 Eliot M. Rosen
Eliot M. Rosen Regina Day2
Regina Day2- 1Departments of Oncology, Biochemistry and Molecular & Cellular Biology, and Radiation Medicine, Lombardi Comprehensive Cancer Center, Georgetown University School of Medicine, Washington, DC, USA
- 2Department of Pharmacology, F. Edward Hébert School of Medicine, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 3Department of Radiation Biology, F. Edward Hébert School of Medicine, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
- 4Radiation Countermeasures Program, Armed Forces Radiobiology Research Institute, Uniformed Services University of the Health Sciences, Bethesda, MD, USA
Radioprotectors are compounds that protect against radiation injury when given prior to radiation exposure. Mitigators can protect against radiation injury when given after exposure but before symptoms appear. Radioprotectors and mitigators can potentially improve the outcomes of radiotherapy for cancer treatment by allowing higher doses of radiation and/or reduced damage to normal tissues. Such compounds can also potentially counteract the effects of accidental exposure to radiation or deliberate exposure (e.g., nuclear reactor meltdown, dirty bomb, or nuclear bomb explosion); hence they are called radiation countermeasures. Here, we will review the general principles of radiation injury and protection and describe selected examples of radioprotectors/mitigators ranging from small-molecules to proteins to cell-based treatments. We will emphasize agents that are in more advanced stages of development.
Introduction
Medical countermeasures (MCM) is a term utilized by the Departments of Defense and Health and Human Services that refers to agents used to prevent (protectors and mitigators) or treat (therapeutics) radiation injury. We will not discuss agents that are solely used to treat established radiation injury (therapeutics). Because of the threat of nuclear terrorism or nuclear accidents (e.g., Chernobyl or Fukushima nuclear reactor meltdowns), several governmental agencies [Department of Defense, National Institute of Allergic and Infectious Diseases (NIAID), Biomedical Advanced Research and Development Authority (BARDA), and Defense Advanced Research Projects Agency (DARPA)] have been interested in the development of agents that can protect against the effects of ionizing radiation (IR), increase survival, and/or decrease morbidity. As an additional benefit, some MCMs may be useful as radioprotectors in the radiation therapy clinic provided that they do not equally render tumors more resistant to IR.
Radiation and Normal Tissue Complications
Most recent advances in radiation oncology related are due to methods to make the radiation beam better conform to the shape of the tumor and thereby reduce the volume of normal tissue within the radiation beam and the dose to normal tissues. These approaches include intensity modulated radiation therapy (IMRT), stereotactic radiosurgery (e.g., using the Gamma Knife or CyberKnife), and proton beam therapy. However, it is not possible to exclude all normal tissues from the radiation field; and normal tissue damage remains a dose-limiting factor in the treatment of some tumor types (e.g., locally advanced cancers of the cervix, lung, head and neck, and brain). Thus, normal tissue radioprotection is a promising strategy to prevent damage to radiosensitive tissues and organs.
Initial studies of radioprotectors and mitigators typically involve investigation of the acute effects of total-body irradiation (TBI) in rodents, using survival as the end-point. While TBI affects multiple organ systems, death in humans and rodents in the first 30 days is mainly due to two mechanisms: (1) gastrointestinal (GI) syndrome, which often leads to death within 10–12 days after exposure to 8–20 Gy of γ-rays, due to fluid and electrolyte imbalance and bacterial translocation (sepsis); and (2) hematopoietic syndrome, which leads to death within 30 days after exposure to 3–8 Gy, due to neutropenia and thrombocytopenia (1–6). The effects of radiation within the first 30 days are called “acute radiation syndrome (ARS)” or “radiation sickness.” ARS follows a similar pattern in humans and rodents, except that the LD50/30 values (dose of whole body exposure required to reduce survival to 50% by day 30, without medical support) are lower in humans (ca. 3.5–4 Gy) than in rodents (ca. 7–9 Gy) (7).
An effective radioprotector/mitigator should improve a 30-day survival in rodents by protecting against GI syndrome, hematopoietic syndrome, or both. It should also have a convenient mode of delivery (e.g., by oral, subcutaneous, or intramuscular routes). For hematopoietic syndrome, it is thought that death within the first 30 days is due to depletion of hematopoietic progenitor cells (HPCs) for white blood cell and megakaryocyte lineages, leading to neutropenia and thrombocytopenia (1, 2). HPCs are more radiosensitive than pluripotent stem cells (HSCs) (8–10). However, irradiated HSCs take a long time (30 days or so) to be recruited into the cell cycle and reconstitute neutrophils and platelets. Thus, if an individual survives for 30 days, HSCs will have sufficient time to reconstitute the various bone marrow lineages, and further hematological support is not required.
Gastrointestinal syndrome is due to depletion of intestinal stem cells (ISCs) located at or near the base of the intestinal crypts (11, 12). These cells die rapidly after exposure to a high dose of radiation by apoptosis. PUMA (p53 up-regulated modulator of apoptosis) appears to be a crucial mediator of apoptosis in ISCs. Crypts become progressively denuded as apical cells are shed and ISCs die or enter cell cycle arrest due to DNA damage. The villus length, number of villi per circumference, and mitotic index decrease starting about four days after irradiation (13). Death due to GI syndrome in mice usually occurs within 10-15 days, depending upon the mouse strain and radiation dose. However, in surviving animals (e.g., due to treatment with a radioprotector), crypts begin to regenerate (as indicated by an increase in DNA synthesis) by day 15 or so.
