 Stefanie B. Marquez
Stefanie B. Marquez Kenneth W. Thompson
Kenneth W. Thompson Li Lu2
Li Lu2 David Reisman
David Reisman- 1Department of Medicine, Division of Hematology/Oncology, University of Florida, Gainesville, FL, USA
- 2Department of Pathology, University of Florida, Gainesville, FL, USA
SWI/SNF is a major regulator of gene expression. Its role is to facilitate the shifting and exposure of DNA segments within the promoter and other key domains to transcription factors and other essential cellular proteins. This complex interacts with a wide range of proteins and does not function within a single, specific pathway; thus, it is involved in a multitude of cellular processes, including DNA repair, differentiation, development, cell adhesion, and growth control. Given SWI/SNF’s prominent role in these processes, many of which are important for blocking cancer development, it is not surprising that the SWI/SNF complex is targeted during cancer initiation and progression both by mutations and by non-mutational mechanisms. Currently, the understanding of the types of alterations, their frequency, and their impact on the SWI/SNF subunits is an area of intense research that has been bolstered by a recent cadre of NextGen sequencing studies. These studies have revealed mutations in SWI/SNF subunits, indicating that this complex is thus important for cancer development. The purpose of this review is to put into perspective the role of mutations versus other mechanisms in the silencing of SWI/SNF subunits, in particular, BRG1 and BRM. In addition, this review explores the recent development of synthetic lethality and how it applies to this complex, as well as how BRM polymorphisms are becoming recognized as potential clinical biomarkers for cancer risk. Significance: Recent reviews have detailed the occurrence of mutations in nearly all SWI/SNF subunits, which indicates that this complex is an important target for cancer. However, when the frequency of mutations in a given tumor type is compared to the frequency of subunit loss, it becomes clear that other non-mutational mechanisms must play a role in the inactivation of SWI/SNF subunits. Such data indicate that epigenetic mechanisms that are known to regulate BRM may also be involved in the loss of expression of other SWI/SNF subunits. This is important since epigenetically silenced genes are inducible, and thus, the reversal of the silencing of these non-mutationally suppressed subunits may be a viable mode of targeted therapy.
Introduction
SWI/SNF is a chromatin-remodeling complex first described in the early 1990s. These studies showed that this complex opens up the chromatin by shifting the position of histones and making the DNA accessible to transcription factors and key cellular proteins. In yeast, there is only a single complex, but in mammals there are at least three different complexes. Each complex is composed of one of two ATPase subunits, Brahma (BRM/SMARCA2) or Brahma related gene 1 (BRG1/SMARCA4) and 8–10 other subunits that results in a 2-MDa complex. In yeast, this complex regulates about 5–6% of the genome, while in mammalian organisms, it is not known how many genes are regulated by this complex and how many protein interactions occur in conjunction with this complex. However, SWI/SNF does not interact with one type of transcription factor, but rather, it regulates the function of many diverse genes as well as the function of many signaling pathways. SWI/SNF has been found to function in DNA repair, growth control, differentiation, development, cellular adhesion, and immunity. In most cases, SWI/SNF functions in general to maintain homeostasis and normal cellular function. The function and composition of SWI/SNF is detailed in many other reviews and is not the focus of this review. As SWI/SNF subunits are frequently found to be mutated in different tumors, we compared published mutation data (COSMIC and Atlas databases) with published IHC (when available) to determine how frequently mutations underlie the loss of expression of different SWI/SNF units. As detailed below, in many cases, mutations cannot account for why certain SWI/SNF subunits are silenced in cancer, which suggests that other mechanisms likely underlie the loss of these proteins in cancer.
Differing Roles for SWI/SNF in Cancer, Depending on the Context
SWI/SNF functions as an epigenetic regulator that opens and closes the chromatin, thereby helping to turn gene expression on and off. In this capacity, the complex serves as both an oncogene and a tumor suppressor, depending on the protein with which it is interacting (1). Abrogation of SWI/SNF can impact the normal functions of DNA repair, cellular differentiation, development, and cell adhesion (2). Its diversity in function has been recently demonstrated by contrasting sets of publications about the role of one of its two catalytic subunits, Brahma (BRM, or SMARCA2), in cancer. Traditionally, BRM has been viewed as a tumor susceptibility or tumor suppressor protein because its loss can potentiate the development of tumors in murine cancer models (3, 4) as well as portend worse clinical survival in lung cancer patients (5, 6). In addition, BRM loss in non-transformed cell lines has been shown to prevent cells from entering a canonical quiescent state (7), which further attests to the growth-inhibitory functions ascribed to BRM. Several labs have shown that BRM is necessary for RB function (8–10): it is known to bind to RB (11), and together, RB and BRM suppress the activity of E2F (12). A constitutively active form of RB fails to inhibit growth when introduced into BRM-deficient cell lines, as well as in cells that are deficient in its homolog, Brahma-related gene 1 (BRG1, or SMARCA4), which is the second catalytic subunit of SWI/SNF (10). In fact, re-expression of BRM can inhibit growth and induces differentiation in a variety of BRG1/BRM-deficient cell lines (8), a process that is disrupted by viral proteins that bind to RB, such as E1A or E7 (8, 13). Moreover, BRM re-expression has been shown to antagonize KRAS-transformed cells by partially reversing the transformed phenotype and substantially slowing growth; based on these data, these authors concluded that increased BRM expression promotes the entry of cells into G1/G0, while the loss of BRM expression may foster oncogenic transformation (14). BRM has been shown to promote exit from the cell cycle and cell differentiation in retinal ganglion cells (15). Re-activation of BRM and SWI/SNF appear to foster the active, hypophosphorylated form of RB through the inhibition of cyclin D1 by directly binding to its promoter and stimulating the downregulation of cyclin D1 as well as the induction of p16 (16, 17). Thus, numerous studies have clearly defined a growth-inhibitory role for BRM and SWI/SNF.
Apparently contrasting sets of observations, however, suggest a dichotomous function for BRM. These findings imply that BRM and SWI/SNF may not solely function in growth-inhibitory pathways but may also serve in a net stimulatory capacity. Thus, the function of BRM depends on the context. This concept has been directly observed in BRG1-deficient cell lines where the knockdown of BRM actually slows growth. This has been described as synthetic lethality – a term, which describes the situation where the loss of function of two or more genes leads to cell death, but where the loss of function in only one of the genes is not lethal. This finding of synthetic lethality after BRM knockdown has also been found to occur in xenograft models (18–20). These data indicate that BRM is necessary for stimulatory pathways – or, alternatively, that BRM may act as an antagonist to growth-inhibitory pathways (i.e., it acts as a dominant negative). Potentially important to these observations is the fact that BRM has been shown to be regulated post-translationally, where the acetylation of BRM blocks BRM-dependent gene expression (16) and conversely, the removal of BRM acetylation causes growth arrest (16). In at least a subset of the cell lines used to demonstrate synthetic lethality, BRM is actually known to be acetylated, which suggests that its growth-inhibitory functions are likely turned off (unpublished observations) (16, 21).
In this particular context, which in this case is in BRG1-deficient genotypes, experimental silencing of BRM has been observed to significantly slow cancer cell growth rather than promote it (19). At least two rationales might explain this observed phenomenon. First, if BRM and SWI/SNF function in the growth-promoting pathway, then their suppression would diminish their respective contributions to this pathway and would likely cause slower growth, as observed. To this end, SWI/SNF and BRM have been shown to be cofactors for such oncogenes as C-Jun and C-Myc (22, 23). Second, at least a subset of the cells that have demonstrated synthetic lethality harbor a form of BRM that is acetylated at the C-terminus (16), which means that BRM is likely inactivated in these cell lines (21). The frequency of BRM acetylation in primary tumors is not yet known, but BRM acetylation is estimated to occur in 65–70% of cancer cell lines, which suggests that BRM acetylation also occurs frequently in primary tumors (21). We have shown that the deacetylation of BRM promptly induces growth arrest (21). In this capacity, BRM may be functioning as a dominant negative, much like p53 does when mutated. Alternatively, the role of BRM may be dictated by its acetylation state, where its unacetylated form binds to RB and RB2 and enhances growth inhibition, and its acetylated form antagonizes RB and also functions in growth pathways such as those driven by oncogenes (e.g., C-Myc and C-Jun). We favor the latter hypothesis, as it explains the majority of experimental observations and minimizes the apparent contradictions in the data, for example, where BRM re-expression as well as BRM knockout can both result in growth inhibition in certain cellular contexts, as outlined in our recent Cancer Research editorial (24). In addition, there are at least three different acetylation sites. Whether each acetylation site governs a different activity of BRM or all three are required to inactivate BRM is not yet known. Nevertheless, these new data demonstrate that BRM, BRG1, and SWI/SNF have potentially different roles depending on the context.
Synthetic Lethality and the Clinical Silencing of BRM
Based on the observation that the knockdown of BRM in BRG1-deficient cell lines induces synthetic lethality, the clinical silencing of BRM has been suggested as a therapeutic strategy. Yet, this approach may not take into account the other anti-cancer functions of BRM and SWI/SNF. SWI/SNF and BRM are known to regulate cellular adhesion, DNA repair, differentiation, and expression of MHC class 1 and 2 proteins (2). Hence, while their suppression in certain circumstances might promote growth inhibition, it is equally possible such a strategy could have adverse clinical consequences by accelerating de-differentiation and promoting metastasis by inhibiting the expression of adhesion proteins such as E-cadherin, CEACAM1, integrins, and CD44, which are known to be regulated by SWI/SNF (2, 25). Moreover, the loss of both BRG1 and BRM would favor the inhibition of DNA repair (2, 25), and in particular, such key DNA repair genes as GADD45 (26), which in turn could enhance tumor progression. As SWI/SNF is reported to regulate MHC class 1 and 2 proteins (27–29), the silencing of BRM might result in the loss of these proteins, impairing the normal function of the immune system (30). BRM and the SWI/SNF complex have roles in the maintenance of genomic integrity and are required for the correct segregation of chromatids (7). BRM loss might also impact gene expression by changing the splicing pattern of genes such as CD44, Cyclin D1, and BIM (31). There are many positive functions governed by SWI/SNF that are yet to be well defined, and they might also be compromised if BRM is clinically silenced.
It might be a better approach to understand the mechanism that underlies synthetic lethality in this context before pursuing it as a mode of clinical therapy. For example, if BRM acetylation is truly a component of this phenomenon, the reversal of acetylation might be a prudent and feasible approach to affect the growth control function of BRM as well as the restoration of BRM’s other anti-cancer functions. This is because deacetylation in vitro results in growth inhibition and in the restoration of the transcriptional ability of BRM (16, 21). To this end, we know that BRM acetylation is governed by the functional balance of at least three proteins: HDAC2, KAT2B, and KAT8 (21). It is possible that aberrant KAT activity could be driving the acetylation of BRM in cancer, as KATs are known in certain cancer types to be hyperactive due to overexpression and the formation of hybrid/fusion proteins (32–34). In particular, as synthetic lethality has been observed in lung cancer-derived cell lines, it is interesting to note that KAT2B (GCN5, PCAF) is overexpressed in lung cancer – and this same protein has been shown to drive the expression of the cell cycle genes E2F1, cyclin D1, and cyclin E1 (35). The targeting and inhibition of KAT2B also inhibit cancer by a variety of mechanisms (36–38), besides restoring BRM function. Hence, it may be more clinically appropriate to restore BRM function and its ability to inhibit growth by simply targeting KAT2B activity, rather than to silence BRM and risk the loss of BRM’s other anti-cancer functions. Further studies into the mechanism of BRM synthetic lethality and acetylation are needed to complete this complex picture before the best clinical avenues can be pursued.
The RB Pathway and SWI/SNF
The SWI/SNF complex has been implicated in the function of the RB pathway ever since it was identified in mammalian cells. In two papers published in the mid-1990s (8, 39), Dr. Goff’s research group was one of the first to show this functional interaction between SWI/SNF and RB. They first demonstrated the interaction with a yeast two-hybrid system and showed that RB bound specifically to BRG1 and BRM and that the RB-dependent growth inhibition was dependent on the binding of RB to these SWI/SNF subunits (39). Next, they showed that the re-introduction of BRG1 or BRM caused growth inhibition that was dependent on members of the RB family, including RB, RB2 (p130), and to a lesser degree, p107 (8). This clearly established the functional interaction and RB dependence on the SWI/SNF complex. Several labs thereafter demonstrated that the growth inhibition induced by the ectopic expression of p16 or by the constitutively active isoform of RB required a functional SWI/SNF complex (9, 10, 40–42). Specifically, neither transduction with p16 nor transient transfection with RB could effectively induce growth arrest in BRG1/BRM-deficient cell lines (e.g., SW13, C33A, H522, etc.); at the same time, the re-expression of either BRG1 or BRM in conjunction with the re-expression of either p16 or constitutively active RB were both sufficient to restore RB-dependent growth inhibition. Interestingly, growth inhibition can also occur in the absence of RB in cell lines such as C33A and Saos-2, which harbor a functional p130 protein (43, 44). This is important because unlike RB, p130 is rarely mutated in cancer and retains its function in the control of the G1 to G0 transition, which is necessary for cancer stem cells to become quiescent (45). This fact coupled with the need for p130 to bind and cooperate with SWI/SNF might explain why BRG1 and BRM are not frequently mutated. The epigenetic regulation of BRG1 and BRM would allow cancer cells to become quiescent and thereby escape the cytotoxicity of standard chemotherapy as well as maintain their ability to subsequently re-enter the cell cycle. This mechanism could also allow cancer cells to delay re-entry into the cell cycle, which could explain why cancers can recur at various times after their initial discovery and treatment.
