 Kim De Veirman1†Els Van Valckenborgh1†
Kim De Veirman1†Els Van Valckenborgh1† Qods Lahmar2,3
Qods Lahmar2,3 Xenia Geeraerts2,3Elke De Bruyne1Eline Menu1Ivan Van Riet1Karin Vanderkerken1
Xenia Geeraerts2,3Elke De Bruyne1Eline Menu1Ivan Van Riet1Karin Vanderkerken1 Jo A. Van Ginderachter2,3*
Jo A. Van Ginderachter2,3*- 1Laboratory of Hematology and Immunology, Myeloma Center Brussels, Vrije Universiteit Brussel, Brussels, Belgium
- 2Laboratory of Cellular and Molecular Immunology, Vrije Universiteit Brussel, Brussels, Belgium
- 3Laboratory of Myeloid Cell Immunology, VIB, Brussels, Belgium
Myeloid-derived suppressor cells (MDSC) are a heterogeneous population of immature myeloid cells that accumulate during pathological conditions such as cancer and are associated with a poor clinical outcome. MDSC expansion hampers the host anti-tumor immune response by inhibition of T cell proliferation, cytokine secretion, and recruitment of regulatory T cells. In addition, MDSC exert non-immunological functions including the promotion of angiogenesis, tumor invasion, and metastasis. Recent years, MDSC are considered as a potential target in solid tumors and hematological malignancies to enhance the effects of currently used immune modulating agents. This review focuses on the characteristics, distribution, functions, cell–cell interactions, and targeting of MDSC in hematological malignancies including multiple myeloma, lymphoma, and leukemia.
Introduction
It has been widely accepted that the bone marrow (BM) microenvironment becomes immunosuppressive and plays a crucial role in cancer development and progression (1, 2). Immune suppression is caused by inhibition of activated immune cells, and generation or expansion of immunosuppressive cell types. Regulatory T cells, myeloid-derived suppressor cells (MDSC) and tumor-associated macrophages (TAM) all contribute to an immunologically permissive microenvironment for cancer cells (2–4). MDSC are generally defined as a heterogeneous cell population that arises from myeloid progenitor cells in the BM. These progenitor cells have the capacity to differentiate into macrophages, dendritic cells (DC), or granulocytes, or remain in an undifferentiated state (defined as MDSC) (5, 6). In healthy mice, immature myeloid cells are present in the BM and in small numbers in the spleen; however, they highly accumulate in spleen, lymph nodes, and tumor tissue of tumor-bearing mice (6–8). MDSC are increased in solid tumors (e.g., breast cancer, colorectal cancer) as well as hematological malignancies [e.g., multiple myeloma (MM), leukemia, and lymphoma] (9–14). MDSC expansion and activation is promoted by tumor cells as well as their microenvironment, mainly by the secretion of cytokines and growth factors (8). For mice, MDSC are characterized based on dual expression of CD11b and GR1. Furthermore, MDSC are subdivided into Ly6Glow (monocyte morphology, MO-MDSC) and Ly6Ghigh cells (polymorphonuclear morphology, PMN-MDSC, or G-MDSC) (5, 6, 15). Human MDSC express the myeloid markers CD11b and CD33. CD14+HLA-DRlow/− MDSC are mainly described as the monocytic subpopulation, while human granulocytic MDSC are mostly defined to be CD15+ (16). MDSC inhibit innate and adaptive immunity by regulatory T cell activation and secretion of nitric oxide (NO), reactive oxygen species (ROS), and immunosuppressive cytokines (e.g., IL-10) (6, 17). They not only affect the immune system but also promote tumor angiogenesis, tumor cell invasion, and metastasis. A more extensive description of the phenotype and functional mechanisms of MDSC in general is available in previous reviews (8, 18–20). In the last years, efforts have been made to unravel the characteristics, distribution, and function of murine and human MDSC in hematological malignancies as illustrated in Table 1 and Figure 1. This will be summarized in this review.
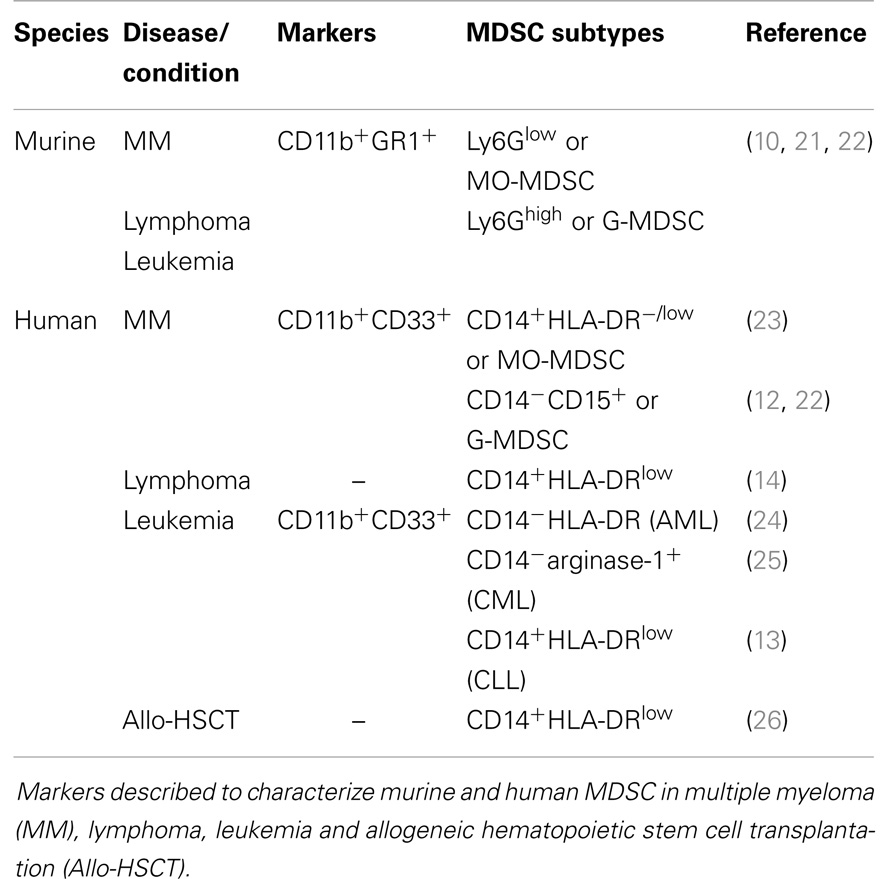
Table 1. General overview of MDSC phenotype.
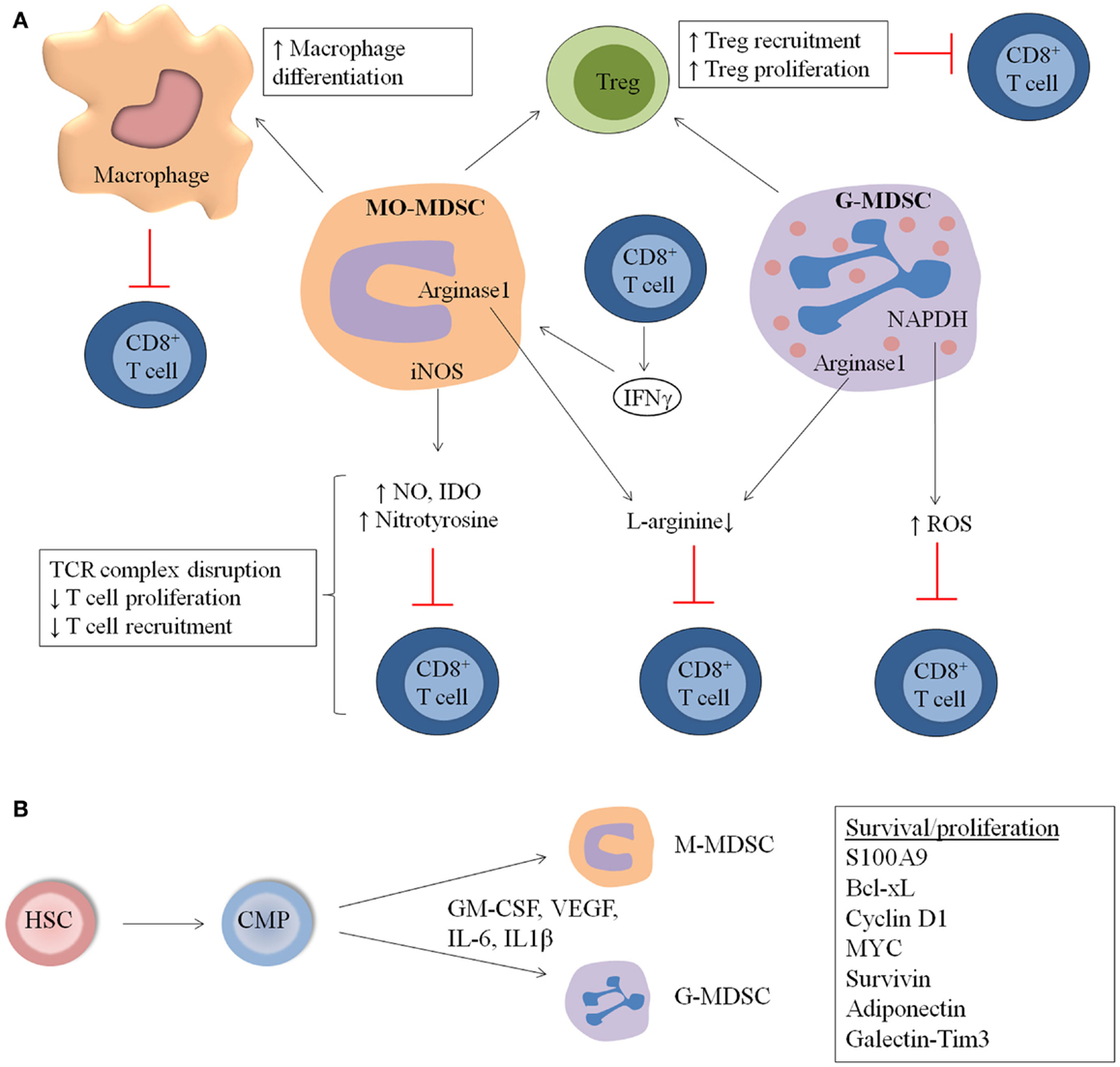
Figure 1. General overview of MDSC immunosuppressive mechanisms and expansion in hematological malignancies and during stem cell transplantation (SCT). (A) MDSC suppress the immune system by distinct mechanisms including increased macrophage differentiation and regulatory T cell (Treg) proliferation, direct actions of MDSC on T cells by increased NO, nitrotyrosine and ROS secretion, and decreased l-arginine production. (B) MDSC originate from common myeloid progenitors (CMP), which arise from hematopoeitic stem cells (HSC). M-MDSC and G-MDSC are formed and proliferate in the presence of distinct factors including GM-CSF, IL-6, VEGF, and IL-1β. Factors involved in the proliferation and survival of MDSC are S100A9, Cyclin D1, Bcl-xL Myc, and Survivin.