Although the GI system and bone marrow are rapidly reacting systems that contribute to ARS following TBI, high dose partial body radiation that includes the lungs can result in delayed toxicity that occurs 3-10 months after exposure. This syndrome is related to repeated cycles of inflammation, eventually resulting in pulmonary fibrosis and death, depending on the dose and volume of lung irradiated (14–17). The skin and kidneys are also “radiosensitive” tissues in which severe effects can be observed in individuals who receive high dose partial body irradiation. ARS is the best understood consequence of TBI. Less is known about the later effects of high dose partial body irradiation and the late consequences of ARS. Much of what we know about the sensitivity of specific tissues and organs to radiation comes from early experience with radiation therapy, before the radiation tolerances of these tissues and organs were established and before the introduction of skin sparing megavoltage radiation.
Radiation therapy is usually delivered as fractionated treatments using small dose increments (1.8-3 Gy) delivered five days per week to the tumor site(s). Total doses may vary from 30 to 80 Gy, depending upon the intent of treatment (i.e., curative vs. palliative) and the type and location of the tumor. Side effects from radiation have been well-studied and are classified as acute, intermediate, or late effects (18–26). Acute effects occur during a course of radiotherapy and are resolved within 4 weeks after the last treatment. Examples include epidermitis and mucositis due to injury to the skin and mucosal membranes, respectively. Intermediate effects are less common and occur within 8-12 weeks after the end of radiation. An example is radiation pneumonitis, which reflects inflammation of the lung and is typically confined to the radiation portals. Late effects occur at least 9 months after the end of radiation and are usually the dose-limiting factor in clinical radiotherapy. Late effects include injury to specific tissues and organs within the radiation field or in the entrance or exit paths of the radiation beam. Other types of late effects due to irradiation include carcinogenesis (second tumors caused by radiation), teratogenesis (malformation of fetus, which is very rare because pregnant women are rarely treated with radiation), and effects on growth and development due to irradiation in childhood.
The likelihood of a late effect depends on the total dose of radiation, the fraction size, the volume of tissue being treated, and other treatments (e.g., chemotherapy). Late effects also depend upon prior or subsequent surgery, genetic factors unique to the individual patient, pre-existing vascular damage (e.g., diabetes), hypertension, age, and other pre-existing conditions (e.g., inflammatory bowel disease in patients who receive abdominal irradiation). The dose of radiation and/or volume of irradiated tissue is limited due to late effects: e.g., tumors of the brain and spinal cord and locally advanced cancers of the lung, cervix, breast, and head and neck. Here, a selective normal tissue protector could allow a higher dose, a larger treatment volume, and/or reduced late normal tissue injury, thus increasing the therapeutic ratio.
A reduction in early effects (e.g., epidermitis, mucositis, cystitis, and proctitis) due to a radioprotector could increase patient comfort. Although these effects usually resolve by themselves, they sometimes require a treatment break that delays the completion of radiation. Concurrent chemotherapy and radiotherapy can cause severe acute effects (e.g., debilitating mucositis and weight loss); and here a normal tissue protector could be beneficial (27–32). Normal tissue protection could be particularly useful in young children undergoing cranial irradiation by protecting a central nervous system that is not fully developed (33, 34). Effects on the growth of bones (before epiphyseal closure) and the possibility of a second tumor due to radiation must be considered whenever children are treated with radiation alone or in combination which chemotherapy.
A relatively recently recognized late consequence of thoracic and chest wall irradiation (e.g., treatment of Hodgkin’s disease or post-operative radiotherapy for breast cancer) is radiation-induced heart disease (RIHD), which is usually observed at least several years after treatment and is characterized by accelerated atherosclerosis, cardiac fibrosis, valvular damage, and an increased risk of cardiac-related mortality (35, 36). RIHD can occur when part or all of the heart is included in the radiation field. This condition is usually progressive; and its incidence increases with time after treatment. A significantly increased risk of neurovascular events (e.g., stroke or transient ischemic attack) has been observed after cranial irradiation for brain tumors in children (37). Neurocognitive decline after whole brain irradiation in adults (“radiotherapy brain”) is common, particularly in individuals who have also received chemotherapy. Since there is no specific treatment for these complications, a prevention strategy is required.
Mechanisms of Radiation Injury and Repair
Although IR can directly target critical cellular macromolecules such as DNA, water (H2O) is by far the most abundant molecule within cells and is thus the most likely target for radiolysis by high energy photons (38–41). As shown in Figure 1, molecular oxygen (O2) is a central component involved in the formation of highly reactive free radicals; and so it is not surprising that high concentrations of O2 potentiate the effects of IR, while low concentrations of O2 (hypoxia) protect cells and tissues from IR, the so-called “oxygen effect” (42–44). The most damaging species of free radical is the hydroxyl radical (OH) (45, 46). DNA is the most critical target for cell survival, but significant damage to other cellular molecules such as proteins and lipids is also produced (47, 48). These oxidative radicals produce two major forms of DNA damage, double-strand breaks (DSBs) (the most lethal form of damage) and base lesions (which are repaired by the base excision repair pathway) (49–52). During the processing of base lesions, single-strand DNA breaks (SSBs) are generated, which are then repaired by one of several mechanisms that involves a scaffolding protein, DNA polymerase, and a DNA ligase. If two base lesions on opposite strands are close enough, the result can be a DSB.