Interestingly, in most reviews and papers, SWI/SNF inactivation is infrequently considered as an alternative method of inactivating the RB pathway. Since the loss of BRG1, BRM, or both is commonly observed in many cancer types, the disruption of the RB pathway likely occurs from a loss of SWI/SNF activity in a subset of tumors (6, 19). Post-translational modifications via phosphorylation are also known to occur in both BRG1 and BRM (46). These modifications shift these proteins from the nucleus to the cytoplasm as cells enter M-phase, but this process is reversed when cells enter G1 phase (46). Whether cancer can highjack this regulatory mechanism as a means to inactivate either protein is not currently known. Some tumors, however, do display prominent cytoplasmic staining, which suggests that BRG1 or BRM must translocate to the cytoplasm (3, 5, 6). Further research is required to determine the scope of these epigenetic mechanisms and their implications for cancer development, progression, and recurrence.
BRM Silencing and BRM Polymorphisms
The role of BRM in cancer initially appeared to be different from that of BRG1. In particular, a BRM knockout in animal models generates a phenotype that does not precipitate cancer de novo, but rather acts synergistically with carcinogens to yield tumors (3, 47). Hence, BRM loss would appear to foster cancer development when partnered with other changes such as exposure to carcinogens, rather than produce cancer by itself akin to driver oncogenes (48). In the pursuit to determine how BRM is silenced, it was found that in cell lines, BRM is epigenetically silenced, and its expression can be pharmacologically restored (49). It has since been found that the loss of BRM expression can be robustly reversed by flavonoids in general, and more specifically, by the synthetic flavonoid flavopiridol (50), which is now being tested in clinical trials (51). Recently, flavopiridol has been shown to inhibit the growth of rhabdoid tumors, which are deficient in BAF47 (SMARCB1; INI1) and commonly deficient in BRM as well (52). The induction of BRM is likely important to the growth-inhibitory mechanism of flavopiridol, as BRM silencing blunts flavopiridol-mediated growth inhibition in rhabdoid cell lines as well as in other cancer cell lines (50, 52). Interestingly, in addition to the ability of flavonoids to induce BRM in BRM-deficient rhabdoid tumor cell lines, the re-expression of BAF47 was also observed to have the modest effect on the restoration of BRM (52), indicating the existence of a cross-regulatory mechanism between SWI/SNF subunits. The re-expression of BAF47 has no effect, however, on HDAC9 or GATA3 expression, which are tightly linked with BRM silencing in cancer cell lines and primary tumors (21, 52). These data suggest that the restoration of BRM could be a mode of targeted therapy, since specific compounds (such as flavopiridol) are available that could be used, in theory, to inhibit cancer growth when applied to BRM-deficient cancers. The use of small molecule inhibitors or antibodies to block or inhibit aberrant activating kinases has become a standard clinical approach for the treatment of cancers. Yet, cancer is not only driven by aberrantly functioning gas pedals (oncogenes) but it also evolves as a result of the loss of brakes (tumor suppressors). Hence, the reversal of BRM silencing is another illustration of how an epigenetic mechanism can be reversed as a means for targeted therapy.
In order to successfully apply targeted therapies, ideally one should have biomarkers to identify those patients who would most likely benefit from a given therapy. To this end, during the pursuit to determine how BRM was silenced, we discovered two germline insertional polymorphisms within the BRM promoter (53). Both of these polymorphisms lie upstream of the transcription start sites within the first ~1300bp of the BRM promoter. The −741 polymorphic site is a triplicate repeat of the sequence TATTTTT, while the wild-type genotype is a duplicate repeat of this sequence (Figure 1). Similarly, the −1321 polymorphic site is a duplicate of the sequence TTTTAA, whereas the wild-type sequence contains this sequence only once (Figure 1) (53). Upon the discovery of these polymorphisms, two characteristics were readily observed. First, these two insertional polymorphisms have a remarkably high homology with MEF2 binding sites (54). In Caucasians, each of these polymorphisms is in Hardy–Weinberg equilibrium and occurs at a frequency of ~20% individually, while the frequency of the combined polymorphisms is roughly 6%. Second, the presence of either one or both polymorphisms statistically correlated with the loss of BRM expression in cancer cell lines (53). This statistical correlation was also found to be true in primary lung cancers (53). These polymorphisms would appear to be good surrogate biomarkers for BRM silencing in cancer given their strong correlation with BRM loss.
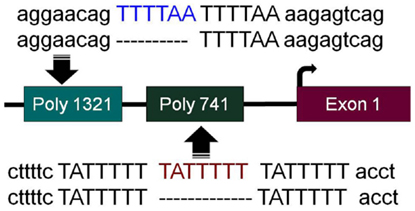
Figure 1. BRM promoter polymorphisms. The two insertion polymorphisms located in the promoter of the BRM gene are illustrated here. The −1321 polymorphic site (poly 1321) (rs3832613 or rs5925917) is located 1321 bp upstream of the transcription start site and contains an additional 6 bp insertion, TTTTAA, to yield a duplicate repeated sequence “TTTTAA-TTTTAA,” while the wild-type allele only has a single “TTTTAA” sequence present. The −741 polymorphic site (poly 741) (rs34480940) is located 741 bp from the transcription start site and contains an additional 7 bp insertion, TATTTTT, to yield a triplicate “TATTTTT-TATTTTT-TATTTTT” sequence, while the wild-type sequence consists of the duplicate sequence “TATTTTT-TATTTTT”.
Mechanism of the Epigenetic Silencing of BRM
As these polymorphisms were observed to be more prevalent in cancer cells that lack BRM expression, this suggested that they could be involved in BRM silencing. The high homology of these polymorphic sites with a known MEF2 binding site indicates that one of the MEF2 family members may be involved in BRM silencing. MEF2 transcription factors are known to silence genes by the recruitment of histone deacetylases (HDACs) (55, 56), and pan-HDAC inhibitors robustly restore BRM expression (57). Unfortunately, these pan-HDAC inhibitors also cause BRM acetylation and the subsequent inactivation of BRM. As such, these compounds cannot be practically used to restore BRM expression (16). This observation, however, indicated that HDACs must at least partially underlie BRM silencing. To this end, the shRNA-mediated knockdown of HDAC3 and HDAC9 robustly induced BRM, thereby showing that these specific HDACs are directly involved in BRM silencing (21). Similarly, the knockdown of both MEF2D and GATA3 also induced BRM, implying that these transcription factors are also part of the BRM silencing mechanism. While none of these proteins was found to be mutated in BRM-deficient cancer cell lines, HDAC9 and GATA3 were overexpressed in both lung cancer-derived cell lines and primary lung tumors, as well as in rhabdoid cell lines and primary tumors (21, 52). GATA3 and MEF2D, moreover, are known to bind to the HDAC9 promoter and drive HDAC9 overexpression (58). Just why HDAC9 overexpression appears to selectively occur in BRM-deficient cancer cells is not known. As HDACs are counter balanced by the activity of lysine acetyltransferases (KATs), it is not surprising that in BRM-deficient cell lines, ectopic expression of KAT6A, 6B, and/or 7 also induces BRM (21). Although KATs are often mutated or rearranged to form hybrid fusion proteins in many cancer types (32, 59), no alterations or mutations were observed in KAT6A, 6B, or 7 in BRM-deficient cancer cell lines (21).
Screening BRM-deficient cell lines with a library of kinase inhibitors led to the finding that the MAP kinase pathway also regulates BRM upstream of HDAC9 and GATA3 (52). Why an activated MAPK pathway leads to BRM silencing in only some circumstances is not known but appears to be linked to the presence of the BRM polymorphisms. The involvement of BRM polymorphisms has been further connected to BRM silencing by ChIP experiments showing that HDAC9 and MEF2D bind to the BRM promoter only when the BRM polymorphisms are present (52). Interestingly, MEF2D recruits HDAC9 to promoter regions, where it silences specific genes (56, 60). Hence, it appears that the activation of the MAP kinase pathway activates MEF2D, which in turn recruits HDAC9 to the BRM promoter by binding to the BRM polymorphisms. We surmise that this then leads to the removal of acetylated chromatin marks, such as H3K9 and H3K14 that control gene expression. The removal of these chromatin marks then prompts the closure of the chromatin and silencing of the BRM gene. Based on these data, the clinical targeting of BRM could be accomplished by inhibition of the MAP kinase pathway via FDA-approved inhibitors of B-raf such as vemurafenib and dabrafenib. Alternatively, as HDAC9 is a class 2 HDAC, its expression is relatively tissue-specific, and because it is highly overexpressed in BRM-deficient cancer cells, it might be a viable target for therapy. It is important to note that while BRM is silenced by this mechanism, it is unlikely that BRM is the only gene regulated by this aberrant gene silencing mechanism. Thus, targeting this mechanism may have broad anti-cancer effects, unrelated to BRM.
BRM Polymorphisms, Cancer Risk and Clinical Outcome
While the loss of BRM per se does not independently drive cancer development in mice, BRM loss appears to tip the balance toward cancer development. The data from murine models indicate that BRM is not a tumor suppressor gene like RB or p53, but rather that it can function as a tumor susceptibility gene (3). Since the BRM polymorphisms appear to be surrogate markers for BRM loss (53), we surmised that these polymorphisms would correlate with cancer development. To investigate this possibility, a series of case control studies was undertaken, and each showed a statistical correlation with lung cancer risk with odds ratios between two and three (53, 61, 62). BRM polymorphisms have not only predicted lung cancer in repeated studies, but these markers also appear to be predictive of the cancer risk for other tumor types, including head/neck cancer (63). Loss of BRM protein expression in primary lung tumors has also been associated with a worse clinical outcome in patients (5, 6). BRM polymorphisms also have a statistical correlation with a worse outcome in patients with liver cancer (64). Based on these data, the BRM polymorphisms are emerging as predictive biomarkers in a variety of tumor types. Given the link between the BRM polymorphisms and BRM expression, it may be feasible to reverse the predictive trends of this biomarker by the induction of BRM or by the prevention of BRM silencing. To this end, flavonoids (42/42) from each of the six structural groups were observed to readily induce BRM, which suggests that BRM might play a part in the anti-cancer properties of flavonoids (50). It is intriguing to speculate that the role of BRM silencing in cancer risk and cancer development could be counter balanced by the dietary consumption of flavonoids, which readily restore BRM and foster the activation of RB (50).
Mutations in SWI/SNF Subunits
Since BRM is reversibly epigenetically silenced in cell lines, it could be considered a target for therapy in tumors. For such therapies to be developed, however, it must be proven that BRM is not frequently silenced by mutations. As this axiom holds true for other SWI/SNF subunits, it is informative to determine the rate of a particular subunit’s mutation frequency versus its frequency of loss in a given tumor type. Mutations within the different SWI/SNF subunits have been well documented and described by a number of important reviews (65, 66). To ascertain the relative importance and role of each mechanism, it is useful to compare the frequency of mutations to other forms of gene alterations and inactivation. Four major sources have recently been mined to reveal such information about SWI/SNF: The Cancer Genome Atlas (TCGA) database, the Catalog of Somatic Mutation in Cancer (Cosmic) database, Sanger Sequencing publications, and available IHC data. In this review, we will focus on the comparison of the data in the Atlas database with published IHC data, and when possible, we will include mutation data from the Cosmic database. The Cosmic data will be denoted by percentage of mutations enclosed by brackets “(%)” and the TCGA data will be denoted by percentage of mutations enclosed by parentheses “(%).” In discussing mutations, it is prudent to acknowledge that mutations that change a single amino acid residue via missense might have a functional consequence, but this is not always easy to determine, and in most cases, functional data that explore the variable impact of such mutations are lacking. In contrast, gene alterations such as aberrant splicing, nonsense mutations (stop codons), or indels (frame shifts), which block the expression of a SWI/SNF subunit or result in the expression of a truncated protein, would be expected to have a much greater impact on the overall function of the mutated subunit, the SWI/SNF complex, and the pathways that are functionally dependent on this complex. NextGen sequencing studies have revealed a gamut of missense mutations that impact an unknown percentage of cancer cells within tumors, as summarized in a recent review (65). In general, current sequence data have not yet demonstrated specific hotspots in SWI/SNF subunits, making mutational analyses harder to interpret. Moreover, specific mutations or deletions in certain regions may fail to abrogate protein function. For example, the C-terminal truncation that causes the loss of the bromodomain of BRG1 has been reported to not block its ability to cooperate with RB and inhibit growth (10).
