 Carole Grasso1
Carole Grasso1 Marie-Sophie Fabre2
Marie-Sophie Fabre2 Sarah V. Collis2
Sarah V. Collis2 M. Leticia Castro2
M. Leticia Castro2 Cameron S. Field1,2
Cameron S. Field1,2 Nanette Schleich3
Nanette Schleich3 Melanie J. McConnell1,2
Melanie J. McConnell1,2 Patries M. Herst1,3*
Patries M. Herst1,3*- 1Malaghan Institute of Medical Research, Wellington, New Zealand
- 2School of Biological Sciences, Victoria University, Wellington, New Zealand
- 3Department of Radiation Therapy, University of Otago, Wellington, New Zealand
Pharmacological ascorbate is currently used as an anti-cancer treatment, potentially in combination with radiation therapy, by integrative medicine practitioners. In the acidic, metal-rich tumor environment, ascorbate acts as a pro-oxidant, with a mode of action similar to that of ionizing radiation; both treatments kill cells predominantly by free radical-mediated DNA damage. The brain tumor, glioblastoma multiforme (GBM), is very resistant to radiation; radiosensitizing GBM cells will improve survival of GBM patients. Here, we demonstrate that a single fraction (6 Gy) of radiation combined with a 1 h exposure to ascorbate (5 mM) sensitized murine glioma GL261 cells to radiation in survival and colony-forming assays in vitro. In addition, we report the effect of a single fraction (4.5 Gy) of whole brain radiation combined with daily intraperitoneal injections of ascorbate (1 mg/kg) in an intracranial GL261 glioma mouse model. Tumor-bearing C57BL/6 mice were divided into four groups: one group received a single dose of 4.5 Gy to the brain 8 days after tumor implantation, a second group received daily intraperitoneal injections of ascorbate (day 8–45) after implantation, a third group received both treatments and a fourth control group received no treatment. While radiation delayed tumor progression, intraperitoneal ascorbate alone had no effect on tumor progression. Tumor progression was faster in tumor-bearing mice treated with radiation and daily ascorbate than in those treated with radiation alone. Histological analysis showed less necrosis in tumors treated with both radiation and ascorbate, consistent with a radio-protective effect of ascorbate in vivo. Discrepancies between our in vitro and in vivo results may be explained by differences in the tumor microenvironment, which determines whether ascorbate remains outside the cell, acting as a pro-oxidant, or whether it enters the cells and acts as an anti-oxidant.
Introduction
The use of high (pharmacological) doses of ascorbate for the treatment of cancer is highly controversial [reviewed by Ref. (1)]. Although most mammals make their own ascorbate from glucose in the liver, humans and other primates (as well as guinea pigs and bats) obtain ascorbate from their diet as they lack a functional gulonolactone oxidase necessary for the last step of ascorbate synthesis (2). Regardless of source, systemic ascorbate levels are very tightly regulated by controlled uptake into (1) the blood stream through the intestinal epithelium (via high affinity sodium-dependent vitamin C transporters, SVCT1) and (2) into body tissues (via SVCT2), followed by (3) renal clearance (via SVCT1) to plateau at 40–60 μM (3). Intravenous/intraperitoneal injection of high-dose ascorbate (0.5–1.25 g/kg in humans; 1–4 g/kg in mice) can bypass regulation to generate serum levels of 30 mM for about 2 h (4, 5). Such high concentrations of ascorbate have been reported to generate extracellular hydrogen peroxide, particularly in the acidic metal-rich environment of solid tumors [reviewed by Ref. (1)]. Hydrogen peroxide diffuses into cells where it generates reactive oxygen species (ROS), such as hydroxyl radicals (6), which overwhelm the anti-oxidant defense system, resulting in DNA damage (7, 8). The extracellular addition of catalase abrogates the pro-oxidant effect of high-dose ascorbate (6–9), verifying the role of extracellular hydrogen peroxide in its mechanism of action.
High-dose ascorbate is well tolerated in cancer patients who have normal renal function, do not have glucose-6-phosphate dehydrogenase deficiency or iron overload (4, 5) and has been shown to delay tumor growth in rodent xenograft models (10). However, high-dose ascorbate as a single agent has never been reported to cure cancer in animals or humans. Recent preclinical research has therefore focused on combining high-dose ascorbate with chemotoxic anticancer drugs such as Carboplatin and Paclitaxel for ovarian cancer (11), Gemcitabine for pancreatic cancer (12), Epigallocatechin-3-gallate plus Gemcitabine for malignant mesothelioma (13), Vincristine (14), and glycolytic inhibitors for small cell lung cancer (15). Potentiation of the hydrogen peroxide generating effect of ascorbate in vitro has been reported for inhibitors of hydrogen peroxide metabolism in pancreatic cancer (16), alpha-tocopherol succinate in prostate cancer (17), manganoporphyrins in breast cancer (18) and pancreatic cancer (19), and phenylaminonaphthoquinones in bladder cancer (20). Increased sensitivity to Docetaxel, Epirubicin, Irinotecan, and 5-FU after ascorbate exposure has been attributed to G0/G1 arrest in prostate carcinoma cells (21).
Only three small clinical trials have so far been published that have evaluated the effect of high-dose ascorbate combination therapy in metastatic ovarian cancer [with carboplatin plus paclitaxel; n = 25 (11)] and metastatic pancreatic cancer [with gemcitabine; n = 9 (22) and with gemcitabine plus erlotinib; n = 14 (23)]. All three trials noted that patients on the ascorbate plus chemotherapy arm lived longer than those taking conventional chemotherapy alone. However, the increase in survival time was not statistically significant due to the small patient numbers typical of stage I trials (11, 22, 23). With respect to side-effects, all trials demonstrated a lack of ascorbate-related toxicities. In fact, two trials (11, 22) reported fewer grade 1 and 2 chemotherapy-related toxicities with the addition of ascorbate to standard chemotherapy.
Radiation resistance of the primary adult brain cancer, glioblastoma multiforme (GBM), is a key factor in poor patient outcomes (24). As the blood–brain barrier limits chemotherapy options for brain tumors radiation is a primary treatment. Patient outcomes can be improved by either escalation of radiation dose to the tumor (25) or by increasing the sensitivity of GBMs to radiation. Because ionizing radiation kills cells mainly by generating free radicals that cause double stranded DNA breaks (DSB) (26), one approach to radio-sensitization would be to combine radiation therapy with compounds that also generate free radicals. As many cancers have inferior radical scavenging and DNA repair capability (27), a combination of treatments that increase free radical production should theoretically affect cancer cells more than normal cells.