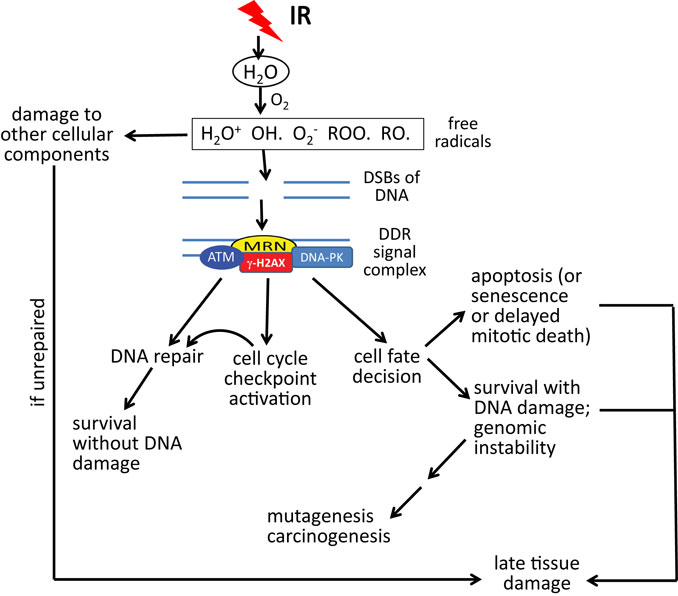
Figure 1. DNA damage response (DDR) to double-strand DNA breaks (DSBs) in relation to acute radiation syndrome and late effects. DSBs caused by oxidative radicals are sensed by the MRN complex (MRE11–RAD50–NBS1), resulting in an ATM (ataxia-telangiectasia, mutated)-driven DDR. Gamma-H2AX (phosphorylated histone H2AX protein) is both a participant in the DDR and a marker of DSBs. Depending upon the dose of radiation, the type of radiation, the volume of tissue irradiated, and other factors, the DDR may lead to some combination of DNA repair, permanent cell cycle arrest (senescence), cell death, or survival with DNA damage. As a result of these processes, acute and late radiation effects may ensue, resulting in survival, death, or survival with late tissue damage. Note that “acute radiation syndrome” refers to the consequences of whole body radiation exposure. Acute effects of radiation may be limited to specific tissues or organs in the case of partial body radiation exposures or radiotherapy treatment to tumor-bearing tissue.
In DSB repair, a DNA-damaging signaling/repair complex accumulates at and around the DSB site. The “MRN” complex of three proteins (MRE11–RAD50–NBS1) senses the damage and binds to the broken DNA ends (53). Following MRN, ataxia telangiectasia mutated (ATM), a nuclear serine/threonine protein kinase, is recruited to the MRN complex and activated through autophosphorylation, after which it phosphorylates a number of substrate proteins on SQ/TQ motifs. The eventual result is the coating of DNA surrounding the break with a set of proteins that orchestrates the DNA repair process. These events are reviewed elsewhere (54). DSB repair can proceed by two pathways: (1) homology-directed repair (HDR) (orchestrated by ATM/BRCA1/BRCA2 signaling), which is an error-free process; and (2) non-homologous end joining (NHEJ), which can be accurate or can lead to significant sequence deletions and translocations [orchestrated by DNA-dependent protein kinase (DNA-PK)] (55, 56). HDR occurs only in S-phase and G2, since it requires a sister chromatid as a template for DNA repair synthesis, while NHEJ can occur in any phase of the cell cycle, but preferably occurs during G1.
In addition to mediating DNA repair, ATM signaling also results in activation of DNA damage-dependent cell cycle checkpoints (e.g., S and G2/M), which allows time for damaged cells to repair their damage, so that it is not passed on to daughter cells (Figure 1). ATM also orchestrates the “cell fate” decision (57). Cells that have too much damage to repair are pushed into rapid death by apoptosis or, alternatively, permanent cell cycle arrest (“senescence”) or delayed death through mitotic catastrophe. ATM can also stimulate cell survival pathways (e.g., the anti-apoptotic transcription factor NF-κB) (58, 59). If cells protected by NF-κB signaling have not fully repaired their DNA damage, this can result in cells with genomic instability, which can result in the accumulation of mutations and, eventually, carcinogenesis, a late effect that usually occurs at a minimum of 3–5 years after radiation exposure (60, 61).
Depending on the dose and proportion of the body exposed to radiation, the relative apoptotic vs. surviving GI and hematopoietic stem/progenitor cell populations may result in ARS (described above), which can lead to death or survival and recovery. In the case of partial body radiation exposure, high dose clinical radiotherapy, or even in survivors of ARS, late complications of radiation may ensue, the seriousness of which depends upon the specific tissue, the radiation dose, and the volume of tissue irradiated. The mechanism(s) of late tissue damage is not fully understood, but may result from damage to parenchymal stem/progenitor cells, blood vessels, inflammation, and/or ongoing oxidative stress due to generation of reactive oxygen species (ROS) (62). Repeated cycles of inflammation may lead to fibrosis [e.g., in lung (62–65)]; and ROS can cause additional DNA damage by causing oxidation of DNA bases, creating a vicious cycle. Possible outcomes include death, survival with permanent late tissue damage of different degrees of severity, or tissue recovery with little or no functional deficit.