Other Considerations in the Silencing of BRG1 and BRM
While it is frequently assumed that abrogating mutations such as indels and nonsense mutations (and probably missense mutations) are ubiquitous and homogeneous throughout tumors, immunohistochemical staining of tumors for BRG1 and BRM has shown that loss of expression has a variable penetrance in a given tumor (6). Such staining can vary by gene expression intensity and/or by the pattern of gene expression loss from a uniform to a mosaic or intermittent pattern as seen in Figure 2. In the absence of complete and homogeneous loss of gene expression, the impact of these other modes of gene expression alterations will invariably impact SWI/SNF and cancer to varying degrees. To complicate this matter, since SWI/SNF is a chromatin-remodeling complex, and thus a catalyst for opening and closing DNA to modulate gene expression, the level of expression of its subunits may not have a linear impact as is seen with other genes. Rather, there may be threshold effects where, despite a lower level of expression of a given subunit, SWI/SNF may have relatively preserved levels of function, and only when the subunit levels drop below a critical level does the SWI/SNF complex become functionally impaired. To this end, lower levels of BRG1 and BRM have been frequently observed in different tumor types (6). Therefore, the sub-threshold levels of a given subunit could be another mechanism by which SWI/SNF function might be perturbed. In support of this assertion is the observation that heterozygous loss of BRG1 and BAF47 predispose mice to tumor development (67–71). As many mutations in SWI/SNF subunits are missense, it is not known if these missense mutations function as dominant negatives or simply impair or abrogate function, causing a relative state of haploinsufficiency. In the setting where a given mutation (e.g., indel or nonsense) causes a loss of function of a given SWI/SNF subunit, these mutations usually only occur in one of the two alleles. The impact of the mutated allele depends in part on the status of the other allele. Such abrogating mutations in SWI/SNF subunits could have an impact, for example, if haploinsufficiency in a given subunit portends cancer development. A single allelic loss of BAF47 or BRG1 has been shown to promote a low but discernible rate of tumor formation in mice (71). Since excess SWI/SNF subunits are generally degraded (72), the protein expression from one allele could generate the required number of functional SWI/SNF complexes to avoid cancer development. If true, this mechanism could diminish the impact of allelic loss of a given subunit on the function of the complex and thus be considered a safeguard against transformation. However, when abrogating mutations (e.g., in ARID1A, ARID1B, and PBRM1) occur, they are frequently accompanied by loss of heterozygosity, leading to the complete loss of expression (see section below).
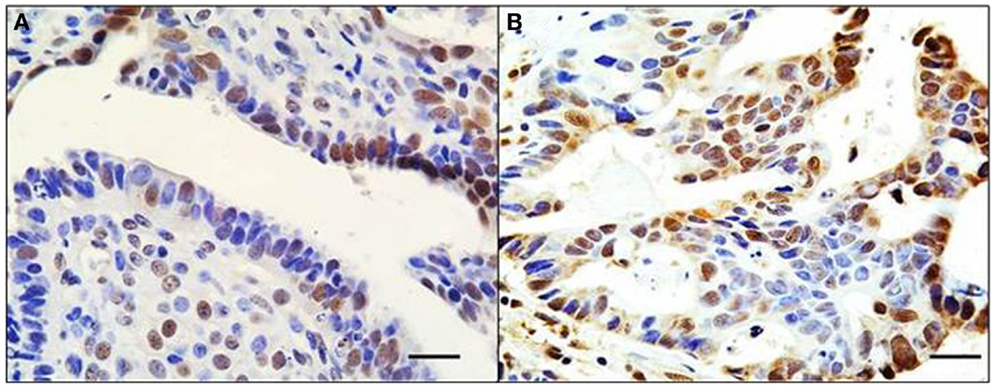
Figure 2. Mosaic pattern of BRG1 expression in human tumors. An ovarian (A) and liver (B) tumor both show an overtly mosaic pattern of BRG1 staining activity. BRG1-positive cells (brown) are observed adjacent to BRG1-negative cells (blue). This demonstrates the heterogeneous nature of these tumors with respect to loss of expression of the SWI/SNF subunits, in this case, BRG1. These pictures illustrate the problems in deciding whether a tumor is actually negative for expression of a given subunit. Original magnification: 63x; scale bar = 20 μm.
Another important consideration is the possibility of the functional redundancy of subunits, in particular BRM and BRG1, which are catalytic homologs within SWI/SNF (73, 74). While certain studies distinguish a functional difference between BRG1- and BRM-containing complexes, including differential interactions with specific transcription factors (75), other studies indicate that these subunits and their respective complexes can also substitute for one another if one subunit is suppressed or inactivated (2). This is most apparent in the studies of RB-mediated growth inhibition, where both BRG1 and BRM were shown to equally interact with RB and foster RB-mediated growth inhibition (8–10, 39). Given these findings, it is not surprising that in some tumors, such as lung cancer, the concomitant loss of BRG1 and BRM occurs in a significant portion (8–10%), compared with the loss of expression of either subunit alone, which occurs in 20–30% (5, 6, 76). Clearly, based on in vitro experiments, the value of inactivating both subunits eliminates any possibility of complementation and residual functionality of the SWI/SNF complex, in comparison with the silencing of either BRG1 or BRM alone. In addition to BRG1 and BRM, functional redundancy may exist among other subunits (e.g., BAF60A, BAF60B, and BAF60C; or BAF155 and BAF170; or ARID1A and ARID1B), which suggests a mechanism by which the SWI/SNF complex may be further protected from cancer-driven subunit loss (2, 25).
Loss of Heterozygosity and the SWI/SNF Subunits
The BRG1 locus is known to be an area of loss of heterozygosity and lies adjacent to LKB1 at 19q13.3 (77, 78). Similarly, the BRM locus is distal to p16 on the short arm of chromosome 9 at 9p23–24, which is another area of loss of heterozygosity (79–81). Both the BRG1 and BRM loci are therefore areas where loss of heterozygosity is known to occur in primary tumors (2, 82). In many cell lines that lack BRG1 or BRM, one allele has been lost (unpublished data). ARID1A lies at 1q25.3, which is another area of loss of heterozygosity (83, 84). Interestingly, ARID1B is located at 6q25, an area of known loss of heterozygosity that has been linked to a documented genomic loss that drives inherited diseases of intellectual disability as well as multiple spinal meningiomas (85, 86). Moreover, the 6q25 locus is one of several regions that is frequently lost in central nervous system tumors, particularly in childhood neuroblastoma (87), where ARID1B and ARID1A are involved in 11% of these cases (88). ARID2 maps to 12q12, an area that drives adenoid cystic carcinoma (89). PBRM1 (BAF180) is located at 3p21.1, which is frequently lost in a variety of tumors (90). While 3p21 is frequently lost in lung cancer (91), PBRM1 does not seem to be targeted in this cancer (92). In contrast, PBRM1 involvement in 3p21 loss of heterozygosity in renal cell carcinoma is well documented and correlates with frequent mutations in PBRM1 in this tumor (93). While loss of heterozygosity is frequently coupled to mutations that result in the inactivation of a target gene, loss of heterozygosity can also be coupled to epigenetic silencing. For example, most if not all BRM-deficient cell lines exhibit loss of heterozygosity (3, 52). Together, these observations regarding the loss of heterozygosity suggest that these genes can be more easily silenced when either mutational or non-mutational mechanisms occur in the remaining allele.
Identification of BRG1 Mutations in Cell Lines Versus a Lack of Mutations in Primary Tumors
Defining BRG1-inactivating mechanisms in primary tumors has been hampered because of the initial discovery of BRG1 mutations in cell lines (94, 95). A number of groups have sequenced BRG1 in both BRG1-positive and BRG1-negative cell lines, and both missense and abrogating mutations have been reported in the vast majority of BRG1-deficient cell lines. In a minority of BRG1-deficient cell lines, such as C33A, H1573, and Panc-1, no detectible mutations have been found (unpublished data) (94). Interestingly, qPCR experiments by our lab have revealed that the levels of BRG1 mRNA in cell lines are much lower if BRG1 is silenced by mutations compared to BRG1-deficient cell lines that lack mutations. We have also found that BRG1 is inducible in those cell lines that lack definable mutations (52). These findings give credence to the possibility that BRG1 is epigenetically regulated in a subset of cancer cells. At least in cell lines, qPCR could serve as a preliminary screening mechanism to determine how BRG1 might be silenced.
Some reports have implied that because BRG1 is mutated in cell lines, it must also be mutated to the same degree in primary tumors (95), and BRG1 has been shown to be silenced by mutations in >65–70% of human cancer cell lines in a number of studies (94–96). These cell line data have driven speculation that mutations must be the primary mechanisms by which cancer targets and inactivates BRG1. Yet, the data from the NIH Atlas database demonstrate that the average weighted frequency (Table 1) and the average unweighted (Table 2A) frequency of abrogating mutations for BRG1 in 23 [13] human tumor types is 0.4 and 0.21%, respectively, out of a total mutation rate of 3.75% (unweighted) and 2.41% (weighted)-which is well below the rate of BRG1 silencing found in many human tumors (15–30%) according to published IHC data (5, 97). For example, according to the Atlas database, the rates of total mutations for BRG1 in melanoma, pancreatic cancer, and head/neck cancer are 7.83, 7.69, and 5.88%, respectively, but the rate of abrogating mutations is <0.3, 1.1, and 0.33%, respectively (Table 2A); in comparison, the loss of BRG1 by IHC in these two tumors is far higher, ~10–27, ~10–25, and ~18%, respectively (98–100) (Marquez et.al. submitted Oncotarget 2015). Similarly, the rate of abrogating mutations of BRG1 in colon and breast cancers is (0.0%) [1.28%] and (0.71%) [0.39%], respectively, while BRG1 is silenced in >25% of these cancers as determined by IHC (Table 2A). In fact, the weighted average of abrogating and non-abrogating mutations as well as the total mutation rate for BRG1 from the Atlas database from 23 tumor types is 0.21, 2.20, and 2.41%, respectively (Table 1). The data from the Atlas database are consistent with Sanger sequencing studies of BRG1 in lung cancer, where abrogating BRG1 mutations (squamous and adenocarcinoma) are relatively low at 0.56 and 3.31% of tumors, while the loss of BRG1 expression occurred in ~15–30% (6, 19, 76, 101). Moreover, no abrogating mutations in BRM have been found in lung cancer. BRM is not significantly mutated (0.0%) as indicated by the Atlas data, which is clearly below the frequency at which its expression is lost in lung cancer (3, 5, 19).
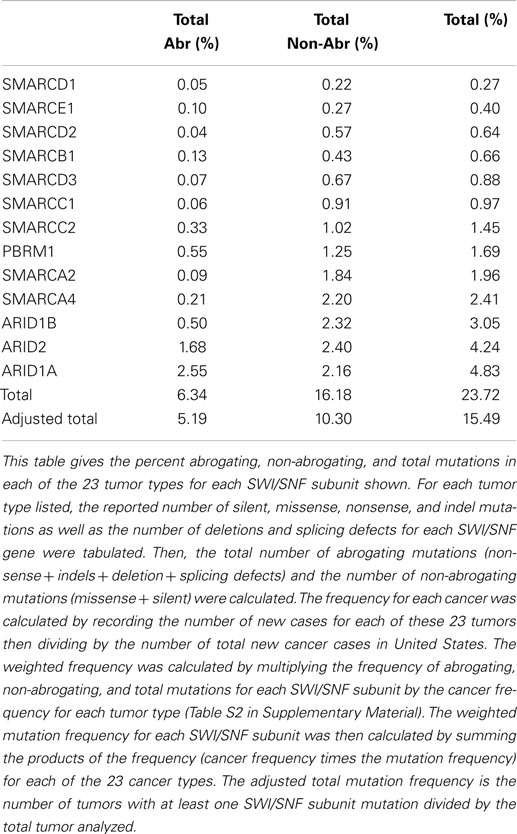
Table 1. Weighted averaged mutations of SWI/SNF subunits.
Table Notes
Abrogating, non-abrogating, and total mutation rates from the TCGA and the Cosmic Databases for SMARCA4 (BRG1): Table 2A, SMARCA2 (BRM): Table 2A, ARID1A (BAF250a): Table 3A, ARID1B (BAF250b): Table 3B, PBRM1 (BAF180): Table 4A, and ARID2 (BAF200): Table 4B. A total of 23 tumors are listed in the first column. The second column lists the total number of tumors that were analyzed in TCGA database for each tumor type. Next, the percentages of non-abrogating (Nabr), abrogating (Abr) mutations, and total (Nabr + Abr) mutation for each tumor type are given. Similar data are then provided for the 13 tumors in the Cosmic database. The penultimate column lists the range of percent loss of SMARCA4 according to immunohistochemical studies, and the final column provides the references for the IHC studies that were included. The final row gives the unweighted average percentage of Nabr, Abr, and total mutations for all 23 tumor types listed, for TCGA database and the 13 tumors listed for Cosmic database.
Table 2A. SMARCA4 (BRG1).
A weighted frequency by cancer incidence shows that all mutations in SWI/SNF subunits occur in ~16% of these 23 human tumor types, where 15.5% of those tumors that harbor SWI/SNF mutations have more than one mutation. In comparison, the mutation rate of SWI/SNF subunits was reported as 20% in a recent review, which reported an unweighted average based on the number of tumors analyzed (65), rather than on the incidence and/or prevalence of these cancers; the latter more accurately reflects their true frequency in a population of cancer patients. In comparison, the mutational data on BRM in cell lines are consistent with experimental observed mutation rates in primary tumors; we did not observe any BRM mutations after we sequenced 20 primary lung cancers (3). Indeed, few if any cell line studies have shown that BRM is silenced by mechanisms other than epigenetic ones (3). These findings have been supported by the low levels of abrogating mutation rates (0.10%) [0.08%] detected when BRM has been sequenced in primary tumors, according to the Atlas database (Cosmic database) (Table 2B). This fact is critically important, as it shows that other mechanisms are at play that inactivate and silence BRM, and likely BRG1, in lung cancer. Given the available IHC data for BRM and BRG1 in other cancers, this observation also appears to be true in other cancer types, and similarly, other mechanisms likely underlie how BRM and BRG1 are silenced in a variety of cancers.
