 Sourik S. Ganguly
Sourik S. Ganguly Xiaohong Li
Xiaohong Li Cindy K. Miranti
Cindy K. Miranti- 1Program for Skeletal Disease and Tumor Metastasis, Laboratory of Tumor Microenvironment and Metastasis, Center for Cancer and Cell Biology, Van Andel Research Institute, Grand Rapids, MI, USA
- 2Program for Skeletal Disease and Tumor Metastasis, Laboratory of Integrin Signaling and Tumorigenesis, Center for Cancer and Cell Biology, Van Andel Research Institute, Grand Rapids, MI, USA
Prostate cancer (PCa) is the second leading cause of cancer death in men worldwide. Most PCa deaths are due to osteoblastic bone metastases. What triggers PCa metastasis to the bone and what causes osteoblastic lesions remain unanswered. A major contributor to PCa metastasis is the host microenvironment. Here, we address how the primary tumor microenvironment influences PCa metastasis via integrins, extracellular proteases, and transient epithelia-mesenchymal transition (EMT) to promote PCa progression, invasion, and metastasis. We discuss how the bone-microenvironment influences metastasis; where chemotactic cytokines favor bone homing, adhesion molecules promote colonization, and bone-derived signals induce osteoblastic lesions. Animal models that fully recapitulate human PCa progression from primary tumor to bone metastasis are needed to understand the PCa pathophysiology that leads to bone metastasis. Better delineation of the specific processes involved in PCa bone metastasize is needed to prevent or treat metastatic PCa. Therapeutic regimens that focus on the tumor microenvironment could add to the PCa pharmacopeia.
Foreward
Prostate cancer (PCa), one of the most common non-skin cancers, results in the death of over a quarter million men annually world-wide (1) and 2.7% American men are estimated to die from this disease in their lifetime (2). The majority of PCa deaths are due to the development of metastatic disease, 80% of which is primarily localized in the bone (3). Furthermore, PCa induces an osteoblastic reaction within the bone, which is rarely observed with other bone-metastatic cancers.
For the patient who presents with metastatic disease, as evidenced by bone lesions detected by X-ray or it is suspected based on a high Gleason score, the first line of treatment after surgical removal of the primary tumor is androgen-deprivation therapy (ADT). The majority of prostate tumors require androgen for their growth and survival (4). Thus, the initial metastatic tumor burden in a patient can be essentially eliminated and they appear to enter remission. But the unfortunate fact is that some subpopulation of these cells either harbor or develop resistance to ADT and the tumor rapidly regrows.
Despite lack of evident dependence on circulating androgen, these castration-resistant tumors are still highly addicted to androgen and/or its cognate receptor, AR (5–8). Evidence for this is provided by several observations. First, second generation enhanced anti-androgen therapies, such as Enzalutamide, are effective, even if only for a while, in patients who failed the first round of ADT (5). Second, recent evidence indicates the tumor itself turns on androgen synthesis, so it no longer needs circulating androgen (9, 10). The successful use of drugs, such as Abiraterone (11), that target enzymes in the androgen synthetic pathway are also effective, again albeit for a short time, in patients who failed ADT. Third, the retention, mutation, and amplification of AR that accompanies ADT-resistant tumors indicate a heavy dependence on AR for the survival and continued persistence of these tumors. Several mutations in AR are known to confer enhanced function and include binding to other steroids or deletion of the ligand binding domain resulting in constitutive activation (12, 13). Fourth, AR could independently enhance invasion and metastasis through non-classical steroid receptor signaling mechanisms (14, 15). Currently, there are no approved therapies available that address these latter two events.
Development of additional therapeutic regimes to target metastatic tumors remains severely limited by the lack of knowledge about (1) what triggers PCa metastasis in the first place, (2) why it displays such a predilection for the bone, and (3) why it induces an osteoblastic bone phenotype. The molecular events thought to be involved in these three processes share a common theme; i.e., interactions with the host, often referred to as the tumor microenvironment. Current approved therapies are highly focused on targeting events occurring intrinsically in the tumor and do not fully consider the contributions of the host. Thus, better understanding of the host and tumor interactions that trigger and drive metastatic processes could provide additional avenues for therapeutic intervention. In this review, we will discuss various possible strategies by which PCa cell interactions with the surrounding tumor microenvironment influence the development of metastases, homing toward the bone, and the development of osteoblastic lesions.
Tumor Microenvironment in Promoting Prostate Cancer Metastasis
Introduction
It is widely accepted that the tumor microenvironment, or stromal compartment, is biologically heterogeneous, consisting of various cell types, such as fibroblasts, endothelial cells, and immune cells, along with growth factors and cytokines, and numerous extracellular matrix (ECM) components. Paracrine signals from these factors released by the tumor activate signaling and gene expression in the neighboring cells and vice versa, ultimately setting up a cycle of reinforcement and continued signal propagation. Interactions between the cancer cells and this stromal compartment are required for invasion, angiogenesis, and metastasis of cancer cells to ectopic sites (16–18). The factors thought to drive the metastatic progression of PCa and to play an important role in the interaction of the tumor with its microenvironment are discussed below.
Olumi et al. (18), was the first to demonstrate the dependency of PCa development on the underlying fibroblasts. It was recognized that fibroblasts found near tumors, i.e., carcinoma-associated fibroblasts (CAFs), were fundamentally different from those in non-tumorigenic samples (19, 20). When CAFs isolated from human PCa patients were mixed with initiated human non-tumorigenic prostate epithelial cells, this was sufficient to initiate tumorigenesis. Normal fibroblasts lacked this capacity, implicating the importance of tumor-induced fibroblast effects feeding back on the initiated tumor cells. In another study, prostate stromal cells could replace Matrigel in LNCaP subcutaneous xenografts to promote tumor growth. One effect of the stromal cells was to promote angiogenesis (21). The stromal compartment of the normal prostate gland is full of smooth muscle cells. However, in PCa lesions, there was a dramatic loss of smooth muscle cells that were replaced by cells displaying myofibroblast characteristics, i.e., expression of Vimentin and increased production of matrix remodeling enzymes like Collagen I and Tenascin (22). This remodeling of the ECM and invasion of tumor cells into the surrounding stromal compartment define a cancerous lesion, as opposed to benign disease. The interaction of the prostate tumor cells with the remodeled matrix and the contribution of the tumor cells themselves to this process are critical first steps in the movement of tumor cells out of their normal niche.
Integrins in PCa Progression
Integrins are a large family of cell-surface glycoproteins, which form heterodimeric adhesion receptors. Integrins bind to a number of ECM components and regulate cytoskeletal organization to maintain cell shape and facilitate migration. These interactions also regulate cell survival, proliferation, adhesion, migration, and invasion (23, 24). PCa initiation and progression is accompanied by preferential expression of integrin α6β1, reduction in integrin α3β1, and complete loss of integrin β4 (25). Integrin α6, a laminin receptor is associated with poor patient prognosis and increased metastasis in a wide range of cancers (26, 27). In the normal prostate, integrin α6 complexed with integrin β4 is present at the basal cell/stromal interface; however, loss of integrin polarity occurs during progression of PIN to invasive cancer where basal cells are lost and integrin α6 complexed with integrin β1 abnormally appears in the luminal-like compartment (28).
Integrin α6β1 was shown to play two major roles in PCa, promoting cell survival and facilitating invasion and metastasis (28–30). Within the normal prostate epithelium, integrin expression is limited to the basal cells, being absent from the AR-expressing luminal cells. However, during PCa development, integrin α6β1 becomes co-expressed with AR in the tumor cells (31). AR directly binds the integrin α6 promoter and induces the expression of integrin α6β1, while simultaneously decreasing integrin β4 expression (29). Adhesion of PCa tumor cells to laminin to engage α6β1 promoted AR-dependent survival of the cells. Survival was mediated through AR-induced integrin α6β1 and subsequent activation of NF-κB and Bcl-xL expression (29). This AR/α6β1 pathway was active in metastatic cell lines, and elevated in castration-resistant cells. Laminins are abundant in the bone microenvironment, with Laminin-10 being the most highly expressed (32, 33), thus providing a mechanism for activating the integrin α6β1 survival pathway in the bone. Elevated NF-κB activity plays a critical role in PCa progression (34, 35). Another study demonstrated that secretory proteins from prostate neuroendocrine cells activate NF-κB signaling in the tumor cells, which in turn transcriptionally activates AR in the tumor cells to promote castration-resistant cell growth (36). The potential role of integrin α6β1 in this process remains to be determined. It was previously shown that the integrin β1a variant is expressed in PCa tumors, while the β1c variant is present in normal tissue (37). Signaling through the β1c variant was shown to suppress p27kip, a major negative cell cycle regulator and tumor suppressor known to be disregulated in PCa. Thus, a potential α6β1a variant may contribute to PCa proliferation.
Integrin α6 remains the primary integrin expressed in lymph node and soft tissue metastases, indicating high retention and selection for this integrin during metastasis (38). Cleavage of integrin α6 by uPA is associated with invasive PCa, the cleavage product is detected in tumors but not in normal prostate tissue, and promotes PCa cell invasion and migration on laminin (39, 40). Furthermore, inhibiting integrin α6-mediated adhesion or cleavage delayed experimental lung metastasis (28, 41), reduced bone growth in mouse femurs, and increased responses of metastatic PCa cells to ionizing radiation (42, 43).
Laminin integrins like α6β1 and α3β1 associate with transmembrane scaffold and membrane organizing molecules called tetraspanins (44). Tetraspanin KAI/CD82 was first identified as a metastasis suppressor in PCa (45). Loss of CD82 in human PCa correlates with poor prognosis, but by itself is not sufficient to predict metastasis (46). Studies in PCa cell lines in vitro demonstrate that CD82 is capable of suppressing integrin-based functions including signaling, migration, and invasion (47–50). CD82 regulates the internalization and turnover of integrins and suppress integrin signaling through Met and Src (47, 48). Its role in suppressing integrin functions was further validated in KO mice. However, the dependency on integrins for its metastasis suppressive functions did not prove to be valid (CKM, unpublished data). Furthermore, while restoration of CD82 to metastatic cells suppresses metastasis, it loss in primary PCa is not sufficient to induce metastasis in genetically engineered mice (CKM, unpublished data). Thus, additional factors or as yet unidentified mechanism is involved in CD82 suppression of metastasis. A more likely target is its role in promoting cell–cell adhesion (51), discussed further in the EMT section.
The importance of integrin α2β1 in PCa metastasis is also emerging (31). Integrin α2β1 binds collagen, another major component of the prostate basement membrane and bone microenvironment. Manipulation of integrin α2 expression in LNCaP cells demonstrated a direct correlation between integrin α2 expression and the ability to grow in the bone (52). RANKL expression in PCa cells enhances bone metastasis. Integrin α2 integrin expression and function was stimulated in PCa cells overexpressing RANKL (53).
Thus, many studies support the importance of integrins in PCa development and progression by promoting, survival, proliferation, invasion, and metastasis. Targeting specific integrins and their matrix interactions may provide a way to prevent metastatic bone PCa.
Proteases in PCa Tumor Invasion and Metastasis
Breakdown of the basement membrane surrounding the prostatic ducts and invasion of prostate cells into the stromal compartment defines the pathology of prostate adenocarcinoma. Proteases that mediate basement membrane and stromal ECM degradation are crucial for this process and represent the first steps toward metastatic dissemination (Figure 1). Penetration into and out of the vasculature and lymphatics similarly requires proteases (54, 55).
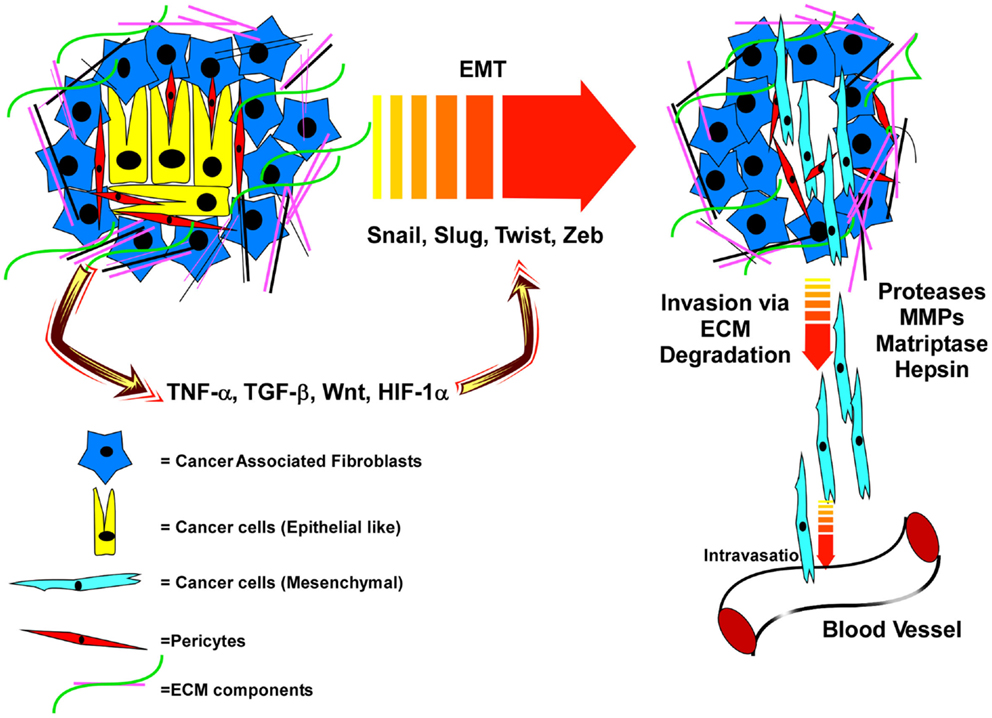
Figure 1. Interactions of PCa cells with an extracellular matrix that is remodeled by cancer-associated fibroblasts, and soluble factors and proteases released within the tumor microenvironment induce EMT and subsequent invasion and dissemination of cancer cells. In the primary tumor microenvironment, the epithelial cancer cells are surrounded by the cancer-associated fibroblasts (CAF), pericytes, and various extracellular matrix (ECM) proteins. This tumor microenvironment produces various factors like TNFα, TGFβ, Wnt, and HIF-1α which promote EMT via up-regulation of specific transcription factors. EMT programing leads to a mesenchymal phenotype of the cancer cells and with the help of various proteases (MMPs, Matriptase, Hepsin), the cancer cells cleave the ECM, break away from the tumor microenvironment and intravasate into the blood vesicles to travel to distal organs.
