 Caroline Kuiper
Caroline Kuiper Margreet C. M. Vissers
Margreet C. M. Vissers- Department of Pathology, Centre for Free Radical Research, University of Otago, Christchurch, New Zealand
Ascorbate is a specific co-factor for a large family of enzymes known as the Fe- and 2-oxoglutarate-dependent dioxygenases. These enzymes are found throughout biology and catalyze the addition of a hydroxyl group to various substrates. The proline hydroxylase that is involved in collagen maturation is well known, but in recent times many new enzymes and functions have been uncovered, including those involved in epigenetic control and hypoxia-inducible factor (HIF) regulation. These discoveries have provided crucial mechanistic insights into how ascorbate may affect tumor biology. In particular, there is growing evidence that HIF-1-dependent tumor progression may be inhibited by increasing tumor ascorbate levels. However, rigorous clinical intervention studies are lacking. This review will explore the physiological role of ascorbate as an enzyme co-factor and how this mechanism relates to cancer biology and treatment. The use of ascorbate in cancer should be informed by clinical studies based on such mechanistic hypotheses.
In 1747, James Lind, a surgeon with the British Royal Navy, performed what is considered the first human clinical trial in an attempt to treat scurvy, administering various potential remedies, such as cider, vinegar, or sea water to a group of 12 scurvy-stricken sailors. The only patients to recover were those who received lemons and oranges, but almost 200 years would pass before the curative compound was identified as hexuronic acid, later named ascorbic acid (ascorbate) for its anti-scorbutic (i.e., anti-scurvy) properties (1). This is commonly known today as vitamin C.
Many of the symptoms of scurvy – gum disease, bleeding, and poor wound healing – are considered to be the result of defective collagen production. It was found that the enzyme responsible for the formation of hydroxy-proline, collagen prolyl-4-hydroxylase (C-P4H), specifically required ascorbate as a co-factor for its activity (2). Hence, under conditions of ascorbate-deficiency, C-P4H loses activity, collagen cannot cross-link sufficiently, and connective tissues can deteriorate.
Collagen prolyl-4-hydroxylase belongs to the family of enzymes known as the Fe- and 2-oxoglutarate dependent dioxygenases (2-OGDDs) that have a wide range of biological functions (3) (Table 1). In mammalian cells, members of this family modify hypoxia-inducible factor (HIF) (4), convert dopamine to noradrenaline, are involved in the α-amidation of numerous pro-hormones (5) and carnitine biosynthesis (6, 7). It has more recently been discovered that 2-OGDDs are epigenetic erasers; these enzymes hydroxylate methyl-lysine residues on histones [Jumonji-C domain-containing histone demethylases (JHDMs)] (8), and ten-eleven translocases (TETs) hydroxylate 5-methyl-cytosine (9). In addition, RNA and ribosomal hydroxylases have very recently been characterized (10, 11).
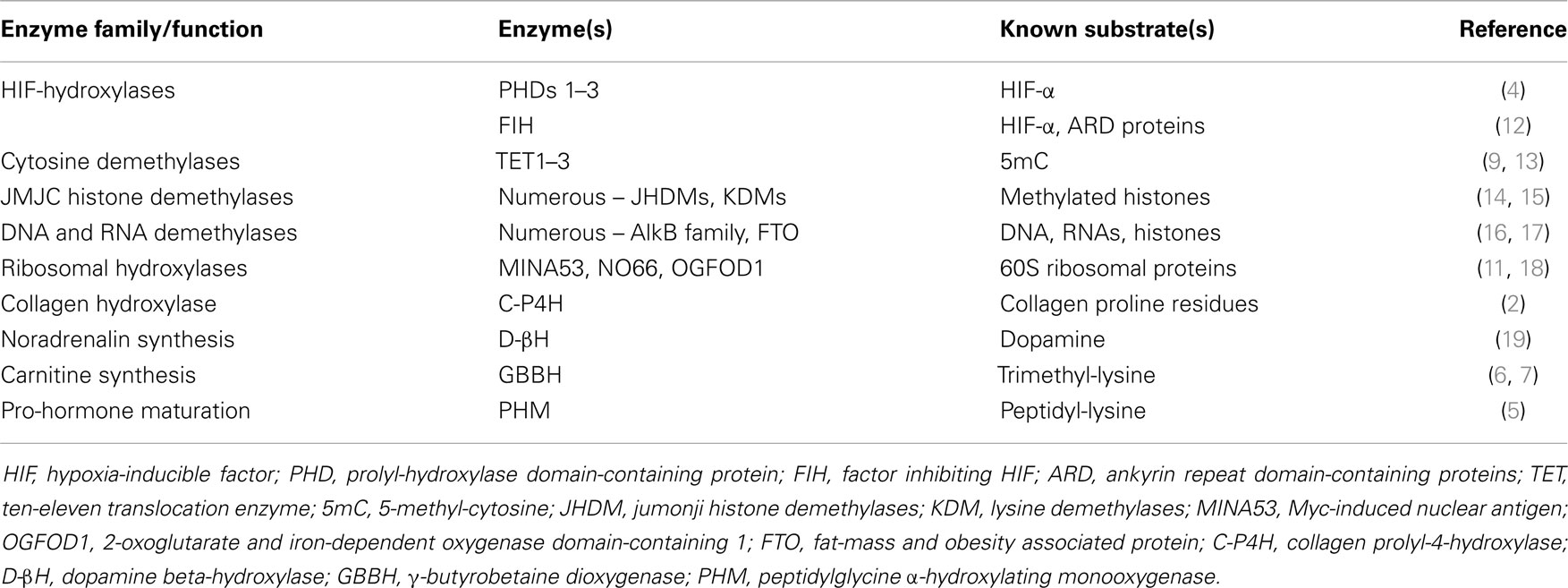
Table 1. Various members of the 2-oxoglutarate dioxygenase family in mammalian cells, their known substrates, and biological functions.
Clearly, the hydroxylation of diverse proteins, DNA and RNA is proving to be a widespread phenomenon. By the addition of a hydroxyl group to their respective substrates, 2-OGDDs are responsible for altering key protein–protein interactions and nucleic acid structure and function that can result in dramatic effects on cell signaling, gene expression, and tissue function. As ascorbate is a specific co-factor for these enzymes, it will be of great interest to determine how ascorbate availability affects their function and the consequences for tumor biology. The following sections will detail the biochemistry of the 2-OGDDs and the activity of ascorbate as a co-factor, and consider whether this activity has any relevance to tumor progression.
Fe- and 2-Oxoglutarate-Dependent Dioxygenases
Despite such large substrate diversity, the catalytic cycle of 2-OGDDs is thought to be highly conserved (3). The enzymes are characterized by a core structural motif consisting of eight β-strands arranged in a “jelly roll” or double-stranded β-helix, surrounded by α-helices (20). The active site within the double-stranded β-helix contains a non-haem iron that is coordinated by a “facial triad” of two histidines and one aspartate/glutamate, with the remaining three coordination sites occupied by labile water molecules (20). This is thought to give the catalytic iron a relatively exposed and flexible arrangement compared to haem oxygenases, allowing for a wide range of catalytic oxidations. However, it may also make the active site more prone to auto-oxidation (21).
The catalytic cycle (Figure 1A) starts with 2-OG binding, allowing entry of the prime substrate and displacing a water molecule from Fe2+. Subsequent binding of molecular O2 to Fe2+ catalyzes the oxidative decarboxylation of 2-OG to succinate and generates a highly reactive ferryl-oxo species (22) that hydroxylates the prime substrate, regenerating Fe3+, which is reverted back to Fe2+ followed by release of the substrates and CO2. One atom of the O2 is incorporated into the hydroxylated substrate and the other into succinate. The CO2 is derived from the oxidative decarboxylation of 2-OG to succinate (3, 20). Ascorbate is a necessary co-factor for this reaction, and although its role remains to be determined, it is thought to be required to maintain Fe2+ in its reduced state.