MDSC in Multiple Myeloma
Multiple myeloma is a malignant plasma cell disorder characterized by the accumulation of neoplastic plasma cells in the BM and the presence of monoclonal immunoglobulins in the blood and/or urine. Clinical features of this disease include anemia, bone pain, renal failure, frequent occurrence of infections, and hypercalcemia (27). A hallmark of MM is the reciprocal interaction between tumor cells and the BM microenvironment, resulting in accelerating bone loss, increased blood vessel formation, and progressive cancer growth (28). MM is the second most common hematological malignancy and constitutes 1% of all cancers and 13% of all hematological cancers. It affects mostly elderly patients (the median age of diagnosis is 67 years), though 3% of all MM patients are under the age of 40 years. The survival of patients has been improved by new agents like the proteasome inhibitor bortezomib and the immune modulating drug lenalidomide in combination with the alkylating agent melphalan and dexamethasone (29). Despite this evolution, patients still relapse or become refractory to treatment, indicating the need for new therapies.
Immune Dysfunction in MM
Immune dysfunction is an important feature of MM patients and leads to infections and increased tumor growth. A variety of immune defects are observed in MM including cellular abnormalities (e.g., B cells, T cells, DC), secretion of immunosuppressive cytokines (e.g., TGF-β, VEGF, HGF), and increased frequencies of immunosuppressive cell types (including regulatory T cells and MDSC) (30, 31). B-cells derived from MM patients showed impaired immunoglobulin synthesis and secretion (32). Therefore, intravenous administration of immunoglobulin has been used to restore the antibody-mediated immunity in patients with hypogammaglobulinemia (33). Besides B-cell defects, the T cell-mediated immunity is also impaired in MM patients. CD4+ T cells and in particular T-helper 2 cells (Th2) were significantly decreased in MM patients, while no difference in CD8+ T cell number could be observed (34, 35). Furthermore, MM cells are able to directly target T cells by Fas/Fas ligand interaction (36).
Dendritic cells are antigen-presenting cells (APC) responsible for the induction of T cell responses. Although controversy remains about the number of DC in MM patients, it has been clear that they are functionally impaired. They show a lower expression of human leukocyte antigen (HLA) molecules and a reduced capacity in stimulating T cell proliferation and cytokine production (37). Furthermore, MM cells inhibit DC function by secretion of distinct cytokines including IL-6, IL-10, and TGF-β (38, 39).
Natural killer T cells (NKT) belong to the innate immune system and show important defects in MM patients. They not only decrease in cell number at the end-stage of the disease but it has been hypothesized that tumor-derived glycolipids also lead to a deficiency in IFNγ production and NKT dysfunction (40, 41).
Immune responses are also hampered by immunosuppressive cell types including regulatory T cells and TAM. TAM (CD68+) are increased in the BM of MM patients and promote cancer cell proliferation, angiogenesis, and drug resistance. In addition, TAM show a poor antigen-presenting capacity and inhibit T cell proliferation (42). Increased frequencies of regulatory T cells (CD4+CD25highFoxP3+) could also be observed in peripheral blood and BM of MM patients (43).
The development of immune modulating agents (IMiDs) including thalidomide, and its more potent and less toxic derivatives lenalidomide and pomalidomide, have made an important impact on treatment of relapsed/refractory MM patients (44). The exact molecular mechanism of IMiDs is not fully understood but broadly they affect the immune response by increasing T cell proliferation, NK cell-mediated lysis, and reducing regulatory T cells. Furthermore, IMiDs decrease bone resorption and angiogenesis (45, 46). Cellular immunotherapy, which is based on a graft-versus-myeloma effect (e.g., donor lymphocyte infusions, dendritic cell vaccines) showed promising results if utilized for relapsed or persistent MM after allogeneic stem cell transplantation (SCT) (47). However, immunosuppressive cell types limit the effectiveness of immunotherapy, indicating the importance of regulatory T cell, TAM and MDSC targeting in combination with current anti-myeloma drugs.
MDSC Characterization and Distribution in MM
In our lab, MDSC were investigated using the 5T2 and 5T33MM mouse models. These models are derived from elderly C57BL/KaLwRij mice that spontaneously developed MM and are maintained by intravenous transfer of cancer cells into young syngeneic mice. Both models are immunocompetent and resemble the human disease closely as MM cells are able to grow in the surrounding BM microenvironment. The 5T33MM model has a more rapid growth (3 weeks), while the 5T2MM model is characterized by a moderate growth (12 weeks) and osteolytic bone lesions (48). In the BM, a clear shift toward a CD11b+Ly6Glow population, typically considered to be the phenotype of MO-MDSC, was observed at the end-stage of the disease in the 5T2 and 5T33MM mice (10). In addition, in this MO-MDSC population, we could discriminate different MDSC subsets based on differential Ly6C expression: inflammatory monocytes (Ly6ChighSSClow), eosinophils (Ly6CintermediateSSChigh), and immature myeloid cells (Ly6CintermediateSSClow). All of these populations may contribute to T cell suppression, with the monocytes being the most potent. Especially, a major increase in immature myeloid cells could be observed in the BM of the 5T33MM mouse model, indicating a differentiation block in the presence of MM cells (10). Of note, circulating CD11b+ cells increased only at the end-stage of the disease (data unpublished). In the similar 5TGM1 model, MDSC expansion in the blood, BM and spleen could also be observed up to 28 days after MM cell inoculation (21). In another immunocompetent mouse model, tumor cell lines derived from transgenic Bcl-xl/Myc mice (ATLN and DP42) were intravenously injected into syngeneic mice (22). A clear increase in BM MDSC (CD11b+GR1+) was shown the first week after MM cell inoculation with a similar increase in both MO-MDSC and G-MDSC subsets. The BM is the primary tumor site for MM cells and also the site where MDSC are generated. The direct contact of the cancer cells with myeloid progenitor cells might explain the early MDSC conversion and accumulation. During disease progression, MM cells take over the BM microenvironment and disturb and replace normal hematopoiesis. Therefore, the absolute number of MDSC in the BM initially increases, but then gradually decreases at later stages of disease, as observed in the 5T33MM model (data unpublished) and in the ATLN and DP42 models (22).
Some controversy exists about the MDSC characterization markers in MM patients. Brimnes and colleagues were the first to describe an increase of CD14+HLA-DR−/low MO-MDSC in peripheral blood of MM patients at diagnosis compared to healthy donors (23). In contrast, recent studies demonstrated no difference in the MO-MDSC population, but a significant increase in G-MDSC, defined as CD11b+CD14−CD33+CD15+ (22) or CD11b+CD14−CD33+CD15+HLA-DRlow (12), in BM and peripheral blood of MM patients. Different flow cytometry analyses, limited patient numbers and distinct treatment regimens of MM patients could explain the discrepancy of the results (Table 1).
MDSC Expansion and Mechanisms in MM
Little is known about the mechanisms of MDSC expansion, differentiation, and activation in MM. Although no specific studies in MM are conducted, distinct factors present in the MM microenvironment or secreted by MM cells are able to regulate MDSC expansion and activation. Possible factors include IL-6, GM-CSF, VEGF, and IL1β, which are mainly secreted by the myeloma BM microenvironment (49–51). STAT3 has been identified as the main transcription factor for MDSC expansion in distinct cancer models and the expression of the STAT3 target genes cyclin D1, MYC, survivin, and Bcl-xL, resulted in increased survival and proliferation. In addition, the myeloid-related proteins Arginase-1, S100A8, and S100A9 are upregulated by STAT3 (8, 52). S100A9 knockout (KO) mice showed a decrease in immature myeloid cells and a potent anti-tumor immune response (53). Importantly, a delay in the development of ovalbumin-expressing MM tumors was observed in S100A9KO mice compared to wild-type (WT) mice. Adoptive transfer of MDSC derived from WT-MM-bearing mice into S100A9KO MM mice resulted in a reduced survival (22), clearly demonstrating the importance of MDSC-derived S100A9 in MM (Figure 1B).
