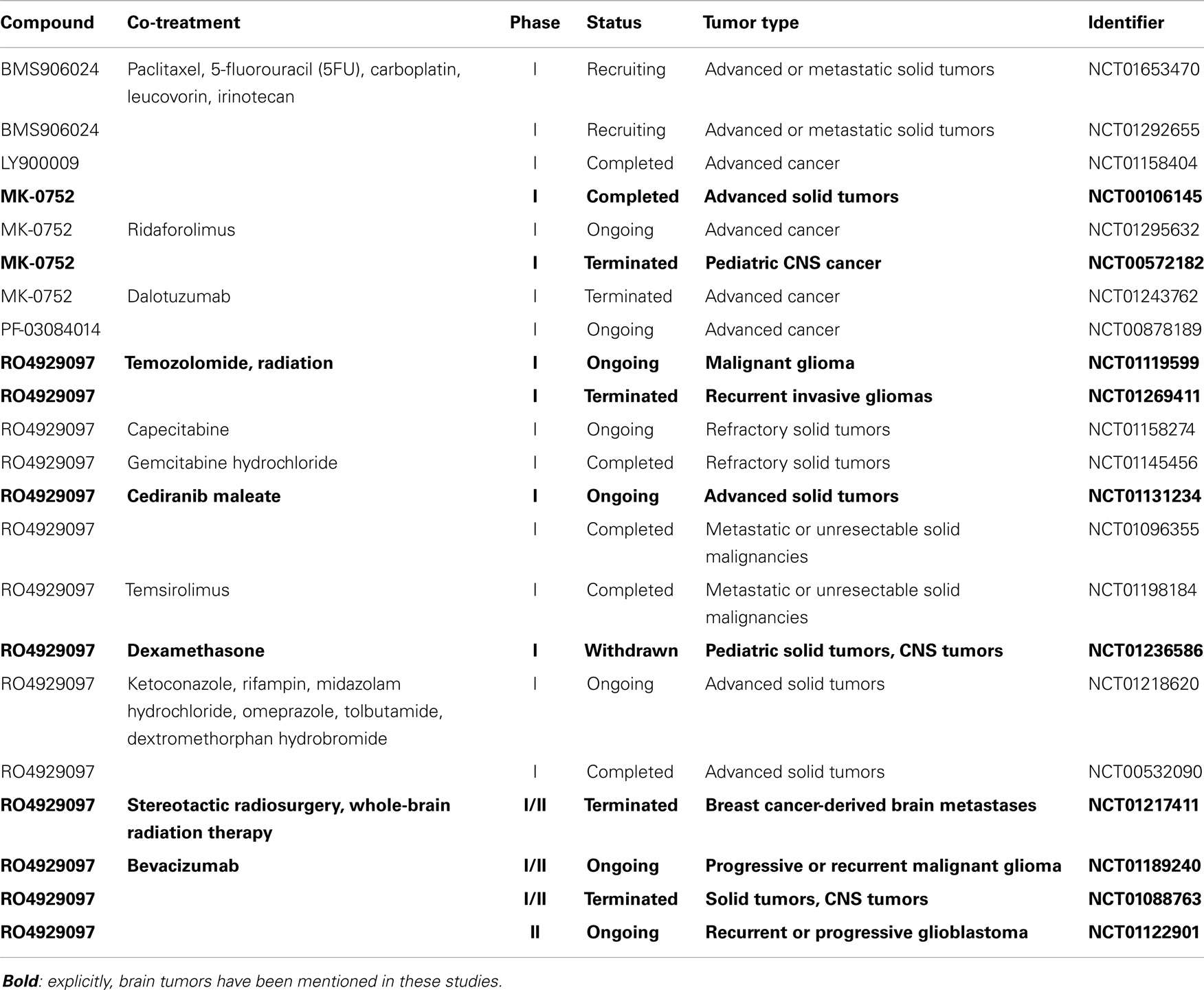
Table 1. Clinical trials involving GSIs and relevant for brain tumor therapy.
MK-0752 is a GSI designed by Merck and has been tested in several phase I studies. It was applied to a cohort of 103 patients with solid tumors. Weekly dosing was well tolerated and led to the modulation of the expression of nine Notch-related genes referred to as Notch gene signature. This study generated promising results as one patient with anaplastic astrocytoma had a complete remission lasting for more than 1 year in addition to a stable disease progression lasting for more than 4 months in 12 patients. All 12 patients were affected by brain malignancies and 10 of them were diagnosed with glioma (24% of all glioma patients in the study) (160). Further, MK-0752 was well tolerated by children with recurrent central nervous system malignancies (161). GBM patients were also included in an ongoing phase I trial involving the combination of MRK-0752 and ridaforolimus, a small-molecule mTOR inhibitor (NCT01295632). The last clinically tested GSI is PF-03084014 from Pfizer, currently in phase I for a wide range of solid and leukemic tumors (NCT00878189) and in the recruiting phase for several other trials but no results have been published so far.
It should be noted that administering GSIs raises several concerns. First of all, γ-secretases are involved in the cleavage of a multitude of proteins (162, 163). Furthermore, animal studies and phase I clinical trials have shown that systemic drug distribution leads to gastrointestinal toxicity, caused by the accumulation of secretory goblet cells in the intestine and due to the inactivation of Notch signaling (164–166). However, animal experiments indicate that these side-effects may be ameliorated by the application of glucocorticoids (167).
Blocking Antibodies
Several antibodies blocking Notch activity have been developed for the treatment of brain tumors due to their higher specificity as compared to GSI inhibitors. Furthermore, they can be used to block individual Notch receptors and ligands. Treatments using specific antibodies offer the advantage of fewer side-effects as compared to GSIs. Accordingly, agents neutralizing Notch1, Notch2, or Dll4 did not affect intestinal goblet cell differentiation in mice (168, 169).
Blocking antibodies targeting Notch receptors can be divided into two groups (Figure 1). Members of the first group are directed against the NRR block the receptor conformation that allows ADAM cleavage. Such antibodies have been raised against Notch1 (NRR1), Notch2 (NRR2), and Notch3 (NRR3) and have been tested in pre-clinical as well as in vitro studies (169–171). The other approach is to block Notch receptor–ligand interactions by hindering EGF repeats required for binding (170). The antibodies of this group are also effective against receptors carrying NRR mutations that destabilize the auto-inhibited receptor conformation and cause constitutive ligand-independent Notch signaling. Based on the success of pre-clinical studies, a humanized antibody targeting Notch1 (OMP-52M51) and Notch2/Notch3 (OMP-59R5; both OncoMed Pharmaceuticals) have entered a dose escalation phase I study in patients with solid tumors (NCT01778439, NCT01277146) followed by phase Ib/II trials in pancreatic and lung cancer patients in case of OMP-59R5 (Table 2).