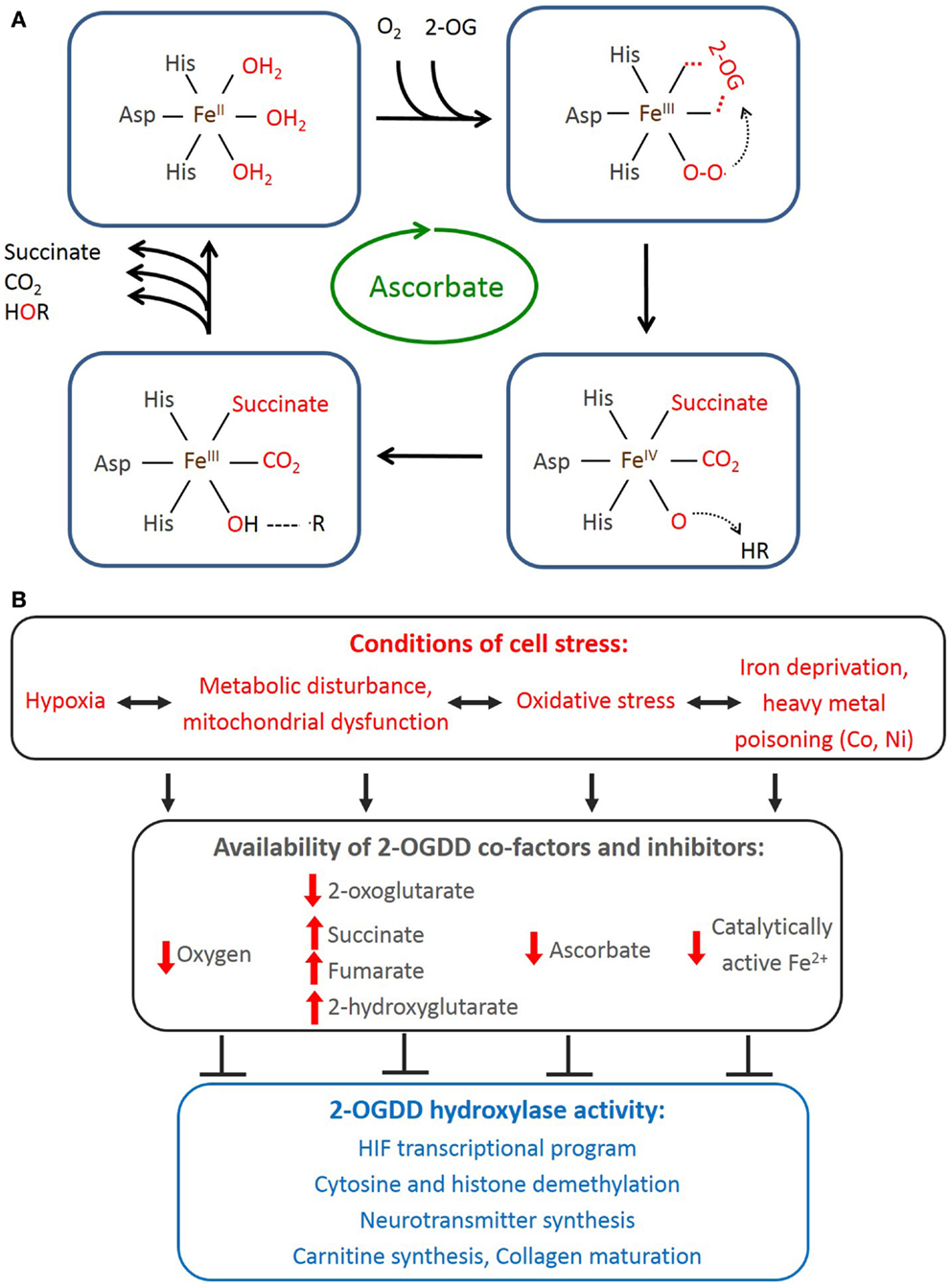
Figure 1. 2-Oxoglutarate-dependent dioxygenase reaction cycle and factors affecting their activity in mammalian cells. (A) Representation of 2-OGDD catalytic cycle. One atom of molecular oxygen is incorporated into the hydroxylated substrate and the other into succinate. 2-OG is converted to succinate, releasing CO2. Ferrous iron and ascorbate are specific co-factors for this reaction. (B) Cellular stressors deprive 2-OGDDs of their required co-factors resulting in inhibition of multiple potential pathways.
Well-characterized members of the 2-OGDD family now include the HIF-hydroxylases (20). These enzymes are thought to have particular relevance to tumor biology due to their direct effects on HIF activation. HIF-1 is a transcription factor known to up-regulate hundreds of genes involved in maintaining oxygen and energy homeostasis under conditions of cell stress, such as hypoxia – a common feature of solid tumors (23). The activation state of HIF-1 is under dual control, with HIF-1α protein stability and transcriptional activity being regulated by hydroxylation reactions. HIF-1α protein is constantly synthesized in most cells in the body, and under normal, physiological conditions is rapidly degraded (24) following hydroxylation of proline residues 402 and 564 by prolyl-hydroxylases (PHD) 1–3 (25, 26) (Table 1). This modification initiates ubiquitination and proteasomal degradation via binding of the tumor-suppressor von Hippel–Lindau protein (pVHL) and an E3 ubiquitin ligase complex (27, 28).
A further hydroxylation event on asparagine 803 of HIF-1α by factor inhibiting HIF (FIH; Table 1) prevents co-activation with p300 and transcriptional activation (29). These events combine to enable a rapid response to cell stressors, where a decrease in hydroxylation activity immediately halts HIF-1α degradation, both allowing the protein to accumulate and also to activate a transcriptional response (20). The activation of HIF-1 is, therefore, dependent upon the activity of the hydroxylases responsible for modification of the HIF-1α subunit.
2-OGDD Regulation and Co-Factor Dependence
The requirements for the 2-OGDD catalytic cycle (O2, Fe2+, 2-oxoglutarate, and ascorbate) provide clues as to the physiological conditions that might affect 2-OGDD activity (Figure 1B).
Although the HIF-hydroxylases clearly respond to changes in cellular O2 levels and are considered direct cellular oxygen sensors, it is currently unknown whether other 2-OG dioxygenases are similarly sensitive. An example of this has recently been described: in tumorigenic neuroblastoma cells, TET1 was shown to be up-regulated under hypoxia via HIF-1, and to cause increased 5hmC levels specifically at HIF-binding sites, suggesting a synergistic relationship (30). This is somewhat counter-intuitive, as hypoxia would also be expected to inhibit TET activity and 5hmC formation. However, whether the TET catalytic cycle is sensitive to O2 changes is yet to be shown. This highlights the complexity of 2-OGDD biology, with each enzyme and isoform having distinct activity dynamics despite sharing a highly conserved active site.
Iron is also a necessary co-factor for 2-OGDD activity, and its substitution/depletion can disrupt HIF-hydroxylase activity and activate the HIF-1 response. Iron chelators such as desferrioxamine (DFO), and Co2+ and Ni2+ ions robustly induce HIF-1, and this is thought to be through poisoning of the hydroxylases via removal of enzyme-bound iron (4, 26, 31). The apparent Km values for Fe2+ are low, with 0.03 μM for PHD1 and 2, 0.1 μM for PHD3 (32) and 0.5 μM for FIH (33). This suggests tight binding of Fe2+ to the HIF-hydroxylase active site, despite the coordination chemistry predicting a labile arrangement (21). However, whether Fe3+ produced during enzymatic cycling has the same tight binding as Fe2+ is unknown.
2-OG (also known as α-ketoglutarate) is an intermediary metabolite of the TCA cycle. Other 2-oxoacids from the TCA cycle, such as succinate and fumarate, can compete with 2-OG to inhibit HIF-hydroxylase activity and induce HIF-1 in vitro (34). Pyruvate, oxaloacetate, and malate have also been shown to have similar HIF-1-inducing effects, and may cause significant basal HIF-1 activation under normoxic conditions (35, 36). HIF-1 activity can be increased by mutations in the TCA cycle enzymes succinate dehydrogenase and fumarate dehydrogenase, which cause a build-up of succinate and fumarate, respectively (37). Furthermore, mutated forms of isocitrate dehydrogenase-1 generate 2-hydroxyglutarate (2-HG) that acts as a competitive inhibitor of the HIF-hydroxylases and activates HIF-1 (38, 39). These mutations have the potential to drive non-hypoxic HIF-1 activity and tumorigenesis (40), and have been clinically associated with the susceptibility to renal cancer and paragangliomas (38). The availability of glycolytic and energy intermediates may confer a complex metabolic-sensing role to the HIF-hydroxylases. However, exactly which 2-oxoacids are relevant under physiological conditions, and to what degree, is unknown.