Immunosuppressive Function of MDSC Subsets in MM
In the 5TMM as well as in the ATLN model, MDSC derived from MM-bearing mice showed clear immunosuppressive activity compared to immature myeloid cells from naïve mice (10, 22). In the 5TMM model, MO-MDSC were more potent to inhibit T cell proliferation compared to G-MDSC. Different subsets in the MO-MDSC population (inflammatory monocytes, eosinophils, and immature myeloid cells) contribute to immunosupression, with inflammatory monocytes being described as the most potent inhibitors. Moreover, myeloma-derived MDSC showed an upregulation of iNOS, arginase-1, and IL-10, compared to MDSC derived from naïve mice. The immunosuppressive action of MO-MDSC was partially abrogated by inhibition of iNOS by l-NMMA, while an arginase-1 inhibitor (norNOHA) did not affect T cell proliferation (10) (Figure 1A).
In human experiments, both MO-MDSC (CD11b+CD14+HLA-DR−/low) and G-MDSC (CD11b+CD14−CD33+CD15+) were sorted from BM of MM patients and significantly suppressed T cell activity in vitro. In contrast, immature myeloid cells derived from healthy donors did not exert any immunosuppressive activity (22). Görgün et al. described strong suppressive activities of MDSC on CD8+ T cells and NKT. As a control, they demonstrated that APC (CD11b+CD14+HLA-DR+) were able to increase T cell proliferation in vitro. However, this was in contrast with a recent study where both CD14+HLA-DRlow and CD14+HLA-DRhigh cells derived from MM patients exerted immunosuppressive capacities (54). As observed in mice, specific inhibitors of arginase-1 and iNOS partially abrogated the immunosuppressive function of human MDSC (12).
MDSC as Osteoclast Progenitors in MM
Besides their role in immune suppression, MDSC also play a pivotal role in bone disease. MDSC are macrophage progenitors which are able to differentiate into osteoclasts. MDSC derived from myeloma-bearing mice had a higher potential to differentiate into mature and functional osteoclasts in vitro and in vivo compared to MDSC from control mice. Co-inoculation of 5TGM1 MM cells and MDSC resulted in increased tumor burden and bone lesions. In addition, in vivo treatment with zoledronic acid, a potent nitrogen-containing bisphophonate, was able to induce a 30% reduction in MDSC (CD11b+GR1+) number, associated with a decrease in osteoclastogenesis to control levels (21). Interestingly, it has been shown that not all MDSC populations were able to differentiate into osteoclasts. Sawant et al. studied breast cancer MDSC derived from lung, blood, spleen, and lymph nodes and observed no osteoclastogenesis when cells were derived from these organs. However, BM MDSC from tumor-bearing mice underwent osteoclast differentiation in contrast to BM MDSC of naïve mice. Although factors responsible for this phenomenon need to be identified, a variety of osteoclastogenic growth factors including RANTES and MCP-1 are secreted by breast cancer cells (55). Although the early idea of MDSC is that they are blocked in their differentiation potential, it seems that in cancers involving bone disease, MDSC can differentiate into osteoclasts.
MDSC in Lymphoma
MDSC Characterization and Distribution in Lymphoma
Lymphoma originates in the lymphatic system and is characterized by abnormal proliferation of B cells and T cells, mostly classified in Hodgkin and non-Hodgkin lymphoma. EG7 and EL4 are two well-characterized subcutaneous lymphoma models that are frequently used to investigate the MDSC subpopulations and functions. MO-MDSC (Ly6G−SSClow) and G-MDSC (Ly6G+SSChigh) accumulated equally in the spleen of EL4 and EG7 murine models (5, 6). Furthermore, the majority of Ly6G− cells showed increased F4/80 expression. Interestingly, three markers were differentially expressed in naïve and tumor-induced monocytes including CD71, CD115, and CD80, indicating a distinct MDSC phenotype in tumor-bearing mice compared to naïve mice (5, 6). Shlecker et al. investigated MDSC distribution in RMA-S lymphoma-bearing mice and found that MO-MDSC as well as G-MDSC accumulated in blood, spleen, and tumor tissue (56).
Little is known about the presence and characteristics of MDSC in human lymphoma patients. In B-cell non-Hodgkin lymphoma (NHL), peripheral blood mononuclear cells (PBMC) showed a reduced Th1-response as determined by IFNγ production compared to healthy controls. Furthermore, less T cell proliferation was observed after coincubation of PBMC with monocytes derived from NHL patients. Importantly, monocyte depletion by anti-CD14 immunomagnetic beads resulted in restored T cell proliferation. It has been shown that NHL monocytes had impaired STAT1 phosphorylation and IFNα production upon CpG oligodeoxynucleotides stimulation and defects in dendritic cell differentiation. No difference in the percentage of monocytes in peripheral blood of NHL patients could be detected compared to healthy controls; however, a clear shift in HLA-DR expression was observed. CD14+ monocytes in NHL patients showed a significant decrease in HLA-DR expression, which was correlated with suppressed immune functions and a more aggressive disease. In addition, elevated arginase-1 levels could be detected in plasma of NHL patients. Furthermore, NHL PBMC proliferation was increased by exogenous l-arginine administration in vitro, indicating that the arginine metabolism is at least partially responsible for the immunosuppressive effects (14) (Table 1).
MDSC Expansion and Differentiation in Lymphoma
Different factors contribute to MDSC expansion and differentiation in lymphoma models. Adiponectin has been identified as an important regulator of MDSC expansion in EL4 T cell lymphoma-bearing mice. Tumor-bearing adiponectin knockout (APNKO) mice showed reduced levels of splenic MDSC compared to WT mice and a higher amount of NK and CD8+ T cells. These effects were associated with reduced lymphoma growth in APNKO mice. In addition, this study demonstrated that G-CSF was lower expressed in tumor-bearing APNKO mice and that this factor played a key role in MDSC differentiation (57). Dardalhon and colleagues described a role for the Tim3-Galectin-9 pathway in MDSC proliferation. EL4 mice treated with anti-Tim3 antibodies showed a delayed tumor progression and a lower frequency of CD11b+GR1+ cells. The authors hypothesized that Tim3 on IFNγ-secreting T cells interact with galectin-9 expressed by MDSC and that this ligand/receptor interaction is involved in MDSC expansion and function (58). Interestingly, galectins have been identified as important modulators of monocyte and macrophage function. Galectin-1 positively correlated with the M2 macrophage marker CD163, while galectin-3 was highly secreted by activated M2 macrophages (59, 60). TAM predict a poor clinical outcome in classical Hodgkin’s lymphoma patients and galectin-1 has been identified as a potential biomarker for relapsed/refractory disease (60, 61).
Besides the ability of immature myeloid cells to rapidly differentiate into macrophages and DC in vitro, Youn and colleagues observed a preferential differentiation of MO-MDSC toward G-MDSC in EL4 tumor-bearing mice (62). MO-MDSC acquire morphological, phenotypical, and functional features of G-MDSC in tumor-bearing mice including a high ROS production and myeloperoxidase activity. Acquisition of the granulocytic phenotype was mediated by epigenetic silencing of the retinoblastoma gene by HDAC2. Schlecker et al. characterized tumor-infiltrating MDSC subpopulations in the RMA-S T cell lymphoma model. Interestingly, tumor-derived MO-MDSC showed increased levels of CCR5 ligands (CCL3, CCL4, and CCL5), which were also associated with regulatory T cell recruitment (56). Serafini and colleagues demonstrated a role for MDSC in regulatory T cell proliferation in the A20 B-cell lymphoma mouse model. They characterized MDSC as APC, which were able to expand regulatory T cells (FOXP3+CD4+) from a preexisting regulatory pool and not by T cell conversion. Furthermore, they demonstrated IL4Rα is expressed on MDSC and correlates with tumor progression. In vivo treatment with sildenafil reduced regulatory T cell expansion and prevented T cell anergy (63).
As observed in MM models, S100A9 protein has been described as an important regulator of MDSC expansion. Tumor-derived conditioned medium induced accumulation of MDSC and reduced dendritic cell differentiation. This was accompanied by increased S100A8 and S100A9 expression. S100A9KO mice injected with EL4 lymphoma cells resulted in a smaller tumor size or even tumor rejection. T cells derived from S100A9KO mice showed higher cytotoxicity against EL4 compared to T cells derived from WT mice. In addition, S100A9 overexpression in hematopoietic stem cells resulted in reduced dendritic cell and macrophage differentiation and accumulation of immature myeloid cells (53). Kälberg et al. demonstrated that the interaction between S100A9 and toll like receptor 4 (TLR4) promoted tumor growth (64). Quinoline-3-carboxamides or Q compounds (e.g., Tasquinimod) were able to block this interaction and inhibited tumor proliferation (65).