Two in vitro studies have shown that high-dose ascorbate radiosensitized primary GBM cells isolated from tumors of GBM patients by generating extracellular hydrogen peroxide and inducing S/G2 arrest, interfering with DNA repair (7, 8). Interestingly, both ionizing radiation and high-dose ascorbate were shown to increase labile iron levels in pancreatic tumor homogenates from athymic nude mice (28). Catalytic metals can accelerate ascorbate oxidation, leading to increased generation of hydrogen peroxide (29). Radio-sensitization of highly aggressive radio-resistant GBMs has significant clinical implications. The addition of ascorbate to radiation protocols would allow for superior tumor control at lower radiation doses, resulting in less severe acute and chronic brain toxicities. The first report of ascorbate chemoradiation in mice was published in 1996 when Taper and colleagues showed that pretreating intramuscularly transplanted liver tumors with ascorbate and menadione (Vitamin K3) potentiated the effect of a single dose of 20–50 Gy of X-ray irradiation (30). Here, we investigate the effects of high-dose ascorbate and radiation on tumor progression and tumor histology in an intracranial mouse glioma model, where GL261 mouse glioma cells are injected directly into the brain of immunocompetent C57BL/6 mice.
Materials and Methods
Materials
Unless otherwise noted, tissue plasticware was purchased from Nunc (ThermoFisher Scientific, Auckland, New Zealand); all cell culture reagents were from Gibco BRL (Invitrogen, Auckland, New Zealand). Sodium ascorbate and all other chemicals and reagents were from Sigma Chemical Company (St. Louis, MO, USA).
Cell Lines
The mouse glioma cell line, GL261 was obtained from the NCI tumor cell line repository (Fredrick, MD). GL261 cells were grown in RPMI-1640 supplemented with 20% (v/v) FBS, GlutaMAX-1 (2 mM), 25 μg/mL penicillin, 25 μg/mL streptomycin and maintained in a humidified incubator at 37°C/5% CO2.
Irradiation of GL261 Cells
GL261 cells were seeded 24 h prior to treatment in six well plates (3 to 5 × 104/well). Cells were 30–40% confluent and growing exponentially on the day of treatment. Cells were irradiated with 1, 3, 6, or 9 Gy using Cesium-137 γ-rays (Gammacell 3000 Elan, Best Theratronics) for viability assays and with 6 Gy for clonogenicity assays. After irradiation, the cells were re-incubated in fresh medium.
Ascorbate Treatment of GL261 Cells
Exponentially growing 30–40% confluent GL261 cells were seeded 24 h prior to treatment in six well plates (3 to 5 × 104/well). Cells were exposed to 5 mM ascorbate in media for 1 h, washed in Dulbecco’s Phosphate Buffer Saline (PBS, 1.4 M NaCl, 27 mM KCl, 170 mM NaH2PO4, 17.6 mM KH2PO4) and re-incubated in fresh medium. Cells that received radiation were irradiated in the presence of ascorbate.
Viability of GL261 Cells
GL261 cells were collected 48 h after radiation (1, 3, 6, and 9 Gy) by trypsinization, washed in PBS, and resuspended in 1 μg/mL propidium iodide, for cell count and dye exclusion using flow cytometry using a BD FACSort (Becton Dickinson, San Jose, CA, USA). All viability assays were completed at least three times in triplicate.
Clonogenicity of GL261 Cells
Colony-forming ability was determined as described by Franken et al. (31). GL261 cells at 30–40% confluency in 100 mm dishes were treated with 6 Gy, 5 mM ascorbate or both 6 Gy and 5 mM ascorbate, trypsinized, counted, and re-plated at varying dilutions, and incubated for 12–14 days. Plates were fixed and stained in 0.5% (w/v) methylene blue solution in 50% (v/v) methanol for 1 h, and colonies consisting of at least 50 cells were counted. The surviving fraction of cells was calculated against the plating efficiency of GL261 cells for each experiment. All clonogenic assays were completed at least three times in triplicate.
Annexin V/Propidium Iodide Staining
Annexin V/propidium iodide staining was used to distinguish between viable cells (A−/PI−), early apoptotic cells (A+/PI−) and dead cells (A+/PI+). Cells were centrifuged at 130 × g at room temperature for 5 min, washed in phosphate buffered saline solution, pH 7.3 (PBS), and resuspended in Annexin V binding buffer. Small aliquots (1 × 106 cells) were transferred to 1.5 mL tubes and spun at 2000 rpm for 2 min in a Biofuge fresco (Kendro, Auckland). Most supernatant was removed and 5 μL PI and 5 μL FITC-labeled AV was added to the wet pellets, which were vortexed. After 30 min in the dark on ice, 500 μL of Annexin V binding buffer was added; the cells were centrifuged for 2 min at 2000 rpm, washed once with Annexin V binding buffer, and resuspended in 1 mL of binding buffer in FACS tubes. Staining was analyzed by flow cytometry using a BD FACSort (Becton Dickinson, San Jose, CA, USA). AV/PI staining was completed three times.
Double Stranded DNA Break Analysis
Double stranded DNA breaks were determined at 2 and 24 h after treatments by the extent of phosphorylation of the histone variant protein, H2AX, using FITC-labeled anti-γH2AX antibodies in permeabilized cells (32). Cells were washed in PBS buffer and distributed in 96 well plates (5 × 105 cells/well), washed in 200 μL FACS buffer (PBS + 1% BSA), pelleted at 2000 rpm (in a Megafuge 2.0R, Heraeus Germany centrifuge) for 2 min, and fixed in 200 μL BD Cytofix/Cytoperm solution. After 15 min incubation at 4°C, cells were washed twice in 200 μL 1× BD Perm/Wash buffer, incubated at 4°C for 15 min, pelleted and resuspended in antibody solution (50 μL anti-γH2AX antibody or isotype control diluted 1: 500 in 1× BD Perm/Wash) at 4°C for 1 h, washed twice in 1× BD Perm/Wash, and resuspended in 400 μL FACS buffer. Stained and unstained control cells were analyzed by flow cytometry.
Intracranial Mouse Model
All experiments using mice were conducted in accordance with the New Zealand Animal Welfare Act 19991 and were approved by the Victoria University Animal Ethics Committee. Mouse glioma GL261 cells were cultured to sub-confluence, washed in PBS, and live cells implanted into the brain of 8- to 12-week old (approx. 30 g) male C57BL/6 mice, following the procedure described by Hunn et al. (33). Briefly, animals were anesthetized by intraperitoneal (i.p.) injection of xylazine (100 mg/kg) and ketamine (10 mg/kg) (Phoenix Pharm), and Lacri-Lube (Allergan) applied across the cornea of the eye. A burr hole was drilled in the skull 0.1 mm posterior to the bregma and 2.3 mm lateral to the midline. Live cells (5 × 103 in 2 μL of PBS) were administered stereotactically (Stoelting Apparatus), into the burr hole, using a Hamilton syringe with a 32-gauge needle. The needle was advanced to a depth of 2.3 mm from the brain surface and the cell suspension delivered slowly over the course of 2–3 min. Following injection, the needle was left in place for 2 min, after which time, it was raised to a depth of 1.5 mm below the brain surface and left in place for an additional 1 min. Upon withdrawal of the needle, the burr hole was sealed with bone wax and the incision sutured. Animals received sub-cutaneous analgesics [Carprofen (5 mg/kg), Norbrook Laboratories, and Buprenorphine (0.1 mg/kg), Renckitt Benckiser Pharmaceuticals], to control post-operative pain. Animals were randomly assigned into control and treatment groups (n = 5/group). Animals were weighed daily and humanely sacrificed by cervical dislocation when weight loss occurred (>10% of body weight) or until neurological signs of disease were evident, whichever came first.