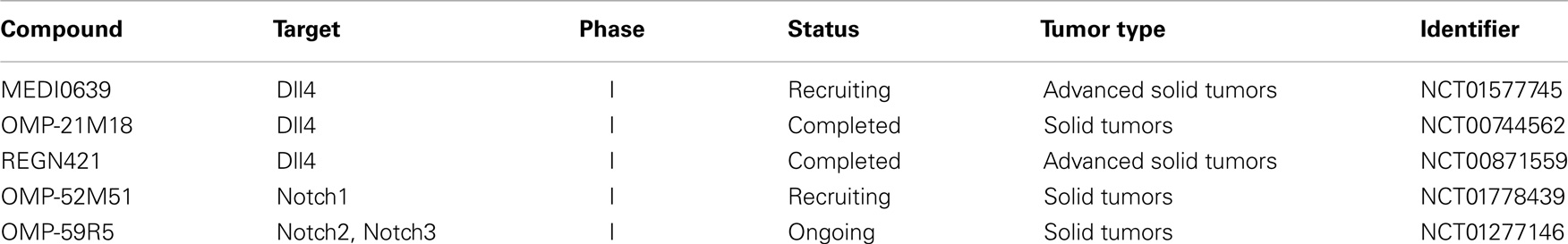
Table 2. Clinical trials involving Notch signaling-specific antibodies and relevant for brain tumor therapy.
Out of all Notch ligands, the blocking of Dll4 has been most thoroughly investigated. This protein plays an important role in angiogenesis and blocking the Dll4-Notch interaction (by overexpression of a soluble Dll4-Fc decoy peptide) enhanced vascular density and angiogenic sprouting in tumors derived from the rat glioma line C6. Surprisingly, this vasculature was rendered non-functional as subcutaneous xenografts of Dll4-Fc-treated cells grew smaller in nude mice. A similar response in C6 tumors was induced upon systemic delivery of Dll4-Fc using an adenoviral overexpression system. Further, the authors developed the first pharmacological model for anti-Dll4 treatment, i.e., using defined amounts of systematically delivered recombinant agent. Administration of recombinant Dll4-Fc or anti-Dll4 polyclonal antibody in an HT1080-RM (generated from a bevacizumab-resistant human fibrosarcoma) tumor model caused an increase in vessel density and smaller tumors volumes (172). These results were recapitulated by Li et al. in the human glioma cell line U87 subcutaneously implanted in nude mice. The authors showed that the overexpression of dominant negative soluble Dll4ECD-Fc enhanced the number of blood vessel and reduced tumor growth in vivo. Accordingly, the ectopic expression of Dll4 caused the opposite response. Moreover, examination of 20 surgical GBM specimens yielded an upregulation of Dll4 both in GBM tumor cells as well as endothelial cells (173). The same tumor model was applied to identify Dll4 as a mediator of tumor resistance to anti-VEGF therapy. According to data presented, the Dll4-mediated formation of larger vessels insensitive to anti-VEGF treatment was responsible for the resistance observed (174). Anti-Dll4 antibodies have already entered clinical trials (Table 2). The pharmacokinetics of three of them, MEDI0639 (175) (Astrazeneca, NCT01577745), OMP-21M18 (OncoMed Pharmaceuticals, NCT00744562), and REGN421 (Regeneron Pharmaceuticals, NCT00871559) have been tested in solid tumors in phase I studies but the results have not yet been published. However, it should be noted that side-effects of manipulating Dll4-induced Notch signaling have been reported in animal studies. One study showed that the treatment with anti-Dll4 antibody led to pathological changes in rat livers and the formation of vascular neoplasms, resembling hemangioblastomas (176). Another group reported chimeric Dll4 to affect hematopoiesis by inhibition of megakaryocyte differentiation (177).
Decoys
Soluble extracellular domains of Notch receptors and ligands offer an alternative to blocking antibodies (Figure 1). A Notch1 decoy has been shown to act as a pan-ligand antagonist, which can be applied to block Notch signaling in endothelial cells. Even though the decoy did not affect mouse breast cancer or human neuroblastoma cells in vitro, its overexpression in engrafted cells decreased tumor viability and disrupted vessel formation in vivo (178, 179). Soluble forms of Dll1 (180), Dll4 (172, 181), and Jagged1 (182) have also been used to block Notch signaling. Endogenous, soluble Notch antagonists like Dll1ECD (183) are either the product of metalloproteinases or are bona fide secreted proteins such as EGFL7, a novel non-canonical soluble Notch ligand with established neurovascular implications. The protein has been shown to act as an antagonist of Jagged1 and inhibited Notch activity in NSCs (34) and primary endothelial cells (184), thus offering a promising tool for the manipulation of glioma formation. In a genetically engineered mouse model of lung cancer, blocking ECM-associated EGFL7 with the antibody m18F7 (Genentech) enhanced progression-free and overall survival induced by anti-VEGF therapy (185). Moreover, silencing of EGFL7 by siRNA in vitro has been shown to inhibit the adherence of endothelial cells to a collagen-coated semipermeable membrane in a co-culture system with U251 glioma cells (186).
Other Agents
Certain natural compounds have been shown to affect the Notch signaling pathway and, even though their mode of action is not entirely known, they have been tested for therapeutic purposes. Curcumin, for example, has been shown to hinder proliferation and invasion of osteosarcoma cells in vitro by inhibiting Notch signaling (187). Several publications report enhanced apoptosis in glioma cell lines and decreased tumor growth in vivo. In most of these reports, the effect on Notch pathway activation has either not been mentioned at all (188) or has not been observed (189). Regardless of the mode of action a phase I study that measured curcumin bioavailability in glioblastoma patients has been completed (NCT01712542). Further natural compounds such as resveratrol and inducing apoptosis in glioma cells have been discussed elsewhere (190). The last group of compounds to be discussed here includes peptides affecting intracellular Notch signaling and small RNA molecules. A stapled peptide that mimics MAML1 binding to NICD–RBPJ has been shown to disrupt the ternary transcription complex and to pass through the cell membrane (191). Although RNA interference is still mostly a tool for basic research, it should be noted that siRNA targeting mutant KRAS in pancreatic cancer passed phase I (192) and entered phase II of clinical trials (NCT01676259).