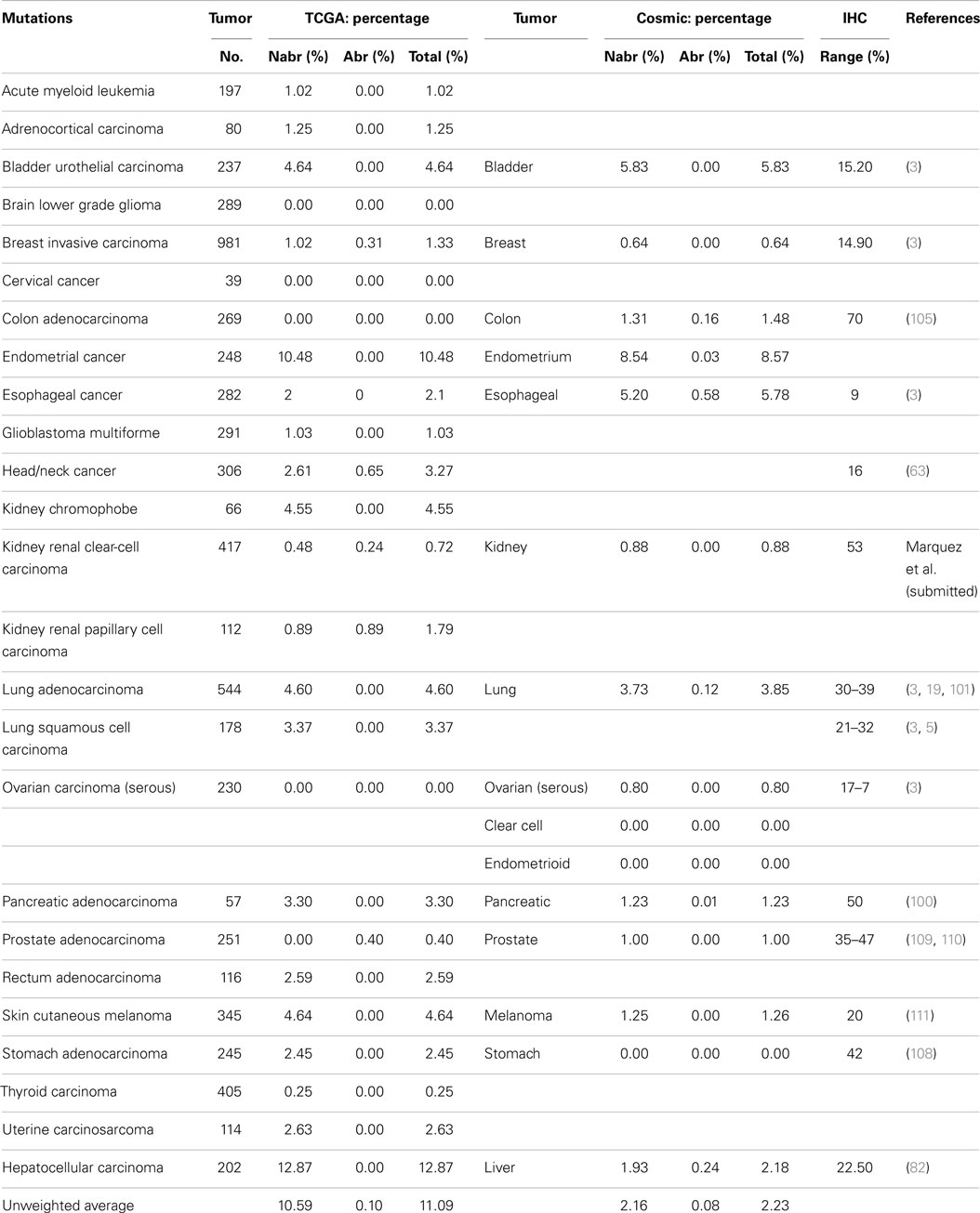
Table 2B. SMARCA2 (BRM).
Other Mechanisms that Silence BRG1?
A comparison of IHC data with NextGen sequencing data clearly shows that non-mutational mechanisms must also underlie most of the BRG1 silencing in at least a subset of human cancers. Even before NextGen sequencing became a major experimental technique, Sanger sequencing in BRG1-negative tumors (as defined by IHC) showed that the frequency of abrogating mutations likely occurred in only a minority of these BRG1-negative tumors. For example, Oike et al. sequenced 16 BRG1-deficient lung cancers for BRG1 mutations and found only one tumor with molecular alterations (missense mutation) (19). A limited analysis of gastric tumors also demonstrated a lack of BRG1 mutations (112). Rodriguez-Nieto et al. analyzed 122 tumors, of which 46 (37%) were found to have low to null levels of BRG1; of these, only 5 missense but no abrogating mutations were found (97). Valdman et al. analyzed 21 prostate tumors by Sanger sequencing and found no detectable BRG1 mutations (113), whereas BRG1 expression is lost in 18–32% of prostate tumors (Table 2A). Endo et al. analyzed 36 hepatocellular carcinomas, of which only four non-abrogating BRG1 mutations were identified in two tumors (82). A combination of a loss of heterozygosity and mutations is a frequent mechanism by which genes are silenced in cancer, but despite the discovery of loss of heterozygosity at the BRG1 locus, Gunduz et al. did not identify any mutations in BRG1 in primary oral cancers (78). Yet BRG1 is lost in 18% of these tumors according to IHC data as illustrated in Table 2A.
In each of these cases, BRG1 mutations occurred in a subset of tumors, but these observed mutational rates cannot explain why or how BRG1 is silenced, given the higher rate of BRG1 expression loss. Hence, other mechanisms are likely occurring that silence BRG1. Whether silencing occurs by biallelic deletion of BRG1, aberrant BRG1 splicing, microRNA regulation, some other epigenetic mechanism or some combination of these mechanisms that has not yet been experimentally defined, is unknown. Indeed, there is clear evidence that each of these mechanisms can be involved in BRG1 silencing. As noted above, both BRM and BRG1 loci have been reported to be sites of loss of heterozygosity (76, 77, 79, 81). Given the frequent loss of one allele, it is possible that the second allele could also be lost, which could result in the silencing of these genes. In addition, microRNA (specifically miR-21), which is elevated in a variety of tumors and is functionally linked to cell growth and migration, has been reported to regulate BRG1 (114). A number of BRG1-deficient cell lines harbor aberrant BRG1 splicing defects where the BRG1 wild-type transcript is replaced by a transcript in which one or more BRG1 exons are omitted, which typically results in a frame-shifted protein. Epigenetic regulation of BRG1 akin to the reported regulation of BRM has also been recently observed in BRG1-deficient cell lines. Understanding the mechanisms that drive BRG1 silencing in primary tumors could be used to delineate which patients might benefit from BRG1-directed or other targeted therapies.
ARID1A, ARID1B, ARID2, and PBRM1 Can be Frequently Mutated in Specific Tumor Types
A series of NextGen sequencing studies have revealed that other SWI/SNF subunits are targeted by mutations. These studies have shown that ARID1A (BAF250A), ARID1B (BAF250B), PBRM1 (BAF180), and ARID2 (BAF200) have a weighted average mutation rate of 4.83, 3.05, 1.69, and 4.24%, respectively, in 23 tumors from the publicly available TCGA database (Table 1). In comparison, all the other SWI/SNF subunits have <2.4% weighted mutation rate. ARID1A is mutated in 7.3% of stomach, 11.9% of colon, 8.8% of pancreatic, 23.1% of cervical, 44.4% of endometrial, 18.1% of bladder, and 9.0% of adenocarcinoma lung cancers (TCGA database) (Table 3A). In those tumors where ARID1A is most frequently mutated, such as endometrial, bladder, pancreatic, and cervical cancers (66), more than ~60–80% of these mutations are abrogating and likely cause the disruption of protein expression. Similarly, the homolog of ARID1A, ARID1B, is frequently mutated in endometrial cancer (9.7%) and in hepatocellular carcinoma (10.4%) (Table 3B) (115), but the abrogation mutation rate is <1%, which indicates that these are missense (i.e., silent) mutations which do not effect ARID1B expression. In contrast, the abrogating mutation rate of ARID1A is 36.3% in endometrial cancer and 8.91% in hepatocellular cancer (Table 3A). Likewise, the rate of abrogating mutations in PBRM1 is (21.1%) [22.5%] out of a total mutation rate of (26.6%) [27.5%] in renal cell carcinomas as indicated in the TCGA and Cosmic databases (Table 4A). For published studies where N > 220 cases, the mutational frequency for PBRM1 in renal cell carcinomas has been reported to be 24, 38, and 41% by Kapur et al., Pena-Llopis et al., and Varela et al., respectively (116–118). In two of these studies, the delineation of mutation types was not given so the impact on expression could not be clarified. Nevertheless, the abrogating mutation rate can be estimated from the TCGA and Cosmic databases, where abrogating mutations are found to be 75–80% of the total mutations. Hence, applying this frequency of abrogating mutations indicates that the loss of PBRM1 occurs in about 20–33% of renal cell tumors. In the study by Pena-Llopis et al. (118), the abrogating mutational rate was 36%. In comparison, the loss of PBRM1 is between 53 and 70% from the published IHC data (119), which suggests that mutations are not the only mechanism underlying PBRM1 silencing in renal cell carcinomas (119, 120).
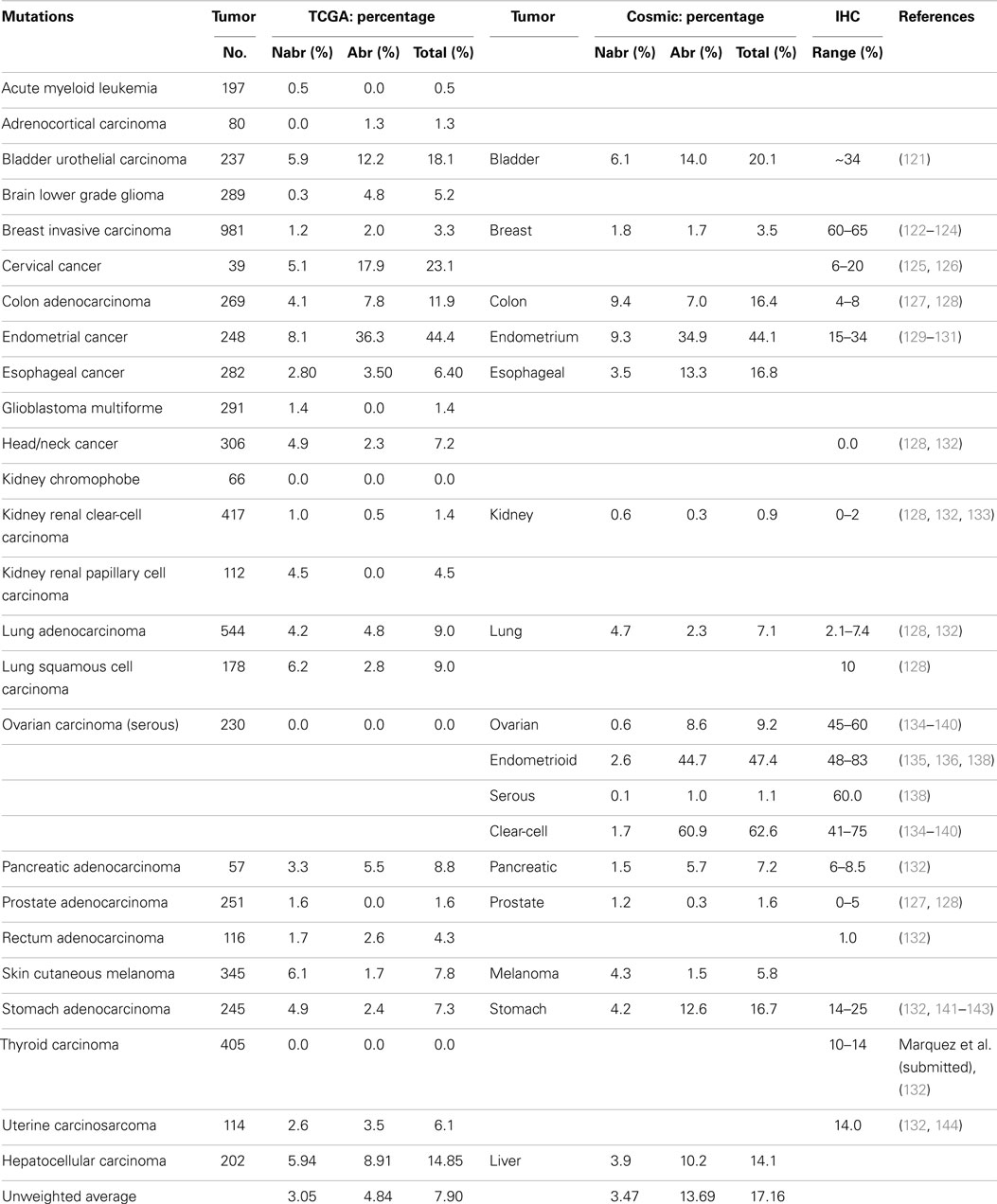
Table 3A. ARID1A (BAF250a).