Whole Brain Irradiation of Mice
Mice received a single dose of 4.5 Gy to the brain (and ≤1 Gy to the shielded body) on day 8 after surgery, based on previous work by Newcomb et al. (34). The Gammacell 3000 Elan irradiator irradiates the content of a steel cylinder with γ-rays from a sealed Cesium-137 line source (length 27 cm, diameter 1.7 cm; nominal activity 48 TBq). Tumor-bearing mice were anesthetized and placed in an upright position, stabilized by tissue paper, in a 50 mL Falcon tube without a tip to facilitate breathing. The tube was placed inside 2 cm thick custom-built lead shielding, which exposes the head to radiation while shielding the body from the ears down. The shielding was placed in the center of the steel cylinder (Figure 1).
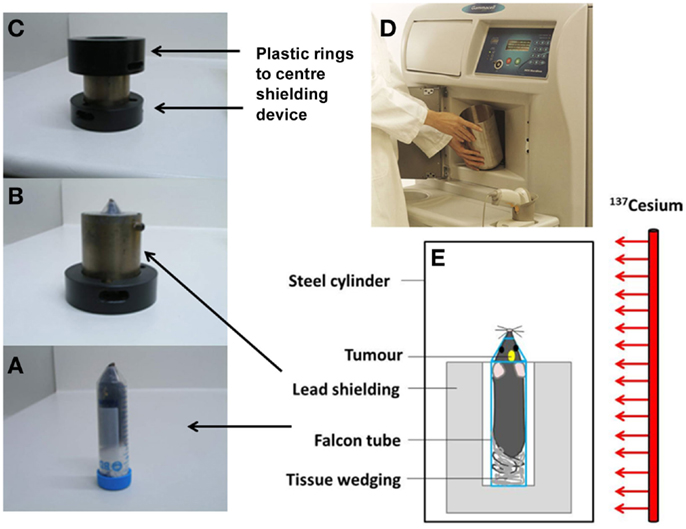
Figure 1. Irradiation setup for whole brain irradiation of mice. The mouse is positioned in a Falcon tube (A), inside a 2 cm thick lead shielding device (B) inside plastic rings (C) which are placed inside the aluminum cylinder in the irradiator (D). Diagram of the irradiation setup (E).
The radiation dose received by the head and the rest of the body was verified using the thermoluminescence dosimetry system located at the Blood and Cancer Center, Wellington Hospital. The system utilizes thermoluminescent dosimeters (TLDs) based on lithium fluoride crystals doped with magnesium and titanium (“TLD-100”), in chip form with dimensions of 3.2 mm × 3.2 mm × 0.9 mm (35, 36). Prior to use, the dosimeters were reset by annealing at high temperatures in an oven (PTW-Freiburg TLDO) and sealed into plastic envelopes as rows of separate dosimeters. In order to ensure the same radiation absorption and scatter properties as during the treatment of live mice, the dosimeters were positioned between a culled mouse and a Falcon tube, along its axial length from the pelvis to the brain region. The Falcon tube including mouse and dosimeters was placed inside the lead shielding, and exposed for a set time in the gamma irradiator. The time required to deliver approximately 4.5 Gy to the mouse head using our shielding – which blocks a fraction of the radiation emitted by the source – was approximately 1.5 min. After irradiation, the TLDs underwent a pre-read anneal cycle, followed by the readout in an automatic TLD reader (Harshaw QS 5500). The calibration of the dosimeters was performed by exposing TLDs to a known radiation dose delivered by a 6 MV X-ray beam from a linear accelerator at Wellington Hospital. This dose was independently measured with a cylindrical ionization chamber following local procedures based on absorbed dose to water and calibration factors traceable to the Australian primary standard for absorbed dose.
The measurements were subject to corrections for measured variations in sensitivity for individual TLD chips, and the difference in beam energies for calibration (6 MV X-ray beam with mean energy near 2 MeV) and measurements (662 keV gamma rays for Cesium-137) (35–38). The reproducibility of the results was verified by repeating the TLD measurements for a small selection of positions in the head region and body region, finding variations not exceeding ±4%. Analysis of the results, including an estimate of the uncertainties, indicates an absorbed dose of 4.5 ± 0.3 Gy delivered to the brain region and a mean of 0.6 ± 0.04 Gy along the mouse body within the shielding. Based on these results, an acute whole body dose of 1 Sv is estimated, which is considered to be non-lethal.
Treatment of Mice with High-Dose Ascorbate
Tumor-bearing mice received an i.p. injection of high-dose ascorbate, 2 h prior to irradiation and after that once daily (Mon–Fri) from day 8 to 45 after surgery, at a concentration of 1 or 2 g/kg bodyweight of a sterile neutralized ascorbate solution (Sigma) in 1 mL of PBS buffer, similar to the procedure used by Chen et al. (10).
Histological Analysis of Mouse Brains
The auto-fluorescent property of H&E stained blood was used to quantify the amount of intravascular and extravascular blood in each section. An Olympus 1 × 51 inverted microscope and Olympus DP72 camera were used to take fluorescent images of the entire tumor at 4× using a mercury bulb and a band-pass filter to measure wavelengths between 604 and 644 nm. ImageJ 1.48v2 was used to measure the percentage of blood in the tumor. Fluorescent images were converted into 8-bit grayscale images. The total area of the tumor was measured by tracing around the tumor border with the freehand selection tool. The threshold tool was adjusted to select and measure only the auto-fluorescent blood in the image. The following formula was used to convert these measurements into the percentage of blood in the tumor:
Necrosis was measured using brightfield images of the same H&E stained sections. Areas of necrosis were traced around and measured with the freehand selection tool. The following formula was used to convert these measurements into the percentage of necrosis in the tumor:
Statistical Analysis
Statistical significance of differences in 48 h survival and clonogenicity of GL261 cells between different treatments were calculated using unpaired two-tailed student t tests (Microsoft v 2010; Redmond Campus, Redmond, Washington, DC, USA). The Mantel–Cox log-rank test (Prism 5.0 Graph Pad Software, Inc., La Jolla, CA, USA) was performed to determine the statistical significance between Kaplan–Meier survival curves. In all instances, p < 0.05 was considered statistically significant.
Results
Sensitivity of GL261 Cells to Radiation, Ascorbate, and Combined Treatments
GL261 is a mouse glioma cell line, which forms tumors in immunocompetent C57BL/6 mice. We first tested the sensitivity of GL261 cells in vitro to a single exposure of different doses of radiation (Gy) (Figure 2A) and a 1 h exposure to different concentrations of ascorbate (AA) (Figure 2B). We have previously shown that our GBM cell lines do not produce many DSB after a single exposure of 1 or 3 Gy. All our in vitro research therefore has used a single dose of 6 Gy to determine the effect of a radiation alone and in combination with 5 mM ascorbate on viability, clonogenicity, generation, and repair of DSBs and cell cycle progression (7, 8). Concentrations of 5 mM ascorbate have been used by a number of authors in the field [reviewed in Ref. (1)]. Similar to our previous research with a panel of GBM cell lines (7, 8), GL261 cells sustained a substantial amount of DNA damage, as shown by the percentage of cells with DSBs 2 h after treatments. Initial DNA damage as well as DNA damage repair was significantly worse after combined treatment compared with single treatments (Figure 2C). We determined the percentage of GL261 cell survival after 48 h (Figure 2D) and colony-forming ability after 10–12 days (Figure 2E), in response to both single and combined treatments. Combining both treatments decreased 48 h survival and clonogenicity compared with radiation treatment alone. GBM cells are very resistant to apoptosis (7, 39). GL261 cells also did not undergo apoptosis when exposed to single and combined treatments in vitro (Figure 2F).