Type II transmembrane serine proteases, like Matriptase and Hepsin, which are important for normal epithelial tissue development and repair, contribute to the breakdown of the basement membrane in PCa (56). Hepsin is dramatically up-regulated in PCa and represents one of the most highly overexpressed proteins in PCa microarrays (57). Elevated Hepsin expression is associated with high Gleason score and poor clinical outcome (55, 58, 59). Hepsin overexpression in the prostate epithelium causes disorganization of the basement membrane and its overexpression in PCa cell lines promotes lymph node metastasis (57). Hepsin reportedly cleaves and activates pro-HGF produced by activated fibroblasts within the stroma, thereby activating the receptor tyrosine kinase Met signaling pathway; a known activator of epithelial cell scattering, migration, and invasion (60). However, it was also reported that another Hepsin target is Laminin-5, a major component of the prostatic basement membrane, which is lost during PCa development (61).
Matriptase is expressed in many cancers and increased expression is seen in primary prostate tumors and metastatic lesions (62–64). One of its reported targets is Laminin-5, deposited in the prostatic duct basement membrane primarily by the basal epithelial cells. Laminin-5 expression is lost in PCa, coinciding with the loss of its primary receptor, integrin β4 (65). Active degradation combined with loss of secretion within the emerging PCa cells likely accounts for the lack of Laminin-5 in PCa tumors. Whether Laminin-10, another component of the prostate basement membrane, which is not lost during PCa development, is also cleaved by Matriptase or Hepsin is not known.
While Hepsin expression is apparently not controlled by AR, both Matriptase expression and its cleavage is highly controlled by androgen signaling (66). Rapid cleavage within minutes of androgen stimulation, mediated by Src signaling, is followed by a more long term increase in new Matriptase mRNA, thus providing a mechanism for replenishing depleted pools. Because Matriptase is essential for detachment of epithelial suprabasal cells during skin differentiation (67), and the AR-positive luminal cells also must detach upon differentiation, it is highly likely that Matriptase is also important in prostate epithelial differentiation. That AR, within the newly emerging luminal cells, can control integrin expression and a protease that degrades basement membrane has interesting implications about how AR contributes to preserving a luminal phenotype and may promote loss of basal cells during PCa development (68).
Two other proteases, which are known direct AR transcriptional targets, PSA and TMPRSS2, are normally secreted into the lumens of prostatic ducts (69, 70). However, due to loss of epithelial polarization and invasion into stromal areas, these enzymes are now also present within the tumor microenvironment (71). Their relative importance in PCa development or progression has remained largely undetermined.
The most extensively studied proteases linked with invasion and metastasis are the matrix-metalloproteinases (MMP). MMPs are involved in the degradation of the stromal ECM components such as Collagen and Fibronectin (72, 73). In normal tissues, MMPs play a major role in ECM remodeling involved in development and tissue repair. Their misregulation contributes to many disease states, including rheumatoid arthritis, pulmonary emphysema, and tumor invasion and metastasis (74–77). Active MMPs are secreted mainly from the cells in the tumor microenvironment, such as connective tissue, fibroblasts, endothelial cells, osteoblasts, macrophages, and neutrophils but also by cancer cells. Active MMPs are used by cancer cells to invade the stromal compartment both at the primary site and at metastatic sites (75, 76).
In PCa, MMP-2 and -9 are considered useful prognostic markers and these MMPs promote invasion and metastasis of PCa cells (78, 79). Elevated levels of MMP-2/9 in serum or plasma are correlated with high Gleason score (78–80). Overexpression of MMP-1 promoted PCa cell invasion and experimental metastasis, and inhibition of MMP-1 activity decreased PCa tumor growth in mice, indicating the importance of MMP-1 in regulating PCa invasion and metastasis (81). Induction of PCa invasion by MMP-9 is mediated through cleavage and subsequent inactivation of the serpin protease nexin-1 (PN-1). PN-1 is known to inhibit urokinase plasminogen activator (uPA) and thus inhibits PCa progression and metastasis (82). uPA and its receptor (uPAR) promote PCa metastasis, as down regulation of uPA or uPAR inhibited PCa cell invasion and metastasis (83, 84). When this is coupled with the reported role of uPA in cleaving integrin α6 (39), it becomes apparent how concerted efforts of proteases and integrin-based cell adhesion work together to promote invasion and metastasis.
Attempts to therapeutically target the MMPs, as a whole class, failed in clinical trials; resulting in worse outcomes (85). The lack of specificity to specific MMPs and the unforeseen protective role of some MMPs are thought to have contributed to the failure. Thus, there has been much resistance to trying to identify specific MMP inhibitors. On the other hand, preclinical testing of a small molecule Hepsin inhibitor demonstrated it blocked PCa metastasis in a genetic mouse model (57). Curcumin has the capacity to inhibit androgen induced Matriptase activation and displays anti-metastatic properties (86). Antibodies that block Matriptase cleavage have been reported (87), and a natural protein product produced by bacteria, Ecotin, is a natural Matriptase inhibitor (88), either of which may offer a therapeutic advantage. More work in defining the protease targets and mechanisms for inducing invasion and metastasis is clearly warranted.
Epithelial-Mesenchymal Transition in Promoting PCa Metastasis
The steps that lead to PCa metastasis (Figure 1) include degradation of the ECM, detachment of the cancer cells from the ECM as well as their detachment from each other, migration toward and subsequent entry into the blood or lymphatic system (89–91). The machinery and signaling pathways used in these invasive events are part of the normal wound healing response of epithelial tissues. Upon tissue damage, a host of growth factors and cytokines are released from the blood stream that activate the stromal fibroblasts (PDGF, TGF-β), endothelial cells (VEGF, FGF, IL8), and epithelial cells (HGF, TGF-β) to repair the tissue and fill in the wound (92). Through these signals epithelial cells are forced to loosen their matrix adhesions via activation of proteases and integrin signaling, and are induced to migrate across matrix being remodeled by the stroma. Some may even detach from each other to facilitate filling in the wound. It is this latter event, loss of cell–cell adhesion and depolarization of epithelial cells that is thought to trigger the final conversion of primary cancer cells into metastatic ones. Once loosened from the matrix and from each other, these cells are now free to roam if they have acquired the proper mutations that allow them to survive as non-adherent cells.
This process is referred to as epithelial–mesenchymal transition (EMT), where the loosened epithelial cells take on the physical properties of mesenchymal fibroblast-like cells (92). The classical marker of EMT is cadherin switching; where E-Cadherin expression is lost and N-Cadherin is gained (89, 90). E-cadherin, which promotes homotypic binding between two adjacent epithelial cells, prevents the cancer cells from breaking away from each other and reinforces tight junctions to preserve epithelial barrier function and apical/basal polarity (90, 93). The metastasis suppressor, tetraspanin CD82, promotes E-cadherin-based cell–cell adhesion, and suppresses integrin-based migration (51). Loss of this crucial regulator reduces cell–cell adhesion, while at the same time promoting enhanced migration; thus, its loss would strongly facilitate an EMT-like phenotype. Decreased CD82, E-Cadherin, or β-catenin (the anchoring protein for E-cadherin) is associated with poor PCa prognosis (23, 46, 94, 95). TGF-β is the most classical inducer of EMT, signaling through Smad family transcription factors to induce the expression of the EMT-regulating transcription factors Snail, Slug, Zeb-1, and/or Twist (96–100). These EMT-associated transcription factors, through interactions with epigenetic regulators, control expression of genes involved in cell polarity, cell–cell contact, cytoskeleton structure, and ECM degradation (Figure 1), including repression of the E-cadherin gene (101).
While EMT is well-established in several other epithelial cancer types, its specific role in PCa remains controversial. This is complicated by the histological data demonstrating that PCa remains very epithelial-like in both primary tumors and in metastatic tissues; expressing E-cadherin and other classical prostate epithelial markers (94, 102). Yet, a few isolated tumor cell lines, especially those that have lost AR expression, readily adapt EMT phenotypes and are relatively invasive and metastatic (102–104). The necessary presence of AR in PCa tumors, which drives a luminal differentiation phenotype may make the conversion to EMT a relatively difficult process (103). It might explain the long latency for conversion of primary PCa to metastatic disease (105). The heterogeneous display of EMT phenotypes in PCa and other human tumor samples, has fueled the hypothesis that EMT is a transient process necessary for escape and dissemination, that reverses back, i.e., MET (mesenchymal epithelial transition) when cells reach their distant sites. A study that tested this hypothesis using a Tet-inducible model, demonstrated that transient induction of EMT was required for the migration of squamous carcinoma cells out of the skin into the blood stream, and subsequent shut off was required to establish metastatic tumors in the lung (106). If this happens in PCa is not clear, and the factors that drive it are even less clear.
The soluble factors that are secreted into a wound by physical disruption are the same factors present in the tumor microenvironment. Transforming growth factor-β (TGF-β) is a potent inducer of EMT and is released by many cellular components of the tumor microenvironment. However, the ability of primary tumors to respond to TGF-β is hindered by the normal growth inhibitory effects of TGF-β signaling, and thus the tumor cells typically block this pathway. So the cells must find ways to reactive some aspect of TGF-β signaling that doesn’t cause growth suppression or find other indirect ways to stimulate EMT. One study suggested that direct cell–cell contact between tumor cells and platelets synergistically cooperated with TGF-β, to directly activate NF-κB signaling in the tumor cell to promote EMT and metastasis (107).
Inflammation induced during wound healing and in tumors also impacts the behavior of tumor cells. Many studies have demonstrated that wounding is a tumor-promoting event, especially chronic wounding where inflammation is high (92). The role of inflammation in PCa initiation was originally identified histologically by the presence of prostate inflammatory atrophy (PIA) in tumor samples (108). Recent mouse studies demonstrate that prostatic inflammation induced by prostitis, enhances basal-to-luminal differentiation and accelerates the initiation of PCa (109). Two sources for inflammatory signaling in PCa have been proposed, the stromal cancer-associated fibroblasts (110) or mesenchymal stem cells (111). A recent study highlighted the importance of adipocytes in inducing inflammatory responses in PCa cells within the bone through the lipid chaperone FABP4, triggering IL-1β expressing and oxidative stress protein HMOX-1 (112). Other studies proposed the inflammatory responses in PCa are mediated through NF-κB signaling (113). Inflammatory cytokines like TNFα and interleukins produced by both tumor cells and surrounding cells (114) activate NF-κB signaling and one consequence of this is the release of TGF-β (115, 116).
Non-TGF-β pathways can also activate EMT. TNFα can act independently of TGF-β to induce EMT by repressing GSK-3β, activating the AKT pathway, and stabilizing Snail (117–119). Wnt signaling also stimulates EMT in PCa cells. Expression of SOX2 induces EMT, and this was shown to be mediated by SOX2 binding to and activating β-catenin (120). The PCa-specific fusion and oncogene, TMPRSS2-Erg, enhances cell invasion (121, 122). Manipulation of the fusion gene in VCaP cells, altered Frizzled4 (Fzd4) expression (123). Fzd4 signaling promoted cell adhesion-related EMT phenotypes in VCaP cells. HIF1α, activated under hypoxic conditions, promotes aggressive tumor cell invasion and metastasis (124, 125). Overexpression of HIF1α in some PCa tumor cell lines promoted EMT that was dependent on β-catenin (126).
Further demonstration that EMT/MET phenotypic conversions are essential for the progression and metastasis of PCa to the bone is highly warranted. While current mouse models have been effective at defining specific molecular events occurring within the primary tumor or bone-resident tumors, the process whereby a confined prostate tumor is converted to a metastatic bone tumor has not been adequately modeled. The models do not currently reflect what is observed in human patients; i.e., AR-positive epithelial cells that migrate to and take up residence in the bone to induce an osteoblastic bone reaction. Understanding the specific genetic and epigenetic alterations that promote EMT-like phenotypes in PCa will be important to understanding the switch between indolent and lethal PCa, improving staging and prognosis of PCa patients and preventing over treatment.
Tumor Microenvironment in Prostate Cancer Bone Homing and Colonization
Secreted Factors in PCa Bone Homing
Different types of cancers develop metastases in very specific organs. Several secreted factors have been proposed to promote organ-specific homing (Figure 2). A few of these chemotactic signals can attract cancer cells toward the bone. Conditioned media from osteoblasts differentiated in vitro served to induce migration and invasion of breast and melanoma cells, indicating osteoblasts secrete potent factors that can induce metastasis to the bone (127, 128). Osteonectin, a purified active factor from the bone, promoted invasion of bone-metastatic cancer cells, but not the non-bone-metastatic cancer cells indicating that bone possess chemotactic factors that can promote tissue-specific homing of cancer cells (129). It was demonstrated that osteoblast conditioned media containing higher amounts of TGF-β promoted chemotaxis and invasion of PC3 cells. Given the abundance of TGF-β in the bone environment, it could act as a chemo-attractant of PCa cells to the bone (130).