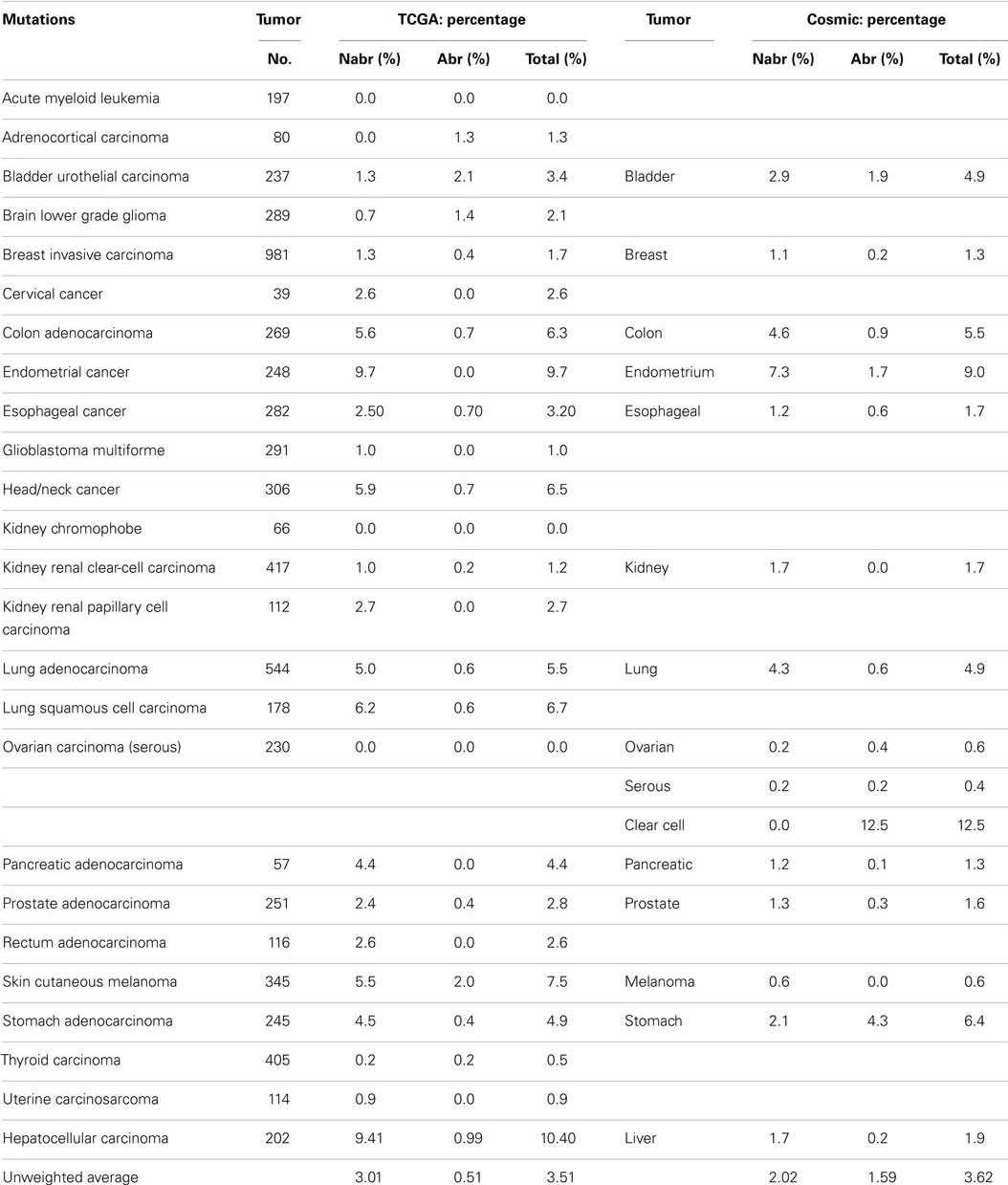
Table 3B. ARID1B (BAF250b).
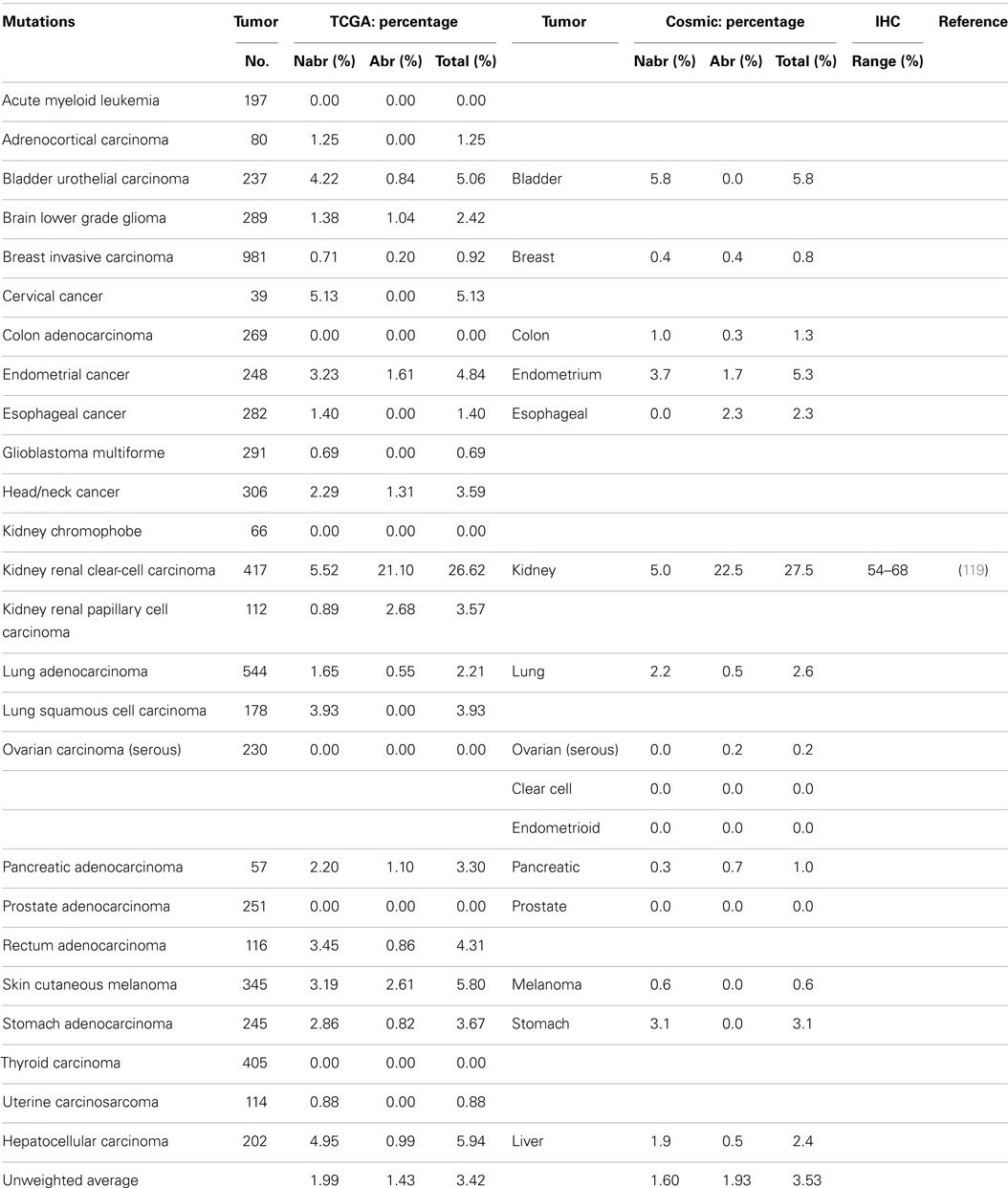
Table 4A. PRBM1 (BAF180).
Despite the apparent high frequency of ARID1A mutations from the TCGA database in some primary tumors such as pancreatic (8.8%), stomach (7.3%), and endometrial (44.4%), BAF250 was initially noted to be infrequently silenced in cancer cell lines (96). ARID2 was discovered by Weidong Wang in 2000 and found to be infrequently lost in cancer cell lines (145) but was recently reported to be mutated in 18% of melanoma and 7.9% of HCV-associated hepatocellular carcinomas (146–148). The TCGA database further shows that ARID2 is also frequently mutated in (10.5%) of endometrial cancers (2.6%) of cervical cancers, and (5.1%) of bladder cancers (Table 4B). Similarly PBRM1 was found to be infrequently silenced by mutations in human cancer cell lines (96); in contrast, PBRM1 has been found to be silenced in 3.6–7% of stomach, endometrial, melanoma, pancreatic, cervical, bladder, and head/neck cancers based on the TCGA database. In comparison, PBRM1 is 3.8–7.4 times more frequently mutated in (26.62%) [27.5%] renal cell carcinomas than in these other tumor types.
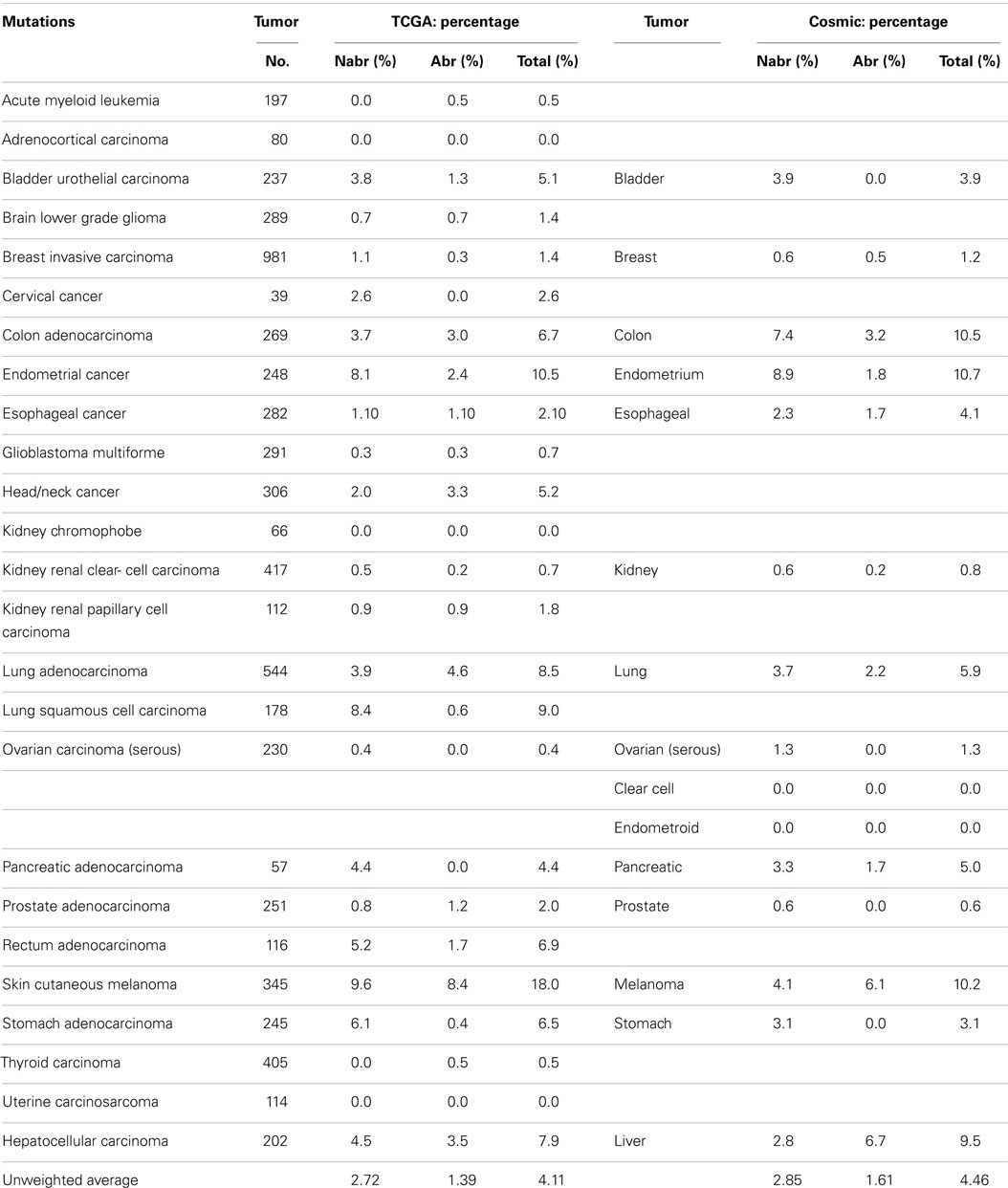
Table 4B. ARID2 (BAF200).
To determine the role of mutations in the silencing of a given gene, one must compare the mutation rates and loss of expression data (IHC) for that gene. It should be noted that the validity of such an analysis is dependent on the accuracy of the reported mutational rates, which can vary depending on the study or database used. For this reason, we have chosen to standardize our comparison using the TCGA database. To this end, IHC data are primarily only available for ARID1A, BRG1, and BRM. The loss of ARID1A expression as determined by IHC and predicted by abrogating mutation rates as designated by “(%)” is illustrated in the following tumor types: ~14–25% (2.4%) of gastric cancers (141), 15–34% (36.3%) of endometrial carcinomas (129, 132, 149), 0–2% (0.5%) of renal cell cancers (121, 133), 60–65% (2.0%) of breast cancers (122–124, 150, 151), and 10% (2.8%) of squamous cell lung cancers (128). Hence, mutations do appear to account for the loss of ARID1A in certain tumor types, as the abrogating mutation rates are comparable to the loss of expression for endometrial tumors as determined by IHC. In other tumor types, however, the reported abrogating mutation rates are less than the frequency of gene expression. For example, nearly 60% of breast cancers demonstrate low to no ARID1A expression (123, 150), while 2.0% [1.7%] of breast cancers showed ARID1A mutations. Nearly 80% of these breast tumors demonstrated promoter methylation (122), and the heterozygous and homozygous allelic loss of ARID1A occurred in 35.2 and 2% of cases, respectively, in breast cancer (151). These data again show that mutations do occur in ARID1A and likely account for the loss of ARID1A expression in some tumor types, but that in other tumor types, other mechanisms of gene silencing must underlie ARID1A silencing. In addition, when mutations are detected in tumors, it does not necessarily mean that they are ubiquitously distributed in every tumor cell. As immunohistochemical studies accumulate, the mechanism of the silencing/abrogation of the other SWI/SNF subunits will be able to be evaluated.