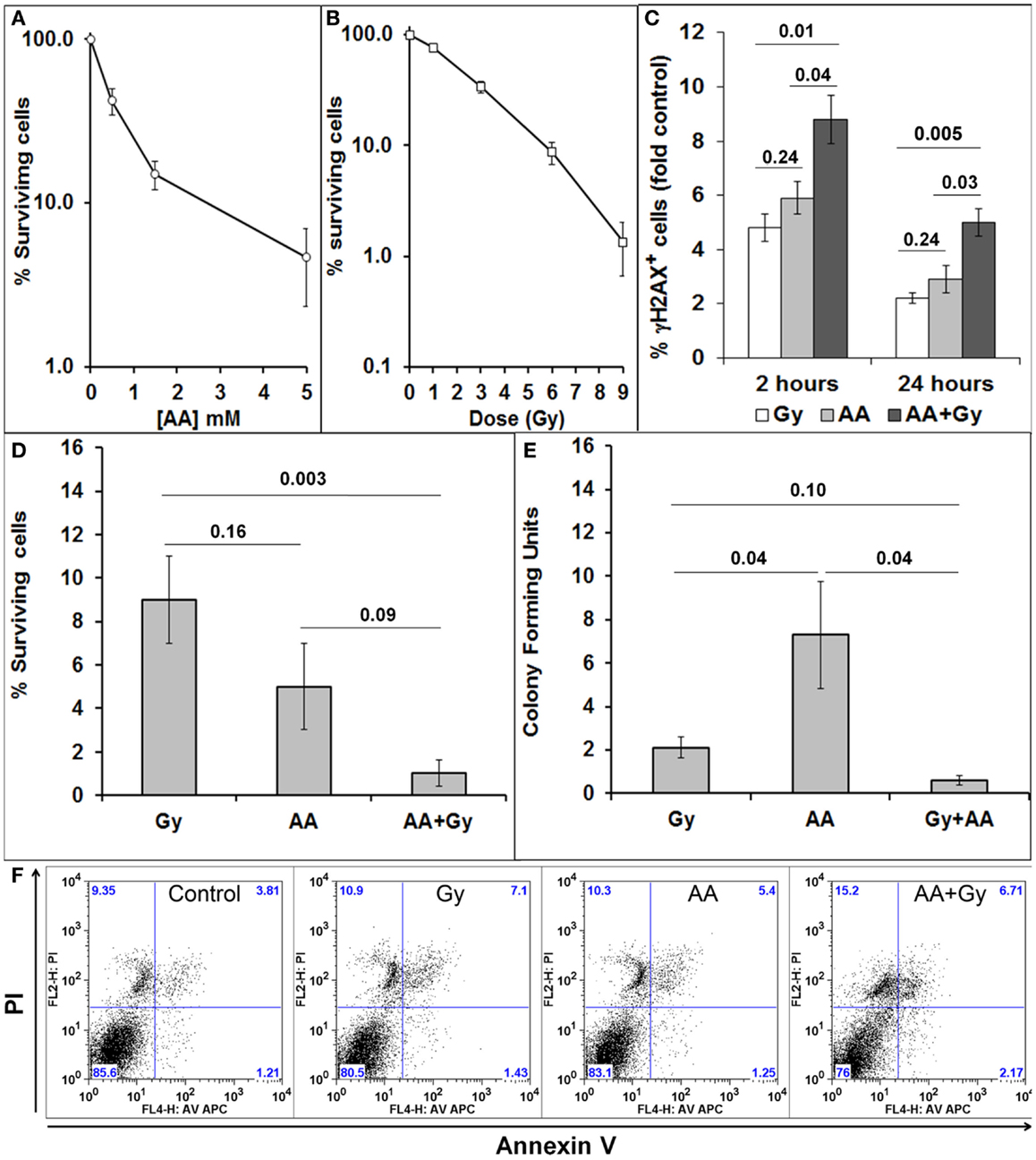
Figure 2. Sensitivity of GL261 to radiation and ascorbate treatments. (A) Cell survival 48 h after a 1 h exposure to different doses of ascorbate (AA), measured by PI exclusion and flow cytometry; (B) cell survival 5 days after irradiation with 1, 3, 6, and 9 Gy, measured by PI exclusion and flow cytometry; (C) DNA damage and repair as fold change (compared to control) of % cells with DSBs was measured as the extent of H2AX phosphorylation (fluorescence intensity) using flow cytometry and anti-γH2AX antibodies at 2 and 24 h after treatments; (D) Cell survival 48 h after 6 Gy, 5 mM AA, or combined treatment as measured by PI exclusion and flow cytometry; (E) clonogenicity (colony-forming units) 10–12 days after 6 Gy, 5 mM AA, or combined treatment. Colonies of 50 cells or more were visualized by methylene blue staining, counted, and numbers are presented as percentage of untreated controls. (F) Lack of apoptosis of GL261 cells 48 h after 6 Gy, 5 mM AA, or combined treatment as measured by FACs analysis of AV/PI staining. Values are averages ± SEM of at least three different experiments conducted in triplicate. Numbers above bars denote p values (unpaired two-tailed student t-test).
Effect of Radiation, Ascorbate, and Combined Treatments on Mouse Survival
Our next step was to validate the in vitro radio-sensitizing effect of high-dose ascorbate on GL261 clonogenicity in an intracranial mouse glioma model. Live GL261 cells (5 × 103 in 2 μL of PBS) were implanted into the brain of 8- to 12-week-old (approximately 30 g) male C57BL/6 mice. Tumor-bearing mice were divided into four groups: one group received a single dose of 4.5 Gy to the whole brain [based on previous work by Newcomb et al. (34)] 8 days after tumor implantation, a second group received daily i.p. injections of ascorbate (1 g/kg AA) from day 8 to 45 after implantation [based on previous work by Chen et al. (10)], a third group received both treatments, and a fourth control group received no treatment at all. Tumor-bearing mice were culled when weight loss due to tumor growth exceeded 10% of body weight or neurological symptoms of disease (40), and survival of animals from all groups are shown in Figure 3. Median survival for untreated animals survived was 26 days (range 23–32). Daily doses of intraperitoneal ascorbate did not affect survival (median 28 days; range 23–30). A single dose of radiation increased survival of tumor-bearing mice significantly (median 43 days; range 31–51; p = 0.002) compared with untreated control animals. However, in sharp contrast with our in vitro data, tumor-bearing mice receiving both a single dose of radiation and daily i.p. ascorbate needed to be culled significantly sooner (median 31 days; range 29–35) than animals treated with a single dose of radiation alone (p = 0.0275). These data suggest that rather than making the tumor more sensitive to radiation, ascorbate protected the tumor from radiation damage. We obtained very similar results when injecting 2 g/kg i.p. ascorbate daily (results not shown).