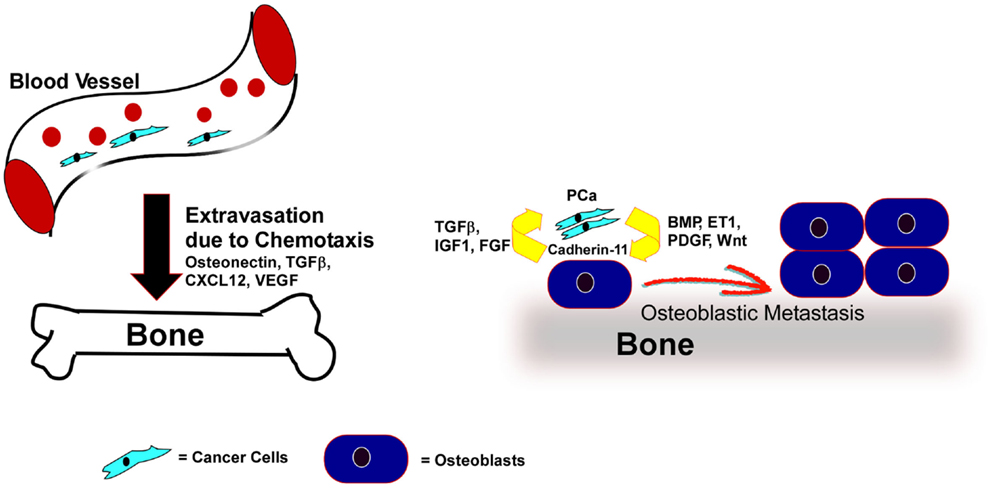
Figure 2. PCa cells home to bone by chemo-attractants and colonize through direct association with osteoblasts, where the PCa cells secrete factors that promote osteoblastic responses and the osteoblasts reinforce tumor cell survival and growth. Expression of various chemo-attractants (Osteonectin, TGFβ, CXCL12, VEGF) guide PCa cells to extravasate and home toward the bone. Once in the bone-microenvironment the cancer cells interact with bone-forming osteoblasts via Cadherin-11. Factors like BMP, ET-1, Wnt, or PDGF, secreted from the cancer cells promote the proliferation and differentiation of osteoblasts. In turn the bone-microenvironment secretes soluble factors like FGF, IGF, and TGFβ to promote tumor cell survival and proliferation.
Chemokines and cytokines have chemo-attractant properties that play an important role in the cancer cell proliferation, survival, and gene transcription. Chemokine receptors are involved in many processes of cancer metastasis (131, 132). Mice bearing autoimmune arthritis have higher incidence of breast cancer metastasis to the bone, which was proposed to be due to the presence of higher amounts of circulating levels of pro-inflammatory cytokines in these autoimmune arthritic mice (133).
The chemokine receptor, CXCR4, and its ligand CXCL12/SDF are widely studied in PCa bone metastasis (134, 135). In bone marrow, CXCL12 is expressed in osteoblasts, fibroblasts, and endothelial cells (136). Blocking CXCR4 in PCa cells using neutralizing antibody inhibited the dissemination and colonization of PCa cells in mice tibia following intra-cardiac injection (135). Akt1 reportedly induces the expression of CXCR4 in PTEN-null PCa cells, and overexpression of Akt-1 promoted intra-tibial tumor growth of PCa cells (137). These results indicate that Akt-1 might be inducing the CXCR4/CXCL12 axis and thus promoting PCa metastasis. PCa cells home toward areas in the bone marrow rich in osteoblasts where the hematopoietic stem cell (HSC) niche resides. In fact, PCa cells can bind to and displace mouse HSCs from the niche. Furthermore, the cancer cells egress out of the HSC niche into the blood when CXCR4 signaling is blocked by AMD3100 (138). These findings suggested that the CXCL12/CXCR4 axis is important for chemotaxis of PCa cells to the bone. However, inhibiting CXCR4 with CTCE-9908, a drug approved by FDA for osteosarcoma, inhibited spleen, liver, and lymph node metastasis of PCa cells, indicating CXCR4 may be a common metastatic factor, rather than one that is bone specific.
Ectopic Site Pre-Remodeling
The famous “Seed and Soil” hypothesis, put forward more than 100 years ago by Dr. Stephen Paget, was used to explain why different types of cancer preferentially metastasize to different specific tissues. The theory proposed that distant organs, like the bone, provide a preferred “fertile soil” for cancer cells, and the cancer cells were preferentially attracted to that tissue. However, Isaiah Fidler’s group demonstrated that tumor cells were present in vasculature of all the organs, yet metastasis only developed in certain organs but not in others (139, 140). David Lyden’s group put forward the pre-metastatic niche model, where remodeling of only the preferred ectopic site(s) for metastasis occurs much earlier, before the cancer cells even break away from the primary tumor (139).
The niche remodeling events, mediated by soluble factors acting on non-cancer cells, govern the route of dissemination of cancer cells to a specific microenvironment. They demonstrated that bone marrow-derived hematopoietic progenitor cells expressing VEGF receptor 1 (VEGFR-1), homed to the specific metastatic sites through integrin α4β1. At the same time, the tumor cells secrete factors that induce the fibroblasts within the pre-metastatic niche to secrete Fibronectin, an α4β1 ECM ligand. The VEGFR1-positive cells then promote chemo-attraction and adherence of circulating tumor cells (141). Consistent with this idea, Hirutsuka et al. (142) demonstrated in a mouse model of melanoma metastasis that VEGF-A, TGF-β, and TNF-α released from the primary tumors induced the expression of chemokines in the lung parenchyma but not in other organs. In another study, persistent STAT3 activation was detected in distant organs such as the lung before tumor cell arrival. S1PR1–STAT3 up-regulation in tumor cells induced S1PR1–STAT3 at these distant sites and in myeloid cells. Ablation of STAT3 in the myeloid compartment inhibited STAT3 activity in the lungs, inhibited formation of pre-metastatic niche, and inhibited lung metastasis (143). Whether a similar pre-metastatic niche remodeling and non-tumor cell signaling governs PCa bone metastasis is not known. However, a past study demonstrated enhanced bone metastasis of orthotopic xenografted human cell lines upon androgen ablation (144). ADT in human PCa patients induces bone loss that is often corrected by bisphosphonates (145). Similarly, castrated mice lose bone mass (146). The full contribution of ADT to pre-metastatic niche conditioning, bone metastasis, osteoblastic reactions, or emergence of castration-resistant disease, as a result of bone-microenvironment interactions needs further investigation.
Adhesion Molecules in PCa Colonization
Several studies have suggested that the choice of metastatic organ is not necessarily dictated at the time tumor cells escape the tumor and end up in the circulation. Circulating tumor cells can be found in patients who do not have or do not develop metastatic disease (147). Furthermore, tumor cell entry is not restricted to specific organs, majority of circulating tumor cells extrude into tissues (147, 148). A PCa study demonstrated that 50% of the patients with primary tumors had circulating tumor cells as well as tumor cells lodged in the bone (149), yet only 12–13% of these patients ever develop metastatic disease. Thus, additional events are required for the tumor cells to grow and colonize the metastatic site.
Cadherin-11, commonly known as osteoblast cadherin, is primarily found in osteoblasts, with very low but detectable expression in brain, testis, and lung (150, 151). It is an adhesion molecule that mediates many steps of osteoblast maturation (150, 151). Cadherin-11 expression is increased in metastatic PCa compared to primary tumors but is not present in normal prostate tissue. Furthermore, elevated Cadherin-11 was found in human PCa bone metastasis relative to lymph node metastasis (152), indicating cadherin-11 is specifically associated with bone metastasis. Cadherin-11 may mediate the binding of cancer cells to osteoblasts (Figure 2). Such binding might also promote cross talk between cancer cells and osteoblasts and induce osteoblastic lesions. Intra-cardiac injection of PC3 cells expressing Cadherin-11-specific shRNA displayed a significant decrease in bone metastasis compared to the control cells (152). PCa cells derived from bone express high levels of cadherin-11, and expression of cadherin-11 in PCa cells promoted PCa cell invasion and migration and increased the adhesion and intercalation between osteoblasts in an in vitro culture model (153). Another group demonstrated that bone-tropic MDA-MB-231 breast cancer cells also express high levels of cadherin-11 compared to brain-tropic MDA-MB-231 cells. Thus, cadherin-11 is likely an important determinant of bone-tropism in cancer cells (154).
Tumor Microenvironment in Promoting Osteoblastic Lesions
Breast cancer metastasis is usually osteolytic (bone degrading); however, PCa is osteoblastic, i.e., leading to new bone formation (155, 156). It has been reported that some colon and cervical cancers are also osteoblastic (157). The exact mechanisms by which osteoblastic versus osteolytic metastases occur is still unclear. However, the differences are likely to reside in the differential interaction of tumor cells with the bone microenvironment. Bone, a dynamic connective tissue is constantly remodeling during an individual’s lifetime. The process of remodeling is dependent on two cells types, osteoblasts and osteoclasts, both of which work in harmony to maintain the normal bone. Osteoblasts derived from mesenchymal stem cells in the bone marrow, make new bone. Whereas osteoclasts, which are modified macropages derived from monocytes, degrade bone (84). During osteoblastic metastasis, the bone remodeling favors bone formation over resorption.
Factors that Induce Osteoblastic Bone Metastasis
The number of osteoblasts surrounding PCa cells is increased in osteoblastic bone metastasis (155). However, the newly formed bone is weak and fragile, lacking mechanical strength. These bones are composed of randomly orientated and loosely packed collagen bundles, resulting in weak bone strength and frequent fracture (158). Furthermore, the excessive bone growth disrupts the bone marrow compartment, reducing immune function. Many factors can induce the growth and differentiation of osteoblasts and their precursors (Figure 2). Tumor cells secrete many of these factors, and thus may actively promote osteoblastogenesis.
Bone morphogenetic proteins
Bone morphogenetic proteins are members of the TGF-β superfamily and various isoforms of BMP promote both prostate and breast cancer metastasis (159, 160). One major source of BMP expression appears to be from the tumor cells. Elevated expression of both mRNA and BMP-6 protein is detected in primary PCa tissues and in PCa cell lines (161, 162). BMPs could influence metastasis by acting directly on the tumor cells or through their effects on the bone microenvironment. For instance, BMP-2 induces resistance to apoptosis due to hypoxia (163) and promotes breast cancer cell invasion and migration (164); whereas, BMP-6 promotes migration and invasion of PCa cells (161). BMP-2 and -7 stimulated cellular migration and invasion of PCa cells (165), and BMP-6 acting through Smad signaling directly induced the transcription of extracellular proteases such as MMP-1 and 9, required for invasion (159).
Findings from genetically modified mice demonstrate that the normal role of BMPs in the bone is to induce the differentiation of osteoblasts (166, 167). For example, BMP-7 knockout mice have smaller skeletons and reduced mineralized bone (168). The ability of BMP-7 to control bone mineralization or osteoblast differentiation can be attributed to the induced expression of crucial differentiation factors, Runx2 and Osterix, in bone stromal cell precursors (169, 170). High BMP-7 expression was detected in PCa-induced bone lesions, while its expression in primary tumors is low (155, 171). However, its exact role in promoting osteoblastic vs. osteolytic lesions remains controversial. Nonetheless, BMP-7 produced by tumor cells has the potential to impact osteoblast differentiation within PCa bone lesions. The most intriguing aspect of BMP-7 is that while its expression is controlled by androgen and it is required for normal prostate development, the most elevated BMP-7 expression was observed in castration-resistant tumors within the bone (171). However, BMP-7 expression in prostate bone tumors appears to be largely growth suppressive and may promote PCa cell dormancy (172).
Elevated BMP-6 expression is also associated with PCa bone metastasis (159). BMP-6 produced by cancer cells was able to induce mineralization of M3T3 pre-osteoblasts, and blocking BMP-6 activity reduced osteolastic lesion formation by LuCaP 23.1 cells in vivo (161). Two studies demonstrated that Wnt5a or Wnt3a, generated by bone stroma cells, induces the expression of BMP-6 in PCa cells (173, 174). This was mediated by non-canonical JNK and canonical β-catenin signaling pathways, respectively (174). The induction of BMP-6 by Wnt5A secretion occurred in the absence of androgen and promoted androgen-independent growth of the tumor cells (173). Interestingly, in a recent patient-derived bone xenograft model, transplantation of human bone-metastatic tumors into the bone, but not in the skin, resulted in castration-resistant tumor growth (175). Thus, the bone microenvironment has a high capacity to influence PCa treatment. The BMPs secreted by PCa cells through Wnt signaling, in turn induce the differentiation of osteoblasts. For instance, BMP-4 produced by PCa cells promoted osteoblast differentiation of mouse stromal cells, as measured by the production of alkaline phosphatase, osteocalcin, and collagen type II. At the same time, BMP-4 also stimulated the production of sonic hedgehog (Shh) by the tumor cells (176). Shh stimulated Smad1 and BMP receptor expression in the mouse stromal cells, which enhanced their response to BMP-4. The ability of Shh to induce osteoblast differentiation was promoted by collagen production and was Gil1-dependent, but Runx2-independent (177, 178). Thus, BMPs and Shh cooperatively provided cues for the growth of PCa cells and the differentiation of bone stromal cells. All these results indicate that BMPs and Shh from the PCa cells play an important role in inducing osteoblast differentiation from bone stromal cells, and thus likely contribute to osteoblastic bone phenotypes.
Endothelin 1
Endothelin 1, a potent vasoconstrictor along with other family members ET-2 and ET-3, is produced by the vascular endothelium. Plasma levels of ET-1 are elevated in several types of cancers, including PCa (179). ET-1 inhibits PCa cell apoptosis via enhanced Bcl-2 family member expression and PI3K/Akt activation (180). In ovarian carcinoma cell lines, ET-1 promotes invasion via the activation of extracellular proteases like MMP-1 (181). In addition to promoting cancer metastasis, ET-1 promotes osteogenic properties; ET-1 null mice exhibit hypoplasia of facial bones (182). ET-1 is a potent mitogenic factor of osteoblasts and patients with osteoblastic bone lesions have increased serum levels of ET-1 (171, 183). Elevated ET-1 might contribute to osteoblast proliferation and differentiation (184–186).