It has been suggested that the HIF-hydroxylases can be inactivated by production of reactive oxygen species produced by mitochondria in response to hypoxia (41). There is evidence that HIF-hydroxylase activity can be inhibited by H2O2, inducing HIF-1 in a process requiring functional mitochondria, although the precise mechanisms behind this are unclear (35, 42–44). Conversely, mitochondrial H2O2 production has been shown to be decreased in response to hypoxia (45) and another study found no association between levels of reactive oxygen species (DCF fluorescence) and HIF-1 target gene expression (46). FIH was shown to be sensitive to H2O2-mediated inactivation, whereas PHD2 was not, providing an explanation for some of these effects (47), where H2O2 could influence transcriptional inactivation of HIF-1 without affecting its protein stability.
Ascorbate as Regulator of 2-OGDD Activity
The Km values for ascorbate for the HIF-hydroxylases and C-P4H are relatively high [140–300 μM (33, 48, 49)], indicating high intracellular requirements and susceptibility to ascorbate loss. However, the exact role of ascorbate in the hydroxylase reaction is unclear. Most data on ascorbate and hydroxylase activity had come from studies in the 1970–1980s on purified C-P4H in the context of collagen proline-hydroxylation that may also be relevant to other 2-OGDDs (22, 50).
Ascorbate was found to be important, if not essential, for C-P4H activity, with an optimal concentration of 1–2 mM (49, 51–55). In its absence, C-P4H could catalyze only ~15–30 reaction cycles over 8 s before the reaction ceased, and addition of 1 mM ascorbate to the reaction mixture rescued 100% of enzyme activity (2). Another study found C-P4H had a 54% lower initial reaction rate in the absence of ascorbate, which decreased to ~8% of total activity within 30 s with subsequent ascorbate addition able to rescue partial activity (51). Others have found no C-P4H activity without ascorbate, demonstrating an absolute requirement for this co-factor (54, 55). Furthermore, ascorbate was needed for activity prior to 2-OG addition, suggesting that under constant turnover conditions, a permanent ascorbate presence is required (51, 54). Although these results may be influenced by variation in co-substrate concentrations or order of addition, it is clear that ascorbate has a significant effect on hydroxylase activity, with recombinant PHD2 activity having also been shown to increase dose-dependently with ascorbate (56).
Several recent independent studies have demonstrated the specific requirement of ascorbate for TET activity, with a consequent widespread effect on DNA demethylation (57–60). TET enzymes catalyze the conversion of 5mC to 5hmC, which can then be further oxidized and converted to an unmodified (demethylated) cytosine. There was a dose-dependent increase in 5hmC levels of up to fourfold above baseline in ascorbate-treated cells (0–1000 μM) (59, 60), which was mediated by the catalytic domain of TET1 and 2 (60). In addition, the reaction rate of TET2 increased eightfold in the presence of ascorbate (60). Ascorbate-treated mouse embryonic stem cells showed dramatic erasure of 5mC marks (32–40% decrease) over 3 days of treatment (60). These studies indicate that intracellular ascorbate availability is likely to have significant implications for cell reprograming and cancer cell biology.
Mechanism of Ascorbate Activity
The mechanism by which ascorbate enhances hydroxylase activity has been thought to be due to the specific reduction of enzyme-bound Fe3+ to Fe2+. Uncoupled reaction cycles may be the primary cause of iron oxidation and although these represented only 0.7 or 1.25% of total C-P4H activity, this was found to be sufficient to oxidize enzyme-bound iron (49, 55). This oxidation could be reversed by ascorbate (51, 53, 55). Ascorbate is not stoichiometrically consumed during hydroxylase activity (54, 55), but one study found that it was stoichiometrically consumed specifically during uncoupled reaction cycles, supporting its involvement in protecting against this mode of enzyme inactivation (53). However, in another study, using recombinant PHD2, there was a significant increase in ascorbate-reversible Fe3+ only when the prime substrate was added, suggesting coupled turnover may also oxidize the iron (61).
We have shown that ascorbate was able to prevent induction of HIF-1α by Co2+, Ni2+, or iron chelation by DFO (62). DFO specifically chelates Fe3+ (63), which is formed during the hydroxylase reaction cycle, and the prevention of which suggests that ascorbate may prevent free loss of enzyme-bound Fe3+ or its substitution with Co2+ or Ni2+ (64). Ascorbate is known to interact with iron by reducing insoluble ferric iron complexes to stable, soluble, ferrous chelates (65, 66) and can chelate iron intracellularly (67). Together, these studies would support a specific role for ascorbate in both chelating and reducing enzyme-bound Fe3+ during hydroxylase reactions to maintain continued enzyme cycling.
This specificity of ascorbate for optimizing hydroxylase activity has been demonstrated; other reducing agents including glutathione, vitamin E, NADPH, dithiothreitol (DTT), L-cysteine, and tetrahydrofolic acid, were unable to substitute for ascorbate for C-P4H or TET activity (2, 54, 55, 59, 60). In addition, ascorbate was specifically necessary for full substrate hydroxylation with the HIF-hydroxylases (purified recombinant PHD2 and FIH), substantially increasing both the initial rate and extent of HIF-1α domain hydroxylation (68). Glutathione and DTT were not able to substitute for ascorbate, although DTT (68) and supra-physiological concentrations of glutathione in combination with ascorbate (69) can partially enhance PHD2 hydroxylase activity. These studies demonstrate that ascorbate is the most effective reducing agent for hydroxylase activity, with a high degree of specificity.
Ascorbate may also be structurally specific to the hydroxylase active site. There is some support for its direct binding to the active site iron in C-P4H, where it may act as an inner-sphere reductant for the iron (49, 52) and a plausible ascorbate binding site within the enzyme–substrate complex has been reported for a plant member of the 2-OGDD family, anthocyanidin synthase (70). This provides a further possible mechanism for ascorbate, where it may stabilize the enzyme–substrate interaction to facilitate hydroxylase activity. Further to this, a recent study on the AlkB protein indicates it has highly dynamic protein folding depending on whether 2-OG and Fe2+ are bound (71), and it has been suggested that the TET enzymes require ascorbate to assist functional protein folding to enhance catalytic activity (60). The effect of ascorbate structural analogs on PHD2 and FIH activity revealed that the enediol reducing moiety of ascorbate is essential for hydroxylase activity (68), supporting its role in maintaining reduced Fe2+. However, the precise binding position of ascorbate in the hydroxylase reaction cycle is still unclear.
In summary, there is a growing number of 2-OGDD family members with various cellular functions, and in those that have been studied (C-P4H, HIF-hydroxylases, and TETs), it is clear that ascorbate is required to maintain their activity. Whether other 2-OGDDs are similarly dependent on ascorbate for activity, and how that affects the relevant cellular pathways, will be of interest to investigate.
Ascorbate Biochemistry and Pharmacokinetics
2-oxoglutarate dependent dioxygenases appear to require above 1 mM intracellular ascorbate for optimal activity, and interestingly, most cells in the body will accumulate these levels under normal, healthy physiological conditions. In most organisms, ascorbate is synthesized from glucose. Animals produce ascorbate in the liver or kidneys, from which it is transported to the plasma for distribution to the rest of the body (72). However, primates, including humans, as well as guinea pigs and some species of bat are unable to synthesize it due to evolutionary loss of the terminal synthetic enzyme, gulonolactone oxidase (72). Therefore, ascorbate is an essential nutrient (vitamin C) that we must obtain from our diet.
Many of the known functions of ascorbate are attributable to its action as an electron donor. Ascorbate has two ionizable hydroxyl groups with pKa values of 4.2 and 11.6, meaning at physiological pH, it is present as the ascorbate monoanion (Figure 2) (73). It readily undergoes two consecutive, reversible, one-electron oxidations, resulting in the ascorbate radical and dehydroascorbate (DHA; Figure 2) (73). Ascorbate is an excellent antioxidant, both thermodynamically and kinetically, is able to neutralize many highly reactive oxidizing species, and is therefore known as a terminal small molecule antioxidant (74).