Radioprotectors, Mitigators, and Candidate Agents
Amifostine as a Radioprotector
No radioprotectors or mitigators are currently approved by the Food and Drug Administration (FDA) for general use in humans for the prevention or treatment of ARS. Although amifostine (EthyolR) is not a new agent, to date, it is the only drug that has been approved by the FDA to reduce the toxicity of radiation therapy in the setting of cancer treatment (66). This agent is also used to protect against renal toxicity due to cis-platinum, a DNA cross-linking agent that is also known to cause oxidative stress (67–69). Amifostine (formerly known as “WR-2721”) was originally developed by the U.S. Army Anti-Radiation Drug Development Program as an MCM. It is a thiol compound that acts as a free radical scavenger to reduce the levels of oxidative radicals that would otherwise attack important cellular targets, such as DNA and other cellular macromolecules (70). Amifostine has been used successfully to prevent xerostomia (dry mouth) due to head and neck irradiation, which can otherwise cause permanent dry mouth due to inclusion of the salivary glands (particularly the parotid glands) within the radiation field (71, 72). Initially, there were some concerns that the widespread usage of amifostine would also protect the tumor against radiation or chemotherapy drugs, but accumulated experience has shown that this is not the case (73).
In a recent report that examined 30 studies utilizing amifostine, no conclusion could be made regarding the efficacy of amifostine in preventing or reducing oral mucositis, because of conflicting and confusing data (74). In a recent meta-analysis that included multiple clinical trials in which amifostine was used to prevent cis-platinum toxicity, there was a trend toward a reduction in the incidence of platinum-induced ototoxicity (hearing loss due to cochlear damage), but the trend did not reach statistical significance (75). In a study of locally advanced non-small cell lung cancers treated with chemoradiotherapy plus or minus amifostine, amifostine conferred a significant reduction in pain and dysphagia (difficulty swallowing). And in a study of patients who received postmastectomy radiation without or with amifostine (at different dose levels), patients who received amifostine had a lower incidence of skin toxicity, and pulmonary and soft tissue fibrosis (76). In a recent meta-analysis of cancer treatment trials that tested amifostine to reduce acute side effects, it was concluded that amifostine did not reduce overall survival or progression-free survival in patients who received radiotherapy plus or chemoradiotherapy plus amifostine (73).
The most commonly accepted explanation for the lack of radioprotection of tumors is that amifostine itself (WR-2721) is an inactive pro-drug, which must be converted to an active drug (WR-1065) by dephosphorylation. The conversion is usually due to alkaline phosphatase in the cell membrane of normal endothelium. Tumors, which have abnormal vasculature which is sparser than in normal tissues and contains lower levels of alkaline phosphatase, are much less efficient at activating amifostine than normal tissue [reviewed in Ref. (66)]. Several other mechanisms were proposed to explain the selective radioprotection by amifostine, including protection of DNA by metabolites of amifostine, causing hypoxia in normal tissues by increasing oxygen consumption, and accelerated recovery of normal endothelial cells [reviewed in Ref. (66)].
Amifostine has several clinically relevant limitations including: (1) the need to administer it within a narrow time window (15–30 min) before each radiation dose; (2) its approval only for intravenous use, although other routes of administration (e.g., subcutaneous) are under investigation (76, 77); and (3) toxicity, including nausea, vomiting, somnolence, and hypotension. Recently, it has been demonstrated that radioprotective doses for amifostine appear to lie between 25 and 50 mg/kg in mice. Mature, lineage-restricted progenitors appear to be more responsive to the protective effects of low doses of amifostine than the more primitive, multipotential progenitors (78).
Palifermin
Palifermin is a recombinant N-terminal truncated form of keratinocyte growth factor [KGF, also known as fibroblast growth factor 7 (FGF7)], a growth factor that is produced by mesenchymal cells and acts in a paracrine manner to stimulate the proliferation of epithelial cells. KGF generally functions in the protection and repair of epithelial tissues through its cognate receptor FGFR2B. Its protective action appears to be due to a combination of stimulation of cell proliferation and protection against apoptosis (79). Oral mucositis is a significant toxicity in patients undergoing radiotherapy and particularly chemoradiotherapy for head and neck cancers. This complication frequently requires a treatment break and reduces the quality of life in patients receiving such treatment. Here, severe oral mucositis can result in weight loss due to reduced oral intake, requirement for pain medicines, increased risk of infections, and, in some cases, the need for hospitalization. Mucositis is due, in part, to an imbalance between death and shedding of oral mucosal lining cells and the ability of cells newly recruited into the cell cycle to replace the lost cells. The result is a partial denudation of the mucosal surface. Oral mucositis has been traditionally managed by supportive care, including basic oral care hygiene, appropriate pain management, and the use of mouthwashes and oral rinses.
A variety of preclinical studies suggested that palifermin could ameliorate the mucosal toxicity due to chemotherapy and/or radiation therapy (79). Palifermin was first approved by the FDA in 2004 for the indication of preventing severe oral mucositis in patients undergoing hematopoietic stem cell transplantation for the treatment of hematological cancers (80, 81). These patients receive high dose chemotherapy without or with total-body radiotherapy prior to transplantation and are at-risk for severe (grades 3–4) mucositis. Beneficial effects of palifermin were documented in patients receiving palifermin for 3 days prior to the preparative regimen and for 3–5 days after transplantation. The use of palifermin has also been shown to reduce the incidence, duration, and severity of oral mucositis in patients treated with chemoradiotherapy for head and neck cancers and in patients receiving chemotherapy using agents that can cause mucositis, including adriamycin and 5-fluorouracil. Palifermin has also been utilized to mitigate dysphagia due to esophagitis in patients treated with chemoradiotherapy for lung carcinoma. Interestingly, in addition to mucositis, palifermin also appears to stimulate immune reconstitution following hematopoietic stem cell transplantation and to reduce graft-vs.-host disease following allogeneic bone marrow transplantation (80). Palifermin is generally well-tolerated but its usage has been associated with skin rash and taste disturbance.