BRM silencing is unlike BRG1 silencing; with respect to BRM, mutations are rarely if ever documented in cancer cell lines (3). In fact, in the majority of BRM-deficient cell lines (20/20), BRM can be readily induced by a variety of agents (50). NextGen sequencing of BRM in numerous tumor types has substantiated this initial observation. The abrogating mutation and total mutation rate for BRM is (<1%) [<1%] and (<11.1%) [<2.6%], respectively, for each of the 23 tumor types available in the TCGA database and the 13 tumor types compiled from the Cosmic database (Table 2B). In comparison, the NextGen sequencing analysis of genetic disorders has revealed BRM germline mutations are higher in some genetic disorders than in cancer. For example, analysis of individuals with Nicolaides–Baraitser syndrome, a rare disease distinguished by sparse hair, unique facial morphology, distal limb abnormalities, and intellectual disability, showed that SMARCA2 (BRM) harbored missense mutations in 36 of 44 cases (~80%). Moreover, these mutations were harbored within conserved catalytic ATPase regions, suggesting that mutations do cluster within hotspots in certain diseases. Similarly, in another study, SMARCA2 (BRM) was mutated in 43% of patients with Coffin–Siris syndrome. Mutations in Coffin–Siris also occur in SMARCB1 (BAF47), SMARCA4 (BRG1), SMARCE1, ARID1A, and ARID1B, which attests to the role of the SWI/SNF complex in this genetic disorder.
Genomic NextGen Sequencing Versus mRNA Splicing
Alternative splicing is a frequent mechanism that leads to aberrant or a complete loss of gene expression. As NextGen sequencing detects deviations in genomic DNA sequences within the coding regions of genes, splicing defects that occur by other mechanisms that do not involve the splicing junctions, but rather in acceptor or donor sites, may not be detected. Genomic DNA changes that affect the composition of branchpoint sequences or their relative position from the 3′ splice acceptor site of the downstream exon are also known to impact splicing, and certain missense mutations might be sufficient to create cryptic sites within the coding region. While non-mutational mechanisms can be inferred when the mutation rates do not match the frequency of gene silencing, other abrogating mechanisms such as aberrant splicing could contribute to this observed difference. Testing in cell lines permits the resolution of this type of discrepancy, but analyses in tumors are much more complicated because tumors are heterogeneous and multiple mechanisms may be active in a single tumor. Moreover, the mRNA levels that result from aberrant splicing are much lower than the mRNA level derived from the normal tissue within most tumor samples; this fact further complicates the detection and analysis of aberrant splicing in tumors compared with cell lines that lack these constraints. Hence, the notion that mutations and non-mutational mechanisms must be mutually exclusive is not true. It is more likely that both mechanisms are operating independently of each other and that the prevalence of each mechanism is driven by the selection force confronted by the tumor in its environment.
Rhabdoid Tumors, Mutations and the SWI/SNF Complex
SMARCB1 (INI1 or BAF47) was the first gene of the SWI/SNF complex that was linked to cancer. The discovery of the role of this gene in the genesis of malignant Rhabdoid pediatric sarcomas was interesting. Mouse models have shown that the inactivation of the BAF47 gene is highly tumorigenic. One might have thought, therefore, that this gene would be targeted and activated in a wide range of other cancer types, given its apparent degree of tumorigenicity. However, this is not the case, as the Atlas database shows that mutations in SMARCB1 are relatively infrequent; the average weighted abrogating and total mutation rate is only 0.13% and 0.66%, respectively, in the TCGA database (Table 1). Several other SWI/SNF subunits are also infrequently mutated: SMARCD1, SMARCD2, SMARCD3, and SMARCE1 have average weighted mutation rates of 0.27, 0.64, 0.88, and 0.40%, respectively, in the TCGA database. These subunits, like SMARCB1, are smaller subunits within the SWI/SNF complex (i.e., the molecular weight of each is under 60 kDa). In comparison, the weighted mutation rates of the large subunits that are in the 180–250 kDa range are generally higher than those of the small subunits, which suggests that this observed difference in the mutation rates may in part be due to the size differences of the subunits. If the mutation rate for each subunit is adjusted for the size difference by dividing the weighted mutation rates by the length/size in 100 kDa, then the size-adjusted-mutation rates for SMARCD1, SMARCD2, SMARCD3, and SMARCE1 become (mutation percentage/100 kDa): 0.43, 1.01, 1.42, and 0.69%, respectively. In comparison, the mutation percentages per 100 kDa for BRG1, BRM, ARID1A, ARID1B, ARID2, and PBRM1 are 1.27, 1.03, 1.93, 1.22, 2.12, and 0.93%, respectively. When corrected for size, most SWI/SNF subunits harbor 1% (±0.4%) mutations per 100 kDa, while the standardized mutational rates of ARID1A and ARID2 are about twice this rate. In most tumors, the mutation rate of SMARCB1 is <1.0%. In cervical carcinomas, squamous cell carcinomas, and adenocarcinomas of the lung, however, the mutation rates are 5.10, 1.7, and 2.8%, respectively (Table S1 in Supplementary Material). Most of these mutations do not affect the expression of SMARCB1 as they are silent or missense mutations. For lung and cervical cancers, the rate of abrogating mutations is 0.6 and 0.0%, respectively. Thus, <1:200 of these tumors harbor a mutation in a SMARCB1 allele.
Abrogation of SMARCB1 in Rhabdoid Tumors Led to the Discovery of SWI/SNF Involvement in Cancer
In 1990s, SMARCB1 mutations and the subsequent inactivation of SMARCB1 in rhabdoid tumors led to the realization that the SWI/SNF complex is often a target in cancer. Analyses of the various subunits were pursued based on these facts, and it was assumed that the SWI/SNF complex requires each of its subunits in order to be functional. Therefore, it was surmised by Bernard Weissman and others that the other SWI/SNF subunits must be involved in cancer akin to the SMARCB1 gene (96). This premise led to the finding that BRG1 and BRM are frequently silenced either alone or concomitantly in lung cancer (6). However, a recent analysis of rhabdoids has shown that BRG1 and BRM are also lost in many Rhabdoid tumors (52). BRM is silenced in 60–70% of primary rhabdoid tumors and in >90% of rhabdoid cell lines (52). Interestingly, the mechanism of silencing of BRM in rhabdoid tumors appears to be identical to that reported in lung cancer (52). Hence, BRM silencing appears to be conserved between these two very different cancer types. Similarly, BRG1 is silenced in about 50% of primary rhabdoid tumors and about 75% of rhabdoid cell lines (unpublished data). In BRM-deficient rhabdoid cell lines and in a subset of BRG1-deficient rhabdoid cell lines, each of these genes is epigenetically silenced, and their expression can be restored (52). Remarkably, both genes can be readily restored by flavopiridol in a subset of lung and rhabdoid BRG1/BRM-deficient cancer cell lines (50, 52). As BRG1 and BRM complement the function of RB and RB2 in growth inhibition, their induction could be a necessary prerequisite for the clinical activity of flavopiridol. The epigenetic silencing of BRM and BRG1 is in concert with the fact that there are few mutated genes in rhabdoid tumors. Hence, the study of the epigenetic mechanism behind BRM and BRG1 silencing in rhabdoid tumors and potentially in other tumor types would seem to be important and a novel avenue for cancer research.
In Summary
In recent reviews of the role of SWI/SNF in cancer, the majority of mutations discussed are missense and not abrogating mutations, yet overwhelming data show that SWI/SNF can be inactivated by both mutations and non-mutational silencing. These findings demonstrate that SWI/SNF function must be negatively impacted even more frequently than suggested by Kadoch et al. (65). The exact frequency of SWI/SNF impairment cannot yet be estimated from mutational analyses alone, but based on currently available data, SWI/SNF is impaired in at least 16% via mutations – and possibly >90% – of cancers, if one considers all mechanisms of inactivation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Dr. Reisman is the major shareholder of Zenagene, Inc.
Supplementary Material
The Supplementary Material for this article can be found online at http://www.frontiersin.org/Journal/10.3389/fonc.2014.00372/abstract
References
1. Peterson CL, Tamkun JW. The SWI-SNF complex: a chromatin remodeling machine? Trends Biochem Sci (1995) 20:143–6. doi: 10.1016/S0968-0004(00)88990-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene (2009) 28:1653–68. doi:10.1038/onc.2009.4
3. Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene (2007) 26:7058–66. doi:10.1038/sj.onc.1210514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Halliday GM, Zhou Y, Sou PW, Huang XX, Rana S, Bugeja MJ, et al. The absence of Brm exacerbates photocarcinogenesis. Exp Dermatol (2012) 21:599–604. doi:10.1111/j.1600-0625.2012.01522.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Fukuoka J, Fujii T, Shih JH, Dracheva T, Meerzaman D, Player A, et al. Chromatin remodeling factors and BRM/BRG1 expression as prognostic indicators in non-small cell lung cancer. Clin Cancer Res (2004) 10:4314–24. doi:10.1158/1078-0432.CCR-03-0489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res (2003) 63:560–6.
7. Coisy-Quivy M, Disson O, Roure V, Muchardt C, Blanchard JM, Dantonel JC. Role for brm in cell growth control. Cancer Res (2006) 66:5069–76. doi:10.1158/0008-5472.CAN-05-0596
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Strober BE, Dunaief JL, Guha S, Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Mol Cell Biol (1996) 16:1576–83.
9. Reisman DN, Strobeck MW, Betz BL, Sciariotta J, Funkhouser W Jr, Murchardt C, et al. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene (2002) 21:1196–207. doi:10.1038/sj.onc.1205188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Strobeck MW, Reisman DN, Gunawardena RW, Betz BL, Angus SP, Knudsen KE, et al. Compensation of BRG-1 function by Brm: insight into the role of the core SWI-SNF subunits in retinoblastoma tumor suppressor signaling. J Biol Chem (2002) 277:4782–9. doi:10.1074/jbc.M109532200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Dahiya A, Gavin MR, Luo RX, Dean DC. Role of the LXCXE binding site in Rb function. Mol Cell Biol (2000) 20:6799–805. doi:10.1128/MCB.20.18.6799-6805.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Trouche D, Le Chalony C, Muchardt C, Yaniv M, Kouzarides T. RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A (1997) 94:11268–73. doi:10.1073/pnas.94.21.11268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Lee D, Lim C, Seo T, Kwon H, Min H, Choe J. The viral oncogene human papillomavirus E7 deregulates transcriptional silencing by Brm-related gene 1 via molecular interactions. J Biol Chem (2002) 277:48842–8. doi:10.1074/jbc.M203583200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Muchardt C, Bourachot B, Reyes JC, Yaniv M. ras transformation is associated with decreased expression of the brm/SNF2alpha ATPase from the mammalian SWI-SNF complex. EMBO J (1998) 17:223–31. doi:10.1093/emboj/17.1.223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Das AV, James J, Bhattacharya S, Imbalzano AN, Antony ML, Hegde G, et al. SWI/SNF chromatin remodeling ATPase Brm regulates the differentiation of early retinal stem cells/progenitors by influencing Brn3b expression and Notch signaling. J Biol Chem (2007) 282:35187–201. doi:10.1074/jbc.M706742200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Bourachot B, Yaniv M, Muchardt C. Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation. EMBO J (2003) 22:6505–15. doi:10.1093/emboj/cdg621
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Zhang ZK, Davies KP, Allen J, Zhu L, Pestell RG, Zagzag D, et al. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol (2002) 22:5975–88. doi:10.1128/MCB.22.16.5975-5988.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Wilson BG, Helming KC, Wang X, Kim Y, Vazquez F, Jagani Z, et al. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol (2014) 34:1136–44. doi:10.1128/MCB.01372-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Oike T, Ogiwara H, Tominaga Y, Ito K, Ando O, Tsuta K, et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res (2013) 43(9):849–55. doi:10.1158/00085472
20. Hoffman GR, Rahal R, Buxton F, Xiang K, McAllister G, Frias E, et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A (2014) 111:3128–33. doi:10.1073/pnas.1316793111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Kahali B, Gramling SJ, Marquez SB, Thompson K, Lu L, Reisman D. Identifying targets for the restoration and reactivation of BRM. Oncogene (2013) 33:653–64. doi:10.1038/onc.2012.613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Ito T, Yamauchi M, Nishina M, Yamamichi N, Mizutani T, Ui M, et al. Identification of SWI/SNF complex subunit BAF60a as a determinant of transactivation potential of Fos/Jun dimers. J Biol Chem (2000) 276(4):2852–7. doi:10.1074/jbc.M009633200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet (1999) 22:102–5. doi:10.1038/8811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Kenneth T, Marquez S, Kahali B, Reisman D. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1 – letter to the editor of cancer research. Cancer Res (2014) 74(17):4946–7. doi:10.1158/0008-5472.CAN-13-2457
25. Gordon V, Rogers C, Reisman D. Alteration to the SWI/SNF complex in human cancers. Oncol Rev (2010) 4:89–99. doi:10.1007/s12156-010-0040-5
26. Gong F, Fahy D, Liu H, Wang W, Smerdon MJ. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell Cycle (2008) 7:1067–74. doi:10.4161/cc.7.8.5647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Mudhasani R, Fontes JD. The class II transactivator requires Brahma-related gene 1 to activate transcription of major histocompatibility complex class II genes. Mol Cell Biol (2002) 22:5019–26. doi:10.1128/MCB.22.14.5019-5026.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Mudhasani R, Fontes JD. Multiple interactions between BRG1 and MHC class II promoter binding proteins. Mol Immunol (2005) 42:673–82. doi:10.1016/j.molimm.2004.09.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Brockmann D, Lehmkuhler O, Schmucker U, Esche H. The histone acetyltransferase activity of PCAF cooperates with the brahma/SWI2-related protein BRG-1 in the activation of the enhancer A of the MHC class I promoter. Gene (2001) 277:111–20. doi:10.1016/S0378-1119(01)00703-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Thibodeau J, Bourgeois-Daigneault MC, Lapointe R. Targeting the MHC Class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology (2012) 1:908–16. doi:10.4161/onci.21205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Batsche E, Yaniv M, Muchardt C. The human SWI/SNF subunit Brm is a regulator of alternative splicing. Nat Struct Mol Biol (2006) 13:22–9. doi:10.1038/nsmb1030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Yang XJ, Ullah M. MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene (2007) 26:5408–19. doi:10.1038/sj.onc.1210609
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Tasaka T, Matsuhashi Y, Uehara E, Tamura T, Kakazu N, Abe T, et al. Secondary acute monocytic leukemia with a translocation t(8;16)(p11;p13): case report and review of the literature. Leuk Lymphoma (2004) 45:621–5. doi:10.1080/10428190310001593058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Moore SD, Herrick SR, Ince TA, Kleinman MS, Dal Cin P, Morton CC, et al. Uterine leiomyomata with t(10;17) disrupt the histone acetyltransferase MORF. Cancer Res (2004) 64:5570–7. doi:10.1158/0008-5472.CAN-04-0050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem (2013) 288:14510–21. doi:10.1074/jbc.M113.458737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squatrito M, Helin K. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res (2013) 73:6323–33. doi:10.1158/0008-5472.CAN-12-4660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Perez RE, Knights CD, Sahu G, Catania J, Kolukula VK, Stoler D, et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J Cell Physiol (2010) 225:394–405. doi:10.1002/jcp.22285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem (2006) 281:26562–8. doi:10.1074/jbc.M605391200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell (1994) 79:119–30. doi:10.1016/0092-8674(94)90405-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Strobeck MW, Knudsen KE, Fribourg AF, DeCristofaro MF, Weissman BE, Imbalzano AN, et al. BRG-1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci U S A (2000) 97:7748–53. doi:10.1073/pnas.97.14.7748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, et al. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell (2000) 101:79–89. doi:10.1016/S0092-8674(00)80625-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Harbour JW, Dean DC. Chromatin remodeling and Rb activity. Curr Opin Cell Biol (2000) 12:685–9. doi:10.1016/S0955-0674(00)00152-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Lacy S, Whyte P. Identification of a p130 domain mediating interactions with cyclin A/cdk 2 and cyclin E/cdk 2 complexes. Oncogene (1997) 14:2395–406. doi:10.1038/sj.onc.1201085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Gao CF, Ren S, Wang J, Zhang SL, Jin F, Nakajima T, et al. P130 and its truncated form mediate p53-induced cell cycle arrest in Rb(-/-) Saos2 cells. Oncogene (2002) 21:7569–79. doi:10.1038/sj.onc.1205933
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Classon M, Dyson N. p107 and p130: versatile proteins with interesting pockets. Exp Cell Res (2001) 264:135–47. doi:10.1006/excr.2000.5135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Muchardt C, Reyes JC, Bourachot B, Leguoy E, Yaniv M. The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J (1996) 15:3394–402.
47. Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha). EMBO J (1998) 17:6979–91. doi:10.1093/emboj/17.23.6979
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature (2009) 458:719–24. doi:10.1038/nature07943
49. Gramling S, Rogers C, Liu G, Reisman D. Pharmacologic reversal of epigenetic silencing of the anticancer protein BRM: a novel targeted treatment strategy. Oncogene (2011) 30:3289–94. doi:10.1038/onc.2011.80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Kahali B, Marquez SB, Thompson KW, Yu J, Gramling SJ, Lu L, et al. Flavonoids from each of the six structural groups reactivate BRM, a possible co-factor for the anti-cancer effects of flavonoids. Carcinogenesis (2014). doi:10.1093/carcin/bgu117
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Barr PM. Flavopiridol: judged by the company one keeps. Leuk Res (2013) 37:1187–8. doi:10.1016/j.leukres.2013.06.015
52. Kahali B, Marquez SB, Thompson KW, Liang SY, Lu L, Reisman D. The silencing of the SWI/SNF subunit and anticancer gene, BRM, in rhabdoid tumors. Oncotarget (2014) 5:3316–32.
53. Liu G, Gramling S, Munoz D, Cheng D, Azad AK, Mirshams M, et al. Two novel BRM insertion promoter sequence variants are associated with loss of BRM expression and lung cancer risk. Oncogene (2011) 30:3295–304. doi:10.1038/onc.2011.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Gregoire S, Xiao L, Nie J, Zhang X, Xu M, Li J, et al. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol Cell Biol (2007) 27:1280–95. doi:10.1128/MCB.00882-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Han A, He J, Wu Y, Liu JO, Chen L. Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor-2. J Mol Biol (2005) 345:91–102. doi:10.1016/j.jmb.2004.10.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Yamamichi N, Yamamichi-Nishina M, Mizutani T, Watanabe H, Minoguchi S, Kobayashi N, et al. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene (2005) 24:5471–81. doi:10.1038/sj.onc.1208716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Haberland M, Arnold MA, McAnally J, Phan D, Kim Y, Olson EN. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol Cell Biol (2007) 27:518–25. doi:10.1128/MCB.01415-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Miremadi A, Oestergaard MZ, Pharoah PD, Caldas C. Cancer genetics of epigenetic genes. Hum Mol Genet (2007) 16(Spec No 1):R28–49. doi:10.1093/hmg/ddm021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol (2001) 79:243–52. doi:10.1139/o01-032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Wong K, Qiu X, Cheng D, Azad AK, Palepu P, Mirshams M, et al. Two BRM promoter polymorphisms and risk of early stage (Stage I-II) upper aerodigestive tract cancer risk. J Clin Oncol (2012) 30(Suppl):abstr10522. doi:10.1002/cam4.201
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Cuffe S, Qiu X, Patel D, Azad AK, Cheng D, Chen Z, et al. The effect of two BRM promoter polymorphisms on the risk of advanced non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC) in smokers lung cancer. World Conference Lung Cancer. (2013).
63. Wang JR, Gramling SJ, Goldstein DP, Cheng D, Chen D, Azad AK, et al. Association of two BRM promoter polymorphisms with head and neck squamous cell carcinoma risk. Carcinogenesis (2013) 34:1012–7. doi:10.1093/carcin/bgt008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Gao X, Huang M, Liu L, He Y, Yu Q, Zhao H, et al. Insertion/deletion polymorphisms in the promoter region of BRM contribute to risk of hepatocellular carcinoma in Chinese populations. PLoS One (2013) 8:e55169. doi:10.1371/journal.pone.0055169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet (2013) 45:592–601. doi:10.1038/ng.2628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov (2013) 3:35–43. doi:10.1158/2159-8290.CD-12-0361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell (2000) 6:1287–95. doi:10.1016/S1097-2765(00)00127-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Bultman SJ, Herschkowitz JI, Godfrey V, Gebuhr TC, Yaniv M, Perou CM, et al. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene (2008) 27:460–8. doi:10.1038/sj.onc.1210664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep (2000) 1:500–6. doi:10.1093/embo-reports/kvd129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol (2001) 21:3598–603. doi:10.1128/MCB.21.10.3598-3603.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci U S A (2000) 97:13796–800. doi:10.1073/pnas.250492697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Chen J, Archer TK. Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol Cell Biol (2005) 25:9016–27. doi:10.1128/MCB.25.20.9016-9027.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Willis MS, Homeister JW, Rosson GB, Annayev Y, Holley D, Holly SP, et al. Functional redundancy of SWI/SNF catalytic subunits in maintaining vascular endothelial cells in the adult heart. Circ Res (2012) 111:E111–34. doi:10.1161/CIRCRESAHA.112.265587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Keenen B, Qi H, Saladi SV, Yeung M, de la Serna IL. Heterogeneous SWI/SNF chromatin remodeling complexes promote expression of microphthalmia-associated transcription factor target genes in melanoma. Oncogene (2010) 29:81–92. doi:10.1038/onc.2009.304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Kadam S, McAlpine GS, Phelan ML, Kingston RE, Jones KA, Emerson BM. Functional selectivity of recombinant mammalian SWI/SNF subunits. Genes Dev (2000) 14:2441–51. doi:10.1101/gad.828000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Medina PP, Carretero J, Fraga MF, Esteller M, Sidransky D, Sanchez-Cespedes M. Genetic and epigenetic screening for gene alterations of the chromatin-remodeling factor, SMARCA4/BRG1, in lung tumors. Genes Chromosomes Cancer (2004) 41:170–7. doi:10.1002/gcc.20068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Rodriguez-Nieto S, Sanchez-Cespedes M. BRG1 and LKB1: tales of two tumor suppressor genes on chromosome 19p and lung cancer. Carcinogenesis (2009) 30:547–54. doi:10.1093/carcin/bgp035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Gunduz E, Gunduz M, Ouchida M, Nagatsuka H, Beder L, Tsujigiwa H, et al. Genetic and epigenetic alterations of BRG1 promote oral cancer development. Int J Oncol (2005) 26:201–10.
79. Neville EM, Stewart M, Myskow M, Donnelly RJ, Field JK. Loss of heterozygosity at 9p23 defines a novel locus in non-small cell lung cancer. Oncogene (1995) 11:581–5.
80. Neville E, Stewart M, Swift A, Liloglou T, Ross H, Gosney J, et al. Allelotype of non-small cell lung cancer. Int J Oncol (1996) 9:533–9.
81. An HX, Claas A, Savelyeva L, Seitz S, Schlag P, Scherneck S, et al. Two regions of deletion in 9p23-24 in sporadic breast cancer. Cancer Res (1999) 59:3941–3.
82. Endo M, Yasui K, Zen Y, Gen Y, Zen K, Tsuji K, et al. Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and BRM in hepatocellular carcinoma. Liver Int (2013) 33:105–17. doi:10.1111/liv.12005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Cheung TH, Chung TK, Poon CS, Hampton GM, Wang VW, Wong YF. Allelic loss on chromosome 1 is associated with tumor progression of cervical carcinoma. Cancer (1999) 86:1294–8. doi:10.1002/(SICI)1097-0142(19991001)86:7<1294::AID-CNCR26>3.0.CO;2-O
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Chen M, Ye Y, Yang H, Tamboli P, Matin S, Tannir NM, et al. Genome-wide profiling of chromosomal alterations in renal cell carcinoma using high-density single nucleotide polymorphism arrays. Int J Cancer (2009) 125:2342–8. doi:10.1002/ijc.24642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet (2012) 90:565–72. doi:10.1016/j.ajhg.2012.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Michelson M, Ben-Sasson A, Vinkler C, Leshinsky-Silver E, Netzer I, Frumkin A, et al. Delineation of the interstitial 6q25 microdeletion syndrome: refinement of the critical causative region. Am J Med Genet A (2012) 158A:1395–9. doi:10.1002/ajmg.a.35361
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Michels E, Vandesompele J, De Preter K, Hoebeeck J, Vermeulen J, Schramm A, et al. ArrayCGH-based classification of neuroblastoma into genomic subgroups. Genes Chromosomes Cancer (2007) 46:1098–108. doi:10.1002/gcc.20496
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, et al. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet (2013) 45:12–7. doi:10.1038/ng.2493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. El-Rifai W, Rutherford S, Knuutila S, Frierson HF Jr, Moskaluk CA. Novel DNA copy number losses in chromosome 12q12 – q13 in adenoid cystic carcinoma. Neoplasia (2001) 3:173–8. doi:10.1038/sj.neo.7900158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kok K, Naylor SL, Buys CH. Deletions of the short arm of chromosome 3 in solid tumors and the search for suppressor genes. Adv Cancer Res (1997) 71:27–92. doi:10.1016/S0065-230X(08)60096-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Pitterle DM, Jolicoeur EM, Bepler G. Hot spots for molecular genetic alterations in lung cancer. In vivo (1998) 12:643–58.
92. Sekine I, Sato M, Sunaga N, Toyooka S, Peyton M, Parsons R, et al. The 3p21 candidate tumor suppressor gene BAF180 is normally expressed in human lung cancer. Oncogene (2005) 24:2735–8. doi:10.1038/sj.onc.1207694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res (2013) 19:3259–67. doi:10.1158/1078-0432.CCR-12-3886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, et al. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res (2000) 60:6171–7.