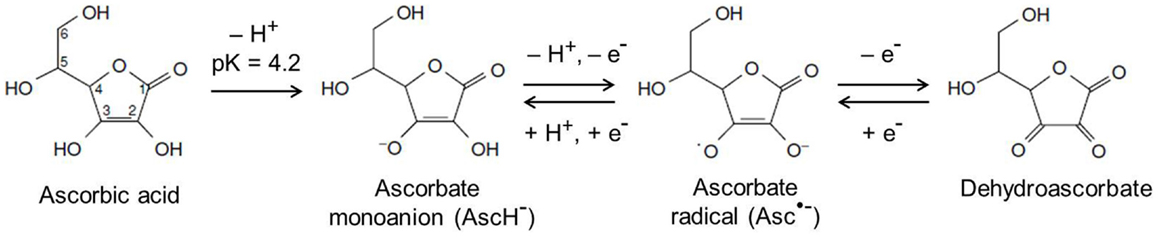
Figure 2. Chemical structures of ascorbate and its oxidation products. At physiological pH, ascorbate exists as the ascorbate monoanion and can undergo two consecutive, reversible, one-electron oxidations to produce the ascorbate radical and dehydroascorbate, respectively. Adapted from Kall (73).
Tissue ascorbate levels vary significantly and this is generally thought to reflect a functional requirement. The highest levels are found in the adrenal medulla where noradrenaline is synthesized and in the pituitary gland where many hormones are produced (75). The brain also has high levels and is the last organ to be depleted during deficiency (76), and many other tissues, including white blood cells, contain high concentrations. Tissue ascorbate levels are maintained by specific active transporters identified as sodium-dependent vitamin C transporters (SVCTs), with two known isoforms – SVCT1 that is specifically involved in gastrointestinal absorption and renal reabsorption, and SVCT2 that expressed in most tissues and is thought to be responsible for whole body cellular uptake (77, 78).
Plasma ascorbate saturation from gastrointestinal absorption is ~100 μM and the SVCTs transport it against a concentration gradient resulting in millimolar intracellular concentrations (79–81). Final intracellular ascorbate levels depend on the circulating plasma concentration available to the cells for uptake. The apparent Km values for the SVCTs have been determined in various cultured human cell lines, and range between 65 and 237 μM for SVCT1 and 8–62 μM for SVCT2, the primary mediator of intracellular uptake (82). An optimal plasma level to achieve tissue saturation is ~70–80 μM, which corresponds to a dietary intake of ~200 mg per day and tissue accumulation becomes significantly impaired if plasma levels fall below ~20 μM (83). This suggests that access to concentrations in the range of those found in plasma will substantially affect intracellular levels. Interestingly, several studies have shown that cancer patients have significantly lower plasma ascorbate levels compared to healthy controls (84–89) and this may limit tumor cell uptake.
Ascorbate in Tumor Tissue
The ascorbate content of tumor tissue has been measured in some studies as early as the 1970s, with variable results. Brain and colorectal tumors contained significantly less ascorbate than normal tissue (90–92) whereas breast (93), oral (87), skin (94), and lung (84) cancers had significantly more ascorbate than corresponding normal tissues.
More recent studies from our lab have shown that both endometrial and colorectal tumors of high histological grade had less ascorbate than matched, adjacent normal tissue (95, 96). This may be due to a disorganized vessel network common in high grade tumors, or may reflect a reduction in expression or activity of SVCT2, which is known to vary among cancer cell lines (97), but to our knowledge, has not been examined in human cancer tissue.
Given the difficulty of accessing the plasma supply, it is possible that delivery of ascorbate to tumor cells may be compromised. Plasma levels are tightly controlled by the SVCTs, limiting absorption and reabsorption at both the intestine and kidney and do not normally exceed ~100 μM with dietary intake (83). Intravenous administration of ascorbate bypasses this tight control and can yield plasma levels up to 100-fold higher with maximum levels of up to 15 mM (98). Whether these supra-physiological concentrations would significantly increase delivery to tumor cells or cellular uptake is unknown, but investigation of this possibility may be of particular relevance to cancer.
Potential 2-OGDD-Mediated Effects of Ascorbate on Tumor Biology
Ascorbate and HIF-1 Regulation
Several in vitro studies have shown that ascorbate (25–1000 μM in culture medium) can suppress HIF-1α protein stabilization and transcriptional activity due to CoCl2 (62, 99, 100), DFO (62, 100), and insulin-like growth factor or insulin (100). The response to hypoxia is less clear, with some of these studies reporting that ascorbate could inhibit HIF-1 at 1–3% O2 (62, 99–101), while other results showed no effect of ascorbate at ≤1% O2 (36, 100). Ascorbate was also able to completely block basal HIF-1α present in oncogenically activated cells that had mutant p53 and PTEN, indicating that it is able to enhance HIF-hydroxylase catalytic capacity in order to cope with the increase in HIF-1α synthesis (100).
One in vivo study has measured xenograft growth in mice of P493 cells that constitutively expressed a mutant, stabilized form of HIF-1α (102). Oral ascorbate supplementation of the animals resulted in significant inhibition of wild-type tumor growth, with no effect on mutant HIF-1α tumors (102). Interestingly, the mice in this study were able to synthesize their own ascorbate, and the control group also had reduced tumor growth compared to the mutant HIF-1α group (102). This indicates that ascorbate can inhibit HIF-1-mediated tumor growth in mice, and that higher concentrations have a greater effect. However, the plasma and tissue concentrations were not reported. To our knowledge, this is the only animal study to determine the HIF-1-dependent effects of ascorbate on tumor growth, and more data measuring HIF-1 and tumor ascorbate levels in a similar model would be particularly valuable.
We have correlated tumor ascorbate content with HIF-1 activation in human tumor tissue and, to our knowledge, these are the only studies to date to have done so. Strikingly similar patterns were seen in endometrial and colorectal cancer, with a significant inverse relationship between tumor ascorbate levels and HIF-1 activation (95, 96). We also measured disease-free survival in the colorectal cancer cohort and found that patients who had low tumor ascorbate content had shorter disease-free survival (95). This relationship was seen in a relatively small group of 50 patients, and was independent of tumor grade and stage, indicating that ascorbate may play a significant role in curbing tumor progression. Interestingly, the ascorbate content of adjacent normal tissue was not related to disease-free survival, suggesting that it is the ascorbate content of the tumor itself that is important (95). The precise mechanism behind this observation requires further clinical investigation including measurement of various 2-OGDD-regulated pathways in relation to changes in tumor ascorbate content.
Ascorbate and Epigenetic Reprograming
The regulation of HIF-1 is likely to contribute to the anti-tumor activity of ascorbate (102). However, the recent discovery of the dependence of the TET enzymes on ascorbate for activity indicates that it may also act to epigenetically reprogram cancer cells. Ascorbate was observed to enhance generation of mouse and human pluripotent stem cells (103). Subsequently, the histone demethylase Jmjd1 was found to be involved in the ascorbate-dependent demethylation of pluripotency genes (104), and furthermore, modulation of TET activity by ascorbate has been implicated in somatic cell reprograming (57, 58). This has become a highly complex field, with dynamic interplay between TET expression levels and ascorbate availability (58), and also TET expression and HIF-1 activity (30). This raises interesting questions about how ascorbate levels in cancer cells could affect the epigenetic phenotype. In addition, as hypoxia may be coincident with ascorbate-deficiency, this could alter the response significantly.
The JHDM and AlkB sub-families of 2-OGDDs are also potential regulators of these processes, and it is unknown how many of them are affected by changes in availability of co-factors. Ascorbate would seem to have broad range of potential targets in a cancer cell, many of which may be currently uncharacterized, where the overall effect could “tip the balance” of signals and push cancer cells toward cell death.