Concluding Remarks
The improved understanding of genetics and molecular biology has increased the number of novel anti-glioblastoma therapies in clinical trials. The Notch signaling pathway is increasingly recognized as a central player in brain tumor formation beyond targeting the “usual suspects” such as EGFR and PI3K. One reason is that the oncogenicity of Notch can be connected to the maintenance of an undifferentiated cell state of brain tumor cells. Altering Notch signaling might lead to the differentiation of cancer cells, while targets such as EGFR or PI3K target cell proliferation only. In the former case, one might get rid of the tumor, while it just grows more slowly in the latter. Consequently, a combination of both strategies could be fruitful. The earliest efforts to modulate the Notch pathway focused on GSIs. Despite concerns regarding side-effects, several compounds have completed phase I of clinical studies and are awaiting further testing. The means to circumvent drawbacks of systemic administration of GSIs are still emerging. One possibility is the targeted delivery of a drug, e.g., via silica nanoparticles (193). Advances in recombinant protein technologies offer further possibilities to modulate Notch signaling, and consequently, reagents such as Notch blocking antibodies are entering clinical trials, while decoys have already been tested in animals. Their specificity might prove advantageous over small molecules and it should be noted that techniques previously considered inapplicable, such as siRNA technology, are now being tested in patients as well. Moreover, blocking Notch transcription factors with cell-permeable peptides offers a promising alternative for brain tumor therapy. The future of cancer treatment lies in personalized regimens and the combination of treatments. The means for molecular diagnostics are rapidly advancing and Notch-targeting drugs are the focus of various clinical trials; therefore, the future is bright for Notch-based brain tumor therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the German Research Foundation DFG via the collaborative research center 1080, project A3, and the grant SCHM 2159/2-1. We thank Darragh O’Neill for proofreading the manuscript.
References
1. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (2007) 114(2):97–109. doi: 10.1007/s00401-007-0278-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med (2008) 359(5):492–507. doi:10.1056/NEJMra0708126
3. Bralten LB, French PJ. Genetic alterations in glioma. Cancers (2011) 3(1):1129–40. doi:10.3390/cancers3011129
4. Yoshida J. Molecular neurosurgery using gene therapy to treat malignant glioma. Nagoya J Med Sci (1996) 59(3–4):97–105.
5. Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol (2006) 7(10):813–20. doi:10.1016/S1470-2045(06)70867-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352(10):987–96. doi:10.1056/NEJMoa043330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455(7216):1061–8. doi:10.1038/nature07385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321(5897):1807–12. doi:10.1126/science.1164382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Teodorczyk M, Martin-Villalba A. Sensing invasion: cell surface receptors driving spreading of glioblastoma. J Cell Physiol (2010) 222(1):1–10. doi:10.1002/jcp.21901
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Blaumueller CM, Qi H, Zagouras P, Artavanis-Tsakonas S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell (1997) 90(2):281–91. doi:10.1016/S0092-8674(00)80336-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Rebay I, Fleming RJ, Fehon RG, Cherbas L, Cherbas P, Artavanis-Tsakonas S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: implications for Notch as a multifunctional receptor. Cell (1991) 67(4):687–99. doi:10.1016/0092-8674(91)90064-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. del Amo FF, Gendron-Maguire M, Swiatek PJ, Jenkins NA, Copeland NG, Gridley T. Cloning, analysis, and chromosomal localization of Notch-1, a mouse homolog of Drosophila Notch. Genomics (1993) 15(2):259–64. doi:10.1006/geno.1993.1055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Weinmaster G, Roberts VJ, Lemke G. Notch2: a second mammalian Notch gene. Development (1992) 116(4):931–41.
14. Lardelli M, Dahlstrand J, Lendahl U. The novel Notch homologue mouse Notch 3 lacks specific epidermal growth factor-repeats and is expressed in proliferating neuroepithelium. Mech Dev (1994) 46(2):123–36. doi:10.1016/0925-4773(94)90081-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Uyttendaele H, Marazzi G, Wu G, Yan Q, Sassoon D, Kitajewski J. Notch4/int-3, a mammary proto-oncogene, is an endothelial cell-specific mammalian Notch gene. Development (1996) 122(7):2251–9.
16. Aster JC, Simms WB, Zavala-Ruiz Z, Patriub V, North CL, Blacklow SC. The folding and structural integrity of the first LIN-12 module of human Notch1 are calcium-dependent. Biochemistry (1999) 38(15):4736–42. doi:10.1021/bi982713o
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Sanchez-Irizarry C, Carpenter AC, Weng AP, Pear WS, Aster JC, Blacklow SC. Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol Cell Biol (2004) 24(21):9265–73. doi:10.1128/MCB.24.21.9265-9273.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Gordon WR, Vardar-Ulu D, Histen G, Sanchez-Irizarry C, Aster JC, Blacklow SC. Structural basis for autoinhibition of Notch. Nat Struct Mol Biol (2007) 14(4):295–300. doi:10.1038/nsmb1227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, et al. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell (2000) 5(2):197–206. doi:10.1016/S1097-2765(00)80416-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Tamura K, Taniguchi Y, Minoguchi S, Sakai T, Tun T, Furukawa T, et al. Physical interaction between a novel domain of the receptor Notch and the transcription factor RBP-J kappa/Su(H). Curr Biol (1995) 5(12):1416–23. doi:10.1016/S0960-9822(95)00279-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Lubman OY, Korolev SV, Kopan R. Anchoring notch genetics and biochemistry; structural analysis of the ankyrin domain sheds light on existing data. Mol Cell (2004) 13(5):619–26. doi:10.1016/S1097-2765(04)00120-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Lieber T, Kidd S, Alcamo E, Corbin V, Young MW. Antineurogenic phenotypes induced by truncated Notch proteins indicate a role in signal transduction and may point to a novel function for Notch in nuclei. Genes Dev (1993) 7(10):1949–65. doi:10.1101/gad.7.10.1949
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Kurooka H, Kuroda K, Honjo T. Roles of the ankyrin repeats and C-terminal region of the mouse notch1 intracellular region. Nucleic Acids Res (1998) 26(23):5448–55. doi:10.1093/nar/26.23.5448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Rechsteiner M. Regulation of enzyme levels by proteolysis: the role of pest regions. Adv Enzyme Regul (1988) 27:135–51. doi:10.1016/0065-2571(88)90014-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Bettenhausen B, Hrabe de Angelis M, Simon D, Guenet JL, Gossler A. Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development (1995) 121(8):2407–18.
26. Dunwoodie SL, Henrique D, Harrison SM, Beddington RS. Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development (1997) 124(16):3065–76.