Superoxide Dismutase
Greenberger and colleagues have been studying the use of a superoxide dismutase (SOD) transgene to protect normal tissues against injury due to IR. Here, over-expression of manganese superoxide dismutase (MnSOD, also called SOD2) by intra-tracheal injection of a replication deficient adenovirus containing the MnSOD transgene conferred protection against lung irradiation and cytokine production (IL-1, TNF-alpha, and TGF-beta) when administered prior to irradiation (82). Interestingly, intra-tracheal administration of the MnSOD transgene protected normal lung but not orthotopic Lewis lung carcinoma against pulmonary irradiation (83). Similarly intraesophageal administration of MnSOD prevented the development of radiation-induced esophagitis and modulated cytokine expression (84, 85). In both the lung and esophageal models, the MnSOD transgene was well-expressed in the respective normal tissues. Incultured cell lines, the MnSOD appeared to work, in part, by protection against radiation-induced apoptosis via stabilization of the mitochondrial membrane (86). In a mouse model of radiation-induced oral mucositis, a significant form of radiation-induced injury in patients receiving head and neck irradiation, intraoral administration of MnSOD caused a decrease in the extent of radiation-induced ulceration (87). As in the case of lung and esophageal irradiation, MnSOD did not confer protection of head and neck carcinoma (87). Here, addition of amifostine to the MDSOD did not confer additional protection beyond that due to MnSOD alone. As Cu/ZnSOD (SOD1) did not protect mice against thoracic irradiation, it appears that mitochondrial localization and prevention of mitochondria-induced apoptosis figure into the mechanism of radiation protection by MnSOD (88).
Together, these findings suggest the possibility of utilizing radioprotective antioxidant gene therapy to prevent or reduce the extent of some forms of radiation injury. Here, it also appears that transgene expression in cells within the microenvironments of protected organs contribute significantly to the protection (89). In this regard, a phase I study of MnSOD administered orally was carried out in patients who received a standard chemoradiation regimen for for stage III unresectable lung carcinoma. In this study, there did not appear to be any dose-limiting additional toxicity due to administration of the MnSOD transgene at any of three doses (up to 30 mg per patient). Interestingly, Greenberger and colleagues also showed that in bone marrow stromal stem cells, doxycycline induced expression of a tetracycline regulated MnSOD conferred radiation resistance, whereas in the absence of doxycycline, the cells showed normal radiation sensitivity (90). Mice fed a diet rich in antioxidants and given MnSOD showed an increased lifespan as compared with MnSOD plus house diet, following TBI to 9.5 Gy (91). These findings suggest that when combined with MnSOD, the antioxidant/chemopreventive diet reduces the extent of radiation-induced life-shortening due to TBI in survivors of the ARS. There was no increase in detectable tumors, or histopathologic evidence of neurodegenerative disease in the increased number of survivors following MnSOD plus irradiation. Intravenous administration of MnSOD also ameliorated the growth retardation in the newborn mice from irradiated mothers (92). And MnSOD when given 24 h prior to irradiation of pregnant mice conferred a significantly increased number of live births. The protection appeared to be due to a remote (bystander) effect, since increased expression of MnSOD in fetal tissues could not be demonstrated by RT-PCR in this study.
Other Radioprotective Agents
Interestingly, in a screen of 13 drugs utilized during bone marrow transplantation, tetracycline, but not other antibiotics and anti-fungal agents appeared to protect cultured HPCs against IR (93). As protection was observed in radiation dose–response assays, these findings suggest that the observed radioprotection of tetracycline is not due directly to its properties as an anti-microbial agent. p53 Up-regulated modulator of apoptosis (PUMA) is a Bcl-2 homology 3 (BH3)-only Bcl-2 family member that has been implicated in radiation-induced apoptosis. Recently, a group of pharmacophiles that inhibit PUMA and radiation-induced apoptosis have been identified (94, 95). These agents appear to work, in part, by disrupting the interaction between PUMA and Bcl-XL. In other studies, a p53/Mdm2/Mdm4 inhibitor, BEB55, protected mice against radiation-induced esophagitis when administered orally (96).
Genistein
Genistein (4′,5,7-trihydroxyisoflavone) is a soy isoflavone with a variety of cellular activities, including selective estrogen receptor activation, protein tyrosine kinase inhibition, antioxidant activity, and free radical scavenging activity (97–100). Genistein has been established as an anti-cancer agent, and has additionally been demonstrated to have anti-microbial and anti-inflammatory activity in vivo (101–105). Genistein was reported in clinical trials to reduce the adverse effects of chemotherapy and radiotherapy (106, 107). The protective effects of genistein for radiation-induced injury to the bone marrow were observed in a murine model of ARS, where neutrophils and platelets were protected (108, 109). Genistein also protected bone marrow progenitor cell populations, thus preventing hematopoietic stem cell pool exhaustion (109, 110). Genistein administration reduced radiation-induced injury in the lung and increase survival from thoracic irradiation in mice (111). Genistein reduced micronuclei in Lin− bone marrow cells and primary lung fibroblasts suggesting a direct reduction of radiation-induced DNA damage (17, 111–113). Several mechanisms have been proposed for radioprotective effects by genistein, including activation of the DNA repair enzyme Gadd45 (114–116), the quiescence of the cell cycle of Lin− cells in the G0/G1 phase in vivo (110, 117), and the suppression of inflammation (14, 105, 118, 119).