Recently, it has been demonstrated that accumulation of MDSC in tumor-bearing EL4 mice was not caused by increased survival of these cells. As a matter of fact, MDSC in tumor-bearing mice have a shorter lifespan than monocytes and neutrophils, but are rapidly replaced by new cells as determined by in vivo BrdU labeling and apoptosis assays. ER stress, present in tumor-bearing mice, causes TNF-related apoptosis-induced ligand receptors (TRAIL-R) upregulation in MDSC. The expression of DR5, a TRAIL-R, was significantly higher in MDSC derived from tumor-bearing mice compared to control mice. Furthermore, MDSC derived from DR5 KO mice showed increased survival compared to WT MDSC. Data clearly demonstrated that inhibition of DR5 improved CD8+ T cell responses in mice bearing TRAIL-insensitive tumors. For cancer patients, a decrease in survival of G-MDSC compared to granulocytic cells was observed. In addition, agonistic DR5 antibodies as well as TRAIL-recombinant protein was able to induce apoptosis of G-MDSC. Although no difference in DR5 expression between G-MDSC and granulocytic cells could be observed in patients, a lower expression of TRAIL decoy receptors 1 and 2 was determined (66) (Figure 1B).
Immunosuppressive Function of MDSC Subsets in Lymphoma
Different lymphoma models have been employed to study MDSC biology. The advantage of the immunogenic mouse BW-Sp3 thymoma is the existence of tumor progressors and regressors within the same model. The majority of the mice develop a tumor-specific CD8+ T cell response resulting in tumor regression but a significant fraction of the tumors ultimately start to progress. By studying mice with different tumor outcomes, a significant contribution of splenic CD11b+ GR1+ cells to the immune dysregulation seen in tumor progressors could be demonstrated. Indeed, while T-cell activating APC were induced in the spleen of regressors, T cell suppressive MDSC were increased in the spleen of progressors. These MDSC, in particular the MO-MDSC fraction, could differentiate into macrophages that had very high arginase levels and expressed high levels of M2-associated genes, suggesting that MDSC could be precursors of M2-oriented and immunosuppressive macrophages (15, 67). Interestingly, stimulation of the nuclear hormone receptor PPARγ, one of the M2-associated markers, diminished the suppressive potential of these MDSC-derived macrophages (15).
Also MO-MDSC and G-MDSC derived from EL4 and EG7 lymphoma models demonstrated immunosuppressive capacity in vitro (5, 6). While G-MDSC showed a higher ROS production, MO-MDSC had increased expression of NO and nitrotyrosine. l-NMMA (NO inhibitor) and arginase inhibitors were able to partially reverse MO-MDSC suppression. Suppressive activities of G-MDSC could be diminished by catalase, a hydroxyl peroxide inhibitor. Employing mice deficient for IFNγR or for the transcription factors downstream of IFNγR – STAT1 and IRF-1, it was demonstrated that EG7 lymphoma-induced MO-MDSC needed to be stimulated by IFNγ to become suppressive and that the suppressive mechanism was mediated by parallel IRF-1/iNOS-dependent and IRF-1/iNOS-independent pathways (68). G-MDSC only minimally depended on IFNγ and iNOS for their suppressive activity (68). Kusmartsev et al. investigated the induction of T cell tolerance by immature myeloid cells from lymphoma-bearing mice in vivo (69). They adoptively transferred transgenic T cells specific for OVA-derived peptide and immature myeloid cells from EG7 tumor-bearing mice into naïve mice. Afterward, lymph node cells were isolated and they observed a dramatically reduced or eliminated antigen-specific T cell response after adoptive transfer of immature myeloid cells. Importantly, GR1+ cells from naive mice and DC from EL4 tumor-bearing mice were not able to inhibit T cell responses. In addition, it has been demonstrated that immature myeloid cells take up soluble protein in vivo, process the protein, and present antigenic epitopes on their surface to induce T cell anergy. Nagaraj et al. subsequently demonstrated that the incubation of MDSC with Ag-specific CD8+ T cells caused nitration of the molecules on the surface of CD8+ T cells that are localized to the site of physical interaction between MDSC and T cells. This process induces CD8+ T cell tolerance that is only specific for the peptide presented by the MDSC. MDSC caused dissociation between the antigen-specific TCR and CD3zeta molecules, disrupting TCR complexes on T cells (70). This is in accordance with the fact that EG7-derived MDSC decreased antigen-driven CD8+ T cell proliferation in vitro, while anti-CD3-driven proliferation was not affected, again indicating an antigen-specific suppression. Our own studies demonstrated that EG7 lymphoma-induced MDSC intricately influence different CD8+ T cell activation events, whereby some parameters are suppressed while others are stimulated (68). For example, while CD8+ T cell proliferative capacity and IL-2 secretion are clearly diminished in the presence of MDSC, the IFNγ production by these cells is actually stimulated on a per cell basis. Complex effects of MO- and G-MDSC on CD8+ T cell adhesiveness to the extracellular matrix and to selectins, on sensitivity to FasL-mediated apoptosis and on cytotoxicity were also noted (Figure 1A).
MDSC in Leukemia
Leukemia is characterized by abnormal proliferation of immature white blood cells that usually starts in the BM. Four main types of leukemia are defined: acute myeloid leukemia (AML), chronic myeloid leukemia (CML), acute lymphoblastic leukemia (ALM), and chronic lymphocytic leukemia (CLL). Studies on MDSC in leukemia are limited. At diagnosis, peripheral blood of AML patients showed a high heterogeneity in MDSC percentage (CD14−HLA-DR−CD33+CD11b+) and a negative correlation with CD34 percentage (24). Christiansson et al. demonstrated increased MDSC numbers, phenotyped by CD11b+CD14−CD33+, and arginase-1 expression in CML patients (25). In untreated CLL patients, the circulating CD14+HLA-DRlow cells were increased compared to healthy controls. In addition, these cells were positive for myeloid markers (CD33, CD11b, CD13, CD11c) and expressed the macrophage colony-stimulating factor receptor CD115 and IL4α receptor, which are associated with MDSC activity. MDSC suppressive capacity in vitro was demonstrated and linked to indoleamine 2,3-dioxygenase (IDO) expression. Furthermore, MDSC promoted regulatory T cell development indicating a cross-talk between CLL cells, MDSC, and regulatory T cells (13). MDSC markers described in literature for MM, lymphoma, and leukemia are summarized in Table 1.
Besides the observations illustrating the importance of MDSC and regulatory T cells in T cell tolerance, it has to be remarked that T cell dysfunction can also be independent of these immunosuppressive cell types. Zhang and colleagues observed T cell dysfunction in a murine model for AML, which was antigen-specific and could not be reversed after MDSC or regulatory T cell depletion. It has been argued that T cell tolerance in that case is regulated by tolerogenic APC. Consequently, T cell activation and prolonged survival could be achieved by the use of agonistic anti-CD40 antibodies, which induces immunogenic DC (71).
MDSC Targeting Strategies in Hematological Malignancies
MDSC could be considered as a valid therapeutic target since they contribute to distinct processes in tumor development, progression and metastasis (18, 72). MDSC targeting can be achieved by distinct strategies including MDSC depletion, MDSC deactivation, induction of maturation/differentiation, and a block in MDSC development (18, 73). Currently used MDSC targeting drugs are listed in Table 2.
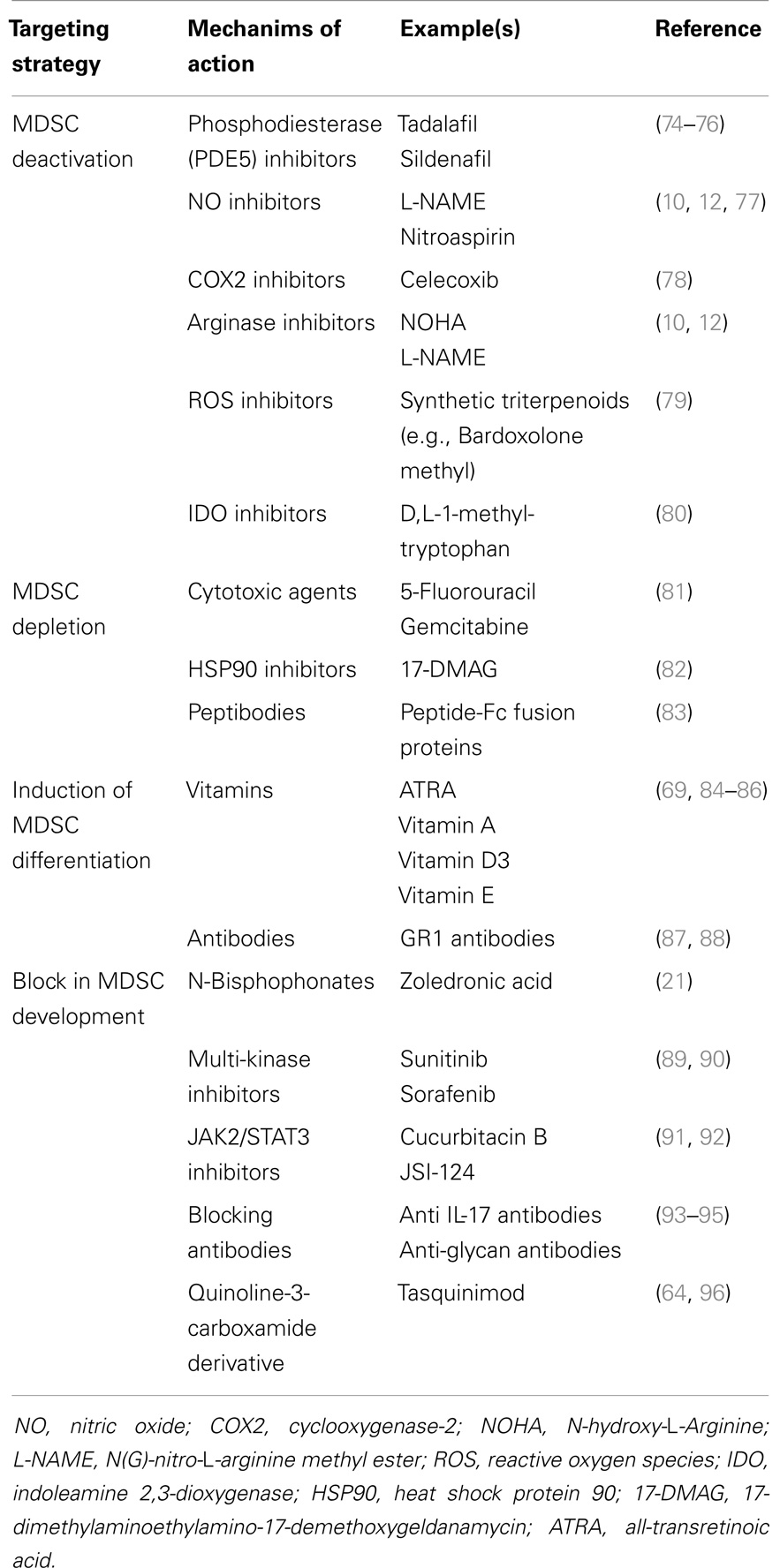
Table 2. General overview of MDSC targeting agents.