Ascorbate and Cancer
Ascorbate has a controversial history in relation to cancer treatment. In the 1970s, Ewan Cameron and Linus Pauling used both intravenous and oral ascorbate to treat 100 advanced cancer patients and found significant improvements in survival time and quality of life (105, 106). Subsequently, the Mayo Clinic sought to further investigate their findings by performing randomized, double-blind, placebo-controlled studies, using only oral ascorbate, initially with 60 advanced cancer patients who had received prior chemotherapy (107), or later in 100 colorectal cancer patients who had not received prior treatment (108). These clinical trials found no difference between ascorbate and the placebo, and ascorbate as a cancer therapy was effectively dismissed.
The recent discovery that intravenous ascorbate administration can provide 100-fold higher plasma levels compared to oral intake has renewed interest in its potential in treating cancer (98). Four credible case studies (including prostate cancer, renal cell carcinoma, bladder cancer, and B-cell lymphoma) have been reported showing intravenous ascorbate had substantial anti-cancer activity in these advanced cancer patients (109, 110), three of which were evaluated in accordance with the National Cancer Institute Best Case Series guidelines (110). However two recent studies, both with 24 patients and no control population, have shown no objective response, with the exception of three patients who had stable disease (111, 112). None of these studies have monitored tissue ascorbate levels or any biological markers other than toxicity parameters, and have been performed in an unselected patient population. Nevertheless, studies have found that high-dose ascorbate is remarkably non-toxic and well-tolerated in patients with normal renal function (111–113), a rare and valuable trait in a potential cancer therapy. This, together with evidence that some cancer patients may benefit from ascorbate treatment, should be grounds for further rigorous clinical investigations, based on sound hypotheses.
The renewed interest for a role for ascorbate in cancer has generated a growing body of in vitro and animal studies to determine its effectiveness against cancer cell survival and tumor growth, particularly using high millimolar doses (79). One hypothesis to support the use of high-dose ascorbate is the finding that at these pharmacological concentrations, it may act as a prodrug to deliver extracellular H2O2 that is selectively toxic to cancer cells (114, 115).
Recent animal studies have investigated the effect of ascorbate and tumor growth, all using different dosing regimens and tumor models (116–120). However, they have consistently shown an anti-tumor effect of ascorbate supplementation in mice. Mouse studies that used pharmacological ascorbate dosing (intravenous or intraperitoneal) showed a reduction in tumor growth rate and volume (116, 119–121). Other studies have used the Gulo−/− mouse model, in which the animals cannot synthesize ascorbate, to investigate the effect of physiological ascorbate levels (117, 118), and have also shown inhibition of tumor growth in orally ascorbate-supplemented mice. A very recent Gulo−/− study showed that physiological ascorbate supplementation dramatically reduced tumor metastases and necrosis, features that were associated with MMP-9 (a HIF-1 target gene) expression and tumor invasiveness (122). Serum VEGF levels were also markedly reduced (122). These mouse studies support a role for ascorbate in inhibiting tumor progression, and although the mechanisms behind some of these effects may need further clarification, the inhibition of HIF-1 is likely to contribute (102) (Figure 3).
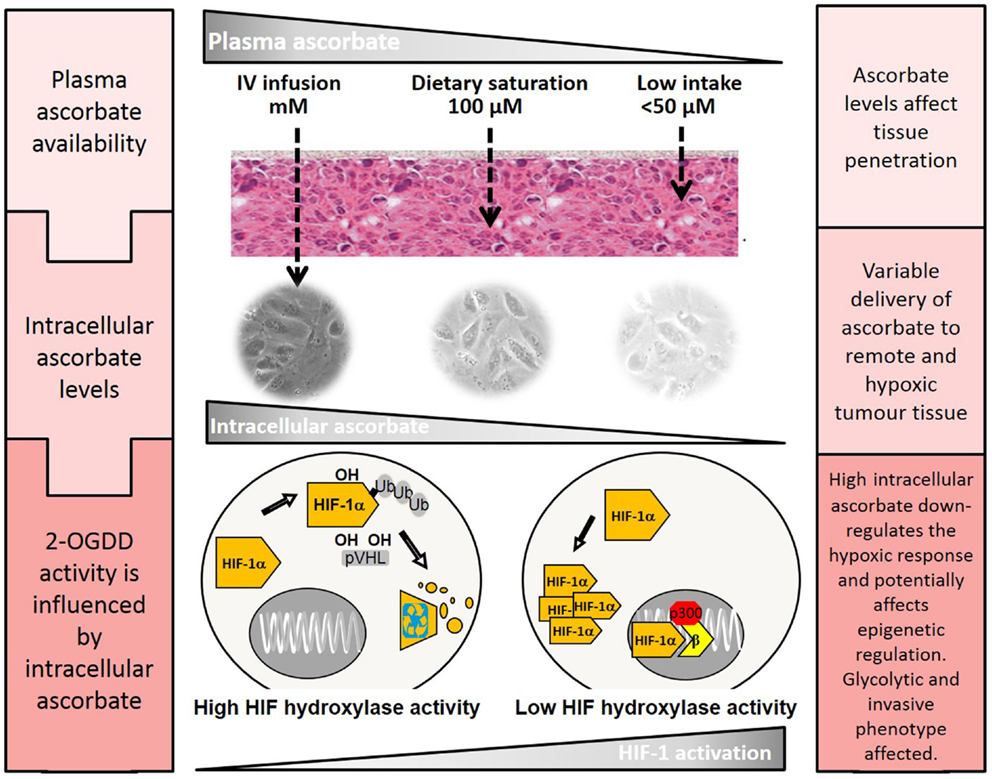
Figure 3. The effect of plasma ascorbate availability on delivery to remote tumor tissue and activity of the HIF-hydroxylases. Higher plasma ascorbate results in increased penetration of remote and hypoxic regions and the ability to down-regulate HIF-1 by promotion of the regulatory hydroxylases. HIF-1 mediated changes that regulate the tumor glycolytic phenotype, cell survival pathways, and angiogenesis could be affected, decreasing tumor viability and improving treatment outcomes. The epigenetic demethylases that also belong to the 2-OGDD family are also likely to be affected but little is known about these processes in cancer cells.
The promise of ascorbate in treating cancer may lie in its combined use with other chemo-therapeutics. HIF-1 is known to be a driver of both chemo- and radio-resistance (123) and boosting intracellular ascorbate levels in the tumor may inhibit this effect and enhance the effectiveness of current treatments. There has been some concern that ascorbate, which is a versatile antioxidant, may in fact counteract the oxidative damage against cancer cells caused by some current cancer therapies, thereby limiting their effectiveness (124). However, recent in vitro and in vivo studies have shown that ascorbate can, in fact, enhance the effectiveness of chemotherapy (120, 125, 126). Pre-treatment of prostate cancer cells with physiological ascorbate concentrations significantly reduced the IC50 values of docetaxel and 5-fluorouracil (126) and pharmacological dosing of ascorbate in mice synergized with gemcitabine resulting in significantly inhibited tumor growth (125). In addition, a recent phase I clinical trial of high-dose intravenous ascorbate with gemcitabine and erlotinib in advanced pancreatic cancer has shown no adverse effects of including ascorbate (127). To our knowledge, no studies have examined the effect of intracellular ascorbate on HIF-1-induced treatment resistance. It would be of interest to study the response of tumor cells with an active HIF-1 response to a range of chemo-therapeutics, and monitor drug effectiveness in ascorbate-deficient or pre-loaded cells.
Conclusion
The control of the 2-OGDDs by ascorbate has the potential to impact tumor growth at all stages of tumor progression. Having a high tissue ascorbate level could help prevent formation of solid tumors, slow tumor growth rates, inhibit aggressive tumor behavior, and even aid in the treatment of established cancers. Despite growing interest in ascorbate as a cancer treatment, there remains a great deal of controversy over its clinical use. However, an objective evaluation of data obtained from systematic inquiry, with an understanding of the underlying mechanisms would provide valuable insights to inform the debate. The studies summarized here clearly indicate that further investigation of the use of ascorbate in enhancing 2-OGDD activity and combating tumor progression is warranted.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Myllylä R, Kuutti-Savolainen E-R, Kivirikko KI. The role of ascorbate in the prolyl hydroxylase reaction. Biochem Biophys Res Commun (1978) 83:441–8. doi:10.1016/0006-291X(78)91010-0
3. Loenarz C, Schofield CJ. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci (2011) 36:7–18. doi:10.1016/j.tibs.2010.07.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol (2004) 5:343–54. doi:10.1038/nrm1366
5. Ball GFM. Ascorbic acid: physiology. In: Caballero B, editor. Encyclopedia of Food Sciences and Nutrition. Oxford: Academic Press (2003). p. 324–32.