Captopril and ACE Inhibitors
Captopril, a sulfhydryl-containing analog of proline, is a competitive inhibitor of the angiotensin converting enzyme (ACE) protease, and reduces systemic blood pressure by blocking both the activation of the vasoconstrictor angiotensin II (Ang II) and the inactivation of the vasodilator bradykinin. Although captopril was initially developed for the treatment of hypertension and heart failure, it was found that captopril was also useful in animal models of radiation-induced renal dysfunction for increasing renal plasma flow and improving glomerular filtration (120, 121). Captopril has been investigated as a radiation countermeasure for the pulmonary, renal, and hematopoietic systems as well as for the brain and skin (122–127). ACE inhibitors and captopril mitigated radiation-induced pulmonary endothelial dysfunction, radiation pneumonitis, and fibrosis in animal models (128, 129). Prophylactic administration of captopril resulted in lower systemic blood pressure and improved renal function following TBI in animal models (121, 130, 131) and reduced chronic renal failure in human patients undergoing clinical radiation (132). Captopril and another ACE inhibitor, perindopril, were demonstrated to block radiation-induced hematopoietic syndrome through accelerated recovery of erythrocytes, reticulocytes, leukocytes, and platelets (122, 133). The improved blood cell recovery was associated with improved survival of specific hematopoietic progenitor populations CFU-GM, CFU-M, and total CFC (122). The mechanism of captopril-induced reduction of radiation injury has not been established. Captopril mitigation of radiation injuries may involve reduced inflammation (134) or the transient quiescence of some cells in vivo (122, 135). However, in vivo effects on radiation-induced DNA damage have not been shown (112).
3,3′-Diindolylmethane
3,3′-Diindolylmethane is a small-molecule compound formed by acid hydrolysis in the stomach of indole-3-carbinol (I3C), a component of cruciferous vegetables (e.g., cabbage, cauliflower, and broccoli) (136). 3,3′-Diindolylmethane (DIM) is a proposed cancer prevention agent that is available as a nutritional supplement and has been administered safely by the oral route to humans in repeated doses in phase I/II clinical trials (137–140). Recently, it was found that administration of DIM in a multidose schedule protected rodents against lethal doses of TBI up to 13 Gy, whether DIM dosing was initiated 24 h before or up to 24 h after irradiation (141). The dose reduction factor (DRF) (i.e., ratio of LD50/30 values in the presence/absence of DIM) was 1.6 when DIM treatment was begun 24 h after irradiation. Low physiologically relevant (submicromolar) concentrations of DIM protected cultured cells against radiation by a novel mechanism. DIM caused rapid activation of ATM and phosphorylation of various ATM substrates, suggesting that DIM induces an ATM-dependent DNA damage response (DDR)-like response, and DIM enhanced radiation-induced ATM signaling and NF-κB activation. Similarly, DIM caused ATM activation and signaling in normal tissues in rodents. However, DIM did not protect human breast cancer xenografts (MDA-MB-231) against radiation. In the tumors, ATM signaling appeared to be defective. The results appear promising, but further work is required to determine if DIM will be a useful radioprotector and/or mitigator.
3,3′-Diindolylmethane was also shown to have cardioprotective properties. Here, subcutaneous administration of DIM decreased the extent of fibrosis due to adriamycin, a DNA-damaging chemotherapy agent by a mechanisms that involves up-regulation of BRCA1 and activation of the antioxidant transcription factor nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) (142). DIM mediated cardioprotection against other stressors including aortic banding, which causes cardiac hypertrophy, due to a mechanism involving 5′-adenosine monophosphate-activated protein kinase-alpha2 (AMPK-α2) and mammalian target of rapamycin (mTOR) (143). Whether DIM will protect the heart against IR has not been described.
Inhibitors of Radiation-Induced Accelerated Senescence
Loss of cellular clonogenic potential following exposure to radiation can be caused by apoptosis, necrosis, autophagy, and accelerated cellular senescence. Recent findings suggest that accelerated cellular senescence may be a primary effect of radiation on normal (non-transformed, non-immortalized) epithelial and endothelial cells and fibroblasts. Cellular senescence results in a range of aberrant biological activities and can influence overall tissue dysfunction (144–150). The blockade of radiation-induced cellular senescence by the pharmacological inhibitors of mTOR was sufficient to prevent mucositis in mice following irradiation of the head and neck area (145). In this study, it was demonstrated that rapamycin blocked radiation-induced senescence, but not apoptosis, in primary keratinocyte in cell cultures and in vivo in a murine model of head/neck irradiation injury.