In clinical trials, atrasentan (endothelin A receptor antagonist) suppressed bone remodeling in castration-resistant metastatic patients (187). However, atrasentan in combination with docetaxel, a chemotherapeutic agent, did not improve progression-free survival in castration-resistant bone-metastatic patients (188). One of the potent pathways by which ET-1 promotes osteoblast activity is through the activation of Wnt signaling via the inhibition of the Wnt suppressor DKK1 (189). ET-1 inhibited DKK-1 expression, but also increased the expression of Type 1 collagen, a predominant protein constituent of bone matrix (185, 189).
Wnt
Wnts constitute a family of 19 secreted glycoproteins, whose dysregulation plays an important role in the progression of many cancers including breast, gastric, prostate, melanoma, and glioblastoma (190, 191). Wnt ligands bind to a seven-pass transmembrane receptor composed of Lrp5/6 and frizzled genes to transduce signals to the cytoplasmic protein Disheveled (Dsh). Dsh blocks GSK-β to inhibit β-catenin phosphorylation, by disrupting the β-catenin/Axin complex, leading to β-catenin stabilization, and its nuclear translocation to interact with TCF/LEF transcription factors (192, 193). Loss of APC activates the Wnt pathway by stabilizing β-catenin. Loss-of-function of APC mutations is common in many cancers (193–195). Increased nuclear β-catenin expression correlated with advanced, metastatic, and hormone-refractory prostate carcinoma (196). Upregulation of the Wnt pathway by means of increased Wnt secretion, decreased expression of inhibitors such as APC, sFRP, DKK1, or Wif1, or constitutive activation of β-catenin, induces the activation of downstream target genes like c-Myc, c-Jun, and various other genes important in both cancer development and metastasis (91, 192, 193). MMP-14, which promotes invasion and metastasis of cancer cells, is also a direct target of β-catenin/TCF signaling (197). The exact mechanisms that lead to elevated β-catenin in PCa are not clear, but it is not usually due to APC mutation (194).
Within the bone, Wnt signaling promotes osteoblast differentiation by directly stimulating Runx2 expression in osteoblasts both in vivo and in vitro (198). Wnt signaling also stimulates BMP-2 expression, inducing the trans-differentiation of non-osteogenic cells into osteoblasts (199). Wnt signaling may contribute to the osteoblastic phenotype. Blocking DDK1 expression in PC3 cells, which releases the block on Wnt signaling, switched the normal osteolytic phenotype induced by PC3 cell to osteoblastic. Conversely, overexpressing DKK-1 in C4-2B cells converted the normal mixed lesion to an osteolytic lesion (200). Thus, Wnt signaling contributes to PCa osteoblastic bone lesions. Further investigations in the specific components of the Wnt pathway involved, and determining if they also contribute to metastasis per se will be important.
Platelet-derived growth factor
Platelet-derived growth factor, a potent growth factor which plays an important role in tumor progression, consists of disulfide-bonded homodimer polypeptide chains of A, B, C, D, and heterodimer AB (201). Aberrant signaling through PDGF receptors promotes progression of many tumors, including PCa. PDGFRβ, which is frequently activated in bone-metastatic PCa patients, is activated both by PDGF-B and PDGF-D (202). PDGF-D, which promotes PCa cell proliferation and tumor growth, is overexpressed in prostate tumors with increasing Gleason score (203). Furthermore, PDGF also stimulates the interaction of PCa cells with bone stromal cells. In the bone microenvironment, PDGF is synthesized by platelets, macrophages, osteoclasts, endothelial cells, and all cells differentiating from mesenchymal stem cells, including pericytes, and osteoblasts (204). Thus, PDGF has the potential to act as a central connector for many interactions within the bone microenvironment that influence tumor growth. Blocking PDGF receptor signaling inhibits the growth of human breast and pancreatic cancer in bones (205, 206), and subsequently reduces bone resorption. However, a phase I clinical trial with a potent PDGFR inhibitor, imatinib mesylate in combination with docetaxel, in castration-resistant PCa patients with bone metastasis did not show any improvement in the median progression-free survival of patients, as compared to docetaxel alone (207). However, imatinib mesylate inhibits many other kinases, like Abl and c-Kit (208). Whether PDGFR inhibitors in combination with the second generation anti-androgens, Enzalutamide or abiraterone, will be more effective should be investigated.
Factors Released from the Bone Microenvironment
Transforming growth factor-β
Transforming growth factor-β is one of the most abundant cytokines which induces osteoblast proliferation but inhibits its differentiation (209, 210). The effects of TGF-β on osteoclasts are controversial. Most of the studies investigating TGF-β in cancer bone metastasis have focused on osteolytic bone metastasis of breast and cancers other than PCa. A “vicious cycle” model has been proposed to explain TGF-β signaling and osteolytic bone metastasis. TGF-β, signaling through Gil2, in a Hedge Hog-independent manner, stimulates the expression of parathyroid hormone-related protein (PTHrP), which activates osteoclasts (211). In turn, more TGF-β is released after bone resorption, which further enhances cancer growth and osteoclast activation, initiating a “vicious cycle” (212, 213). Inhibition of TGF-β decreases osteolytic lesions induced by breast and melanoma cancer cells in mouse tibia (214–216). In the first report on the role of TGF-β in PCa bone metastasis, it was found that loss of TGF-β responsiveness in the fibroblasts induced the up regulation of CXCL16 and CXCL1, which promoted PCa cells adhesion to the bone matrix, and promoted mixed (osteoblastic/osteolytic) metastatic lesions (217). Thus, it will be important to further investigate the cell-specific role of TGF-β in the bone microenvironment on PCa osteoblastic bone metastasis.
Insulin-like growth factor
High serum levels of insulin-like growth factor are associated with higher risk of breast, prostate, and colorectal cancer. Signaling through IGF1R promotes cell proliferation, apoptosis, and invasion of cancer cells; which are all integral steps in cancer metastasis (218). For example, inhibition of IGF-1R diminishes the invasion of PCa cells and also inhibits expression of MMP-2, an extracellular protease necessary for invasion (219). The ability of IGF-1 to induce PCa cell proliferation and survival is dependent on loss of Pten, a tumor suppressor commonly lost in PCa (220). However, IGF is another important coupling factor in the bone, activating both bone formation and resorption. IGF, which promotes proliferation, invasion, and metastasis of cancer cells is released during bone resorption (84). One interesting study showed that neutralizing antibody to IGF, but not antibody to TGF-β, FGF, or PDGF, blocked the breast cancer anchorage-independent growth induced by resorbed bone extract; further supporting a unique role for IGF-1 in bridging the cross talk between the bone microenvironment and the cancer cells (221). Bone-derived IGF promoted bone metastasis of breast cancer cells by stimulating proliferation and inhibiting apoptosis of cancer cells (221). Elevated IGF-1 receptor expression in the stroma surrounding clinical PCa samples correlates with high Gleason score (222). Thus, IGF-1 released from the bone could stimulate the stroma to support PCa growth. Indeed, blocking IGF-1 and IGF-1 receptor inhibits PCa growth in the bone and reduced the osteoblastic bone formation. Thus, IGF-1 signal inhibition could be strategy for limiting PCa bone metastasis.
Fibroblast growth factor
Fibroblast growth factor, a family of ubiquitously expressed and secreted factors, regulates processes like development, wound healing, and neoplastic transformation through mitogenesis and angiogenesis. Increased expression of some growth factors from the FGF family and their receptors are reportedly associated with PCa progression. Aberrant activation of FGF receptors (FGFR) induces the activation of downstream targets like PI3K, MAPK, and STAT3, all of which play an important role in the progression of PCa (223–225). One of the FGF family members, FGF-1 (acidic FGF) is expressed in a majority of PCa and its expression is associated with high Gleason score (226). Levels of FGF-2 (basic FGF) and FGF-6 are also elevated in PCa tissues as compared to normal (227, 228). In one transgenic mouse model, FGFR1 overexpression led to PIN (229), but in another model, activated FGFR1 promoted adenocarcinoma and metastasis to lymph nodes and liver (230). FGF may promote PCa metastasis via regulation of cell survival (231). PCa bone metastases express FGF-8 and/or FGF-9, and both of these FGFs are reported to promote osteoblast differentiation and new bone formation (232–234). FGF-9 expression is also associated with high Gleason score and neutralizing antibody against FGF-9 inhibited bone formation and bone lesions in mice (232, 235). On the other hand, other family members, like FGF-7 and FGF-10, are required for normal differentiation of normal prostate epithelial cells (236). Nonetheless, overexpression of FGF-7 in a transgenic mouse prostate epithelium led to PIN (237), and overexpression of FGF-10 in the mesenchyme in the prostate regeneration mouse model was sufficient to induce multifocal PIN had low-grade PCa (238). Thus, the context and level of FGF signaling may differentially impact PCa development and progression. The mechanisms that underlie the differential expression of FGF members and their respective receptors to promote or inhibit PCa growth in different microenvironments, and the cellular constituents upon which it acts need to be better resolved in PCa.
Challenges That Remain
The high dependence of host environmental factors on metastatic processes necessitates the use of animal models to clarify and demonstrate that dependency. The most commonly used model is the mouse, which has a physiology and prostate organ structure that is significantly different from human. Nonetheless, valuable insight can be gleaned from the proper models. Unfortunately, for PCa researchers many of the current mouse models fail to fully recapitulate human disease progression; i.e., an AR-dependent tumor growing in the prostate gland that spontaneously metastasizes to the bone to from an AR-dependent osteoblastic cancer.
Models using genetic introduction of known PCa-associated mutations into the mouse genome, rarely metastasize to the bone. The few models that do are neuroendocrine or use mutations not reported to be present in human disease. Nonetheless, one unifying theme in the models that do produce significant metastases is the abrogation of p53. While p53 loss is reported in <20% of metastatic PCa tumors, other mutations that indirectly alter p53 function remain a strong possibility. Another tumor suppressor, Pten, whose homozygous loss strongly correlates with metastatic progression in human cancer, but whose loss alone in the mouse does not lead to metastatic bone disease, may also contribute to metastasis. Interestingly, both p53 and Pten impact chromosomal stability (239), disruption of which is thought to be necessary for the selection of genetic variants within the tumor that eventually evolve the capacity to metastasize. Recent studies suggest that EMT-associated transcription factors suppress p53 signaling and impact DNA repair; thus, linking disease progression by the tumor microenvironment with chromosomal instability (106). Identifying the factors that specifically lead to chromosomal instability, the genetic alterations within PCa that promote metastasis, and how the tumor microenvironment influences this, has the potential to significantly increase our understanding of metastatic conversion in PCa.
Several metastatic PCa cell lines isolated from bone metastases, either from human samples or mouse xenografts (VCaP, C4–2, PC3), retain high capacity to grow when implanted directly into the bone, or in a few cases when injected into the heart. Two PCa lines, one isolated from the brain, DU145, and one from ascites, ARCaP, can also grow when implanted in bone; however, the LNCaP line isolated from a lymph node grows poorly in the bone. Most of them, when implanted orthotopically in the prostate, do not metastasize to the bone; though some make it to the lung. ARCaP was reported to metastasize to the bone after orthotopic injection (240). This was accompanied by EMT conversion. Only a few of these, when implanted in the bone, make osteoblastic lesions (DU145, C4–2, ARCaP). Thus, whatever properties these cells once had in the human host that permitted their full metastatic progression in humans has been lost. Furthermore, at least 3 of these lines, PC3, DU145, and ARCaP express AR at such low levels they do not use the AR-regulated pathways seen in over 90% of human PCa metastases. The LuCaP series of human xenografted and SubQ-passaged tumors (241), isolated primarily from soft tissue metastases and still expressing AR, can grow and form osteoblastic lesions when implanted in the bone. Removal of primary LuCaP tumors following orthotopic injection, allowed the development of micrometastases to lymph nodes and soft tissues (242), but not bone. Thus, these studies indicate that many PCa tumor cell lines, whether they originally came from bone or not, have a high capacity to grow in bone. However, they all lack the ability to home to bone from the prostate, and their capacity to induce osteoblastic lesions is variable. Developing human lines or mouse models that can display the full metastatic progression, either through selection or genetic manipulation, remains the Holy Grail for understanding PCa metastasis and having models that can be used for effective therapy development and testing.
Conclusion and Perspectives
Metastasis is the major cause of PCa death. Understanding how cancer cells metastasize toward the bone is needed to design drugs that prevent or interfere with PCa metastasis. Many studies suggest that EMT transcription factors drive the initial phases of PCa metastasis, although the events, either within the tumor or contributed by the tumor microenvironment, that trigger EMT specifically in PCa are still not known. Overexpression of EMT transcription factors, in conjunction with responses to host-derived chemotactic factors, might lead to PCa-specific homing and metastasis to the bone. Once PCa is disseminated in the bone, the cross talk between the bone microenvironmental factors and the PCa tumor cells contribute to the establishment of osteoblastic lesions. The factors mediating this cross talk and their signaling pathways need to be further delineated to ultimately halt the progression of metastatic lesions in the bone. The development of better animal models that fully recapitulate the metastatic process as seen in human disease is paramount to deciphering the molecular events associated with PCa metastasis.
Author Contributions
Dr. Sourik S. Ganguly drafted, helped revise, and designed the figures for the manuscript. Dr. Xiaohong Li revised and provided intellectual content. Dr. Cindy K. Miranti revised, provided intellectual content, and finalized the manuscript.