MDSC Deactivation
MDSC suppressive mechanisms are often associated with the l-arginine metabolism, which has been extensively reviewed (8, 18, 77). Activated MDSC express high amounts of arginase-1 and NOS2, and inhibitors of both enzymes (L-NMMA for NOS2 and norNOHA for arginase-1) reversed MDSC suppressive mechanisms in MM and lymphoma models (10, 12, 68). Phosphodiesterase (PDE5) inhibitors (such as Sildenafil) inhibit cyclic guanosine monophosphate (cGMP) degradation and reduce arginase-1 and NOS2 expression. It has been hypothesized that high levels of cGMP interfere with the IL4Rα expression by MDSC, resulting in reduced levels of arginase-1 and NOS2. Consequently, Sildenafil treatment of peripheral blood mononuclear cells isolated from MM patients resulted in increased T cell proliferation in vitro (74). Another PDE5 inhibitor, Tadalafil, entered a phase II clinical trial to improve the response to dexamethasone and lenalidomide in 13 MM patients who were refractory to lenalidomide. However, the study was stopped due to a lack of response (75). Recently, the group of Borello et al. demonstrated a reduction in M-spike by Tadalafil treatment in an end-stage relapsed/refractory MM patient. BM CD14+ cells decreased over time by Tadalafil treatment. This was associated with a decrease in IL4Rα, iNOS, and arginase-1 expression in MDSC. BM nitrosylation was also decreased and T cell activity enhanced upon Tadalafil administration (76).
The tryptophan-degrading enzyme IDO is synthesized by MDSC and contributes to immune tolerance by mediating T cell suppression. IDO locally depletes tryptophan and generates tryptophan metabolites including kynurenine resulting in reduced proliferation of CD4+ T cells, CD8+ T cells, and NK cells (97). IDO activity has been correlated with increased immunosuppressive activity in MM patients and can be targeted by d,l-1-methyl-tryptophan (80). ROS secretion also contributes to the immunosuppressive action of MDSC and is caused by the increase in NADPH oxidase activity in granulocytic MDSC. Increased ROS levels were observed in EL4 tumor-bearing mice and could be targeted by a STAT3 inhibitor JSI-124 (91). Synthetic triterpenoid Bardoxolone Methyl (CDDO-Me) also reduced ROS and nitrotyrosine levels in EL4 mice, which was accompanied by an increased T cell response and reduced tumor load (79). Although not tested in hematological malignancies, cyclooxygenase-2 (COX2) inhibitors have also been described to reduce MDSC numbers and their immunosuppressive function in mesothelioma (78).
MDSC Depletion
The MDSC-depleting capacity of cytotoxic agents including 5-fluorouracil (5FU) and gemcitabine has been explored in the EL4 murine lymphoma model (81). Both agents were able to deplete MDSC in the spleen and tumor bed, with 5FU being the most potent drug. Similar effects on granulocytic and monocytic subpopulations could be observed. It has been demonstrated that 5FU exerted anti-tumor activities at least in part by elimination of MDSC and the restoration of T cell specific immune responses. Another compound, the HSP90 inhibitor 17-DMAG, was recently tested in a murine sarcoma model and resulted in a reduced number of MDSC and regulatory T cells upon in vivo administration; however, the underlying mechanism remains unknown (82).
Recently, peptide-Fc fusion proteins, named peptibodies, were developed to target the MDSC population in vivo. Via a competitive phage display platform MDSC specific peptibodies were identified. The peptibodies were engineered to express the mouse IgG2b Fc portion to enhance antibody-dependent cell-mediated cytotoxicity (ADCC). Intravenous peptibody injection was able to deplete blood, spleen, and intratumoral MDSC in distinct lymphoma models (A20, EG7, EL4). No effects on other immune cells, including DC and lymphocytes (T, B, and NK cells), could be detected. Importantly, peptibody treatment in EL4 tumor-bearing mice delayed tumor growth in vivo as determined by tumor size and tumor mass. In contrast to anti-GR1 specific antibodies, which predominantly target the granulocytic population, peptibodies were able to deplete both monocytic and granulocytic MDSC (83).
Induction of MDSC Differentiation
Another mechanism to target the MDSC population is the induction of MDSC differentiation into mature myeloid cells with no suppressive activities. MDSC differentiation can be triggered by distinct vitamins including vitamin A, vitamin D3, or vitamin E (84–86, 98). ATRA (all-transretinoic acid), a vitamin A metabolite, induces the differentiation of monocytic MDSC in DC and macrophages, and causes apopotosis of the granulocytic MDSC population. As a consequence, ATRA improved immunotherapy in distinct murine models (98).
Anti-GR1 antibodies are described to bind with a high affinity to Ly6G molecules and have been extensively used to deplete G-MDSC in tumor-bearing mice. In contrast, Ribechini et al. demonstrated that anti-GR1 antibodies induced myeloid expansion and upregulation of macrophage markers (F4/80, CD115) in BM-derived MDSC. In addition, they observed increased STAT phosphorylation (STAT1, STAT3, STAT5) by anti-GR1 antibody treatment of BM cells in vitro. Protection of BM MO-MDSC against apoptosis was correlated with increased expression of the anti-apoptotic factor Mcl1. Despite the incapacity of MDSC depletion, anti-GR1 antibodies transiently decreased the immune-suppressive activity of GR1high and GR1low subpopulations (87). The effect of anti-GR1 antibodies was also investigated in EL4 tumor-bearing mice (88). In this murine lymphoma model, a complete elimination of MDSC in the spleen and peripheral blood was observed, but hepatic MDSC remained. Despite induction of apoptosis of hepatic MDSC by anti-GR1 antibodies, the population is immediately replaced and showed immunosuppressive activities.
Block in MDSC Development
IL-17 has been identified as an important factor in MDSC development. IL-17R−/− EG7-OVA tumor-bearing mice (EL4 transfected with the chicken ovalbumine gene) showed a lower MDSC number compared to WT mice (93). In addition, reduced arginase-1, MMP9, and S100A8/9 expression was observed in IL-17R−/− compared to WT MDSC. IL-17 neutralizing antibodies were able to decrease tumor growth in WT mice indicating the importance of IL-17 targeting strategies in cancer treatment (93). Besides the effect on MDSC expansion, it has been demonstrated that S100A8/S100A9 proteins are also involved in MDSC recruitment and retention. Migration to the tumor site is promoted by the binding of S100 proteins to carboxylated N-glycan receptors. The anti-carboxylated glycan antibody mAbGB3.1 was able to reduce MDSC numbers in the blood and secondary lymphoid organs (99). Furthermore, mAbGB3.1 was able to block tumor cell proliferation in colorectal cancer (94, 95). In addition, Tasquinimod, a quinoline-3-carboxamide derivative, binds to S100A9 and blocks the interaction with its ligands receptor of advanced glycation end products (RAGE) and toll like receptor 4 (TLR4). It has been demonstrated that Tasquinimod reduced MDSC accumulation, modulated local tumor immunity, and reduced tumor growth and metastasis (64, 96).
As previously mentioned, the N-bisphosphonate zoledronic acid reduced MDSC number and osteoclast formation in MM disease (21). JAK2/STAT3 inhibitors (JSI-124 and Cucurbitacin B) (91, 92) and multi-kinase inhibitors (Sunitinib and Sorafenib) were also described to reduce MDSC levels (89, 90). Cucurbitacin B induces differentiation and maturation of DC (92). Sunitinib treatment resulted in 50% reduction of peripheral blood MDSC, reduced regulatory T cell number, and enhanced Th1 response in renal cell cancer patients (89). Sorafenib, another multi-kinase inhibitor, decreased immunosuppressive cell types in hepatocellular carcinoma (90).