Investigation into receptor signaling pathways that contribute to aging-associated cellular senescence revealed the involvement of the insulin-like growth factor-1 receptor (IGF-1R) (151, 152). IGF-1 enhances senescence in primary cell cultures via a mechanism that involves increase in ROS leading to induction of the p53/p21 pathway (153). In mouse embryonic fibroblasts, treatment with IGF-1 inhibits the deacetylase activity of Sirtuin 1 (SIRT1) and promotes stability of p53, ultimately leading to induction of senescence (154). IGF-1R expression levels increase during the development of replicative in vitro senescence in primary cortical neurons (155). In agreement with these findings, a recent study demonstrated that inhibition of IGF-1R, PI3K, and mTor blocked radiation-induced accelerated senescence in primary lung endothelial cells in cell culture (156).
CBLB502/Entolimod™
CBLB502 is a potent and stable agent derived from the flagellin protein of Salmonella bacteria (Salmonella enterica serovar Dublin). Its pharmacologic action is based on binding to toll-like receptor 5 (TLR5) of targeted cells and activating NF-κB signaling. Biologically, purified flagellin protects mice from lethal doses of total-body gamma-irradiation (157). Cleveland BioLabs, Inc. (Buffalo, NY, USA) identified CBLB502 (now known as Entolimod) as a TLR5 ligand that significantly improved the radioprotective efficacy of flagellin while having reduced toxicity and immunogenicity (158).
A single injection of CBLB502 either before lethal TBI (24 h prior) or up to 48 h following irradiation protected mice from both GI and hematopoietic syndromes, with significantly improved survival. CBLB502 also demonstrated radioprotective and radiomitigative potential in lethally irradiated non-human primates (158). A single intramuscular injection of CBLB502 significantly increased the survival of rhesus non-human primates exposed to 6.5 Gy TBI and promoted the regeneration of their small intestine, spleen, thymus, and bone marrow when administered from 1 to 48 h after irradiation. The severity and duration of irradiation-induced thrombocytopenia and neutropenia decreased significantly with CBLB502 treatment. Two cytokines, granulocyte colony-stimulating factor (G-CSF) and interleukin-6 (IL-6) were identified as candidate biomarkers for the radioprotective and radiomitigative efficacy of CBLB502. Induction of both G-CSF and IL-6 by CBLB502 is TLR5-dependent, dose-dependent within its efficacious dose range in both unirradiated and irradiated mammals (including rodents and non-human primates), and critically important for the CBLB502-mediated increased survival of irradiated animals (159). Administration of either G-CSF or IL-6 neutralizing antibody abrogated the radiomitigation by CBLB502. These biomarkers are likely to be useful for the accurate prediction of CBLB502 dose providing radioprotection or radiomitigation in humans. Furthermore, CBLB502 was shown to significantly reduce the severity of dermatitis and oral mucositis caused by local radiation exposure (160). The FDA has granted IND status to CBLB502 as a radiation countermeasure for ARS and it is currently in clinical development.
ON01210/Ex-RAD®
ON01210 (a chlorobenzylsulfone derivative known as Ex-RAD) is a novel, small-molecule kinase inhibitor under development as a radiation countermeasure. Ex-RAD provided significant protection against cobalt-60 gamma-irradiation when administered sc (500 mg/kg) to C3H/HeN mice 24 h and 15 min before irradiation. Ex-RAD’s estimated DRF is 1.16 (161). In another study, Ex-RAD showed a significant survival benefit after prophylactic oral administration of the drug (162).
This drug accelerated the recovery of peripheral blood elements in irradiated mice when administered either subcutaneously (sc) or orally (162, 163). In addition, Ex-RAD-treated mice (either through the oral or sc route) contained higher numbers of granulocyte macrophage-colony forming units (GM-CFUs) than in vehicle-treated mice. Bone marrow obtained from irradiated mice indicated that Ex-RAD protected cells from radiation-induced apoptosis after exposure to cobalt-60 gamma-irradiation (163). Ex-RAD also assists in the recovery of the GI system, with a higher number of surviving intestinal crypts after acute radiation exposure in Ex-RAD-treated mice than untreated irradiated controls (163). These effects may be due in part to signaling pathways that are affected by Ex-RAD. Attenuation of ATM-p53 mediated DDR by Ex-RAD contributes to the mitigation of radiation-induced hematopoietic toxicity (164). Recently, Kang et al. demonstrated that Ex-RAD manifests its protective effects through the up-regulation of phosphatidylinositol-3-kinase/AKT pathways in cells exposed to radiation (165). Ex-RAD has been granted FDA IND status and has demonstrated oral efficacy (162). Oral administration holds better clinical promise as an effective countermeasure for first responder use as well as for at-risk civilian populations in a nuclear accident.
Gamma-Tocotrienol
Gamma-tocotrienol is one of the eight isomers (tocols) of vitamin E. It is a potent inhibitor of HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme A) reductase. Gamma-tocotrienol (GT3) has been shown to increase survival in rodents, through ameliorating the hematopoietic and GI systems (166). When administered 24 h before cobalt-60 gamma-irradiation, GT3 significantly protected mice against radiation doses as high as 11.5 Gy, and its DRF as a radioprotector (24 h before irradiation, 200 mg/kg dose, sc route) was 1.29 in mice. GT3 treatment accelerated hematopoietic recovery in peripheral blood and enhanced recovery of hematopoietic progenitors in bone marrow of irradiated mice (167, 168). GT3 treatment resulted in significant induction of G-CSF and IL-6 in mice (170). Mouse survival studies with GT3 suggested the most efficacious time for drug administration was 24 h prior to irradiation, possibly due to the induction of key hematopoietic cytokines during that time window. Prophylactic GT3 administration caused up-regulation of anti-apoptotic genes and down regulation of pro-apoptotic genes (both at the transcriptional and the protein levels) at 4 and 24 h after irradiation (169). The administration of G-CSF antibody abrogated the radioprotective efficacy of GT3 (170).