6. Hulse JD, Ellis SR, Henderson LM. Carnitine biosynthesis. beta-hydroxylation of trimethyllysine by an alpha-ketoglutarate-dependent mitochondrial dioxygenase. J Biol Chem (1978) 253:1654–9.
7. Lindstedt G, Lindstedt S. Cofactor requirements of γ-butyrobetaine hydroxylase from rat liver. J Biol Chem (1970) 245:4178–86.
8. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature (2006) 439:811–6. doi:10.1038/nature04433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science (2009) 324:930–5. doi:10.1126/science.1170116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Zhao X, Yang Y, Sun B-F, Zhao Y-L, Yang Y-G. FTO and obesity: mechanisms of association. Curr Diab Rep (2014) 14:1–9. doi:10.1007/s11892-014-0486-0
11. Singleton RS, Liu-Yi P, Formenti F, Ge W, Sekirnik R, Fischer R, et al. OGFOD1 catalyzes prolyl hydroxylation of RPS23 and is involved in translation control and stress granule formation. Proc Natl Acad Sci U S A (2014) 111:4031–6. doi:10.1073/pnas.1314482111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science (2002) 295:858–61. doi:10.1126/science.1068592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature (2010) 466:1129–33. doi:10.1038/nature09303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Monfort A, Wutz A. Breathing-in epigenetic change with vitamin C. EMBO Rep (2013) 14:337–46. doi:10.1038/embor.2013.29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science (2009) 325:90–3. doi:10.1126/science.1175865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Zheng G, Dahl JA, Niu Y, Fu Y, Klungland A, Yang YG, et al. Sprouts of RNA epigenetics: the discovery of mammalian RNA demethylases. RNA Biol (2013) 10:915–8. doi:10.4161/rna.24711
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Ma M, Harding HP, O’Rahilly S, Ron D, Yeo GS. Kinetic analysis of FTO (fat mass and obesity-associated) reveals that it is unlikely to function as a sensor for 2-oxoglutarate. Biochem J (2012) 444:183–7. doi:10.1042/BJ20120065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Ge W, Wolf A, Feng T, Ho CH, Sekirnik R, Zayer A, et al. Oxygenase-catalyzed ribosome hydroxylation occurs in prokaryotes and humans. Nat Chem Biol (2012) 8:960–2. doi:10.1038/nchembio.1093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Diliberto EJJ, Daniels AJ, Viveros OH. Multicompartmental secretion of ascorbate and its dual role in dopamine beta-hydroxylation. Am J Clin Nutr (1991) 54:1163S–72S.
20. Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol (2007) 3:144–53. doi:10.1038/nchembio863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Mantri M, Zhang Z, McDonough MA, Schofield CJ. Autocatalysed oxidative modifications to 2-oxoglutarate dependent oxygenases. FEBS J (2012) 279:1563–75. doi:10.1111/j.1742-4658.2012.08496.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Krebs C. Direct spectroscopic detection of a C-H-cleaving high-spin Fe(IV) complex in a prolyl-4-hydroxylase. Proc Natl Acad Sci U S A (2006) 103:14738–43. doi:10.1073/pnas.0604005103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood (2011) 117:e207–17. doi:10.1182/blood-2010-10-314427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J (2003) 22:4082–90. doi:10.1093/emboj/cdg392
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science (2001) 294:1337–40. doi:10.1126/science.1066373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell (2001) 107:43–54. doi:10.1016/S0092-8674(01)00507-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-1α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science (2001) 292:468–72. doi:10.1126/science.1059796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Yu F, White SB, Zhao Q, Lee FS. HIF-1α binding to VHL is regulated by stimulus-sensitive prolyl hydroxylation. Proc Natl Acad Sci U S A (2001) 98:9630–5. doi:10.1073/pnas.181341498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev (2002) 16:1466–71. doi:10.1101/gad.991402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y, et al. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep (2014) 7:1343–52. doi:10.1016/j.celrep.2014.04.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis (1999) 20:1819–23. doi:10.1093/carcin/20.9.1819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hirsilä M, Koivunen P, Xu L, Seeley T, Kivirikko KI, Myllyharju J. Effect of desferrioxamine and metals on the hydroxylases in the oxygen sensing pathway. FASEB J (2005) 19:1308–10. doi:10.1096/fj.04-3399fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Koivunen P, Hirsilä M, Günzler V, Kivirikko KI, Myllyharju J. Catalytic properties of the asparagine hydroxylase (FIH) in the oxygen sensing pathway are distinct from those of its prolyl 4-hydroxylases. J Biol Chem (2004) 279:9899–904. doi:10.1074/jbc.M312254200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Koivunen P, Hirsilä M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem (2007) 282:4524–32. doi:10.1074/jbc.M610415200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, et al. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol (2007) 27:912–25. doi:10.1128/MCB.01223-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Lu H, Dalgard CL, Mohyeldin A, McFate T, Tait AS, Verma A. Reversible inactivation of HIF-1 prolyl hydroxylases allows cell metabolism to control basal HIF-1. J Biol Chem (2005) 280:41928–39. doi:10.1074/jbc.M508718200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Pollard PJ, Brière JJ, Alam NA, Barwell J, Barclay E, Wortham NC, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1α in tumours which result from germline FH and SDH mutations. Hum Mol Genet (2005) 14:2231–9. doi:10.1093/hmg/ddi227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Pollard PJ, Ratcliffe PJ. Puzzling patterns of predisposition. Science (2009) 324:192–4. doi:10.1126/science.1173362
39. Losman J-A, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev (2013) 27:836–52. doi:10.1101/gad.217406.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev (2010) 20:51–6. doi:10.1016/j.gde.2009.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab (2005) 1:393–9. doi:10.1016/j.cmet.2005.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia. J Biol Chem (2000) 275:25130–8. doi:10.1074/jbc.M001914200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Kaelin WG Jr. ROS: really involved in oxygen sensing. Cell Metab (2005) 1:357–8. doi:10.1016/j.cmet.2005.05.006
44. Qutub AA, Popel AS. Reactive oxygen species regulate hypoxia-inducible factor 1α differentially in cancer and ischemia. Mol Cell Biol (2008) 28:5106–19. doi:10.1128/MCB.00060-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Hoffman DL, Salter JD, Brookes PS. Response of mitochondrial reactive oxygen species generation to steady-state oxygen tension: implications for hypoxic cell signaling. Am J Physiol Heart Circ Physiol (2007) 292:H101–8. doi:10.1152/ajpheart.00699.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Salnikow K, Su W, Blagosklonny MV, Costa M. Carcinogenic metals induce hypoxia-inducible factor-stimulated transcription by reactive oxygen species-independent mechanism. Cancer Res (2000) 60:3375–8.
47. Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, et al. The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep (2012) 13:251–7. doi:10.1038/embor.2012.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem (2003) 278:30772–80. doi:10.1074/jbc.M304982200
49. Myllyharju J, Kivirikko KI. Characterization of the iron- and 2-oxoglutarate-binding sites of human prolyl 4-hydroxylase. EMBO J (1997) 16:1173–80. doi:10.1093/emboj/16.6.1173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J Inorg Biochem (2006) 100:644–69. doi:10.1016/j.jinorgbio.2006.01.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. De Jong L, Albracht SPJ, Kemp A. Prolyl 4-hydroxylase activity in relation to the oxidation state of enzyme-bound iron: the role of ascorbate in peptidyl proline hydroxylation. Biochim Biophys Acta (1982) 704:326–32. doi:10.1016/0167-4838(82)90162-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Majamaa K, Gunzler V, Hanauske-Abel HM, Myllylä R, Kivirikko KI. Partial identity of the 2-oxoglutarate and ascorbate binding sites of prolyl 4-hydroxylase. J Biol Chem (1986) 261:7819–23.