Conflict of Interest Statement
The authors declare that the writing of this review article was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank Alexandra Vander Ark for her editorial comments. Writing of this article was supported by the National Cancer Institute of the National Institutes of Health under award number CA154835 (Cindy K. Miranti), Department of Defense Prostate Cancer Research Program Award number W81XWH-12-1-0271 (Xiaohong Li), and by the Van Andel Research Institute and Program for Skeletal Disease and Metastasis. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
1. Center MM, Jemal A, Lortet-Tieulent J, Ward E, Ferlay J, Brawley O, et al. International variation in prostate cancer incidence and mortality rates. Eur Urol (2012) 61(6):1079–92. doi: 10.1016/j.eururo.2012.02.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin (2013) 63(1):11–30. doi:10.3322/caac.21166
3. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res (2006) 12(20 Pt 2):6243s–9s. doi:10.1158/1078-0432.CCR-06-0931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Hoimes CJ, Kelly WK. Redefining hormone resistance in prostate cancer. Ther Adv Med Oncol (2010) 2(2):107–23. doi:10.1177/1758834009356433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene (2013) 32(49):5501–11. doi:10.1038/onc.2013.206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res (2001) 61(9):3550–5.
7. Ruizeveld de Winter JA, Janssen PJ, Sleddens HM, Verleun-Mooijman MC, Trapman J, Brinkmann AO, et al. Androgen receptor status in localized and locally progressive hormone refractory human prostate cancer. Am J Pathol (1994) 144(4):735–46.
8. van der Kwast TH, Schalken J, Ruizeveld de Winter JA, van Vroonhoven CC, Mulder E, Boersma W, et al. Androgen receptors in endocrine-therapy-resistant human prostate cancer. Int J Cancer (1991) 48(2):189–93. doi:10.1002/ijc.2910480206
9. Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res (2006) 66(5):2815–25. doi:10.1158/0008-5472.CAN-05-4000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Azzouni F, Godoy A, Li Y, Mohler J. The 5 α-reductase isozyme family: a review of basic biology and their role in human diseases. Adv Urol (2012) 2012:530121. doi:10.1155/2012/530121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Zobniw CM, Causebrook A, Fong MK. Clinical use of abiraterone in the treatment of metastatic castration-resistant prostate cancer. Res Rep Urol (2014) 6:97–105. doi:10.2147/RRU.S29003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Eisermann K, Wang D, Jing Y, Pascal LE, Wang Z. Androgen receptor gene mutation, rearrangement, polymorphism. Transl Androl Urol (2013) 2(3):137–47. doi:10.3978/j.issn.2223-4683.2013.09.15
13. Li Y, Chan SC, Brand LJ, Hwang TH, Silverstein KA, Dehm SM. Androgen receptor splice variants mediate enzalutamide resistance in castration-resistant prostate cancer cell lines. Cancer Res (2013) 73(2):483–9. doi:10.1158/0008-5472.CAN-12-3630
14. Foradori CD, Weiser MJ, Handa RJ. Non-genomic actions of androgens. Front Neuroendocrinol (2008) 29(2):169–81. doi:10.1016/j.yfrne.2007.10.005
15. Migliaccio A, Castoria G, Auricchio F. Analysis of androgen receptor rapid actions in cellular signaling pathways: receptor/Src association. Methods Mol Biol (2011) 776:361–70. doi:10.1007/978-1-61779-243-4_21
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Corn PG. The tumor microenvironment in prostate cancer: elucidating molecular pathways for therapy development. Cancer Manag Res (2012) 4:183–93. doi:10.2147/CMAR.S32839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Alphonso A, Alahari SK. Stromal cells and Integrins: conforming to the needs of the tumor microenvironment. Neoplasia (2009) 11(12):1264–71. doi:10.1593/neo.91302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res (1999) 59(19):5002–11.
19. Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature (2004) 432(7015):332–7. doi:10.1038/nature03096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Schauer IG, Sood AK, Mok S, Liu J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia (2011) 13(5):393–405. doi:10.1593/neo.101720
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Tuxhorn JA, McAlhany SJ, Dang TD, Ayala GE, Rowley DR. Stromal cells promote angiogenesis and growth of human prostate tumors in a differential reactive stroma (DRS) xenograft model. Cancer Res (2002) 62(11):3298–307.
22. Tuxhorn JA, Ayala GE, Rowley DR. Reactive stroma in prostate cancer progression. J Urol (2001) 166(6):2472–83. doi:10.1097/00005392-200112000-00126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Horvath LG, Henshall SM, Lee CS, Kench JG, Golovsky D, Brenner PC, et al. Lower levels of nuclear β-catenin predict for a poorer prognosis in localized prostate cancer. Int J Cancer (2005) 113(3):415–22. doi:10.1002/ijc.20599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Frank SB, Miranti CK. Disruption of prostate epithelial differentiation pathways and prostate cancer development. Front Oncol (2013) 3:273. doi:10.3389/fonc.2013.00273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Schmelz M, Cress AE, Scott KM, Burger F, Cui H, Sallam K, et al. Different phenotypes in human prostate cancer: α6 or α3 integrin in cell-extracellular adhesion sites. Neoplasia (2002) 4(3):243–54. doi:10.1038/sj.neo.7900223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Danen EH, van Muijen GN, van de Wiel-van Kemenade E, Jansen KF, Ruiter DJ, Figdor CG. Regulation of integrin-mediated adhesion to laminin and collagen in human melanocytes and in non-metastatic and highly metastatic human melanoma cells. Int J Cancer (1993) 54(2):315–21. doi:10.1002/ijc.2910540225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Friedrichs K, Ruiz P, Franke F, Gille I, Terpe HJ, Imhof BA. High expression level of alpha 6 integrin in human breast carcinoma is correlated with reduced survival. Cancer Res (1995) 55(4):901–6.
28. Ports MO, Nagle RB, Pond GD, Cress AE. Extracellular engagement of α6 integrin inhibited urokinase-type plasminogen activator-mediated cleavage and delayed human prostate bone metastasis. Cancer Res (2009) 69(12):5007–14. doi:10.1158/0008-5472.CAN-09-0354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Lamb LE, Zarif JC, Miranti CK. The androgen receptor induces integrin α6β1 to promote prostate tumor cell survival via NF-κB and Bcl-xL Independently of PI3K signaling. Cancer Res (2011) 71(7):2739–49. doi:10.1158/0008-5472.CAN-10-2745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Sroka IC, Anderson TA, McDaniel KM, Nagle RB, Gretzer MB, Cress AE. The laminin binding integrin α6β1 in prostate cancer perineural invasion. J Cell Physiol (2010) 224(2):283–8. doi:10.1002/jcp.22149
31. Bonkhoff H, Stein U, Remberger K. Differential expression of α6 and α2 very late antigen integrins in the normal, hyperplastic, and neoplastic prostate: simultaneous demonstration of cell surface receptors and their extracellular ligands. Hum Pathol (1993) 24(3):243–8. doi:10.1016/0046-8177(93)90033-D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Siler U, Rousselle P, Muller CA, Klein G. Laminin γ2 chain as a stromal cell marker of the human bone marrow microenvironment. Br J Haematol (2002) 119(1):212–20. doi:10.1046/j.1365-2141.2002.03800.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Siler U, Seiffert M, Puch S, Richards A, Torok-Storb B, Muller CA, et al. Characterization and functional analysis of laminin isoforms in human bone marrow. Blood (2000) 96(13):4194–203.
34. Jin R, Yi Y, Yull FE, Blackwell TS, Clark PE, Koyama T, et al. NF-κB gene signature predicts prostate cancer progression. Cancer Res (2014) 74(10):2763–72. doi:10.1158/0008-5472.CAN-13-2543
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Lessard L, Begin LR, Gleave ME, Mes-Masson AM, Saad F. Nuclear localisation of nuclear factor-κB transcription factors in prostate cancer: an immunohistochemical study. Br J Cancer (2005) 93(9):1019–23. doi:10.1038/sj.bjc.6602796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res (2008) 68(16):6762–9. doi:10.1158/0008-5472.CAN-08-0107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Fornaro M, Tallini G, Zheng DQ, Flanagan WM, Manzotti M, Languino LR. p27(kip1) acts as a downstream effector of and is coexpressed with the β1C integrin in prostatic adenocarcinoma. J Clin Invest (1999) 103(3):321–9. doi:10.1172/JCI4585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Pontes-Junior J, Reis ST, Dall’Oglio M, Neves de Oliveira LC, Cury J, Carvalho PA, et al. Evaluation of the expression of integrins and cell adhesion molecules through tissue microarray in lymph node metastases of prostate cancer. J Carcinog (2009) 8:3. doi:10.4103/1477-3163.48453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Demetriou MC, Pennington ME, Nagle RB, Cress AE. Extracellular α6 integrin cleavage by urokinase-type plasminogen activator in human prostate cancer. Exp Cell Res (2004) 294(2):550–8. doi:10.1016/j.yexcr.2003.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Pawar SC, Demetriou MC, Nagle RB, Bowden GT, Cress AE. Integrin alpha6 cleavage: a novel modification to modulate cell migration. Exp Cell Res (2007) 313(6):1080–9. doi:10.1016/j.yexcr.2007.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Landowski TH, Gard J, Pond E, Pond GD, Nagle RB, Geffre CP, et al. Targeting integrin α6 stimulates curative-type bone metastasis lesions in a xenograft model. Mol Cancer Ther (2014) 13(6):1558–66. doi:10.1158/1535-7163.MCT-13-0962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Pawar SC, Dougherty S, Pennington ME, Demetriou MC, Stea BD, Dorr RT, et al. α6 integrin cleavage: sensitizing human prostate cancer to ionizing radiation. Int J Radiat Biol (2007) 83(11–12):761–7. doi:10.1080/09553000701633135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. King TE, Pawar SC, Majuta L, Sroka IC, Wynn D, Demetriou MC, et al. The role of α6 integrin in prostate cancer migration and bone pain in a novel xenograft model. PLoS One (2008) 3(10):e3535. doi:10.1371/journal.pone.0003535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Miranti CK. Controlling cell surface dynamics and signaling: how CD82/KAI1 suppresses metastasis. Cell Signal (2009) 21(2):196–211. doi:10.1016/j.cellsig.2008.08.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Dong JT, Lamb PW, Rinker-Schaeffer CW, Vukanovic J, Ichikawa T, Isaacs JT, et al. KAI1, a metastasis suppressor gene for prostate cancer on human chromosome 11p11.2. Science (1995) 268(5212):884–6. doi:10.1126/science.7754374
46. Dong JT, Suzuki H, Pin SS, Bova GS, Schalken JA, Isaacs WB, et al. Down-regulation of the KAI1 metastasis suppressor gene during the progression of human prostatic cancer infrequently involves gene mutation or allelic loss. Cancer Res (1996) 56(19):4387–90.
47. He B, Liu L, Cook GA, Grgurevich S, Jennings LK, Zhang XA. Tetraspanin CD82 attenuates cellular morphogenesis through down-regulating integrin α6-mediated cell adhesion. J Biol Chem (2005) 280(5):3346–54. doi:10.1074/jbc.M406680200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Sridhar SC, Miranti CK. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene (2006) 25(16):2367–78. doi:10.1038/sj.onc.1209269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Han SY, Lee M, Hong YK, Hwang S, Choi G, Suh YS, et al. Tsp66E, the Drosophila KAI1 homologue, and Tsp74F function to regulate ovarian follicle cell and wing development by stabilizing integrin localization. FEBS Lett (2012) 586(22):4031–7. doi:10.1016/j.febslet.2012.09.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Termini CM, Cotter ML, Marjon KD, Buranda T, Lidke KA, Gillette JM. The membrane scaffold CD82 regulates cell adhesion by altering α4 integrin stability and molecular density. Mol Biol Cell (2014) 25(10):1560–73. doi:10.1091/mbc.E13-11-0660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Abe M, Sugiura T, Takahashi M, Ishii K, Shimoda M, Shirasuna K. A novel function of CD82/KAI-1 on E-cadherin-mediated homophilic cellular adhesion of cancer cells. Cancer Lett (2008) 266(2):163–70. doi:10.1016/j.canlet.2008.02.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Sottnik JL, Daignault-Newton S, Zhang X, Morrissey C, Hussain MH, Keller ET, et al. Integrin α2β1 (α2β1) promotes prostate cancer skeletal metastasis. Clin Exp Metastasis (2013) 30(5):569–78. doi:10.1007/s10585-012-9561-6
53. Ziaee S, Chung LW. Induction of integrin α2 in a highly bone metastatic human prostate cancer cell line: roles of RANKL and AR under three-dimensional suspension culture. Mol Cancer (2014) 13:208. doi:10.1186/1476-4598-13-208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol (2011) 21(4):228–37. doi:10.1016/j.tcb.2010.12.002
55. Klezovitch O, Chevillet J, Mirosevich J, Roberts RL, Matusik RJ, Vasioukhin V. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell (2004) 6(2):185–95. doi:10.1016/j.ccr.2004.07.008
56. Jung H, Lee KP, Park SJ, Park JH, Jang YS, Choi SY, et al. TMPRSS4 promotes invasion, migration and metastasis of human tumor cells by facilitating an epithelial-mesenchymal transition. Oncogene (2008) 27(18):2635–47. doi:10.1038/sj.onc.1210914
57. Tang X, Mahajan SS, Nguyen LT, Beliveau F, Leduc R, Simon JA, et al. Targeted inhibition of cell-surface serine protease Hepsin blocks prostate cancer bone metastasis. Oncotarget (2014) 5(5):1352–62.