27. Shutter JR, Scully S, Fan W, Richards WG, Kitajewski J, Deblandre GA, et al. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes Dev (2000) 14(11):1313–8. doi:10.1101/gad.14.11.1313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Lindsell CE, Shawber CJ, Boulter J, Weinmaster G. Jagged: a mammalian ligand that activates Notch1. Cell (1995) 80(6):909–17. doi:10.1016/0092-8674(95)90294-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Shawber C, Boulter J, Lindsell CE, Weinmaster G. Jagged2: a serrate-like gene expressed during rat embryogenesis. Dev Biol (1996) 180(1):370–6. doi:10.1006/dbio.1996.0310
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Parks AL, Stout JR, Shepard SB, Klueg KM, Dos Santos AA, Parody TR, et al. Structure-function analysis of delta trafficking, receptor binding and signaling in Drosophila. Genetics (2006) 174(4):1947–61. doi:10.1534/genetics.106.061630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Shimizu K, Chiba S, Kumano K, Hosoya N, Takahashi T, Kanda Y, et al. Mouse jagged1 physically interacts with notch2 and other notch receptors. Assessment by quantitative methods. J Biol Chem (1999) 274(46):32961–9. doi:10.1074/jbc.274.46.32961
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hu QD, Ang BT, Karsak M, Hu WP, Cui XY, Duka T, et al. F3/contactin acts as a functional ligand for Notch during oligodendrocyte maturation. Cell (2003) 115(2):163–75. doi:10.1016/S0092-8674(03)00810-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. D’Souza B, Meloty-Kapella L, Weinmaster G. Canonical and non-canonical Notch ligands. Curr Top Dev Biol (2010) 92:73–129. doi:10.1016/S0070-2153(10)92003-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Schmidt MHH, Bicker F, Nikolic I, Meister J, Babuke T, Picuric S, et al. Epidermal growth factor-like domain 7 (EGFL7) modulates Notch signalling and affects neural stem cell renewal. Nat Cell Biol (2009) 11(7):873–80. doi:10.1038/ncb1896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Meister J, Schmidt MHH. miR-126 and miR-126*: new players in cancer. ScientificWorldJournal (2010) 10:2090–100. doi:10.1100/tsw.2010.198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Dikic I, Schmidt MHH. Notch: implications of endogenous inhibitors for therapy. Bioessays (2010) 32(6):481–7. doi:10.1002/bies.200900140
37. de Celis JF, Bray S. Feed-back mechanisms affecting Notch activation at the dorsoventral boundary in the Drosophila wing. Development (1997) 124(17):3241–51.
38. Li Y, Baker NE. The roles of cis-inactivation by Notch ligands and of neuralized during eye and bristle patterning in Drosophila. BMC Dev Biol (2004) 4:5. doi:10.1186/1471-213X-4-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Miller AC, Lyons EL, Herman TG. Cis-inhibition of Notch by endogenous Delta biases the outcome of lateral inhibition. Curr Biol (2009) 19(16):1378–83. doi:10.1016/j.cub.2009.06.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Cordle J, Johnson S, Tay JZ, Roversi P, Wilkin MB, de Madrid BH, et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat Struct Mol Biol (2008) 15(8):849–57. doi:10.1038/nsmb.1457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Zolkiewska A. ADAM proteases: ligand processing and modulation of the Notch pathway. Cell Mol Life Sci (2008) 65(13):2056–68. doi:10.1007/s00018-008-7586-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Glomski K, Monette S, Manova K, De Strooper B, Saftig P, Blobel CP. Deletion of Adam10 in endothelial cells leads to defects in organ-specific vascular structures. Blood (2011) 118(4):1163–74. doi:10.1182/blood-2011-04-348557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Weber S, Niessen MT, Prox J, Lullmann-Rauch R, Schmitz A, Schwanbeck R, et al. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development (2011) 138(3):495–505. doi:10.1242/dev.055210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol (2009) 29(21):5679–95. doi:10.1128/MCB.00406-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development (2012) 139(20):3693–709. doi:10.1242/dev.076398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature (1999) 398(6727):518–22. doi:10.1038/19083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Wu L, Aster JC, Blacklow SC, Lake R, Artavanis-Tsakonas S, Griffin JD. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat Genet (2000) 26(4):484–9. doi:10.1038/82644
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Wallberg AE, Neely KE, Hassan AH, Gustafsson JA, Workman JL, Wright AP. Recruitment of the SWI-SNF chromatin remodeling complex as a mechanism of gene activation by the glucocorticoid receptor tau1 activation domain. Mol Cell Biol (2000) 20(6):2004–13. doi:10.1128/MCB.20.6.2004-2013.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Kurooka H, Honjo T. Functional interaction between the mouse notch1 intracellular region and histone acetyltransferases PCAF and GCN5. J Biol Chem (2000) 275(22):17211–20. doi:10.1074/jbc.M000909200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Iso T, Sartorelli V, Chung G, Shichinohe T, Kedes L, Hamamori Y. HERP, a new primary target of Notch regulated by ligand binding. Mol Cell Biol (2001) 21(17):6071–9. doi:10.1128/MCB.21.17.6071-6079.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Iso T, Sartorelli V, Poizat C, Iezzi S, Wu HY, Chung G, et al. HERP, a novel heterodimer partner of HES/E(spl) in Notch signaling. Mol Cell Biol (2001) 21(17):6080–9. doi:10.1128/MCB.21.17.6080-6089.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature (1995) 377(6547):355–8. doi:10.1038/377355a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Fischer A, Gessler M. Delta-Notch – and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res (2007) 35(14):4583–96. doi:10.1093/nar/gkm477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Kageyama R, Ohtsuka T. The Notch-Hes pathway in mammalian neural development. Cell Res (1999) 9(3):179–88. doi:10.1038/sj.cr.7290016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol (2003) 194(3):237–55. doi:10.1002/jcp.10208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Cheng P, Zlobin A, Volgina V, Gottipati S, Osborne B, Simel EJ, et al. Notch-1 regulates NF-kappaB activity in hemopoietic progenitor cells. J Immunol (2001) 167(8):4458–67. doi:10.4049/jimmunol.167.8.4458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Oswald F, Liptay S, Adler G, Schmid RM. NF-kappaB2 is a putative target gene of activated Notch-1 via RBP-Jkappa. Mol Cell Biol (1998) 18(4):2077–88.