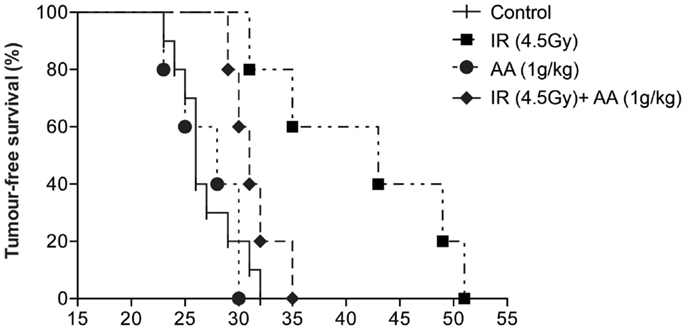
Figure 3. Effect of a single dose of radiation, daily i.p. ascorbate and combined treatment on survival of tumor-bearing C57BL/6 mice. Tumor-bearing C57BL/6 mice were divided into four groups: one group received a single dose of 4.5 Gy to the brain 8 days after tumor implantation, a second group received daily intraperitoneal injections of ascorbate (day 8–45) after implantation, a third group received both treatments, and a fourth control group received no treatment. Animals were sacrificed when they had lost 10% of their weight in 24 h or after neurological symptoms were evident. Results are representative of four experiments (two with 1 g/kg ascorbate and two with 2 g/kg ascorbate).
Effect of Radiation, Ascorbate, and Combined Treatments on Tumor Histology
Tumor-bearing brains were collected from animals at the endpoint of the experiment, which was weight loss of >10% or neurological symptoms of disease. Tumors were approximately the same size upon collection but irradiated only mice took longer to develop these tumors compared with control mice, ascorbate treated mice, and mice treated with both radiation and ascorbate (see section above). Analysis of necrosis and presence of blood in tumors was carried out by H&E staining at multiple sections across the tumors of at least four mice (a representative example is shown in Figure 4). GL261 tumors contained substantial amounts of intravascular and extravascular blood, regardless of whether they were treated or not (Figure 5A). Untreated tumors had variable but low levels of necrosis, making up 0–6% of tumor area. Radiation treatment doubled the incidence of necrosis, up to 12% of tumor area. Treatment with ascorbate alone had no effect on necrosis. Strikingly, addition of ascorbate to radiation prevented the radiation-induced necrosis, even below the level of necrosis in control tumors (Figure 5B). These results were consistent with a radio-protective effect of ascorbate on the tumor, resulting in shorter survival times of tumor-bearing mice receiving both ascorbate and radiation. Fluorescent confocal microscopy using anti-PCNA (Proliferating Cell Nuclear Antigen) antibodies showed a complete lack of proliferation in the normal brain tissues and proliferation in 45–50% of cells throughout the tumors, regardless of whether the mice had been treated or not (results not shown).
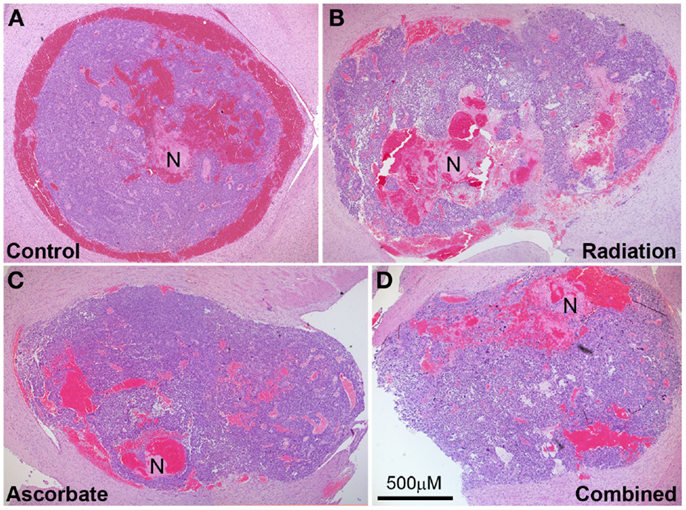
Figure 4. Representative tumor histology of mice that were not treated (A), treated with a single dose of 4.5 Gy (B), daily i.p ascorbate (C), and both treatments (D). Analysis of necrosis and presence of blood in tumors was carried out by H&E staining at multiple sections across the tumors of several mice. N indicates necrosis. Magnification 4×; bar represents scale for all four photos (500 μM).

Figure 5. Percentage necrosis (A) and statistical analysis (B) of tumors treated with daily i.p. ascorbate (AA), a single dose of radiation (Gy), combined treatment (AA + Gy), and no treatment (C). *p < 0.05 is statistically significant.
Discussion
We set out to validate our previous reports on the in vitro radiosensitizing effect of high-dose ascorbate in primary GBM cells (7, 8) in an intracranial mouse glioma model. Although high-dose ascorbate made GL261 cells more sensitive to radiation in vitro, unexpectedly it protected tumors derived from GL261 cells implanted in the brain of mice from radiation injury. To our knowledge, this is the first time the combination of high-dose ascorbate with radiation has been tested in mice. Previous studies have shown that high-dose ascorbate as a single agent (two daily i.p. injections of 4 g/kg) significantly retarded tumor growth of rat 9L gliosarcoma cells implanted subcutaneously in the flank of immunocompromised mice (10). In contrast, our immunocompetent mice with brain tumors treated with 1 or 2 g/kg daily i.p. ascorbate as a single agent developed tumors at the same time as untreated animals. While it may have been useful to examine the 4 g/kg doses used in other studies, our tumor-bearing mice did not cope with the osmotic challenge of buffered ascorbate sodium salt at doses in excess of 2 g/kg, preventing the escalation to 4 g/kg.
We hypothesize that the discrepancies between the in vitro data, the mouse subcutaneous and intracranial data can be explained by differences in tumor microenvironments. The DNA damage and other anti-tumor effects of ascorbate require the extracellular conversion to hydrogen peroxide [reviewed in Ref. (1)]. Laboratory conditions are characterized by optimal access to oxygen, nutrients, and space in the absence of normal stroma, and extracellular ascorbate is readily converted to peroxide in this setting. Although the subcutaneous microenvironment of “flank models” mimics a “real” tumor with access to oxygen, nutrients, growth factors, and cytokines available in the systemic blood circulation, it does not model the conditions in the intracranial interstitial fluid inside the blood–brain barrier, or the highly specialized interactions between glioma cells on one hand and astrocytes and microglial cells on the other hand that may control the fate of ascorbate in the brain tumor microenvironment.