MDSC in Hematopoietic Stem Cell Transplantation
Hematopoietic SCTs have been used as a frequent therapy in blood and BM cancers. However, infections and graft-versus-host disease (GVHD) remain a major cause of mortality. In order to reduce GVHD in allogeneic hematopoietic stem cell transplantation (HSCT), it might be of interest to expand the MDSC population and reduce allogeneic donor T cell activity in patients. Interestingly, it was found that MDSC numbers were higher in SHIP−/− mice displaying a reduced GVHD compared to WT mice (100). In subsequent studies, it was demonstrated that administration of MDSC together with BM transplantation was able to significantly inhibit GVHD lethality and was associated with a decreased proliferation and activation of donor T cells (101, 102). Furthermore, arginase-1 is an important contributor to this effect since administration of a PEGylated form of Arginase-1 could reduce GVHD lethality. In preclinical models, it is described that during BM chimerism monocytic and polymorphonuclear MDSC subsets with alloreactive T cell suppressive capacity have expanded (103). Interestingly, phenotypical and functional studies demonstrated an expansion of similar subsets, MO-MDSC (CD14+CD15−) and G-MDSC (CD14−CD15+), in peripheral blood derived from G-CSF-treated stem cell donors (103). Furthermore, an increased frequency of T cell suppressive CD14+ HLA-DRlow/neg cells was found in allo-HSCT patients after transplantation (26) (Table 1). All these studies indicate that MDSC might have a beneficial role in preventing GVHD and thus can have important clinical implications.
Perspectives
The past years, extensive research has been performed on MDSC in hematological malignancies. Despite the increasing knowledge, many questions remain concerning the role of distinct MDSC subtypes and mechanisms inducing MDSC expansion and/or survival. MDSC differentiation stages toward TAM and tumor-associated neutrophils also remain an unexplored field. In addition, caution in the interpretation of murine experiments is needed. Subcutaneous tumor models (including EG7, EL4, A20) have the major limitation of a localized tumor growth and are less representative because of the lack of an appropriate tumor microenvironment.
Immune modulating agents, especially in combination with other drugs, have significantly improved the outcome of patients with relapsed/refractory MM and lymphoma (104, 105). However, cancer-induced accumulation of immunosuppressive cell types including MDSC, TAM, and regulatory T cells counteract this immune reaction. In order to improve the efficacy of immune modulating drugs and to induce a durable anti-tumor immune response, MDSC targeting could be of great interest. Recently, Tadalafil, a PDE5-inhibitor, induced a long-term anti-myeloma immune and clinical response in a patient with end-stage relapsed/refractory MM (76). In addition, MM-induced osteolytic bone lesions could be reduced by MDSC targeting strategies. Currently, no specific MDSC targeting agents are available and hamper further investigation. Development of new agents and combination studies of MDSC targeting drugs (e.g. 5FU, anti-GR1) with currently used therapy for MM, lymphoma and leukemia are necessary in the future.
Authors Contribution
Kim De Veirman, Els Van Valckenborgh, and Jo A. Van Ginderachter wrote and critically revised the paper. Elke De Bruyne, Eline Menu, Ivan Van Riet, Qods Lahmar, Xenia Geeraerts, and Karin Vanderkerken critically revised the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the European Commission’s Seventh Framework Programme (EU FP7) OVER-MyR and International Myeloma Foundation (IMF). Elke De Bruyne, Eline Menu, and Els Van Valckenborgh are post-doctoral fellows of FWO-VI. Qods Lahmar and Jo A. Van Ginderachter are supported by Stichting tegen Kanker and Vlaamse Liga tegen Kanker.
References
1. Han Z, Tian Z, Lv G, Zhang L, Jiang G, Sun K, et al. Immunosuppressive effect of bone marrow-derived mesenchymal stem cells in inflammatory microenvironment favours the growth of B16 melanoma cells. J Cell Mol Med (2011) 15:2343–52. doi: 10.1111/j.1582-4934.2010.01215.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Bianchi G, Borgonovo G, Pistoia V, Raffaghello L. Immunosuppressive cells and tumour microenvironment: focus on mesenchymal stem cells and myeloid derived suppressor cells. Histol Histopathol (2011) 26:941–51.
3. Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol (2007) 25:267–96. doi:10.1146/annurev.immunol.25.022106.141609
4. Schouppe E, De Baetselier P, Van Ginderachter JA, Sarukhan A. Instruction of myeloid cells by the tumor microenvironment: open questions on the dynamics and plasticity of different tumor-associated myeloid cell populations. Oncoimmunology (2012) 1:1135–45. doi:10.4161/onci.21566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood (2008) 111:4233–44. doi:10.1182/blood-2007-07-099226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Youn J-I, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol (2008) 181:5791–802. doi:10.4049/jimmunol.181.8.5791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Zhao F, Obermann S, von Wasielewski R, Haile L, Manns MP, Korangy F, et al. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology (2009) 128:141–9. doi:10.1111/j.1365-2567.2009.03105.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol (2009) 9:162–74. doi:10.1038/nri2506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother (2009) 58:49–59. doi:10.1007/s00262-008-0523-4
10. Van Valckenborgh E, Schouppe E, Movahedi K, De Bruyne E, Menu E, De Baetselier P, et al. Multiple myeloma induces the immunosuppressive capacity of distinct myeloid-derived suppressor cell subpopulations in the bone marrow. Leukemia (2012) 26:2424–8. doi:10.1038/leu.2012.113
11. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One (2013) 8:e57114. doi:10.1371/journal.pone.0057114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Görgün GT, Whitehill G, Anderson JL, Hideshima T, Maguire C, Laubach J, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood (2013) 121:2975–87. doi:10.1182/blood-2012-08-448548
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood (2014) 124:750–60. doi:10.1182/blood-2013-12-546416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Lin Y, Gustafson MP, Bulur PA, Gastineau DA, Witzig TE, Dietz AB. Immunosuppressive CD14+HLA-DR(low)/− monocytes in B-cell non- Hodgkin lymphoma. Blood (2011) 117:872–81. doi:10.1182/blood-2010-05-283820
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Van Ginderachter JA, Meerschaut S, Liu Y, Brys L, De Groeve K, Hassanzadeh Ghassabeh G, et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands reverse CTL suppression by alternatively activated (M2) macrophages in cancer. Blood (2006) 108:525–35. doi:10.1182/blood-2005-09-3777
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol (2011) 11:802–7. doi:10.1016/j.intimp.2011.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Huang B, Pan P-Y, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res (2006) 66:1123–31. doi:10.1158/0008-5472.CAN-05-1299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Tadmor T, Attias D, Polliack A. Myeloid-derived suppressor cells – their role in haemato-oncological malignancies and other cancers and possible implications for therapy. Br J Haematol (2011) 153:557–67. doi:10.1111/j.1365-2141.2011.08678.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Rodríguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev (2008) 222:180–91. doi:10.1111/j.1600-065X.2008.00608.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Talmadge JE, Gabrilovich DI. History of myeloid-derived suppressor cells. Nat Rev Cancer (2013) 13:739–52. doi:10.1038/nrc3581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Zhuang J, Zhang J, Lwin ST, Edwards JR, Edwards CM, Mundy GR, et al. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS One (2012) 7:e48871. doi:10.1371/journal.pone.0048871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Ramachandran IR, Martner A, Pisklakova A, Condamine T, Chase T, Vogl T, et al. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol (2013) 190:3815–23. doi:10.4049/jimmunol.1203373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Brimnes MK, Vangsted AJ, Knudsen LM, Gimsing P, Gang AO, Johnsen HE, et al. Increased level of both CD4+FOXP3+ regulatory T cells and CD14+HLA-DR-/low myeloid-derived suppressor cells and decreased level of dendritic cells in patients with multiple myeloma. Scand J Immunol (2010) 72:540–7. doi:10.1111/j.1365-3083.2010.02463.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Ansu Abu A, Tartour E, Gey A, Poonkuzhali B, Srivastava V, Rayaz A, et al. Myeloid derived suppressor cells in acute leukemia and its association with conventional cytogenetic and molecular risk factors. Ash Abstr (2012) 1446.