58. Nickoloff BJ, Qin JZ, Chaturvedi V, Denning MF, Bonish B, Miele L. Jagged-1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF-kappaB and PPARgamma. Cell Death Differ (2002) 9(8):842–55. doi:10.1038/sj.cdd.4401036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A (2006) 103(48):18261–6. doi:10.1073/pnas.0606108103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L, et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol (2006) 26(21):8022–31. doi:10.1128/MCB.01091-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C, et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev (2006) 20(15):2096–109. doi:10.1101/gad.1450406
62. Ehm O, Goritz C, Covic M, Schaffner I, Schwarz TJ, Karaca E, et al. RBPJkappa-dependent signaling is essential for long-term maintenance of neural stem cells in the adult hippocampus. J Neurosci (2010) 30(41):13794–807. doi:10.1523/JNEUROSCI.1567-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Kumar JP, Moses K. EGF receptor and Notch signaling act upstream of Eyeless/Pax6 to control eye specification. Cell (2001) 104(5):687–97. doi:10.1016/S0092-8674(01)00265-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Mol Cell Biol (2001) 21(17):5925–34. doi:10.1128/MCB.21.17.5925-5934.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Rangarajan A, Talora C, Okuyama R, Nicolas M, Mammucari C, Oh H, et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J (2001) 20(13):3427–36. doi:10.1093/emboj/20.13.3427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Beatus P, Lendahl U. Notch and neurogenesis. J Neurosci Res (1998) 54(2):125–36. doi:10.1002/(SICI)1097-4547(19981015)54:2<125::AID-JNR1>3.0.CO;2-G
67. Lasky JL, Wu H. Notch signaling, brain development, and human disease. Pediatr Res (2005) 57(5 Pt 2):104R–9R. doi:10.1203/01.PDR.0000159632.70510.3D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science (1999) 284(5415):770–6. doi:10.1126/science.284.5415.770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol (2006) 7(9):678–89. doi:10.1038/nrm2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Gaiano N, Fishell G. The role of notch in promoting glial and neural stem cell fates. Ann Rev Neurosci (2002) 25:471–90. doi:10.1146/annurev.neuro.25.030702.130823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Wang S, Barres BA. Up a notch: instructing gliogenesis. Neuron (2000) 27(2):197–200. doi:10.1016/S0896-6273(00)00028-3
72. Bicker F, Schmidt MHH. EGFL7: a new player in homeostasis of the nervous system. Cell Cycle (2010) 9(7):1263–9. doi:10.4161/cc.9.7.11091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. de la Pompa JL, Wakeham A, Correia KM, Samper E, Brown S, Aguilera RJ, et al. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development (1997) 124(6):1139–48.
74. Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev (1994) 8(6):707–19. doi:10.1101/gad.8.6.707
75. Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD, et al. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell (1991) 66(4):649–61. doi:10.1016/0092-8674(91)90111-B
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell (2014) 25(3):318–34. doi:10.1016/j.ccr.2014.02.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D, et al. Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res (2004) 64(21):7787–93. doi:10.1158/0008-5472.CAN-04-1446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Sivasankaran B, Degen M, Ghaffari A, Hegi ME, Hamou MF, Ionescu MC, et al. Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res (2009) 69(2):458–65. doi:10.1158/0008-5472.CAN-08-2610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Cheung HC, Corley LJ, Fuller GN, McCutcheon IE, Cote GJ. Polypyrimidine tract binding protein and Notch1 are independently re-expressed in glioma. Mod Pathol (2006) 19(8):1034–41. doi:10.1038/modpathol.3800635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Purow BW, Haque RM, Noel MW, Su Q, Burdick MJ, Lee J, et al. Expression of Notch-1 and its ligands, Delta-like-1 and Jagged-1, is critical for glioma cell survival and proliferation. Cancer Res (2005) 65(6):2353–63. doi:10.1158/0008-5472.CAN-04-1890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell (2006) 9(3):157–73. doi:10.1016/j.ccr.2006.02.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Li J, Cui Y, Gao G, Zhao Z, Zhang H, Wang X. Notch1 is an independent prognostic factor for patients with glioma. J Surg Oncol (2011) 103(8):813–7. doi:10.1002/jso.21851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Xu P, Yu S, Jiang R, Kang C, Wang G, Jiang H, et al. Differential expression of Notch family members in astrocytomas and medulloblastomas. Pathol Oncol Res (2009) 15(4):703–10. doi:10.1007/s12253-009-9173-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Boulay JL, Miserez AR, Zweifel C, Sivasankaran B, Kana V, Ghaffari A, et al. Loss of NOTCH2 positively predicts survival in subgroups of human glial brain tumors. PLoS One (2007) 2(6):e576. doi:10.1371/journal.pone.0000576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, et al. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell (2006) 11(6):831–44. doi:10.1016/j.devcel.2006.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Somasundaram K, Reddy SP, Vinnakota K, Britto R, Subbarayan M, Nambiar S, et al. Upregulation of ASCL1 and inhibition of Notch signaling pathway characterize progressive astrocytoma. Oncogene (2005) 24(47):7073–83. doi:10.1038/sj.onc.1208865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell (2010) 17(1):98–110. doi:10.1016/j.ccr.2009.12.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Kanamori M, Kawaguchi T, Nigro JM, Feuerstein BG, Berger MS, Miele L, et al. Contribution of Notch signaling activation to human glioblastoma multiforme. J Neurosurg (2007) 106(3):417–27. doi:10.3171/jns.2007.106.3.417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Zhang XP, Zheng G, Zou L, Liu HL, Hou LH, Zhou P, et al. Notch activation promotes cell proliferation and the formation of neural stem cell-like colonies in human glioma cells. Mol Cell Biochem (2008) 307(1–2):101–8. doi:10.1007/s11010-007-9589-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Koutelou E, Sato S, Tomomori-Sato C, Florens L, Swanson SK, Washburn MP, et al. Neuralized-like 1 (Neurl1) targeted to the plasma membrane by N-myristoylation regulates the Notch ligand Jagged1. J Biol Chem (2008) 283(7):3846–53. doi:10.1074/jbc.M706974200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Nakamura H, Yoshida M, Tsuiki H, Ito K, Ueno M, Nakao M, et al. Identification of a human homolog of the Drosophila neuralized gene within the 10q25.1 malignant astrocytoma deletion region. Oncogene (1998) 16(8):1009–19. doi:10.1038/sj.onc.1201618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Teider N, Scott DK, Neiss A, Weeraratne SD, Amani VM, Wang Y, et al. Neuralized1 causes apoptosis and downregulates Notch target genes in medulloblastoma. Neuro Oncol (2010) 12(12):1244–56. doi:10.1093/neuonc/noq091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. McGill MA, McGlade CJ. Mammalian numb proteins promote Notch1 receptor ubiquitination and degradation of the Notch1 intracellular domain. J Biol Chem (2003) 278(25):23196–203. doi:10.1074/jbc.M302827200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Qiu L, Joazeiro C, Fang N, Wang HY, Elly C, Altman Y, et al. Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J Biol Chem (2000) 275(46):35734–7. doi:10.1074/jbc.M007300200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Nie J, McGill MA, Dermer M, Dho SE, Wolting CD, McGlade CJ. LNX functions as a RING type E3 ubiquitin ligase that targets the cell fate determinant Numb for ubiquitin-dependent degradation. EMBO J (2002) 21(1–2):93–102. doi:10.1093/emboj/21.1.93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Chen J, Xu J, Zhao W, Hu G, Cheng H, Kang Y, et al. Characterization of human LNX, a novel ligand of Numb protein X that is downregulated in human gliomas. Int J Biochem Cell Biol (2005) 37(11):2273–83. doi:10.1016/j.biocel.2005.02.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Hulleman E, Quarto M, Vernell R, Masserdotti G, Colli E, Kros JM, et al. A role for the transcription factor HEY1 in glioblastoma. J Cell Mol Med (2009) 13(1):136–46. doi:10.1111/j.1582-4934.2008.00307.