Astrocytes are a subset of glial cells responsible for maintaining intracranial homeostasis by regulating cerebral blood flow, neuronal activity through neuron–astrocyte metabolic coupling and protect neurons from toxic wastes and chemotherapeutic drugs (41, 42). Small, early stage tumors that develop in this microenvironment can harness the protective effects of astrocytes as shown by Kim and colleagues, who reported that brain metastases from NCI-H358 lung cancer cells were less sensitive to rapamycin-induced apoptosis when co-cultured with astrocytes but not when co-cultured with fibroblasts (41). These authors also showed that co-culture of human MDA-MB-231 breast cancer cells with murine astrocytes protected the cancer cells from apoptosis by vincristine through upregulation of the anti-apoptotic survival genes, BCL2L1, TWIST1, and GSTA5. Protection of tumor cells by astrocytes was mediated by continued physical contact between astrocytes and tumor cells and direct communication via GAP junctions (42). Strong expression of these survival genes was also seen in clinical specimen from breast cancer brain metastases but not in breast cancer metastasized to the lungs (42). With respect to radiation sensitivity, human NSC11 stem cell-like GBM cells grown in vitro were reported to be much more sensitive to radiation than the same cells implanted in mouse brains (43, 44).
The brain microenvironment is also unique with respect to ascorbate metabolism. Ascorbate passes through the blood–brain barrier very slowly but it is actively transported into the cerebrospinal fluid via the high affinity sodium-dependent vitamin C transporter, SVCT2. Ependymal cells that line the choroid plexi and produce cerebrospinal fluid from blood plasma highly express SVCT2 (3, 45–47). Neurons further concentrate intracellular ascorbate to millimolar levels (3, 45, 47), where it acts as a free radicals scavenger (3), assists the cellular hypoxia response through hydroxylation of hypoxia-inducible factor 1α (48), promotes collagen synthesis, and neuronal maturation and transmission (3). Therefore, ascorbate concentrations in the brain are very high (2–10 mM) compared with liver (0.8–1 mM), muscle (400 μM), cerebrospinal fluid and interstitial fluid (160–400 μM), and blood plasma (40–60 μM) (3). These high ascorbate concentrations in the brain are generated through active transport via the high affinity sodium-dependent vitamin C transporter, SVCT2, which is highly expressed on ependymal cells and neurons (3, 45). So, rather than through the blood–brain barrier, ascorbate is actively transported into the cerebrospinal fluid through SVCT2.
Astrocytes and microglial cells (central nervous system resident macrophages) do not normally express SVCT2 but acquire ascorbate in its oxidized form, dehydroascorbate (DHA), imported via the glucose transporter, GLUT1. Once inside the cell DHA is rapidly reduced to ascorbate, resulting in intracellular ascorbate levels in astrocytes and microglial cells similar to those of cerebrospinal and interstitial fluid of 160–400 μM, adding to their anti-oxidant capacity (3, 45).
The mechanism of ascorbate uptake in gliomas and GBM is likely to include both SVCT2 and GLUT1. A recent report suggests that proliferative neural progenitor cells up-regulate SVCT2 in response to ascorbate (49), a mechanism that could also transport ascorbate into GBMs. An extensive analysis of gene expression and DNA copy number in gliomas (50–52) has indicated the SVCT2 locus is amplified in gliomas, and expression is significantly higher than in normal brain tissue. However, data from other expression studies suggest that SVCT2 expression in gliomas may be lost (53, 54). Further, gene expression does not guarantee transporter activity. Rodriguez and colleagues showed that the high SVCT2 expression in the TC620 oligodendroglioma cell line was offset by its lack of activity and intracellular location (55). In vivo, the acidic tumor environment is likely to reduce the activity of the SVCT2 transporters, which have an optimal activity at a slightly alkaline pH (56). For gliomas that lack an active SVCT2, tumor associated microglial cells, which can make up to 30% of tumor mass in high grade gliomas could supply the glioma cells with DHA taken up via GLUT1 (57).
Regardless of the mechanism of uptake, glioma cells may be able to rapidly acquire significant quantities of ascorbate. This will have two effects – first, it will prevent the extracellular conversion of ascorbate to hydrogen peroxide, neutralizing the DNA damage potential. Second, the anti-oxidant activity of ascorbate will protect the cell from radiation-induced oxidative damage. Taken together, our results strongly suggest that the unique brain microenvironment promotes accumulation of ascorbate inside GBM cells to concentrations high enough to facilitate radioprotection. This is in sharp contrast with the reported pro-oxidant effect of extracellular ascorbate in vitro and in flank xenograft models [reviewed in Ref. (1)].
Although our orthotopic glioma model mimics the brain microenvironment and is therefore clinically more relevant than subcutaneous flank models, it has certain limitations that make extrapolation to the clinical setting problematic. GL261 cells represent a stage II–III glioma and not a stage IV GBM, form well-defined tumors rather than the diffuse pattern often seen in GBMs and are a lot more sensitive to radiation and ascorbate than true primary and established GBM cell lines as shown previously (7). Perhaps more importantly, our mice received only a single dose of 4.5 Gy, whereas standard radiation therapy for GBM patients consists of daily fractions of 2 Gy over 10 weeks to a total dose of 60 Gy (24, 58). In addition, most GBM patients will receive the chemotherapy drug temozolomide at the same time as radiation therapy as well as in the adjuvant setting in the form of several cycles after completion of radiation therapy (24). We also did not measure tissue ascorbate levels in this study, which should be done in future studies to determine to what extent the injected ascorbate affects ascorbate levels in both brain and tumor tissue. However, although our findings cannot be directly extrapolated to the clinical setting, the underlying problem with combining radiation and high-dose ascorbate remains the same: does the ascorbate get into the glioma cells and act as an anti-oxidant, protecting the tumor from radiation OR does it remain outside the cells, generating hydrogen peroxide and acting as a pro-oxidant. It may be useful in this context to test individual patient tumors for the presence and activity of SVCT2.
In contrast to combining high-dose ascorbate with radiation in brain cancers, clinical studies combining high-dose ascorbate with chemotherapeutic agents for metastatic ovarian and pancreatic cancer have produced more promising results. Adding high-dose ascorbate to existing chemotherapy regimens decreased chemotherapy-related toxicities and increased patient overall survival even if the latter was not statistically significant in these small phase I trials (11, 22, 23). It is important to note, however, that these clinical studies were not done in patients with brain cancers, and included drugs that do not work primarily by generating free radicals. Ascorbate would not have affected the efficacy of these chemotherapy drugs whether it acted as a pro-oxidant or an anti-oxidant.
Author Contributions
Carole Grasso, Marie-Sophie Fabre, and Cameron S. Field conducted and analyzed the intracranial mouse experiments; M. Leticia Castro conducted and analyzed in vitro GL261 experiments; Sarah V. Collis performed the histological analysis. Nanette Schleich designed shielding device and performed the dosimetry. Melanie J. McConnell contributed to experimental design and data interpretation. Patries M. Herst developed the concept and contributed to the design of the lead shielding device, experiments, and data analysis. All authors contributed to writing of and approved of the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the contributions of Mr. Chris Covich who helped design and who built the lead shielding device. The authors further acknowledge the Wellington Blood and Cancer Centre for access to their TLD system. This research was funded by grants from the University of Otago, Genesis Oncology Trust, Neurological Foundation and the Surgical Research Trust (NZ).