53. Myllylä R, Majamaa K, Günzler V, Hanauske-Abel HM, Kivirikko KI. Ascorbate is consumed stoichiometrically in the uncoupled reactions catalyzed by prolyl 4-hydroxylase and lysyl hydroxylase. J Biol Chem (1984) 259:5403–5.
54. Nietfeld JJ, Kemp A. The function of ascorbate with respect to prolyl 4-hydroxylase activity. Biochim Biophys Acta (1981) 657:159–67. doi:10.1016/0005-2744(81)90139-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Tuderman L, Myllylä R, Kivirikko KI. Mechanism of the prolyl hydroxylase reaction. Eur J Biochem (1977) 80:341–8. doi:10.1111/j.1432-1033.1977.tb11888.x
56. Dao JH, Kurzeja RJ, Morachis JM, Veith H, Lewis J, Yu V, et al. Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology. Anal Biochem (2009) 384:213–23. doi:10.1016/j.ab.2008.09.052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Blaschke K, Ebata KT, Karimi MM, Zepeda-Martínez JA, Goyal P, Mahapatra S, et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature (2013) 500:222–6. doi:10.1038/nature12362
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Chen J, Guo L, Zhang L, Wu H, Yang J, Liu H, et al. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat Genet (2013) 45:1504–9. doi:10.1038/ng.2807
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Minor EA, Court BL, Young JI, Wang G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J Biol Chem (2013) 288:13669–74. doi:10.1074/jbc.C113.464800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Yin R, Mao SQ, Zhao B, Chong Z, Yang Y, Zhao C, et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J Am Chem Soc (2013) 135:10396–403. doi:10.1021/ja4028346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. McNeill LA, Flashman E, Buck MR, Hewitson KS, Clifton IJ, Jeschke G, et al. Hypoxia-inducible factor prolyl hydroxylase 2 has a high affinity for ferrous iron and 2-oxoglutarate. Mol Biosyst (2005) 1:321–4. doi:10.1039/b511249b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Kuiper C, Dachs GU, Currie MJ, Vissers MCM. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Rad Biol Med (2014) 69:308–17. doi:10.1016/j.freeradbiomed.2014.01.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Goodwin JF, Whitten CF. Chelation of ferrous sulphate solutions by desferrioxamine B. Nature (1965) 205:281–3. doi:10.1038/205281b0
64. Kaczmarek M, Cachau RE, Topol IA, Kasprzak KS, Ghio A, Salnikow K. Metal ions-stimulated iron oxidation in hydroxylases facilitates stabilization of HIF-1α protein. Toxicol Sci (2009) 107:394–403. doi:10.1093/toxsci/kfn251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Hallberg L, Brune M, Rossander L. The role of vitamin C in iron absorption. Int J Vitam Nutr Res Suppl (1989) 30:103–8.
66. Lynch SR, Cook JD. Interaction of vitamin C and iron. Ann N Y Acad Sci (1980) 355:32–44. doi:10.1111/j.1749-6632.1980.tb21325.x
67. May JM, Qu Z-C. Chelation of intracellular iron enhances endothelial barrier function: a role for vitamin C? Arch Biochem Biophys (2010) 500:162–8. doi:10.1016/j.abb.2010.05.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Flashman E, Davies SL, Yeoh KK, Schofield CJ. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem J (2010) 427:135–42. doi:10.1042/BJ20091609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Nytko KJ, Maeda N, Schläfli P, Spielmann P, Wenger RH, Stiehl DP. Vitamin C is dispensable for oxygen sensing in vivo. Blood (2011) 117:5485–93. doi:10.1182/blood-2010-09-307637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Wilmouth RC, Turnbull JJ, Welford RWD, Clifton IJ, Prescott AG, Schofield CJ. Structure and mechanism of anthocyanidin synthase from Arabidopsis thaliana. Structure (2002) 10:93–103. doi:10.1016/S0969-2126(01)00695-5
71. Bleijlevens B, Shivarattan T, van den Boom KS, de Haan A, van der Zwan G, Simpson PJ, et al. Changes in protein dynamics of the DNA repair dioxygenase AlkB upon binding of Fe2+ and 2-oxoglutarate. Biochemistry (2012) 51:3334–41. doi:10.1021/bi201699e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Linster CL, Van Schaftingen E. Vitamin C: biosynthesis, recycling and degradation in mammals. FEBS J (2007) 274:1–22. doi:10.1111/j.1742-4658.2006.05607.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Kall MA. Ascorbic acid: properties and determination. In: Caballero B, editor. Encyclopedia of Food Sciences and Nutrition. Oxford: Academic Press (2003). p. 316–24.
74. Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res (1996) 145:532–41. doi:10.2307/3579271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Hornig D. Distribution of ascorbic acid, metabolites and analogues in man and animals. Annal N Y Acad Sci (1975) 258:103–18. doi:10.1111/j.1749-6632.1975.tb29271.x
76. May JM. Vitamin C transport and its role in the central nervous system. In: Stanger O, editor. Water Soluble Vitamins. (Vol. 56), Netherlands: Springer (2012). p. 85–103.
77. Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang Y, et al. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature (1999) 399:70–5. doi:10.1038/19986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch Biochem Biophys (2010) 500:107–15. doi:10.1016/j.abb.2010.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta (2012) 1826:443–57. doi:10.1016/j.bbcan.2012.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. May JM. The SLC23 family of ascorbate transporters: ensuring that you get and keep your daily dose of vitamin C. Br J Pharmacol (2011) 164:1793–801. doi:10.1111/j.1476-5381.2011.01350.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Evans RM, Currie L, Campbell A. The distribution of ascorbic acid between various cellular components of blood, in normal individuals, and its relation to the plasma concentration. Br J Nutr (1982) 47:473–82. doi:10.1079/BJN19820059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Savini I, Rossi A, Pierro C, Avigliano L, Catani MV. SVCT1 and SVCT2: key proteins for vitamin C uptake. Amino Acids (2008) 34:347–55. doi:10.1007/s00726-007-0555-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A (1996) 93:3704–9. doi:10.1073/pnas.93.8.3704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Anthony HM, Schorah CJ. Severe hypovitaminosis C in lung-cancer patients: the utilization of vitamin C in surgical repair and lymphocyte-related host resistance. Br J Cancer (1982) 46:354–67. doi:10.1038/bjc.1982.211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Caliskan-Can E, Firat H, Ardiç S, Simsek B, Torun M, Yardim-Akaydin S. Increased levels of 8-hydroxydeoxyguanosine and its relationship with lipid peroxidation and antioxidant vitamins in lung cancer. Clin Chem Lab Med (2008) 46:107–12. doi:10.1515/CCLM.2008.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Choi M-A, Kim B-S, Yu R. Serum antioxidative vitamin levels and lipid peroxidation in gastric carcinoma patients. Cancer Lett (1999) 136:89–93. doi:10.1016/S0304-3835(98)00312-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Fiaschi AI, Cozzolino A, Ruggiero G, Giorgi G. Glutathione, ascorbic acid and antioxidant enzymes in the tumour tissue and blood of patients with oral squamous cell carcinoma. Eur Rev Med Pharamcol Sci (2005) 9:361–7.
88. Lima de Araújo L, Maciel Barbosa J, Gomes Ribeiro AP, Oliveira dos Santos AC, Pedrosa F. Nutritional status, dietary intake and serum levels of vitamin C upon diagnosis of cancer in children and adolescents. Nutr Hosp (2012) 27:496–503. doi:10.1590/S0212-16112012000200022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Mayland CR, Bennett MI, Allan K. Vitamin C deficiency in cancer patients. Palliat Med (2005) 19:17–20. doi:10.1191/0269216305pm970oa
90. Dudek H, Farbiszewski R, Rydzewska M, Michno T, Kozłowksi A. Evaluation of antioxidant enzymes activity and concentration of non-enzymatic antioxidants in human brain tumours. Wiad Lek (2004) 57:16–9.