58. Holt SK, Kwon EM, Lin DW, Ostrander EA, Stanford JL. Association of hepsin gene variants with prostate cancer risk and prognosis. Prostate (2010) 70(9):1012–9. doi:10.1002/pros.21135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Kim HJ, Han JH, Chang IH, Kim W, Myung SC. Variants in the HEPSIN gene are associated with susceptibility to prostate cancer. Prostate Cancer Prostatic Dis (2012) 15(4):353–8. doi:10.1038/pcan.2012.17
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Kirchhofer D, Peek M, Lipari MT, Billeci K, Fan B, Moran P. Hepsin activates pro-hepatocyte growth factor and is inhibited by hepatocyte growth factor activator inhibitor-1B (HAI-1B) and HAI-2. FEBS Lett (2005) 579(9):1945–50. doi:10.1016/j.febslet.2005.01.085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Tripathi M, Nandana S, Yamashita H, Ganesan R, Kirchhofer D, Quaranta V. Laminin-332 is a substrate for hepsin, a protease associated with prostate cancer progression. J Biol Chem (2008) 283(45):30576–84. doi:10.1074/jbc.M802312200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature (2001) 412(6849):822–6. doi:10.1038/35090585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Wu SR, Cheng TS, Chen WC, Shyu HY, Ko CJ, Huang HP, et al. Matriptase is involved in ErbB-2-induced prostate cancer cell invasion. Am J Pathol (2010) 177(6):3145–58. doi:10.2353/ajpath.2010.100228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Takeuchi T, Shuman MA, Craik CS. Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc Natl Acad Sci U S A (1999) 96(20):11054–61. doi:10.1073/pnas.96.20.11054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Davis TL, Cress AE, Dalkin BL, Nagle RB. Unique expression pattern of the α6β4 integrin and laminin-5 in human prostate carcinoma. Prostate (2001) 46(3):240–8. doi:10.1002/1097-0045(20010215)46:3<240::AID-PROS1029>3.3.CO;2-S
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Kiyomiya K, Lee MS, Tseng IC, Zuo H, Barndt RJ, Johnson MD, et al. Matriptase activation and shedding with HAI-1 is induced by steroid sex hormones in human prostate cancer cells, but not in breast cancer cells. Am J Physiol Cell Physiol (2006) 291(1):C40–9. doi:10.1152/ajpcell.00351.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Miller GS, List K. The matriptase-prostasin proteolytic cascade in epithelial development and pathology. Cell Tissue Res (2013) 351(2):245–53. doi:10.1007/s00441-012-1348-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Abate-Shen C, Shen MM. Molecular genetics of prostate cancer. Genes Dev (2000) 14(19):2410–34. doi:10.1101/gad.819500
69. Chen YW, Lee MS, Lucht A, Chou FP, Huang W, Havighurst TC, et al. TMPRSS2, a serine protease expressed in the prostate on the apical surface of luminal epithelial cells and released into semen in prostasomes, is misregulated in prostate cancer cells. Am J Pathol (2010) 176(6):2986–96. doi:10.2353/ajpath.2010.090665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Balk SP, Ko YJ, Bubley GJ. Biology of prostate-specific antigen. J Clin Oncol (2003) 21(2):383–91. doi:10.1200/JCO.2003.02.083
71. Lucas JM, True L, Hawley S, Matsumura M, Morrissey C, Vessella R, et al. The androgen-regulated type II serine protease TMPRSS2 is differentially expressed and mislocalized in prostate adenocarcinoma. J Pathol (2008) 215(2):118–25. doi:10.1002/path.2330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol (2011) 3(12):1–24. doi:10.1101/cshperspect.a005058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Westermarck J, Kähäri VM. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J (1999) 13(8):781–92.
74. Loo WT, Cheung MN, Chow LW. Production of matrix metalloproteinases in specific subpopulations of human-patient breast cancer invading in three dimensional cultural system. Life Sci (2004) 76(7):743–52. doi:10.1016/j.lfs.2004.06.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol (2000) 18(5):1135–49. doi:10.1016/j.lfs.2004.06.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Benaud C, Dickson RB, Thompson EW. Matrix metalloproteinases in mammary gland development and cancer. Breast Cancer Res Treat (1998) 50(2):97–116. doi:10.1023/A:1006061115909
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Ganguly SS, Fiore LS, Sims JT, Friend JW, Srinivasan D, Thacker MA, et al. c-Abl and Arg are activated in human primary melanomas, promote melanoma cell invasion via distinct pathways, and drive metastatic progression. Oncogene (2012) 31(14):1804–16. doi:10.1038/onc.2011.361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Morgia G, Falsaperla M, Malaponte G, Madonia M, Indelicato M, Travali S, et al. Matrix metalloproteinases as diagnostic (MMP-13) and prognostic (MMP-2, MMP-9) markers of prostate cancer. Urol Res (2005) 33(1):44–50. doi:10.1007/s00240-004-0440-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Wood M, Fudge K, Mohler JL, Frost AR, Garcia F, Wang M, et al. In situ hybridization studies of metalloproteinases 2 and 9 and TIMP-1 and TIMP-2 expression of human prostate cancer. Clin Cancer Res (1997) 15(3):246–58.
80. Incorvaia L, Badalamenti G, Rini G, Arcara C, Fricano S, Sferrazza C, et al. MMP-2, MMP-9 and activin A blood levels in patients with breast cancer or prostate cancer metastatic to the bone. Anticancer Res (2007) 27(3B):1519–25.
81. Pulukuri SM, Rao JS. Matrix metalloproteinase-1 promotes prostate tumor growth and metastasis. Int J Oncol (2008) 32(4):757–65. doi:10.3892/ijo.32.4.757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Xu D, McKee CM, Cao Y, Ding Y, Kessler BM, Muschel RJ. Matrix metalloproteinase-9 regulates tumor cell invasion through cleavage of protease nexin-1. Cancer Res (2010) 70(17):6988–98. doi:10.1158/0008-5472.CAN-10-0242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Pulukuri SM, Gondi CS, Lakka SS, Jutla A, Estes N, Gujrati M, et al. RNA interference-directed knockdown of urokinase plasminogen activator and urokinase plasminogen activator receptor inhibits prostate cancer cell invasion, survival, and tumorigenicity in vivo. J Biol Chem (2005) 280(43):36529–40. doi:10.1074/jbc.M503111200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Jin JK, Dayyani F, Gallick GE. Steps in prostate cancer progression that lead to bone metastasis. Int J Cancer (2011) 128(11):2545–61. doi:10.1002/ijc.26024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science (2002) 295(5564):2387–92. doi:10.1126/science.1067100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Cheng TS, Chen WC, Lin YY, Tsai CH, Liao CI, Shyu HY, et al. Curcumin-targeting pericellular serine protease matriptase role in suppression of prostate cancer cell invasion, tumor growth, and metastasis. Cancer Prev Res (Phila) (2013) 6(5):495–505. doi:10.1158/1940-6207.CAPR-12-0293-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Ganesan R, Eigenbrot C, Kirchhofer D. Structural and mechanistic insight into how antibodies inhibit serine proteases. Biochem J (2010) 430(2):179–89. doi:10.1042/BJ20100634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Ihara S, Miyoshi E, Ko JH, Murata K, Nakahara S, Honke K, et al. Prometastatic effect of N-acetylglucosaminyltransferase V is due to modification and stabilization of active matriptase by adding beta 1-6 GlcNAc branching. J Biol Chem (2002) 277(19):16960–7. doi:10.1074/jbc.M200673200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
90. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15(3):178–96. doi:10.1038/nrm3758
91. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133(4):704–15. doi:10.1016/j.cell.2008.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Rybinski B, Franco-Barraza J, Cukierman E. The wound healing, chronic fibrosis, and cancer progression triad. Physiol Genomics (2014) 46(7):223–44. doi:10.1152/physiolgenomics.00158.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Adams CL, Chen YT, Smith SJ, Nelson WJ. Mechanisms of epithelial cell-cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J Cell Biol (1998) 142(4):1105–19. doi:10.1083/jcb.142.4.1105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Umbas R, Isaacs WB, Bringuier PP, Schaafsma HE, Karthaus HF, Oosterhof GO, et al. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res (1994) 54(14):3929–33.
95. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev (2010) 24(18):1967–2000. doi:10.1101/gad.1965810
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Chaudhry P, Fabi F, Singh M, Parent S, Leblanc V, Asselin E. Prostate apoptosis response-4 mediates TGF-β-induced epithelial-to-mesenchymal transition. Cell Death Dis (2014) 5:e1044. doi:10.1038/cddis.2014.7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet (2001) 29(2):117–29. doi:10.1038/ng1001-117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Garg M. Epithelial-mesenchymal transition – activating transcription factors – multifunctional regulators in cancer. World J Stem Cells (2013) 5(4):188–95. doi:10.4252/wjsc.v5.i4.188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat Cell Biol (2009) 11(8):943–50. doi:10.1038/ncb1905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Zavadil J, Bottinger EP. TGF-β and epithelial-to-mesenchymal transitions. Oncogene (2005) 24(37):5764–74. doi:10.1038/sj.onc.1208927
101. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol (2014) 16(6):488–94. doi:10.1038/ncb2976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Putzke AP, Ventura AP, Bailey AM, Akture C, Opoku-Ansah J, Celiktas M, et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am J Pathol (2011) 179(1):400–10. doi:10.1016/j.ajpath.2011.03.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Jacob S, Nayak S, Fernandes G, Barai RS, Menon S, Chaudhari UK, et al. Androgen receptor as a regulator of ZEB2 expression and its implications in epithelial-to-mesenchymal transition in prostate cancer. Endocr Relat Cancer (2014) 21(3):473–86. doi:10.1530/ERC-13-0514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. He H, Yang X, Davidson AJ, Wu D, Marshall FF, Chung LW, et al. Progressive epithelial to mesenchymal transitions in ARCaP E prostate cancer cells during xenograft tumor formation and metastasis. Prostate (2010) 70(5):518–28. doi:10.1002/pros.21086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Etzioni R, Cha R, Feuer EJ, Davidov O. Asymptomatic incidence and duration of prostate cancer. Am J Epidemiol (1998) 148(8):775–85. doi:10.1093/oxfordjournals.aje.a009698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell (2012) 22(6):725–36. doi:10.1016/j.ccr.2012.09.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell (2011) 20(5):576–90. doi:10.1016/j.ccr.2011.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Vral A, Magri V, Montanari E, Gazzano G, Gourvas V, Marras E, et al. Topographic and quantitative relationship between prostate inflammation, proliferative inflammatory atrophy and low-grade prostate intraepithelial neoplasia: a biopsy study in chronic prostatitis patients. Int J Oncol (2012) 41(6):1950–8. doi:10.3892/ijo.2012.1646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Kwon OJ, Zhang L, Ittmann MM, Xin L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc Natl Acad Sci U S A (2014) 111(5):E592–600. doi:10.1073/pnas.1318157111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell (2014) 26(1):121–35. doi:10.1016/j.ccr.2014.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Brennen WN, Denmeade SR, Isaacs JT. Mesenchymal stem cells as a vector for the inflammatory prostate microenvironment. Endocr Relat Cancer (2013) 20(5):R269–90. doi:10.1530/ERC-13-0151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Herroon MK, Rajagurubandara E, Hardaway AL, Powell K, Turchick A, Feldmann D, et al. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget (2013) 4(11):2108–23.
113. Nguyen DP, Li J, Yadav SS, Tewari AK. Recent insights into NF-κB signalling pathways and the link between inflammation and prostate cancer. BJU Int (2014) 114(2):168–76. doi:10.1111/bju.12488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Charles KA, Kulbe H, Soper R, Escorcio-Correia M, Lawrence T, Schultheis A, et al. The tumor-promoting actions of TNF-α involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J Clin Invest (2009) 119(10):3011–23. doi:10.1172/JCI39065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Sullivan DE, Ferris M, Nguyen H, Abboud E, Brody AR. TNF-α induces TGF-β1expression in lung fibroblasts at the transcriptional levelviaAP-1 activation. J Cell Mol Med (2009) 13(8b):1866–76. doi:10.1111/j.1582-4934.2009.00647.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Bates RC, Mercurio AM. Tumor necrosis factor-α stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell (2003) 14(5):1790–800. doi:10.1091/mbc.E02-09-0583
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta-induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci (2008) 121(Pt 20):3317–24. doi:10.1242/jcs.028282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Naber HP, Drabsch Y, Snaar-Jagalska BE, ten Dijke P, van Laar T. Snail and Slug, key regulators of TGF-β-induced EMT, are sufficient for the induction of single-cell invasion. Biochem Biophys Res Commun (2013) 435(1):58–63. doi:10.1016/j.bbrc.2013.04.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Wang H, Wang HS, Zhou BH, Li CL, Zhang F, Wang XF, et al. Epithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. PLoS One (2013) 8(2):e56664. doi:10.1371/journal.pone.0056664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Li X, Xu Y, Chen Y, Chen S, Jia X, Sun T, et al. SOX2 promotes tumor metastasis by stimulating epithelial-to-mesenchymal transition via regulation of WNT/beta-catenin signal network. Cancer Lett (2013) 336(2):379–89. doi:10.1016/j.canlet.2013.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Furusato B, Tan SH, Young D, Dobi A, Sun C, Mohamed AA, et al. ERG oncoprotein expression in prostate cancer: clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis (2010) 13(3):228–37. doi:10.1038/pcan.2010.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science (2005) 310(5748):644–8. doi:10.1126/science.1117679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Gupta S, Iljin K, Sara H, Mpindi JP, Mirtti T, Vainio P, et al. FZD4 as a mediator of ERG oncogene-induced WNT signaling and epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res (2010) 70(17):6735–45. doi:10.1158/0008-5472.CAN-10-0244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Michieli P. Hypoxia, angiogenesis and cancer therapy: to breathe or not to breathe? Cell Cycle (2009) 8(20):3291–6. doi:10.4161/cc.8.20.9741
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell (2003) 3(4):347–61. doi:10.1016/S1535-6108(03)00085-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Zhao JH, Luo Y, Jiang YG, He DL, Wu CT. Knockdown of β-Catenin through shRNA cause a reversal of EMT and metastatic phenotypes induced by HIF-1α. Cancer Invest (2011) 29(6):377–82. doi:10.3109/07357907.2010.512595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Boyce BF, Yoneda T, Guise TA. Factors regulating the growth of metastatic cancer in bone. Endocr Relat Cancer (1999) 6(3):333–47. doi:10.1677/erc.0.0060333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Giunciuglio D, Cai T, Filanti C, Manduca P, Albini A. Effect of osteoblast supernatants on cancer cell migration and invasion. Cancer Lett (1995) 97(1):69–74. doi:10.1016/0304-3835(95)03955-V
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Jacob K, Webber M, Benayahu D, Kleinman HK. Osteonectin promotes prostate cancer cell migration and invasion: a possible mechanism for metastasis to bone. Cancer Res (1999) 59:4453.