25. Christiansson L, Söderlund S, Svensson E, Mustjoki S, Bengtsson M, Simonsson B, et al. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS One (2013) 8:e55818. doi:10.1371/journal.pone.0055818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Mougiakakos D, Jitschin R, von Bahr L, Poschke I, Gary R, Sundberg B, et al. Immunosuppressive CD14+HLA-DRlow/neg IDO+ myeloid cells in patients following allogeneic hematopoietic stem cell transplantation. Leukemia (2013) 27:377–88. doi:10.1038/leu.2012.215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Caers J, Vande Broek I, De Raeve H, Michaux L, Trullemans F, Schots R, et al. Multiple myeloma – an update on diagnosis and treatment. Eur J Haematol (2008) 81:329–43. doi:10.1111/j.1600-0609.2008.01127.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Menu E, Asosingh K, Van Riet I, Croucher P, Van Camp B, Vanderkerken K. Myeloma cells (5TMM) and their interactions with the marrow microenvironment. Blood Cells Mol Dis (2004) 33:111–9. doi:10.1016/j.bcmd.2004.04.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Bergsagel PL, Mateos M-V, Gutierrez NC, Rajkumar SV, San Miguel JF. Improving overall survival and overcoming adverse prognosis in the treatment of cytogenetically high-risk multiple myeloma. Blood (2013) 121:884–92. doi:10.1182/blood-2012-05-432203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Romano A, Conticello C, Cavalli M, Vetro C, La Fauci A, Parrinello NL, et al. Immunological dysregulation in multiple myeloma microenvironment. Biomed Res Int (2014) 2014:198539. doi:10.1155/2014/198539
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Pratt G, Goodyear O, Moss P. Immunodeficiency and immunotherapy in multiple myeloma. Br J Haematol (2007) 138:563–79. doi:10.1111/j.1365-2141.2007.06705.x
32. Carter A, Silvian I, Tatarsky I, Spira G. Impaired immunoglobulin synthesis in multiple myeloma: a B-cell dysfunction. Am J Hematol (1986) 22:143–54. doi:10.1002/ajh.2830220205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Raanani P, Gafter-Gvili A, Paul M, Ben-Bassat I, Leibovici L, Shpilberg O. Immunoglobulin prophylaxis in hematological malignancies and hematopoietic stem cell transplantation. Cochrane Database Syst Rev (2008) 4:CD006501. doi:10.1002/14651858.CD006501.pub2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Ogawara H, Handa H, Yamazaki T, Toda T, Yoshida K, Nishimoto N, et al. High Th1/Th2 ratio in patients with multiple myeloma. Leuk Res (2005) 29:135–40. doi:10.1016/j.leukres.2004.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Miguel JFS, González MM, Gascón A, Moro J, Hernández JM, Ortega F, et al. Lymphoid subsets and prognostic factors in multiple myeloma. Br J Haematol (1992) 80:305–9. doi:10.1111/j.1365-2141.1992.tb08137.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Villunger A, Egle A, Marschitz I, Kos M, Bock G, Ludwig H, et al. Constitutive expression of Fas (Apo-1/CD95) ligand on multiple myeloma cells: a potential mechanism of tumor-induced suppression of immune surveillance. Blood (1997) 90:12–20.
37. Brimnes MK, Svane IM, Johnsen HE. Impaired functionality and phenotypic profile of dendritic cells from patients with multiple myeloma. Clin Exp Immunol (2006) 144:76–84. doi:10.1111/j.1365-2249.2006.03037.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Ratta M, Fagnoni F, Curti A, Vescovini R, Sansoni P, Oliviero B, et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood (2002) 100:230–7. doi:10.1182/blood.V100.1.230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Brown RD, Pope B, Murray A, Esdale W, Sze DM, Gibson J, et al. Dendritic cells from patients with myeloma are numerically normal but functionally defective as they fail to up-regulate CD80 (B7-1) expression after huCD40LT stimulation because of inhibition by transforming growth factor-beta1 and interleukin-10. Blood (2001) 98:2992–8. doi:10.1182/blood.V98.10.2992
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Dhodapkar MV, Geller MD, Chang DH, Shimizu K, Fujii S-I, Dhodapkar KM, et al. A reversible defect in natural killer T cell function characterizes the progression of premalignant to malignant multiple myeloma. J Exp Med (2003) 197:1667–76. doi:10.1084/jem.20021650
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Nur H, Fostier K, Aspeslagh S, Renmans W, Bertrand E, Leleu X, et al. Preclinical evaluation of invariant natural killer T cells in the 5T33 multiple myeloma model. PLoS One (2013) 8:e65075. doi:10.1371/journal.pone.0065075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood (2009) 114:3625–8. doi:10.1182/blood-2009-05-220285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Beyer M, Kochanek M, Giese T, Endl E, Weihrauch MR, Knolle PA, et al. In vivo peripheral expansion of naive CD4+CD25high FoxP3+ regulatory T cells in patients with multiple myeloma. Blood (2006) 107:3940–9. doi:10.1182/blood-2005-09-3671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. McCurdy AR, Lacy MQ. Pomalidomide and its clinical potential for relapsed or refractory multiple myeloma: an update for the hematologist. Ther Adv Hematol (2013) 4:211–6. doi:10.1177/2040620713480155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Busch A, Zeh D, Janzen V, Mügge L-O, Wolf D, Fingerhut L, et al. Treatment with lenalidomide induces immunoactivating and counter-regulatory immunosuppressive changes in myeloma patients. Clin Exp Immunol (2014) 177:439–53. doi:10.1111/cei.12343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Chanan-Khan AA, Swaika A, Paulus A, Kumar SK, Mikhael JR, Rajkumar SV, et al. Pomalidomide: the new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J (2013) 3:e143. doi:10.1038/bcj.2013.38
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Rutella S, Locatelli F. Targeting multiple-myeloma-induced immune dysfunction to improve immunotherapy outcomes. Clin Dev Immunol (2012) 2012:196063. doi:10.1155/2012/196063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Asosingh K, Radl J, Van Riet I, Van Camp B, Vanderkerken K. The 5TMM series: a useful in vivo mouse model of human multiple myeloma. Hematol J (2000) 1:351–6. doi:10.1038/sj.thj.6200052
49. Zhang XG, Bataille R, Jourdan M, Saeland S, Banchereau J, Mannoni P, et al. Granulocyte-macrophage colony-stimulating factor synergizes with interleukin-6 in supporting the proliferation of human myeloma cells. Blood (1990) 76:2599–605.
50. Lechner MG, Liebertz DJ, Epstein AL. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J Immunol (2010) 185:2273–84. doi:10.4049/jimmunol.1000901
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Alexandrakis MG, Passam FH, Sfiridaki A, Pappa CA, Moschandrea JA, Kandidakis E, et al. Serum levels of leptin in multiple myeloma patients and its relation to angiogenic and inflammatory cytokines. Int J Biol Markers (2004) 19:52–7.
52. Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, et al. STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest (2013) 123:1580–9. doi:10.1172/JCI60083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med (2008) 205:2235–49. doi:10.1084/jem.20080132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. De Keersmaecker B, Fostier K, Corthals J, Wilgenhof S, Heirman C, Aerts JL, et al. Immunomodulatory drugs improve the immune environment for dendritic cell-based immunotherapy in multiple myeloma patients after autologous stem cell transplantation. Cancer Immunol Immunother (2014) 63:1023–36. doi:10.1007/s00262-014-1571-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Sawant A, Deshane J, Jules J, Lee CM, Harris BA, Feng X, et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res (2013) 73:672–82. doi:10.1158/0008-5472.CAN-12-2202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J Immunol (2012) 189:5602–11. doi:10.4049/jimmunol.1201018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Han S, Jeong AL, Lee S, Park JS, Kim KD, Choi I, et al. Adiponectin deficiency suppresses lymphoma growth in mice by modulating NK cells, CD8 T cells, and myeloid-derived suppressor cells. J Immunol (2013) 190:4877–86. doi:10.4049/jimmunol.1202487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, et al. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol (2010) 185:1383–92. doi:10.4049/jimmunol.0903275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Novak R, Dabelic S, Dumic J. Galectin-1 and galectin-3 expression profiles in classically and alternatively activated human macrophages. Biochim Biophys Acta (2012) 1820:1383–90. doi:10.1016/j.bbagen.2011.11.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kamper P, Ludvigsen M, Bendix K, Hamilton-Dutoit S, Rabinovich GA, Møller MB, et al. Proteomic analysis identifies galectin-1 as a predictive biomarker for relapsed/refractory disease in classical Hodgkin lymphoma. Blood (2011) 117:6638–49. doi:10.1182/blood-2010-12-327346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med (2010) 362:875–85. doi:10.1056/NEJMoa0905680
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Youn J-I, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol (2013) 14:211–20. doi:10.1038/ni.2526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res (2008) 68:5439–49. doi:10.1158/0008-5472.CAN-07-6621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Källberg E, Vogl T, Liberg D, Olsson A, Björk P, Wikström P, et al. S100A9 interaction with TLR4 promotes tumor growth. PLoS One (2012) 7:e34207. doi:10.1371/journal.pone.0034207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Björk P, Björk A, Vogl T, Stenström M, Liberg D, Olsson A, et al. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol (2009) 7:e97. doi:10.1371/journal.pbio.1000097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Condamine T, Kumar V, Ramachandran IR, Youn J-I, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest (2014) 124:2626–39. doi:10.1172/JCI74056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Liu Y, Van Ginderachter JA, Brys L, De Baetselier P, Raes G, Geldhof AB. Nitric oxide-independent CTL suppression during tumor progression: association with arginase-producing (M2) myeloid cells. J Immunol (2003) 170:5064–74. doi:10.4049/jimmunol.170.10.5064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Schouppe E, Mommer C, Movahedi K, Laoui D, Morias Y, Gysemans C, et al. Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8+ T-cell activation events. Eur J Immunol (2013) 43:2930–42. doi:10.1002/eji.201343349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Kusmartsev S, Nagaraj S, Gabrilovich DI. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J Immunol (2005) 175:4583–92. doi:10.4049/jimmunol.175.7.4583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Nagaraj S, Schrum AG, Cho H-I, Celis E, Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol (2010) 184:3106–16. doi:10.4049/jimmunol.0902661
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Zhang L, Chen X, Liu X, Kline DE, Teague RM, Gajewski TF, et al. CD40 ligation reverses T cell tolerance in acute myeloid leukemia. J Clin Invest (2013) 123:1999–2010. doi:10.1172/JCI63980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Parrinello NL, Di Raimondo F, Romano A. Myeloid derived suppressor cells in multiple myeloma. Exp Rev Immunol Vaccine Informat (2014) 1:4–13.