Footnotes
References
1. Parrow NL, Leshin JA, Levine M. Parenteral ascorbate as a cancer therapeutic: a reassessment based on pharmacokinetics. Antioxid Redox Signal (2013) 19(17):2141–56. doi: 10.1089/ars.2013.5372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Nishikimi M, Yagi K. Molecular basis for the deficiency in humans of gulonolactone oxidase, a key enzyme for ascorbic acid biosynthesis. Am J Clin Nutr (1991) 54(6):1203S–8S.
3. Harrison FE, May JM. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol Med (2009) 46(6):719–30. doi:10.1016/j.freeradbiomed.2008.12.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol (2008) 19(12):1969–74. doi:10.1093/annonc/mdn377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother Pharmacol (2013) 72(1):139–46. doi:10.1007/s00280-013-2179-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A (2007) 104(21):8749–54. doi:10.1073/pnas.0702854104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Herst PM, Broadley KWR, Harper JL, McConnell MJ. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol Med (2012) 52(8):1486–93. doi:10.1016/j.freeradbiomed.2012.01.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Castro ML, McConnell MJ, Herst PM. Radio-sensitisation by pharmacological ascorbate in glioblastoma multiforme cells, human glial cells and HUVECs depends on their antioxidant and DNA repair capabilities and is not cancer specific. Free Radic Biol Med (2014) 74:200–9. doi:10.1016/j.freeradbiomed.2014.06.022
9. Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, et al. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res (2010) 16(2):509–20. doi:10.1158/1078-0432.CCR-09-1713
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A (2008) 105(32):11105–9. doi:10.1073/pnas.0804226105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Ma Y, Chapman J, Levine M, Polireddy K, Drisko J. High dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci Transl Med (2014) 6(222):222ra18. doi:10.1126/scitranslmed.3007154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, Levine M, et al. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic Biol Med (2011) 50(11):1610–9. doi:10.1016/j.freeradbiomed.2011.03.007
13. Martinotti S, Ranzato E, Parodi M, Vitale M, Burlando B. Combination of ascorbate/epigallocatechin-3-gallate/gemcitabine synergistically induces cell cycle deregulation and apoptosis in mesothelioma cells. Toxicol Appl Pharmacol (2014) 274(1):35–41. doi:10.1016/j.taap.2013.10.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Song EJ, Yang VC, Chiang CD, Chao CC. Potentiation of growth inhibition due to vincristine by ascorbic acid in a resistant human non-small cell lung cancer cell line. Eur J Pharmacol (1995) 292(2):119–25.
15. Vuyyuri SB, Rinkinen J, Worden E, Shim H, Lee S, Davis KR. Ascorbic acid and a cytostatic inhibitor of glycolysis synergistically induce apoptosis in non-small cell lung cancer cells. PLoS One (2013) 8(6):e67081. doi:10.1371/journal.pone.0067081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Olney KE, Du J, van’t Erve TJ, Witmer JR, Sibenaller ZA, Wagner BA, et al. Inhibitors of hydroperoxide metabolism enhance ascorbate-induced cytotoxicity. Free Radic Res (2013) 47(3):154–63. doi:10.3109/10715762.2012.755263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Tomasetti M, Nocchi L, Neuzil J, Goodwin J, Nguyen M, Dong L, et al. Alpha-tocopheryl succinate inhibits autophagic survival of prostate cancer cells induced by vitamin K3 and ascorbate to trigger cell death. PLoS One (2012) 7(12):e52263. doi:10.1371/journal.pone.0052263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Evans MK, Tovmasyan A, Batinic-Haberle I, Devi GR. Mn porphyrin in combination with ascorbate acts as a pro-oxidant and mediates caspase-independent cancer cell death. Free Radic Biol Med (2014) 68:302–14. doi:10.1016/j.freeradbiomed.2013.11.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Rawal M, Schroeder SR, Wagner BA, Cushing CM, Welsh JL, Button AM, et al. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res (2013) 73(16):5232–41. doi:10.1158/0008-5472.CAN-13-0470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Felipe KB, Benites J, Glorieux C, Sid B, Valenzuela M, Kviecinski MR, et al. Antiproliferative effects of phenylaminonaphthoquinones are increased by ascorbate and associated with the appearance of a senescent phenotype in human bladder cancer cells. Biochem Biophys Res Commun (2013) 433(4):573–8. doi:10.1016/j.bbrc.2013.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Frömberg A, Gutsch D, Schulze D, Vollbracht C, Weiss G, Czubayko F, et al. Ascorbate exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells towards cytostatic drugs. Cancer Chemother Pharmacol (2011) 67(5):1157–66. doi:10.1007/s00280-010-1418-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Welsh JL, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol (2013) 71(3):765–75. doi:10.1007/s00280-013-2070-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One (2012) 7(1):e29794. doi:10.1371/journal.pone.0029794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10(5):459–66. doi:10.1016/S1470-2045(09)70025-7
25. Larson EW, Peterson HE, Lamoreaux WT, MacKay AR, Fairbanks RK, Call JA, et al. Clinical outcomes following salvage Gamma Knife radiosurgery for recurrent glioblastoma. World J Clin Oncol (2014) 5(2):142–8. doi:10.5306/wjco.v5.i2.142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Facchino S, Abdouh M, Chatoo W, Bernier G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J Neurosci (2010) 30(30):10096–111. doi:10.1523/JNEUROSCI.1634-10.2010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxidants Redox Signal (2009) 11(12):3013–69. doi:10.1089/ARS.2009.2541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Moser JC, Rawal M, Wagner B, Du J, Cullen JJ, Buettner GR. Pharmacological ascorbate and ionizing radiation (IR) increase labile iron in pancreatic cancer. Redox Biol (2013) 2:22–7. doi:10.1016/j.redox.2013.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Baader SL, Bruchelt G, Carmine TC, Lode HN, Rieth AG, Niethammer D. Ascorbic acid mediated iron release from cellular ferritin and its relation to DNA strand break formation in neuroblastoma cells. J Cancer Res Clin Oncol (1994) 120:415–21. doi:10.1007/BF01240141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Taper HS, Keyeux A, Roberfroid M. Potentiation of radiotherapy by nontoxic pretreatment with combined vitamins C and K3 in mice bearing solid transplantable tumor. Anticancer Res (1996) 16(1):499–503.
31. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc (2006) 1:2315–9. doi:10.1038/nprot.2006.339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Tanaka T, Halicka D, Traganos F, Darzynkiewicz Z. Cytometric analysis of DNA damage: phosphorylation of histone H2AX as a marker of DNA double-strand breaks (DSBs). Methods Mol Biol (2009) 523:161–8. doi:10.1007/978-1-59745-190-1_11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Hunn MK, Farrand KJ, Broadley KWR, Weinkove R, Ferguson P, Miller RJ, et al. Vaccination with irradiated tumor cells pulsed with an adjuvant that stimulates NKT cells is an effective treatment for glioma. Clin Cancer Res (2012) 18(23):6446–59. doi:10.1158/1078-0432.CCR-12-0704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Newcomb EW, Lukyanov Y, Kawashima N, Alonso-Basanta M, Wang SC, Liu M, et al. Radiotherapy enhances antitumor effect of anti-CD137 therapy in a mouse Glioma model. Radiat Res (2010) 173(4):426–32. doi:10.1667/RR1904.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Kron T. Thermoluminescence dosimetry and its applications in medicine – part 1: physics, materials and equipment. Australas Phys Eng Sci Med (1994) 17(4):175–99.