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Gaetani P, Hulleman E, Levi D, Quarto M, Scorsetti M, Helins K, et al. Expression of the transcription factor HEY1 in glioblastoma: a preliminary clinical study. Tumori (2010) 96(1):97–102. doi:10.1700/479.5658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Yin D, Xie D, Sakajiri S, Miller CW, Zhu H, Popoviciu ML, et al. DLK1: increased expression in gliomas and associated with oncogenic activities. Oncogene (2006) 25(13):1852–61. doi:10.1038/sj.onc.1209219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Xu P, Zhang A, Jiang R, Qiu M, Kang C, Jia Z, et al. The different role of Notch1 and Notch2 in astrocytic gliomas. PLoS One (2013) 8(1):e53654. doi:10.1371/journal.pone.0053654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Leins A, Riva P, Lindstedt R, Davidoff MS, Mehraein P, Weis S. Expression of tenascin-C in various human brain tumors and its relevance for survival in patients with astrocytoma. Cancer (2003) 98(11):2430–9. doi:10.1002/cncr.11796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, et al. Emerging insights into the molecular and cellular basis of glioblastoma. Genes Dev (2012) 26(8):756–84. doi:10.1101/gad.187922.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell (2002) 1(3):269–77. doi:10.1016/S1535-6108(02)00046-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature (2008) 455(7216):1129–33. doi:10.1038/nature07443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell (2005) 8(2):119–30. doi:10.1016/j.ccr.2005.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell (2011) 146(2):209–21. doi:10.1016/j.cell.2011.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, et al. Non-stem cell origin for oligodendroglioma. Cancer Cell (2010) 18(6):669–82. doi:10.1016/j.ccr.2010.10.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev (2001) 15(15):1913–25. doi:10.1101/gad.903001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells – perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res (2006) 66(19):9339–44. doi:10.1158/0008-5472.CAN-06-3126
110. Jordan CT. Cancer stem cells: controversial or just misunderstood? Cell Stem Cell (2009) 4(3):203–5. doi:10.1016/j.stem.2009.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Li Y, Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res (2012) 72(3):576–80. doi:10.1158/0008-5472.CAN-11-3070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature (2004) 432(7015):396–401. doi:10.1038/nature03128
113. Bayin NS, Modrek AS, Placantonakis DG. Glioblastoma stem cells: molecular characteristics and therapeutic implications. World J Stem Cells (2014) 6(2):230–8. doi:10.4252/wjsc.v6.i2.230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Christensen K, Schroder HD, Kristensen BW. CD133+ niches and single cells in glioblastoma have different phenotypes. J Neurooncol (2011) 104(1):129–43. doi:10.1007/s11060-010-0488-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Kelly PN, Dakic A, Adams JM, Nutt SL, Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science (2007) 317(5836):337. doi:10.1126/science.1142596
116. Pfenninger CV, Roschupkina T, Hertwig F, Kottwitz D, Englund E, Bengzon J, et al. CD133 is not present on neurogenic astrocytes in the adult subventricular zone, but on embryonic neural stem cells, ependymal cells, and glioblastoma cells. Cancer Res (2007) 67(12):5727–36. doi:10.1158/0008-5472.CAN-07-0183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Christensen K, Schroder HD, Kristensen BW. CD133 identifies perivascular niches in grade II-IV astrocytomas. J Neurooncol (2008) 90(2):157–70. doi:10.1007/s11060-008-9648-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell (2012) 149(1):36–47. doi:10.1016/j.cell.2012.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Shih AH, Holland EC. Notch signaling enhances nestin expression in gliomas. Neoplasia (2006) 8(12):1072–82. doi:10.1593/neo.06526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell (2007) 11(1):69–82. doi:10.1016/j.ccr.2006.11.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Dahlrot RH, Hermansen SK, Hansen S, Kristensen BW. What is the clinical value of cancer stem cell markers in gliomas? Int J Clin Exp Pathol (2013) 6(3):334–48.
122. Hitoshi S, Alexson T, Tropepe V, Donoviel D, Elia AJ, Nye JS, et al. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev (2002) 16(7):846–58. doi:10.1101/gad.975202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C, Weinmaster G, et al. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell (2000) 101(5):499–510. doi:10.1016/S0092-8674(00)80860-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron (2001) 31(4):557–68. doi:10.1016/S0896-6273(01)00395-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell (2006) 9(5):391–403. doi:10.1016/j.ccr.2006.03.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, et al. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene (2008) 27(20):2897–909. doi:10.1038/sj.onc.1210949
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Guichet PO, Guelfi S, Teigell M, Hoppe L, Bakalara N, Bauchet L, et al. Notch1 stimulation induces a vascularization switch with pericyte-like cell differentiation of glioblastoma stem cells. Stem Cells (2014). doi:10.1002/stem.1767
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Jiang X, Xing H, Kim TM, Jung Y, Huang W, Yang HW, et al. Numb regulates glioma stem cell fate and growth by altering epidermal growth factor receptor and Skp1-Cullin-F-box ubiquitin ligase activity. Stem Cells (2012) 30(7):1313–26. doi:10.1002/stem.1120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Kargiotis O, Rao JS, Kyritsis AP. Mechanisms of angiogenesis in gliomas. J Neurooncol (2006) 78(3):281–93. doi:10.1007/s11060-005-9097-6
130. Poellinger L, Lendahl U. Modulating Notch signaling by pathway-intrinsic and pathway-extrinsic mechanisms. Curr Opin Genet Dev (2008) 18(5):449–54. doi:10.1016/j.gde.2008.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Bar EE, Lin A, Mahairaki V, Matsui W, Eberhart CG. Hypoxia increases the expression of stem-cell markers and promotes clonogenicity in glioblastoma neurospheres. Am J Pathol (2010) 177(3):1491–502. doi:10.2353/ajpath.2010.091021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell (2005) 9(5):617–28. doi:10.1016/j.devcel.2005.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Pistollato F, Rampazzo E, Persano L, Abbadi S, Frasson C, Denaro L, et al. Interaction of hypoxia-inducible factor-1alpha and Notch signaling regulates medulloblastoma precursor proliferation and fate. Stem Cells (2010) 28(11):1918–29. doi:10.1002/stem.518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Qiang L, Wu T, Zhang HW, Lu N, Hu R, Wang YJ, et al. HIF-1alpha is critical for hypoxia-mediated maintenance of glioblastoma stem cells by activating Notch signaling pathway. Cell Death Differ (2012) 19(2):284–94. doi:10.1038/cdd.2011.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Hu YY, Fu LA, Li SZ, Chen Y, Li JC, Han J, et al. Hif-1alpha and Hif-2alpha differentially regulate Notch signaling through competitive interaction with the intracellular domain of Notch receptors in glioma stem cells. Cancer Lett (2014) 349(1):67–76. doi:10.1016/j.canlet.2014.03.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y, et al. PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol (1998) 57(7):684–9. doi:10.1097/00005072-199807000-00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Wong AJ, Bigner SH, Bigner DD, Kinzler KW, Hamilton SR, Vogelstein B. Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A (1987) 84(19):6899–903. doi:10.1073/pnas.84.19.6899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Taylor TE, Furnari FB, Cavenee WK. Targeting EGFR for treatment of glioblastoma: molecular basis to overcome resistance. Curr Cancer Drug Targets (2012) 12(3):197–209. doi:10.2174/156800912799277557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal. Nature (2010) 467(7313):323–7. doi:10.1038/nature09347
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH. Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J Neurosci (1997) 17(15):5820–9.