73. Wesolowski R, Markowitz J, Carson WE. Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. J Immunother Cancer (2013) 1:10. doi:10.1186/2051-1426-1-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203:2691–702. doi:10.1084/jem.20061104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Ghosh N, Rudraraju L, Ye X, Noonan K, Huff CA. Administration Of An Oral PDE5 Inhibitor, Tadalafil In Conjunction With a Lenalidomide Containing Regimen In Patients With Multiple Myeloma – The Myeloma Beacon. New Orleans (2013).
76. Noonan KA, Ghosh N, Rudraraju L, Bui M, Borrello I. Targeting immune suppression with PDE5 inhibition in end-stage multiple myeloma. Cancer Immunol Res (2014) 2:725–31. doi:10.1158/2326-6066.CIR-13-0213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Schouppe E, Van Overmeire E, Laoui D, Keirsse J, Van Ginderachter JA. Modulation of CD8(+) T-cell activation events by monocytic and granulocytic myeloid-derived suppressor cells. Immunobiology (2013) 218:1385–91. doi:10.1016/j.imbio.2013.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Veltman JD, Lambers MEH, van Nimwegen M, Hendriks RW, Hoogsteden HC, Aerts JGJV, et al. COX-2 inhibition improves immunotherapy and is associated with decreased numbers of myeloid-derived suppressor cells in mesothelioma. Celecoxib influences MDSC function. BMC Cancer (2010) 10:464. doi:10.1186/1471-2407-10-464
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Nagaraj S, Youn J-I, Weber H, Iclozan C, Lu L, Cotter MJ, et al. Anti-inflammatory triterpenoid blocks immune suppressive function of MDSCs and improves immune response in cancer. Clin Cancer Res (2010) 16:1812–23. doi:10.1158/1078-0432.CCR-09-3272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Bonanno G, Mariotti A, Procoli A, Folgiero V, Natale D, De Rosa L, et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity correlates with immune system abnormalities in multiple myeloma. J Transl Med (2012) 10:247. doi:10.1186/1479-5876-10-247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res (2010) 70:3052–61. doi:10.1158/0008-5472.CAN-09-3690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Rao A, Taylor JL, Chi-Sabins N, Kawabe M, Gooding WE, Storkus WJ. Combination therapy with HSP90 inhibitor 17-DMAG reconditions the tumor microenvironment to improve recruitment of therapeutic T cells. Cancer Res (2012) 72:3196–206. doi:10.1158/0008-5472.CAN-12-0538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Qin H, Lerman B, Sakamaki I, Wei G, Cha SC, Rao SS, et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat Med (2014) 20:676–81. doi:10.1038/nm.3560
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Kang TH, Knoff J, Yeh W-H, Yang B, Wang C, Kim YS, et al. Treatment of tumors with vitamin E suppresses myeloid derived suppressor cells and enhances CD8+ T cell-mediated antitumor effects. PLoS One (2014) 9:e103562. doi:10.1371/journal.pone.0103562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Young MRI, Day TA. Immune regulatory activity of vitamin d3 in head and neck cancer. Cancers (Basel) (2013) 5:1072–85. doi:10.3390/cancers5031072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Kuwata T, Wang IM, Tamura T, Ponnamperuma RM, Levine R, Holmes KL, et al. Vitamin A deficiency in mice causes a systemic expansion of myeloid cells. Blood (2000) 95:3349–56.
87. Ribechini E, Leenen PJM, Lutz MB. Gr-1 antibody induces STAT signaling, macrophage marker expression and abrogation of myeloid-derived suppressor cell activity in BM cells. Eur J Immunol (2009) 39:3538–51. doi:10.1002/eji.200939530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Ma C, Kapanadze T, Gamrekelashvili J, Manns MP, Korangy F, Greten TF. Anti-Gr-1 antibody depletion fails to eliminate hepatic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol (2012) 92:1199–206. doi:10.1189/jlb.0212059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Molina AM, Lin X, Korytowsky B, Matczak E, Lechuga MJ, Wiltshire R, et al. Sunitinib objective response in metastatic renal cell carcinoma: analysis of 1059 patients treated on clinical trials. Eur J Cancer (2014) 50:351–8. doi:10.1016/j.ejca.2013.08.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Cao M, Xu Y, Youn J, Cabrera R, Zhang X, Gabrilovich D, et al. Kinase inhibitor Sorafenib modulates immunosuppressive cell populations in a murine liver cancer model. Lab Invest (2011) 91:598–608. doi:10.1038/labinvest.2010.205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol (2009) 182:5693–701. doi:10.4049/jimmunol.0900092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Lu P, Yu B, Xu J. Cucurbitacin B regulates immature myeloid cell differentiation and enhances antitumor immunity in patients with lung cancer. Cancer Biother Radiopharm (2012) 27:495–503. doi:10.1089/cbr.2012.1219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, et al. IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol (2010) 184:2281–8. doi:10.4049/jimmunol.0902574
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Manitz M-P, Horst B, Seeliger S, Strey A, Skryabin BV, Gunzer M, et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol (2003) 23:1034–43. doi:10.1128/MCB.23.3.1034-1043.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis- associated carcinogenesis. Carcinogenesis (2008) 29:2035–43. doi:10.1093/carcin/bgn188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Raymond E, Dalgleish A, Damber J-E, Smith M, Pili R. Mechanisms of action of tasquinimod on the tumour microenvironment. Cancer Chemother Pharmacol (2014) 73:1–8. doi:10.1007/s00280-013-2321-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med (2002) 196:459–68. doi:10.1084/jem.20020121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res (2003) 63:4441–9.
99. Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol (2008) 181:4666–75. doi:10.4049/jimmunol.181.7.4666
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Ghansah T, Paraiso KHT, Highfill S, Desponts C, May S, McIntosh JK, et al. Expansion of myeloid suppressor cells in SHIP-deficient mice represses allogeneic T cell responses. J Immunol (2004) 173:7324–30. doi:10.4049/jimmunol.173.12.7324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Wang L, Chang EWY, Wong SC, Ong S-M, Chong DQY, Ling KL. Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins. J Immunol (2013) 190:794–804. doi:10.4049/jimmunol.1202088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Highfill SL, Rodriguez PC, Zhou Q, Goetz CA, Koehn BH, Veenstra R, et al. Bone marrow myeloid-derived suppressor cells (MDSCs) inhibit graft-versus-host disease (GVHD) via an arginase-1-dependent mechanism that is up-regulated by interleukin-13. Blood (2010) 116:5738–47. doi:10.1182/blood-2010-06-287839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Luyckx A, Schouppe E, Rutgeerts O, Lenaerts C, Fevery S, Devos T, et al. G-CSF stem cell mobilization in human donors induces polymorphonuclear and mononuclear myeloid-derived suppressor cells. Clin Immunol (2012) 143:83–7. doi:10.1016/j.clim.2012.01.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Pan B, Lentzsch S. The application and biology of immunomodulatory drugs (IMiDs) in cancer. Pharmacol Ther (2012) 136:56–68. doi:10.1016/j.pharmthera.2012.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Wang M, Fowler N, Wagner-Bartak N, Feng L, Romaguera J, Neelapu SS, et al. Oral lenalidomide with rituximab in relapsed or refractory diffuse large cell, follicular and transformed lymphoma: a phase II clinical trial. Leukemia (2013) 27:1902–9. doi:10.1038/leu.2013.95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: myeloid-derived suppressor cells, immune system, hematological malignancies, multiple myeloma, leukemia, lymphoma, stem cell transplantations
Citation: De Veirman K, Van Valckenborgh E, Lahmar Q, Geeraerts X, De Bruyne E, Menu E, Van Riet I, Vanderkerken K and Van Ginderachter JA (2014) Myeloid-derived suppressor cells as therapeutic target in hematological malignancies. Front. Oncol. 4:349. doi: 10.3389/fonc.2014.00349
Received: 18 September 2014; Accepted: 23 November 2014;
Published online: 08 December 2014.
Edited by:
Alessandra Romano, University of Catania, ItalyReviewed by:
Nadim Mahmud, University of Illinois at Chicago, USAAlessandra Romano, University of Catania, Italy
Copyright: © 2014 De Veirman, Van Valckenborgh, Lahmar, Geeraerts, De Bruyne, Menu, Van Riet, Vanderkerken and Van Ginderachter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jo A. Van Ginderachter, Laboratory of Cellular and Molecular Immunology, Laboratory of Myeloid Cell Immunology, VIB, Vrije Universiteit Brussel, Pleinlaan 2, Brussels B-1050, Belgium e-mail:anZhbmdpbmRAdnViLmFjLmJl
†Kim De Veirman and Els Van Valckenborgh have contributed equally to this work.