95. Medina PP, Romero OA, Kohno T, Montuenga LM, Pio R, Yokota J, et al. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum Mutat (2008) 29:617–22. doi:10.1002/humu.20730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. DeCristofaro MF, Betz BL, Rorie CJ, Reisman DN, Wang W, Weissman BE. Characterization of SWI/SNF protein expression in human breast cancer cell lines and other malignancies. J Cell Physiol (2001) 186:136–45. doi:10.1002/1097-4652(200101)186:1<136::AID-JCP1010>3.3.CO;2-W
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Rodriguez-Nieto S, Canada A, Pros E, Pinto AI, Torres-Lanzas J, Lopez-Rios F, et al. Massive parallel DNA pyrosequencing analysis of the tumor suppressor BRG1/SMARCA4 in lung primary tumors. Hum Mutat (2011) 32:E1999–2017. doi:10.1002/humu.21415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Dal Molin M, Hong SM, Hebbar S, Sharma R, Scrimieri F, de Wilde RF, et al. Loss of expression of the SWI/SNF chromatin remodeling subunit BRG1/SMARCA4 is frequently observed in intraductal papillary mucinous neoplasms of the pancreas. Hum Pathol (2012) 43:585–91. doi:10.1016/j.humpath.2011.06.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Lin H, Wong RP, Martinka M, Li G. BRG1 expression is increased in human cutaneous melanoma. Br J Dermatol (2010) 163:502–10. doi:10.1111/j.1365-2133.2010.09851.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Numata M, Morinaga S, Watanabe T, Tamagawa H, Yamamoto N, Shiozawa M, et al. The clinical significance of SWI/SNF complex in pancreatic cancer. Int J Oncol (2013) 42:403–10. doi:10.3892/ijo.2012.1723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Matsubara D, Kishaba Y, Ishikawa S, Sakatani T, Oguni S, Tamura T, et al. Lung cancer with loss of BRG1/BRM, shows epithelial mesenchymal transition phenotype and distinct histologic and genetic features. Cancer Sci (2013) 104:266–73. doi:10.1111/cas.12065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Bai J, Mei PJ, Liu H, Li C, Li W, Wu YP, et al. BRG1 expression is increased in human glioma and controls glioma cell proliferation, migration and invasion in vitro. J Cancer Res Clin Oncol (2012). doi:10.1007/s00432-012-1172-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Bai J, Mei P, Zhang C, Chen F, Li C, Pan Z, et al. BRG1 is a prognostic marker and potential therapeutic target in human breast cancer. PLoS One (2013) 8:e59772. doi:10.1371/journal.pone.0059772
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Kuo KT, Liang CW, Hsiao CH, Lin CH, Chen CA, Sheu BC, et al. Downregulation of BRG-1 repressed expression of CD44s in cervical neuroendocrine carcinoma and adenocarcinoma. Mod Pathol (2006) 19:1570–7. doi:10.1038/modpathol.3800687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Watanabe T, Semba S, Yokozaki H. Regulation of PTEN expression by the SWI/SNF chromatin-remodelling protein BRG1 in human colorectal carcinoma cells. Br J Cancer (2011) 104:146–54. doi:10.1038/sj.bjc.6606018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Rodriguez-Garcia A, Sanchez-Martin M, Perez-Losada J, Perez-Mancera PA, Sagrera-Aparisi A, Gutierrez-Cianca N, et al. Selective destruction of tumor cells through specific inhibition of products resulting from chromosomal translocations. Curr Cancer Drug Targets (2001) 1:109–19. doi:10.2174/1568009013334214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Li Y, Shi QL, Jin XZ, Meng K, Zhou XJ, Sun LP. [BRG1 expression in prostate carcinoma by application of tissue microarray]. Zhonghua Nan Ke Xue (2006) 12:629–32.
108. Yamamichi N, Inada K, Ichinose M, Yamamichi-Nishina M, Mizutani T, Watanabe H, et al. Frequent loss of Brm expression in gastric cancer correlates with histologic features and differentiation state. Cancer Res (2007) 67:10727–35. doi:10.1158/0008-5472.CAN-07-2601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Sun A, Tawfik O, Gayed B, Thrasher JB, Hoestje S, Li C, et al. Aberrant expression of SWI/SNF catalytic subunits BRG1/BRM is associated with tumor development and increased invasiveness in prostate cancers. Prostate (2007) 67:203–13. doi:10.1002/pros.20521
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Shen H, Powers N, Saini N, Comstock CE, Sharma A, Weaver K, et al. The SWI/SNF ATPase Brm is a gatekeeper of proliferative control in prostate cancer. Cancer Res (2008) 68:10154–62. doi:10.1158/0008-5472.CAN-08-1794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Becker TM, Haferkamp S, Dijkstra MK, Scurr LL, Frausto M, Diefenbach E, et al. The chromatin remodelling factor BRG1 is a novel binding partner of the tumor suppressor p16INK4a. Mol Cancer (2009) 8:4. doi:10.1186/1476-4598-8-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Sentani K, Oue N, Kondo H, Kuraoka K, Motoshita J, Ito R, et al. Increased expression but not genetic alteration of BRG1, a component of the SWI/SNF complex, is associated with the advanced stage of human gastric carcinomas. Pathobiology (2001) 69:315–20. doi:10.1159/000064638
113. Valdman A, Nordenskjold A, Fang X, Naito A, Al-Shukri S, Larsson C, et al. Mutation analysis of the BRG1 gene in prostate cancer clinical samples. Int J Oncol (2003) 22:1003–7.
114. Schramedei K, Morbt N, Pfeifer G, Lauter J, Rosolowski M, Tomm JM, et al. MicroRNA-21 targets tumor suppressor genes ANP32A and SMARCA4. Oncogene (2011) 30:2975–85. doi:10.1038/onc.2011.15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet (2012) 44:760–4. doi:10.1038/ng.2291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature (2011) 469:539–42. doi:10.1038/nature09639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Kapur P, Pena-Llopis S, Christie A, Zhrebker L, Pavia-Jimenez A, Rathmell WK, et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol (2013) 14:159–67. doi:10.1016/S1470-2045(12)70584-3
118. Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet (2012) 44:751–9. doi:10.1038/ng.2323
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Pawlowski R, Muhl SM, Sulser T, Krek W, Moch H, Schraml P. Loss of PBRM1 expression is associated with renal cell carcinoma progression. Int J Cancer (2013) 132:E11–7. doi:10.1002/ijc.27822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Joseph RW, Kapur P, Serie D, Eckel-Passow J, Ho TH, Brugarolas J, et al. Loss of BAP1 and PBRM1 protein expression and its association with clear cell renal cell carcinoma-specific survival. J Clin Oncol (2014) 32.
121. Balbas-Martinez C, Rodriguez-Pinilla M, Casanova A, Dominguez O, Pisano DG, Gomez G, et al. ARID1A alterations are associated with FGFR3-wild type, poor-prognosis, urothelial bladder tumors. PLoS One (2013) 8:e62483. doi:10.1371/journal.pone.0062483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Zhang X, Sun Q, Shan M, Niu M, Liu T, Xia B, et al. Promoter hypermethylation of ARID1A gene is responsible for its low mRNA expression in many invasive breast cancers. PLoS One (2013) 8:e53931. doi:10.1371/journal.pone.0053931
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Zhao J, Liu C, Zhao Z. ARID1A: a potential prognostic factor for breast cancer. Tumour Biol (2014) 35:4813–9. doi:10.1007/s13277-014-1632-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Mamo A, Cavallone L, Tuzmen S, Chabot C, Ferrario C, Hassan S, et al. An integrated genomic approach identifies ARID1A as a candidate tumor- suppressor gene in breast cancer. Oncogene (2012) 31:2090–100. doi:10.1038/onc.2011.386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Cho H, Kim JS, Chung H, Perry C, Lee H, Kim JH. Loss of ARID1A/BAF250a expression is linked to tumor progression and adverse prognosis in cervical cancer. Hum Pathol (2013). doi:10.1016/j.humpath.2012.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Guan B, Wang TL, Shih Ie M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res (2011) 71:6718–27. doi:10.1158/0008-5472.CAN-11-1562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Kim MS, Je EM, Yoo NJ, Lee SH. Loss of ARID1A expression is uncommon in gastric, colorectal, and prostate cancers. APMIS (2012) 120:1020–2. doi:10.1111/j.1600-0463.2012.02930.x
128. Guan B, Mao TL, Panuganti PK, Kuhn E, Kurman RJ, Maeda D, et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol (2011) 35:625–32. doi:10.1097/PAS.0b013e318212782a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Bosse T, ter Haar NT, Seeber LM, v Diest PJ, Hes FJ, Vasen HF, et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol (2013) 26:1525–35. doi:10.1038/modpathol.2013.96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Samartzis EP, Samartzis N, Noske A, Fedier A, Caduff R, Dedes KJ, et al. Loss of ARID1A/BAF250a-expression in endometriosis: a biomarker for risk of carcinogenic transformation? Mod Pathol (2012) 25:885–92. doi:10.1038/modpathol.2011.217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Rahman M, Nakayama K, Rahman MT, Katagiri H, Katagiri A, Ishibashi T, et al. Clinicopathologic analysis of loss of AT-rich interactive domain 1A expression in endometrial cancer. Hum Pathol (2013) 44:103–9. doi:10.1016/j.humpath.2012.04.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol (2011) 224:328–33. doi:10.1002/path.2911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Lichner Z, Scorilas A, White NM, Girgis AH, Rotstein L, Wiegand KC, et al. The chromatin remodeling gene ARID1A is a new prognostic marker in clear cell renal cell carcinoma. Am J Pathol (2013) 182:1163–70. doi:10.1016/j.ajpath.2013.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Huang HN, Lin MC, Huang WC, Chiang YC, Kuo KT. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Mod Pathol (2014) 27:983–90. doi:10.1038/modpathol.2013.216
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Lowery WJ, Schildkraut JM, Akushevich L, Bentley R, Marks JR, Huntsman D, et al. Loss of ARID1A-associated protein expression is a frequent event in clear cell and endometrioid ovarian cancers. Int J Gynecol Cancer (2012) 22:9–14. doi:10.1097/IGC.0b013e318231f140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Ayhan A, Mao TL, Seckin T, Wu CH, Guan B, Ogawa H, et al. Loss of ARID1A expression is an early molecular event in tumor progression from ovarian endometriotic cyst to clear cell and endometrioid carcinoma. Int J Gynecol Cancer (2012) 22:1310–5. doi:10.1097/IGC.0b013e31826b5dcc
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol (2012) 25:615–24. doi:10.1038/modpathol.2011.189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Yokoyama Y, Matsushita Y, Shigeto T, Futagami M, Mizunuma H. Decreased ARID1A expression is correlated with chemoresistance in epithelial ovarian cancer. J Gynecol Oncol (2014) 25:58–63. doi:10.3802/jgo.2014.25.1.58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Xiao W, Awadallah A, Xin W. Loss of ARID1A/BAF250a expression in ovarian endometriosis and clear cell carcinoma. Int J Clin Exp Pathol (2012) 5:642–50.
140. Maeda D, Mao TL, Fukayama M, Nakagawa S, Yano T, Taketani Y, et al. Clinicopathological significance of loss of ARID1A immunoreactivity in ovarian clear cell carcinoma. Int J Mol Sci (2010) 11:5120–8. doi:10.3390/ijms11125120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ, et al. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis (2014) 35:867–76. doi:10.1093/carcin/bgt398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Wiegand KC, Sy K, Kalloger SE, Li-Chang H, Woods R, Kumar A, et al. ARID1A/BAF250a as a prognostic marker for gastric carcinoma: a study of 2 cohorts. Hum Pathol (2014) 45:1258–68. doi:10.1016/j.humpath.2014.02.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Abe H, Maeda D, Hino R, Otake Y, Isogai M, Ushiku AS, et al. ARID1A expression loss in gastric cancer: pathway-dependent roles with and without Epstein-Barr virus infection and microsatellite instability. Virchows Arch (2012) 461:367–77. doi:10.1007/s00428-012-1303-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Werner HM, Berg A, Wik E, Birkeland E, Krakstad C, Kusonmano K, et al. ARID1A loss is prevalent in endometrial hyperplasia with atypia and low-grade endometrioid carcinomas. Mod Pathol (2013) 26:428–34. doi:10.1038/modpathol.2012.174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, et al. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev (2005) 19:1662–7. doi:10.1101/gad.1323805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Zhao H, Wang J, Han Y, Huang Z, Ying J, Bi X, et al. ARID2: a new tumor suppressor gene in hepatocellular carcinoma. Oncotarget (2011) 2:886–91.
147. Manceau G, Letouze E, Guichard C, Didelot A, Cazes A, Corte H, et al. Recurrent inactivating mutations of ARID2 in non-small cell lung carcinoma. Int J Cancer (2013) 132:2217–21. doi:10.1002/ijc.27900
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell (2012) 150:251–63. doi:10.1016/j.cell.2012.06.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Zhang ZM, Xiao S, Sun GY, Liu YP, Zhang FH, Yang HF, et al. The clinicopathologic significance of the loss of BAF250a (ARID1A) expression in endometrial carcinoma. Int J Gynecol Cancer (2014) 24:534–40. doi:10.1097/IGC.0000000000000092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Zhang X, Zhang Y, Yang Y, Niu M, Sun S, Ji H, et al. Frequent low expression of chromatin remodeling gene ARID1A in breast cancer and its clinical significance. Cancer Epidemiol (2012) 36:288–93. doi:10.1016/j.canep.2011.07.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Brahma, chromatin remodeling, epigenetic BRG1, SMARCA2, SMARCA4
Citation: Marquez SB, Thompson KW, Lu L and Reisman D (2015) Beyond mutations: additional mechanisms and implications of SWI/SNF complex inactivation. Front. Oncol. 4:372. doi: 10.3389/fonc.2014.00372
Received: 22 October 2014; Paper pending published: 25 November 2014;
Accepted: 11 December 2014; Published online: 27 February 2015.
Edited by:
Scott Bultman, University of North Carolina, USAReviewed by:
Luisa Lanfrancone, European Institute of Oncology, ItalySteven De Jong, University Medical Center Groningen, Netherlands
Copyright: © 2015 Marquez, Thompson, Lu and Reisman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Reisman, 2033 Mowry Road, Gainesville, Florida, USA e-mail:ZG5yZWlzbWFuQHVmbC5lZHU=
†Denotes Co-First Author as each person contributed equally to this manuscript.