141. Ayuso-Sacido A, Moliterno JA, Kratovac S, Kapoor GS, O’Rourke DM, Holland EC, et al. Activated EGFR signaling increases proliferation, survival, and migration and blocks neuronal differentiation in post-natal neural stem cells. J Neurooncol (2010) 97(3):323–37. doi:10.1007/s11060-009-0035-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Purow BW, Sundaresan TK, Burdick MJ, Kefas BA, Comeau LD, Hawkinson MP, et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis (2008) 29(5):918–25. doi:10.1093/carcin/bgn079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, Holland EC. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell (2003) 12(4):889–901. doi:10.1016/S1097-2765(03)00395-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444(7120):756–60. doi:10.1038/nature05236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer (2006) 5:67. doi:10.1186/1476-4598-5-67
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells (2010) 28(1):5–16. doi:10.1002/stem.254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, et al. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res (2006) 66(15):7445–52. doi:10.1158/0008-5472.CAN-06-0858
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res (2004) 64(21):7794–800. doi:10.1158/0008-5472.CAN-04-1813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Lewis SJ, Smith AL, Neduvelil JG, Stevenson GI, Lindon MJ, Jones AB, et al. A novel series of potent gamma-secretase inhibitors based on a benzobicyclo[4.2.1]nonane core. Bioorg Med Chem Lett (2005) 15(2):373–8. doi:10.1016/j.bmcl.2004.10.062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Gilbert CA, Daou MC, Moser RP, Ross AH. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res (2010) 70(17):6870–9. doi:10.1158/0008-5472.CAN-10-1378
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Hiddingh L, Tannous BA, Teng J, Tops B, Jeuken J, Hulleman E, et al. EFEMP1 induces gamma-secretase/Notch-mediated temozolomide resistance in glioblastoma. Oncotarget (2014) 5(2):363–74.
152. Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells (2010) 28(1):17–28. doi:10.1002/stem.261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Saito N, Fu J, Zheng S, Yao J, Wang S, Liu DD, et al. A high Notch pathway activation predicts response to gamma secretase inhibitors in proneural subtype of glioma tumor-initiating cells. Stem Cells (2014) 32(1):301–12. doi:10.1002/stem.1528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol (2012) 30(19):2348–53. doi:10.1200/JCO.2011.36.8282
155. Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G, et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer (2013) 109(4):943–9. doi:10.1038/bjc.2013.380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Richter S, Bedard PL, Chen EX, Clarke BA, Tran B, Hotte SJ, et al. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest New Drugs (2014) 32(2):243–9. doi:10.1007/s10637-013-9965-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Diaz-Padilla I, Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M, et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs (2013) 31(5):1182–91. doi:10.1007/s10637-013-0001-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Strosberg JR, Yeatman T, Weber J, Coppola D, Schell MJ, Han G, et al. A phase II study of RO4929097 in metastatic colorectal cancer. Eur J Cancer (2012) 48(7):997–1003. doi:10.1016/j.ejca.2012.02.056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A, et al. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs (2014) 32(4):739–45. doi:10.1007/s10637-014-0083-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J Clin Oncol (2012) 30(19):2307–13. doi:10.1200/JCO.2011.39.1540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol (2011) 29(26):3529–34. doi:10.1200/JCO.2011.35.7806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Beel AJ, Sanders CR. Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci (2008) 65(9):1311–34. doi:10.1007/s00018-008-7462-2
163. Hemming ML, Elias JE, Gygi SP, Selkoe DJ. Proteomic profiling of gamma-secretase substrates and mapping of substrate requirements. PLoS Biol (2008) 6(10):e257. doi:10.1371/journal.pbio.0060257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem (2004) 279(13):12876–82. doi:10.1074/jbc.M311652200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Imbimbo BP. Therapeutic potential of gamma-secretase inhibitors and modulators. Curr Top Med Chem (2008) 8(1):54–61. doi:10.2174/156802608783334015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Milano J, McKay J, Dagenais C, Foster-Brown L, Pognan F, Gadient R, et al. Modulation of notch processing by gamma-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol Sci (2004) 82(1):341–58. doi:10.1093/toxsci/kfh254
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Real PJ, Tosello V, Palomero T, Castillo M, Hernando E, de Stanchina E, et al. Gamma-secretase inhibitors reverse glucocorticoid resistance in T cell acute lymphoblastic leukemia. Nat Med (2009) 15(1):50–8. doi:10.1038/nm.1900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature (2006) 444(7122):1083–7. doi:10.1038/nature05313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, et al. Therapeutic antibody targeting of individual Notch receptors. Nature (2010) 464(7291):1052–7. doi:10.1038/nature08878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Aste-Amezaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, et al. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One (2010) 5(2):e9094. doi:10.1371/journal.pone.0009094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Li K, Li Y, Wu W, Gordon WR, Chang DW, Lu M, et al. Modulation of Notch signaling by antibodies specific for the extracellular negative regulatory region of NOTCH3. J Biol Chem (2008) 283(12):8046–54. doi:10.1074/jbc.M800170200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, et al. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature (2006) 444(7122):1032–7. doi:10.1038/nature05355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Li JL, Sainson RC, Shi W, Leek R, Harrington LS, Preusser M, et al. Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res (2007) 67(23):11244–53. doi:10.1158/0008-5472.CAN-07-0969