36. DeWerd LA, Bartol LJ, Davis SD. Thermoluminescence Dosimetry. Clinical Dosimetry for Radiotherapy. Colorado: AAPM Summer School (2009). p. 815–40.
37. Nunn AA, Davis SD, Micka JA, DeWerd LA. LiF:Mg,Ti TLD response as a function of photon energy for moderately filtered x-ray spectra in the range of 20–250 kVp relative to 60Co. Med Phys (2008) 35(5):1859. doi:10.1118/1.2898137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Haworth A, Butler DJ, Wilfert L, Ebert MA, Todd SP, Hayton AJ, et al. Comparison of TLD calibration methods for 192Ir dosimetry. J Appl Clin Med Phys (2013) 14(1):258–72. doi:10.1120/jacmp.v14i1.4037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Stegh AH, Chin L, Louis DN, DePinho RA. What drives intense apoptosis resistance and propensity for necrosis in glioblastoma? A role for Bcl2L12 as a multifunctional cell death regulator. Cell Cycle (2008) 7(18):2833–9. doi:10.4161/cc.7.18.6759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Broadley KWR, Hunn MK, Farrand KJ, Price KM, Grasso C, Miller RJ, et al. Side population is not necessary or sufficient for a cancer stem cell phenotype in gliobastoma multiforme. Stem Cells (2011) 29:452–61. doi:10.1002/stem.582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Kim SH, Lee JE, Yang S-H, Lee SW. Induction of cytokines and growth factors by rapamycin in the microenvironment of brain metastases of lung cancer. Oncol Lett (2013) 5(3):953–8. doi:10.3892/ol.2013.1135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kim S-J, Kim J-S, Park ES, Lee J-S, Lin Q, Langley RR, et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia (2011) 13(3):286–98.
43. Jamal M, Rath BH, Tsang PS, Camphausen K, Tofilon PJ. The brain microenvironment preferentially enhances the radioresistance of CD133(+) glioblastoma stem-like cells. Neoplasia (2012) 14(2):150–8.
44. Jamal M, Rath BH, Williams ES, Camphausen K, Tofilon PJ. Microenvironmental regulation of glioblastoma radioresponse. Clin Cancer Res (2010) 16(24):6049–59. doi:10.1158/1078-0432.CCR-10-2435
45. Bürzle M, Suzuki Y, Ackermann D, Miyazaki H, Maeda N, Clémençon B, et al. The sodium-dependent ascorbic acid transporter family SLC23. Mol Aspects Med (2013) 34(2–3):436–54. doi:10.1016/j.mam.2012.12.002
46. Agus DB, Gambhir SS, Pardridge WM, Spielholz C, Baselga J, Vera JC, et al. Vitamin C crosses the blood-brain barrier in the oxidized form through the glucose transporters. J Clin Invest (1997) 100(11):2842–8. doi:10.1172/JCI119832
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bürzle M, Hediger M. Functional and physiological role of vitamin C transporters. Curr Top Membr (2012) 70:357–75. doi:10.1016/B978-0-12-394316-3.00011-9
48. Vissers MC, Gunningham SP, Morrison MJ, Dachs GU, Currie MJ. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic Biol Med (2007) 42(6):765–72. doi:10.1016/j.freeradbiomed.2006.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Pastor P, Cisternas P, Salazar K, Silva-Alvarez C, Oyarce K, Jara N, et al. SVCT2 vitamin C transporter expression in progenitor cells of the postnatal neurogenic niche. Front Cell Neurosci (2013) 7(August):119. doi:10.3389/fncel.2013.00119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Sun L, Hui AM, Su Q, Vortmeyer A, Kotliarov Y, Pastorino S, et al. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell (2006) 9(4):287–300. doi:10.1016/j.ccr.2006.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, et al. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res (2005) 65(19):8679–89. doi:10.1158/0008-5472.CAN-05-1204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Shai R, Shi T, Kremen TJ, Horvath S, Liau LM, Cloughesy TF, et al. Gene expression profiling identifies molecular subtypes of gliomas. Oncogene (2003) 22(31):4918–23. doi:10.1038/sj.onc.1206753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol (2008) 26(18):3015–24. doi:10.1200/JCO.2007.15.7164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Liang Y, Diehn M, Watson N, Bollen AW, Aldape KD, Nicholas MK, et al. Gene expression profiling reveals molecularly and clinically distinct subtypes of glioblastoma multiforme. Proc Natl Acad Sci U S A (2005) 102(16):5814–9. doi:10.1073/pnas.0402870102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Rodríguez FS, Salazar KA, Jara NA, García-Robles MA, Pérez F, Ferrada LE, et al. Superoxide-dependent uptake of vitamin C in human glioma cells. J Neurochem (2013) 127(6):793–804. doi:10.1111/jnc.12365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. McCarty MF. Expression and/or activity of the SVCT2 ascorbate transporter may be decreased in many aggressive cancers, suggesting potential utility for sodium bicarbonate and dehydroascorbic acid in cancer therapy. Med Hypotheses (2013) 81(4):664–70. doi:10.1016/j.mehy.2013.07.023
57. Da Fonseca AC, Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol (2013) 2013:264124. doi:10.1155/2013/264124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Badiyan S, Markovina S, Simpson J, Robinson C, DeWees T, Tran D, et al. Radiation therapy dose escalation for glioblastoma multiforme in the era of temozolomide. Int J Radiat Oncol Biol Phys (2014) 90(4):877–85. doi:10.1016/j.ijrobp.2014.07.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: pharmacological ascorbate, radiation, intracranial mouse glioma model, GL261, radioprotection, radiosensitization
Citation: Grasso C, Fabre M-S, Collis SV, Castro ML, Field CS, Schleich N, McConnell MJ and Herst PM (2014) Pharmacological doses of daily ascorbate protect tumors from radiation damage after a single dose of radiation in an intracranial mouse glioma model. Front. Oncol. 4:356. doi: 10.3389/fonc.2014.00356
Received: 03 August 2014; Accepted: 27 November 2014;
Published online: 15 December 2014.
Edited by:
Margreet C. M. Vissers, University of Otago, New ZealandReviewed by:
Justin Lathia, Cleveland Clinic, USAMin Hee Kang, Texas Tech University Health Sciences Center School of Medicine, USA
Garry R. Buettner, The University of Iowa, USA
Copyright: © 2014 Grasso, Fabre, Collis, Castro, Field, Schleich, McConnell and Herst. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patries M. Herst, Department of Radiation Therapy, University of Otago, P. O. Box 7343, Wellington 6242, New Zealand e-mail:cGF0cmllcy5oZXJzdEBvdGFnby5hYy5ueg==