91. Landolt H, Langemann H, Probst A, Gratzl O. Levels of water-soluble antioxidants in astrocytoma and in adjacent tumor-free tissue. J Neurooncol (1994) 21:127–33. doi:10.1007/BF01052896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Skrzydlewska E, Stankiewicz A. Antioxidant status and lipid peroxidation in colorectal cancer. J Toxicol Env Heal A (2001) 64:213–22. doi:10.1080/15287390152543690
93. Langemann H, Torhorst J, Kabiersch A, Krenger W, Honegger CG. Quantitative determination of water- and lipid-soluble antioxidants in neoplastic and non-neoplastic human breast tissue. Int J Cancer (1989) 43:1169–73. doi:10.1002/ijc.2910430634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Moriarty M, Mulgrew S, Malone J, O’Connor M. Results and analysis of tumour levels of ascorbic acid. Ir J Med Sci (1977) 146:74–8. doi:10.1007/BF03030933
95. Kuiper C, Dachs GU, Munn D, Currie MJ, Robinson BA, Pearson JF, et al. Increased tumour ascorbate is associated with extended disease-free survival and decreased hypoxia-inducible factor-1 activation in human colorectal cancer. Front Oncol (2014) 4:10. doi:10.3389/fonc.2014.00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MC. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res (2010) 70:5749–58. doi:10.1158/0008-5472.CAN-10-0263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Hong SW, Lee SH, Moon JH, Hwang JJ, Kim DE, Ko E, et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene (2012) 32(12):1508–17. doi:10.1038/onc.2012.176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, et al. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med (2004) 140:533–7. doi:10.7326/0003-4819-140-7-200404060-00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Vissers MCM, Gunningham SP, Morrison MJ, Dachs GU, Currie MJ. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Rad Biol Med (2007) 42:765–72. doi:10.1016/j.freeradbiomed.2006.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res (2003) 63:1764–8.
101. Qiao H, Li L, Qu Z-C, May JM. Cobalt-induced oxidant stress in cultured endothelial cells: prevention by ascorbate in relation to HIF-1a. Biofactors (2009) 35:306–13. doi:10.1002/biof.43
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Gao P, Zhang H, Dinavahi R, Li F, Xiang Y, Raman V, et al. HIF-dependent antitumorigenic effects of antioxidants in vivo. Cancer Cell (2007) 12:230–8. doi:10.1016/j.ccr.2007.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Esteban MA, Wang T, Qin B, Yang J, Qin D, Cai J, et al. Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell (2010) 6:71–9. doi:10.1016/j.stem.2009.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Wang T, Chen K, Zeng X, Yang J, Wu Y, Shi X, et al. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell (2011) 9:575–87. doi:10.1016/j.stem.2011.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A (1976) 73:3685–9. doi:10.1073/pnas.73.10.3685
106. Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: reevaluation of prolongation of survival times in terminal human cancer. Proc Natl Acad Sci U S A (1978) 75:4538–42. doi:10.1073/pnas.75.9.4538
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Creagan ET, Moertel CG, O’Fallon JR, Schutt AJ, O’Connell MJ, Rubin J, et al. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. N Engl J Med (1979) 301:687–90. doi:10.1056/NEJM197909273011303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Moertel CG, Fleming TR, Creagan ET, Rubin J, O’Connell MJ, Ames MM. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N Engl J Med (1985) 312:137–41. doi:10.1056/NEJM198501173120301
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Dusing RW, Drisko JA, Grado GG, Levine M, Holzbeierlein JM, Van Veldhuizen P. Prostate imaging modalities that can be used for complementary and alternative medicine clinical studies. Urol Clin North Am (2011) 38:343–57. doi:10.1016/j.ucl.2011.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Padayatty SJ, Riordan HD, Hewitt SM, Katz A, Hoffer LJ, Levine M. Intravenously administered vitamin C as cancer therapy: three cases. CMAJ (2006) 174:937–42. doi:10.1503/cmaj.050346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, et al. Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol (2008) 19:1969–74. doi:10.1093/annonc/mdn377
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Riordan HD, Casciari JJ, González MJ, Riordan NH, Miranda-Massari JR, Taylor P, et al. A pilot clinical study of continuous intravenous ascorbate in terminal cancer patients. P R Health Sci J (2005) 24:269–76.
113. Padayatty SJ, Sun AY, Chen Q, Espey MG, Drisko J, Levine M. Vitamin C: intravenous use by complementary and alternative medicine practitioners and adverse effects. PLoS One (2010) 5:e11414. doi:10.1371/journal.pone.0011414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, et al. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A (2005) 102:13604–9. doi:10.1073/pnas.0506390102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A (2007) 104:8749–54. doi:10.1073/pnas.0702854104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Belin S, Kaya F, Duisit G, Giacometti S, Ciccolini J, Fontés M. Antiproliferative effect of ascorbic acid is associated with the inhibition of genes necessary to cell cycle progression. PLoS One (2009) 4:e4409. doi:10.1371/journal.pone.0004409
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Cha J, Roomi MW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M. Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp Oncol (2011) 33:226–30.
118. Kasprzak KS, Diwan BA, Kaczmarek MZ, Logsdon DL, Fivash MJ, Salnikow K. Effects of ascorbic acid on carcinogenicity and acute toxicity of nickel subsulfide, and on tumor transplants growth in gulonolactone oxidase knock-out mice and wild-type C57BL mice. Toxicol Appl Pharmacol (2011) 257:32–7. doi:10.1016/j.taap.2011.08.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Kim J, Lee SD, Chang B, Jin DH, Jung SI, Park MY, et al. Enhanced antitumor activity of vitamin C via p53 in cancer cells. Free Rad Biol Med (2012) 53:1607–15. doi:10.1016/j.freeradbiomed.2012.07.079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Rad Biol Med (2009) 47:32–40. doi:10.1016/j.freeradbiomed.2009.02.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, et al. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A (2008) 105:11105–9. doi:10.1073/pnas.0804226105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Cha J, Roomi MW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int J Oncol (2013) 42:55–64. doi:10.3892/ijo.2012.1712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Update (2011) 14:191–201. doi:10.1016/j.drup.2011.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Wenzel U, Nickel A, Kuntz S, Daniel H. Ascorbic acid suppresses drug-induced apoptosis in human colon cancer cells by scavenging mitochondrial superoxide anions. Carcinogenesis (2004) 25:703–12. doi:10.1093/carcin/bgh079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, Levine M, et al. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Rad Biol Med (2011) 50:1610–9. doi:10.1016/j.freeradbiomed.2011.03.007
126. Frömberg A, Gutsch D, Schulze D, Vollbracht C, Weiss G, Czubayko F, et al. Ascorbate exerts anti-proliferative effects through cell cycle inhibition and sensitizes tumor cells towards cytostatic drugs. Cancer Chemoth Pharm (2011) 67:1157–66. doi:10.1007/s00280-010-1418-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, et al. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One (2012) 7:e29794. doi:10.1371/journal.pone.0029794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: ascorbate, cancer, hydroxylation, hypoxia-inducible factor-1, TET enzymes, tumor microenvironment, vitamin C
Citation: Kuiper C and Vissers MCM (2014) Ascorbate as a co-factor for Fe- and 2-oxoglutarate dependent dioxygenases: physiological activity in tumor growth and progression. Front. Oncol. 4:359. doi: 10.3389/fonc.2014.00359
Received: 05 September 2014; Paper pending published: 31 October 2014;
Accepted: 27 November 2014; Published online: 10 December 2014.
Edited by:
Ruggero De Maria, Istituto Superiore di Sanità, ItalyReviewed by:
Michael Wayne Epperly, University of Pittsburgh Cancer Institute, USAMaria Rosa Ciriolo, University of Rome Tor Vergata, Italy
Copyright: © 2014 Kuiper and Vissers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caroline Kuiper, Department of Pathology, Centre for Free Radical Research, University of Otago, Christchurch, PO Box 4345, Christchurch 8140, New Zealand e-mail:Y2Fyb2xpbmUua3VpcGVyQG90YWdvLmFjLm56
†Present address: Caroline Kuiper, Nuffield Department of Medicine, Centre for Cellular and Molecular Physiology, University of Oxford, Oxford, UK e-mail:Y2Fyb2xpbmUua3VpcGVyQHdlbGwub3guYWMudWs=