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Li JL, Sainson RC, Oon CE, Turley H, Leek R, Sheldon H, et al. DLL4-Notch signaling mediates tumor resistance to anti-VEGF therapy in vivo. Cancer Res (2011) 71(18):6073–83. doi:10.1158/0008-5472.CAN-11-1704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Jenkins DW, Ross S, Veldman-Jones M, Foltz IN, Clavette BC, Manchulenko K, et al. MEDI0639: a novel therapeutic antibody targeting Dll4 modulates endothelial cell function and angiogenesis in vivo. Mol Cancer Ther (2012) 11(8):1650–60. doi:10.1158/1535-7163.MCT-11-1027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Yan M, Callahan CA, Beyer JC, Allamneni KP, Zhang G, Ridgway JB, et al. Chronic DLL4 blockade induces vascular neoplasms. Nature (2010) 463(7282):E6–7. doi:10.1038/nature08751
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Poirault-Chassac S, Six E, Catelain C, Lavergne M, Villeval JL, Vainchenker W, et al. Notch/Delta4 signaling inhibits human megakaryocytic terminal differentiation. Blood (2010) 116(25):5670–8. doi:10.1182/blood-2010-05-285957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Funahashi Y, Hernandez SL, Das I, Ahn A, Huang J, Vorontchikhina M, et al. A notch1 ectodomain construct inhibits endothelial notch signaling, tumor growth, and angiogenesis. Cancer Res (2008) 68(12):4727–35. doi:10.1158/0008-5472.CAN-07-6499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Hernandez SL, Banerjee D, Garcia A, Kangsamaksin T, Cheng WY, Anastassiou D, et al. Notch and VEGF pathways play distinct but complementary roles in tumor angiogenesis. Vasc Cell (2013) 5(1):17. doi:10.1186/2045-824X-5-17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Varnum-Finney B, Wu L, Yu M, Brashem-Stein C, Staats S, Flowers D, et al. Immobilization of Notch ligand, Delta-1, is required for induction of notch signaling. J Cell Sci (2000) 113(Pt 23):4313–8.
181. Scehnet JS, Jiang W, Kumar SR, Krasnoperov V, Trindade A, Benedito R, et al. Inhibition of Dll4-mediated signaling induces proliferation of immature vessels and results in poor tissue perfusion. Blood (2007) 109(11):4753–60. doi:10.1182/blood-2006-12-063933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Small D, Kovalenko D, Kacer D, Liaw L, Landriscina M, Di Serio C, et al. Soluble Jagged 1 represses the function of its transmembrane form to induce the formation of the Src-dependent chord-like phenotype. J Biol Chem (2001) 276(34):32022–30. doi:10.1074/jbc.M100933200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Six E, Ndiaye D, Laabi Y, Brou C, Gupta-Rossi N, Israel A, et al. The Notch ligand Delta1 is sequentially cleaved by an ADAM protease and gamma-secretase. Proc Natl Acad Sci U S A (2003) 100(13):7638–43. doi:10.1073/pnas.1230693100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Nichol D, Shawber C, Fitch MJ, Bambino K, Sharma A, Kitajewski J, et al. Impaired angiogenesis and altered Notch signaling in mice overexpressing endothelial Egfl7. Blood (2010) 116(26):6133–43. doi:10.1182/blood-2010-03-274860
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Johnson L, Huseni M, Smyczek T, Lima A, Yeung S, Cheng JH, et al. Anti-EGFL7 antibodies enhance stress-induced endothelial cell death and anti-VEGF efficacy. J Clin Invest (2013) 123(9):3997–4009. doi:10.1172/JCI67892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Huang C, Yuan X, Li Z, Tian Z, Zhan X, Zhang J, et al. VE-statin/Egfl7 siRNA inhibits angiogenesis in malignant glioma in vitro. Int J Clin Exp Pathol (2014) 7(3):1077–84.
187. Li Y, Zhang J, Ma D, Zhang L, Si M, Yin H, et al. Curcumin inhibits proliferation and invasion of osteosarcoma cells through inactivation of Notch-1 signaling. FEBS J (2012) 279(12):2247–59. doi:10.1111/j.1742-4658.2012.08607.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Lee WH, Loo CY, Bebawy M, Luk F, Mason RS, Rohanizadeh R. Curcumin and its derivatives: their application in neuropharmacology and neuroscience in the 21st century. Curr Neuropharmacol (2013) 11(4):338–78. doi:10.2174/1570159X11311040002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Lim KJ, Bisht S, Bar EE, Maitra A, Eberhart CG. A polymeric nanoparticle formulation of curcumin inhibits growth, clonogenicity and stem-like fraction in malignant brain tumors. Cancer Biol Ther (2011) 11(5):464–73. doi:10.4161/cbt.11.5.14410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacol Ther (2013) 139(2):95–110. doi:10.1016/j.pharmthera.2013.02.003
191. Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Direct inhibition of the NOTCH transcription factor complex. Nature (2009) 462(7270):182–8. doi:10.1038/nature08543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Zorde Khvalevsky E, Gabai R, Rachmut IH, Horwitz E, Brunschwig Z, Orbach A, et al. Mutant KRAS is a druggable target for pancreatic cancer. Proc Natl Acad Sci U S A (2013) 110(51):20723–8. doi:10.1073/pnas.1314307110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Mamaeva V, Rosenholm JM, Bate-Eya LT, Bergman L, Peuhu E, Duchanoy A, et al. Mesoporous silica nanoparticles as drug delivery systems for targeted inhibition of Notch signaling in cancer. Mol Ther (2011) 19(8):1538–46. doi:10.1038/mt.2011.105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: brain tumor therapy, clinical trials, glioma, medulloblastoma, Notch signaling, stem-like brain tumor-propagating cells
Citation: Teodorczyk M and Schmidt MHH (2015) Notching on cancer’s door: Notch signaling in brain tumors. Front. Oncol. 4:341. doi: 10.3389/fonc.2014.00341
Received: 27 October 2014; Accepted: 14 November 2014;
Published online: 05 January 2015.
Edited by:
Aleksandra Filipovic, Imperial College London, UKReviewed by:
Justin Lathia, Lerner Research Institute, USAVerdon Taylor, University of Sheffield, UK
Copyright: © 2015 Teodorczyk and Schmidt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirko H. H. Schmidt, Molecular Signal Transduction Laboratories, Institute for Microscopic Anatomy and Neurobiology, Johannes Gutenberg University of Mainz School of Medicine, Langenbeckstr. 1, Mainz D-55131, Germany e-mail:bWlya28uc2NobWlkdEB1bmltZWRpemluLW1haW56LmRl