130. Festuccia C, Bologna M, Gravina GL, Guerra F, Angelucci A, Villanova I, et al. Osteoblast conditioned media contain TGF-beta1 and modulate the migration of prostate tumor cells and their interactions with extracellular matrix components. Int J Cancer (1999) 81(3):395–403. doi:10.1002/(SICI)1097-0215(19990505)81:3<395::AID-IJC13>3.3.CO;2-M
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Teicher BA, Fricker SP. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin Cancer Res (2010) 16(11):2927–31. doi:10.1158/1078-0432.CCR-09-2329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Salazar N, Castellan M, Shirodkar SS, Lokeshwar BL. Chemokines and chemokine receptors as promoters of prostate cancer growth and progression. Crit Rev Eukaryot Gene Expr (2013) 23(1):77–91. doi:10.1615/CritRevEukaryotGeneExpr.2013006905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Das Roy L, Pathangey LB, Tinder TL, Schettini JL, Gruber HE, Mukherjee P. Breast-cancer-associated metastasis is significantly increased in a model of autoimmune arthritis. Breast Cancer Res (2009) 11(4):R56. doi:10.1186/bcr2345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Uygur B, Wu WS. SLUG promotes prostate cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol Cancer (2011) 10:139. doi:10.1186/1476-4598-10-139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Sun YX, Schneider A, Jung Y, Wang J, Dai J, Wang J, et al. Skeletal localization and neutralization of the SDF-1(CXCL12)/CXCR4 axis blocks prostate cancer metastasis and growth in osseous sites in vivo. J Bone Miner Res (2005) 20(2):318–29. doi:10.1359/JBMR.041109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res (2002) 16(6):1832–7.
137. Conley-LaComb MK, Saliganan A, Kandagatla P, Chen YQ, Cher ML, Chinni SR. PTEN loss mediated Akt activation promotes prostate tumor growth and metastasis via CXCL12/CXCR4 signaling. Mol Cancer (2013) 12(1):85. doi:10.1186/1476-4598-12-85
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Shiozawa Y, Pienta KJ, Taichman RS. Hematopoietic stem cell niche is a potential therapeutic target for bone metastatic tumors. Clin Cancer Res (2011) 17(17):5553–8. doi:10.1158/1078-0432.CCR-10-2505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nat Rev Cancer (2009) 9(4):285–93. doi:10.1038/nrc2621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Langley RR, Fidler IJ. The seed and soil hypothesis revisited – the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer (2011) 128(11):2527–35. doi:10.1002/ijc.26031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature (2005) 438(7069):820–7. doi:10.1038/nature04186
142. Hiratsuka S, Watanabe A, Aburatani H, Maru Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol (2006) 8(12):1369–75. doi:10.1038/ncb1507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell (2012) 21(5):642–54. doi:10.1016/j.ccr.2012.03.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Zhau HE, Li CL, Chung LW. Establishment of human prostate carcinoma skeletal metastasis models. Cancer (2000) 88(12 Suppl):2995–3001. doi:10.1002/1097-0142(20000615)88:12+<2995::AID-CNCR15>3.3.CO;2-P
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Amir E, Tannock IF. Prostate cancer: androgen deprivation therapy and bone loss. Nat Rev Urol (2009) 6(12):642–4. doi:10.1038/nrurol.2009.218
146. Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. The effects of androgen deficiency on murine bone remodeling and bone mineral density are mediated via cells of the osteoblastic lineage. Endocrinology (1997) 138(9):4013–21. doi:10.1210/endo.138.9.5359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Economos C, Morrissey C, Vessella RL. Circulating tumor cells as a marker of response: implications for determining treatment efficacy and evaluating new agents. Curr Opin Urol (2012) 22(3):190–6. doi:10.1097/MOU.0b013e3283519b58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Koop S, MacDonald IC, Luzzi K, Schmidt EE, Morris VL, Grattan M, et al. Fate of melanoma cells entering the microcirculation: over 80% survive and extravasate. Cancer Res (1995) 55(12):2520–3.
149. Wikman H, Vessella R, Pantel K. Cancer micrometastasis and tumour dormancy. APMIS (2008) 116(7–8):754–70. doi:10.1111/j.1600-0463.2008.01033.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Okazaki M, Takeshita S, Kawai S, Kikuno R, Tsujimura A, Kudo A, et al. Molecular cloning and characterization of OB-cadherin, a new member of cadherin family expressed in osteoblasts. J Biol Chem (1994) 269(16):12092–8.
151. Kawaguchi J, Kii I, Sugiyama Y, Takeshita S, Kudo A. The transition of cadherin expression in osteoblast differentiation from mesenchymal cells: consistent expression of cadherin-11 in osteoblast lineage. J Bone Miner Res (2001) 16(2):260–9. doi:10.1359/jbmr.2001.16.2.260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Chu K, Cheng CJ, Ye X, Lee YC, Zurita AJ, Chen DT, et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol Cancer Res (2008) 6(8):1259–67. doi:10.1158/1541-7786.MCR-08-0077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Huang CF, Lira C, Chu K, Bilen MA, Lee YC, Ye X, et al. Cadherin-11 increases migration and invasion of prostate cancer cells and enhances their interaction with osteoblasts. Cancer Res (2010) 70(11):4580–9. doi:10.1158/0008-5472.CAN-09-3016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Tamura D, Hiraga T, Myoui A, Yoshikawa H, Yoneda T. Cadherin-11 mediated interactions with bone marrow stromal/osteoblastic cells support selective colonization of breast cancer cells in bone. Int J Oncol (2008) 33(1):17–24.
155. Logothetis CJ, Lin SH. Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer (2005) 5(1):21–8. doi:10.1038/nrc1528
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Mishra A, Shiozawa Y, Pienta KJ, Taichman RS. Homing of cancer cells to the bone. Cancer Microenviron (2011) 4(3):221–35. doi:10.1007/s12307-011-0083-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Yin JJ, Pollock CB, Kelly K. Mechanisms of cancer metastasis to the bone. Cell Res (2005) 15(1):57–62. doi:10.1038/sj.cr.7290266
158. Keller ET, Brown J. Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem (2004) 91(4):718–29. doi:10.1002/jcb.10662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Darby S, Cross SS, Brown NJ, Hamdy FC, Robson CN. BMP-6 over-expression in prostate cancer is associated with increased Id-1 protein and a more invasive phenotype. J Pathol (2008) 214(3):394–404. doi:10.1002/path.2292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Katsuno Y, Hanyu A, Kanda H, Ishikawa Y, Akiyama F, Iwase T, et al. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene (2008) 27(49):6322–33. doi:10.1038/onc.2008.232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Dai J, Keller J, Zhang J, Lu Y, Yao Z, Keller ET. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res (2005) 65(18):8274–85. doi:10.1158/0008-5472.CAN-05-1891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Bentley H, Hamdy FC, Hart KA, Seid JM, Williams JL, Johnstone D, et al. Expression of bone morphogenetic proteins in human prostatic adenocarcinoma and benign prostatic hyperplasia. Br J Cancer (1992) 66(6):1159–63. doi:10.1038/bjc.1992.427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Raida M, Clement JH, Ameri K, Han C, Leek RD, Harris AL. Expression of bone morphogenetic protein 2 in breast cancer cells inhibits hypoxic cell death. Int J Oncol (2005) 26(6):1465–70.
164. Clement JH, Raida M, Sanger J, Bicknell R, Liu J, Naumann A, et al. Bone morphogenetic protein 2 (BMP-2) induces in vitro invasion and in vivo hormone independent growth of breast carcinoma cells. Int J Oncol (2005) 27(2):401–7. doi:10.3892/ijo.27.2.401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Feeley BT, Gamradt SC, Hsu WK, Liu N, Krenek L, Robbins P, et al. Influence of BMPs on the formation of osteoblastic lesions in metastatic prostate cancer. J Bone Miner Res (2005) 20(12):2189–99. doi:10.1359/JBMR.050802
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Abe E, Yamamoto M, Taguchi Y, Lecka-Czernik B, O’Brien CA, Economides AN, et al. Essential requirement of BMPs-2/4 for both osteoblast and osteoclast formation in murine bone marrow cultures from adult mice: antagonism by noggin. J Bone Miner Res (2000) 15(4):663–73. doi:10.1359/jbmr.2000.15.4.663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Gori F, Thomas T, Hicok KC, Spelsberg TC, Riggs BL. Differentiation of human marrow stromal precursor cells: bone morphogenetic protein-2 increases OSF2/CBFA1, enhances osteoblast commitment, and inhibits late adipocyte maturation. J Bone Miner Res (1999) 14(9):1522–35. doi:10.1359/jbmr.1999.14.9.1522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Jena N, Martin-Seisdedos C, McCue P, Croce CM. BMP7 null mutation in mice: developmental defects in skeleton, kidney, and eye. Exp Cell Res (1997) 230(1):28–37. doi:10.1006/excr.1996.3411
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Nishimura R, Hata K, Matsubara T, Wakabayashi M, Yoneda T. Regulation of bone and cartilage development by network between BMP signalling and transcription factors. J Biochem (2012) 151(3):247–54. doi:10.1093/jb/mvs004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell (2002) 108(1):17–29. doi:10.1016/S0092-8674(01)00622-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Morrissey C, Brown LG, Pitts TE, Vessella RL, Corey E. Bone morphogenetic protein 7 is expressed in prostate cancer metastases and its effects on prostate tumor cells depend on cell phenotype and the tumor microenvironment. Neoplasia (2010) 12(2):192–205.
172. Kobayashi A, Okuda H, Xing F, Pandey PR, Watabe M, Hirota S, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J Exp Med (2011) 208(13):2641–55. doi:10.1084/jem.20110840
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Lee GT, Kang DI, Ha YS, Jung YS, Chung J, Min K, et al. Prostate cancer bone metastases acquire resistance to androgen deprivation via WNT5A-mediated BMP-6 induction. Br J Cancer (2014) 110(6):1634–44. doi:10.1038/bjc.2014.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Dai J, Hall CL, Escara-Wilke J, Mizokami A, Keller JM, Keller ET. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res (2008) 68(14):5785–94. doi:10.1158/0008-5472.CAN-07-6541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Godebu E, Muldong M, Strasner A, Wu C, Park S, Woo JR, et al. PCSD1, a new patient-derived model of bone metastatic prostate cancer, is castrate-resistant in the bone-niche. J Transl Med (2014) 12(1):275. doi:10.1186/s12967-014-0275-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Nishimori H, Ehata S, Suzuki HI, Katsuno Y, Miyazono K. Prostate cancer cells and bone stromal cells mutually interact with each other through bone morphogenetic protein-mediated signals. J Biol Chem (2012) 287(24):20037–46. doi:10.1074/jbc.M112.353094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Zunich SM, Douglas T, Valdovinos M, Chang T, Bushman W, Walterhouse D, et al. Paracrine sonic hedgehog signalling by prostate cancer cells induces osteoblast differentiation. Mol Cancer (2009) 8:12. doi:10.1186/1476-4598-8-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Zunich SM, Valdovinos M, Douglas T, Walterhouse D, Iannaccone P, Lamm ML. Osteoblast-secreted collagen upregulates paracrine Sonic hedgehog signaling by prostate cancer cells and enhances osteoblast differentiation. Mol Cancer (2012) 11:30. doi:10.1186/1476-4598-11-30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Rosano L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer (2013) 13(9):637–51. doi:10.1038/nrc3546
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Nelson JB, Udan MS, Guruli G, Pflug BR. Endothelin-1 inhibits apoptosis in prostate cancer. Neoplasia (2005) 7(7):631–7. doi:10.1593/neo.04787
181. Rosano L, Varmi M, Salani D, Di Castro V, Spinella F, Natali PG, et al. Endothelin-1 induces tumor proteinase activation and invasiveness of ovarian carcinoma cells. Cancer Res (2001) 61(22):8340–6.
182. Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature (1994) 368(6473):703–10. doi:10.1038/368703a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med (1995) 1(9):944–9. doi:10.1038/nm0995-944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Yin JJ, Mohammad KS, Kakonen SM, Harris S, Wu-Wong JR, Wessale JL, et al. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci U S A (2003) 100(19):10954–9. doi:10.1073/pnas.1830978100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Rahim F, Hajizamani S, Mortaz E, Ahmadzadeh A, Shahjahani M, Shahrabi S, et al. Molecular regulation of bone marrow metastasis in prostate and breast cancer. Bone Marrow Res (2014) 2014:405920. doi:10.1155/2014/405920
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Chiao JW, Moonga BS, Yang YM, Kancherla R, Mittelman A, Wu-Wong JR, et al. Endothelin-1 from prostate cancer cells is enhanced by bone contact which blocks osteoclastic bone resorption. Br J Cancer (2000) 83(3):360–5. doi:10.1054/bjoc.2000.1261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Nelson JB, Nabulsi AA, Vogelzang NJ, Breul J, Zonnenberg BA, Daliani DD, et al. Suppression of prostate cancer induced bone remodeling by the endothelin receptor A antagonist atrasentan. J Urol (2003) 169(3):1143–9. doi:10.1097/01.ju.0000042162.08938.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Quinn DI, Tangen CM, Hussain M, Lara PN, Goldkorn A, Moinpour CM, et al. Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): a randomised phase 3 trial. Lancet Oncol (2013) 14(9):893–900. doi:10.1016/S1470-2045(13)70294-8
189. Clines GA, Mohammad KS, Bao Y, Stephens OW, Suva LJ, Shaughnessy JD Jr., et al. Dickkopf homolog 1 mediates endothelin-1-stimulated new bone formation. Mol Endocrinol (2007) 21(2):486–98. doi:10.1210/me.2006-0346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer (2013) 13(1):11–26. doi:10.1038/nrc3419
191. Kypta RM, Waxman J. Wnt/beta-catenin signalling in prostate cancer. Nat Rev Urol (2012) 9(8):418–28. doi:10.1038/nrurol.2012.116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Hall CL, Kang S, MacDougald OA, Keller ET. Role of Wnts in prostate cancer bone metastases. J Cell Biochem (2006) 97(4):661–72. doi:10.1002/jcb.20735
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Yardy GW, Brewster SF. Wnt signalling and prostate cancer. Prostate Cancer Prostatic Dis (2005) 8(2):119–26. doi:10.1038/sj.pcan.4500794
194. Watanabe M, Kakiuchi H, Kato H, Shiraishi T, Yatani R, Sugimura T, et al. APC gene mutations in human prostate cancer. Jpn J Clin Oncol (1996) 26(2):77–81. doi:10.1093/oxfordjournals.jjco.a023188
195. Majid S, Saini S, Dahiya R. Wnt signaling pathways in urological cancers: past decades and still growing. Mol Cancer (2012) 11:7. doi:10.1186/1476-4598-11-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Chen G, Shukeir N, Potti A, Sircar K, Aprikian A, Goltzman D, et al. Up-regulation of Wnt-1 and β-catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer (2004) 101(6):1345–56. doi:10.1002/cncr.20518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Boissan M, De Wever O, Lizarraga F, Wendum D, Poincloux R, Chignard N, et al. Implication of metastasis suppressor NM23-H1 in maintaining adherens junctions and limiting the invasive potential of human cancer cells. Cancer Res (2010) 70(19):7710–22. doi:10.1158/0008-5472.CAN-10-1887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem (2005) 280(39):33132–40. doi:10.1074/jbc.M500608200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Cho YD, Yoon WJ, Kim WJ, Woo KM, Baek JH, Lee G, et al. Epigenetic modifications and canonical wingless/int-1 Class (WNT) signaling enable trans-differentiation of nonosteogenic cells into osteoblasts. J Biol Chem (2014) 289(29):20120–8. doi:10.1074/jbc.M114.558064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Hall CL, Bafico A, Dai J, Aaronson SA, Keller ET. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res (2005) 65(17):7554–60. doi:10.1158/0008-5472.CAN-05-1317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun Signal (2013) 11:97. doi:10.1186/1478-811X-11-97
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. Ustach CV, Huang W, Conley-LaComb MK, Lin CY, Che M, Abrams J, et al. A novel signaling axis of matriptase/PDGF-D/ss-PDGFR in human prostate cancer. Cancer Res (2010) 70(23):9631–40. doi:10.1158/0008-5472.CAN-10-0511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Ustach CV, Taube ME, Hurst NJ Jr., Bhagat S, Bonfil RD, Cher ML, et al. A potential oncogenic activity of platelet-derived growth factor d in prostate cancer progression. Cancer Res (2004) 64(5):1722–9. doi:10.1158/0008-5472.CAN-03-3047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
204. Caplan AI, Correa D. PDGF in bone formation and regeneration: new insights into a novel mechanism involving MSCs. J Orthop Res (2011) 29(12):1795–803. doi:10.1002/jor.21462
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Lev DC, Kim SJ, Onn A, Stone V, Nam DH, Yazici S, et al. Inhibition of platelet-derived growth factor receptor signaling restricts the growth of human breast cancer in the bone of nude mice. Clin Cancer Res (2005) 11(1):306–14.
206. Hwang RF, Yokoi K, Bucana CD, Tsan R, Killion JJ, Evans DB, et al. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin Cancer Res (2003) 9(17):6534–44.
207. Mathew P, Thall PF, Bucana CD, Oh WK, Morris MJ, Jones DM, et al. Platelet-derived growth factor receptor inhibition and chemotherapy for castration-resistant prostate cancer with bone metastases. Clin Cancer Res (2007) 13(19):5816–24. doi:10.1158/1078-0432.CCR-07-1269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Iqbal N, Iqbal N. Imatinib: a breakthrough of targeted therapy in cancer. Chemother Res Pract (2014) 2014:357027. doi:10.1155/2014/357027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Erlebacher A, Derynck R. Increased expression of TGF-β2 in osteoblasts results in an osteoporosis-like phenotype. J Cell Biol (1996) 132(1–2):195–210. doi:10.1083/jcb.132.1.195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Filvaroff E, Erlebacher A, Ye J, Gitelman SE, Lotz J, Heillman M, et al. Inhibition of TGF-beta receptor signaling in osteoblasts leads to decreased bone remodeling and increased trabecular bone mass. Development (1999) 126(19):4267–79.
211. Johnson RW, Nguyen MP, Padalecki SS, Grubbs BG, Merkel AR, Oyajobi BO, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res (2011) 71(3):822–31. doi:10.1158/0008-5472.CAN-10-2993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Buijs JT, Stayrook KR, Guise TA. TGF-beta in the Bone Microenvironment: Role in Breast Cancer Metastases. Cancer Microenviron (2011) 4(3):261–81. doi:10.1007/s12307-011-0075-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
213. Kingsley LA, Fournier PG, Chirgwin JM, Guise TA. Molecular biology of bone metastasis. Mol Cancer Ther (2007) 6(10):2609–17. doi:10.1158/1535-7163.MCT-07-0234
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
214. Ehata S, Hanyu A, Fujime M, Katsuno Y, Fukunaga E, Goto K, et al. Ki26894, a novel transforming growth factor-beta type I receptor kinase inhibitor, inhibits in vitro invasion and in vivo bone metastasis of a human breast cancer cell line. Cancer Sci (2007) 98(1):127–33. doi:10.1111/j.1349-7006.2006.00357.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
215. Mohammad KS, Javelaud D, Fournier PG, Niewolna M, McKenna CR, Peng XH, et al. TGF-β-RI kinase inhibitor SD-208 reduces the development and progression of melanoma bone metastases. Cancer Res (2011) 71(1):175–84. doi:10.1158/0008-5472.CAN-10-2651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
216. Yin JJS, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser RM, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest (1999) 103(2):197–206. doi:10.1172/JCI3523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
217. Li X, Sterling JA, Fan KH, Vessella RL, Shyr Y, Hayward SW, et al. Loss of TGF-β responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol Cancer Res (2012) 10(4):494–503. doi:10.1158/1541-7786.MCR-11-0506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
218. Furstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol (2002) 3(5):298–302. doi:10.1016/S1470-2045(02)00731-3
219. Grzmil M, Hemmerlein B, Thelen P, Schweyer S, Burfeind P. Blockade of the type I IGF receptor expression in human prostate cancer cells inhibits proliferation and invasion, up-regulates IGF binding protein-3, and suppresses MMP-2 expression. J Pathol (2004) 202(1):50–9. doi:10.1002/path.1492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
220. Zhao H, Dupont J, Yakar S, Karas M, LeRoith D. PTEN inhibits cell proliferation and induces apoptosis by downregulating cell surface IGF-IR expression in prostate cancer cells. Oncogene (2004) 23(3):786–94. doi:10.1038/sj.onc.1207162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
221. Hiraga T, Myoui A, Hashimoto N, Sasaki A, Hata K, Morita Y, et al. Bone-derived IGF mediates crosstalk between bone and breast cancer cells in bony metastases. Cancer Res (2012) 72(16):4238–49. doi:10.1158/0008-5472.CAN-11-3061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
222. Ryan CJ, Haqq CM, Simko J, Nonaka DF, Chan JM, Weinberg V, et al. Expression of insulin-like growth factor-1 receptor in local and metastatic prostate cancer. Urol Oncol (2007) 25(2):134–40.
223. Kwabi-Addo B, Ozen M, Ittmann M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr Relat Cancer (2004) 11(4):709–24. doi:10.1677/erc.1.00535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
224. Abate-Shen C, Shen MM. FGF signaling in prostate tumorigenesis – new insights into epithelial-stromal interactions. Cancer Cell (2007) 12(6):495–7. doi:10.1016/j.ccr.2007.11.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
225. Corn PG, Wang F, McKeehan WL, Navone N. Targeting fibroblast growth factor pathways in prostate cancer. Clin Cancer Res (2013) 19(21):5856–66. doi:10.1158/1078-0432.CCR-13-1550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
226. Dorkin TJ, Robinson MC, Marsh C, Neal DE, Leung HY. aFGF immunoreactivity in prostate cancer and its co-localization with bFGF and FGF8. J Pathol (1999) 189(4):564–9. doi:10.1002/(SICI)1096-9896(199912)189:4<564::AID-PATH480>3.0.CO;2-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
227. Giri D, Ropiquet F, Ittmann M. Alterations in expression of basic fibroblast growth factor (FGF) 2 and its receptor FGFR-1 in human prostate cancer. Clin Cancer Res (1999) 5(5):1063–71.
228. Ropiquet F, Giri D, Kwabi-Addo B, Mansukhani A, Ittmann M. Increased expression of fibroblast growth factor 6 in human prostatic intraepithelial neoplasia and prostate cancer. Cancer Res (2000) 60(15):4245–50.
229. Freeman KW, Welm BE, Gangula RD, Rosen JM, Ittmann M, Greenberg NM, et al. Inducible prostate intraepithelial neoplasia with reversible hyperplasia in conditional FGFR1-expressing mice. Cancer Res (2003) 63(23):8256–63.
230. Acevedo VD, Gangula RD, Freeman KW, Li R, Zhang Y, Wang F, et al. Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell (2007) 12(6):559–71. doi:10.1016/j.ccr.2007.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
231. Ozen M, Giri D, Ropiquet F, Mansukhani A, Ittmann M. Role of fibroblast growth factor receptor signaling in prostate cancer cell survival. J Natl Cancer Inst (2001) 93(23):1783–90. doi:10.1093/jnci/93.23.1783
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
232. Li ZG, Mathew P, Yang J, Starbuck MW, Zurita AJ, Liu J, et al. Androgen receptor-negative human prostate cancer cells induce osteogenesis in mice through FGF9-mediated mechanisms. J Clin Invest (2008) 118(8):2697–710. doi:10.1172/JCI33093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
233. Valta MP, Hentunen T, Qu Q, Valve EM, Harjula A, Seppanen JA, et al. Regulation of osteoblast differentiation: a novel function for fibroblast growth factor 8. Endocrinology (2006) 147(5):2171–82. doi:10.1210/en.2005-1502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
234. Valta MP, Tuomela J, Bjartell A, Valve E, Vaananen HK, Harkonen P. FGF-8 is involved in bone metastasis of prostate cancer. Int J Cancer (2008) 123(1):22–31. doi:10.1002/ijc.23422
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
235. Teishima J, Shoji K, Hayashi T, Miyamoto K, Ohara S, Matsubara A. Relationship between the localization of fibroblast growth factor 9 in prostate cancer cells and postoperative recurrence. Prostate Cancer Prostatic Dis (2012) 15(1):8–14. doi:10.1038/pcan.2011.48
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
236. Lamb LE, Knudsen BS, Miranti CK. E-cadherin-mediated survival of androgen-receptor-expressing secretory prostate epithelial cells derived from a stratified in vitro differentiation model. J Cell Sci (2010) 123(Pt 2):266–76. doi:10.1242/jcs.054502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
237. Foster BA, Evangelou A, Gingrich JR, Kaplan PJ, DeMayo F, Greenberg NM. Enforced expression of FGF-7 promotes epithelial hyperplasia whereas a dominant negative FGFR2iiib promotes the emergence of neuroendocrine phenotype in prostate glands of transgenic mice. Differentiation (2002) 70(9–10):624–32. doi:10.1046/j.1432-0436.2002.700915.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
238. Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell (2007) 12(6):572–85. doi:10.1016/j.ccr.2007.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
239. Liu W, Zhou Y, Reske SN, Shen C. PTEN mutation: many birds with one stone in tumorigenesis. Anticancer Res (2008) 28(6A):3613–9.
240. Xu J, Wang R, Xie ZH, Odero-Marah V, Pathak S, Multani A, et al. Prostate cancer metastasis: role of the host microenvironment in promoting epithelial to mesenchymal transition and increased bone and adrenal gland metastasis. Prostate (2006) 66(15):1664–73. doi:10.1002/pros.20488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
241. Corey E, Quinn JE, Buhler KR, Nelson PS, Macoska JA, True LD, et al. LuCaP 35: a new model of prostate cancer progression to androgen independence. Prostate (2003) 55(4):239–46. doi:10.1002/pros.10198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
242. Corey E, Quinn JE, Vessella RL. A novel method of generating prostate cancer metastases from orthotopic implants. Prostate (2003) 56(2):110–4. doi:10.1002/pros.10235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: prostate, cancer, tumor microenvironment, bone metastasis, EMT
Citation: Ganguly SS, Li X and Miranti CK (2014) The host microenvironment influences prostate cancer invasion, systemic spread, bone colonization, and osteoblastic metastasis. Front. Oncol. 4:364. doi: 10.3389/fonc.2014.00364
Received: 01 October 2014; Paper pending published: 17 November 2014;
Accepted: 29 November 2014; Published online: 15 December 2014.
Edited by:
Shidong Jia, Genentech Inc., USAReviewed by:
Andrea Morrione, Thomas Jefferson University, USAMarco Falasca, Queen Mary University of London, UK
Copyright: © 2014 Ganguly, Li and Miranti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cindy K. Miranti, Van Andel Research Institute, 333 Bostwick Avenue NE, Grand Rapids, MI 49503, USA e-mail:Y2luZHkubWlyYW50aUB2YWkub3Jn