δ-Tocotrienol
δ-Tocotrienol has demonstrated antioxidant activity greater than that of γ- and α-tocotrienol in the membrane system while protecting primary neuronal cells against glutamate toxicity (166). A single sc injection of δ-tocotrienol before or after cobalt-60 γ-irradiation significantly protected mice in a 30-day survival experiment. δ-Tocotrienol was effective at a wide dose range of 19–400 mg/kg (171, 172). The DRF values for radioprotective treatment (24 h before irradiation) with 150 and 300 mg/kg were 1.19 and 1.27, respectively. For radiomitigation treatment with 150 mg/kg of δ-tocotrienol administered 2 h after irradiation, the DRF was 1.1. When δ-tocotrienol was administered at 300 mg/kg dose 24 h before irradiation, it significantly reduced radiation-induced cytopenia, suggesting its stimulatory effects on hematopoietic recovery (171). Similar to countermeasures mentioned above, we have demonstrated that the administration of G-CSF antibody abrogates the radioprotective efficacy of δ-tocotrienol (173, 174). Recently, it was demonstrated that δ-tocotrienol reduces activation of caspases 3, 7, and 8 while increasing autophagy-related beclin-1 expression in irradiated bone marrow cells (175). δ-Tocotrienol has been reported to increase cell survival and regeneration of hematopoietic microfoci and lineage−/Sca-1+/c-Kit+ stem and progenitor cells in irradiated mouse bone marrow cells. δ-Tocotrienol also protected CD34+ cells from radiation-induced damage (172).
R-Spondin1
Human R-spondin1 (Rspo1), a 29 kDa, 263 amino acid protein acts as a mitogenic factor for ISCs and it was hypothesized that its systemic administration would amplify intestinal crypt cells, accelerate regeneration of irradiated intestine and ameliorate radiation-induced GI syndrome. Mice receiving recombinant adenovirus expressing human R-spondin1 (a potent Wnt signal enhancer and one of the four analogs of R-spondin) before potentially lethal TBI or local abdominal irradiation had higher survival than the control group (176). Rspo1 promoted radioprotection against radiation-induced GI syndrome and improved survival of mice. The mechanism was likely related to induction of the Wnt/β-catenin pathway and promotion of ISC regeneration. Rspo1 has a protective effect only on normal intestinal tissue but not in tumors and thereby may increase the therapeutic ratio of chemoradiation therapy in patients undergoing abdominal irradiation for GI malignancies.
Transforming Growth Factor-β3
Radiation-induced pulmonary fibrosis is a frequently occurring complication from radiotherapy of thoracic tumors. The transforming growth factor-β superfamily plays a key regulatory role in pulmonary fibrosis. A single thoracic irradiation of 20 Gy was applied in mice to establish the model of radiation-induced pulmonary fibrosis and the mice were treated by intraperitoneal injections of recombinant transforming growth factor-β3 weekly after irradiation (177). Transforming growth factor-β3 decelerated the progress of radiation-induced pulmonary fibrosis and hindered the recruitment of fibrocytes to lung. In addition, Th1 response was suppressed as shown by diminished interferon-γ in transforming growth factor-β3 after irradiation, and enhancement of Th2 response was marked by increased interleukin in transforming growth factor-β3. These data suggest that TGF-β3 might be involved in the regulatory mechanism for attenuation of radiation-induced pulmonary fibrosis.
Injury-Mitigating, Therapeutic Cell Transplants: Cellular Therapy
Mesenchymal Stem Cells
There has been an explosion of interest in adult stem/progenitor cells that have the potential to repair tissues to treat individuals for a broad range of clinical indications (178). These cells attracted attention because of their stem-cell-like properties, but the cells frequently repair injured tissues without much evidence of either engraftment or differentiation. These cells have been shown to secrete a large numbers of cytokines and chemokines (179, 180). The pattern of secreted cytokines changes after the cell engraftment into new microenvironments suggesting that these cells could enhance repair by stimulating the regeneration of damaged cells. These cells also suppressed the mixed-lymphocyte reaction in culture indicating tissue repair by suppressing immune reaction.
Mesenchymal stem cells have been reported to repair various tissues damaged by radiation exposure when injected intravenously (180). As stated above, the stemness of these cells was probably not relevant to their efficacy in such indications, and it may even be a drawback when possible complications associated with the use of such cells are considered (181, 182). In such cases, cells with low antigenicity and with minimal differentiation potential but with adequate secretion of key modulators of inflammation and immunity such as prostaglandin E2, tumor necrosis factor-stimulated gene 6, and stanniocalcin-1 may profile more optimal candidates. Furthermore, intravenous administration of mesenchymal stem cells (MSCs) genetically modified with extracellular superoxide dismutase improved survival in irradiated mice (183).
Bone Marrow Stromal Cells
There is a report suggesting that mitigation of lethal intestinal injury can be achieved by intravenous transplantation of marrow-derived stromal cells (including mesenchymal, endothelial, and macrophage cell population) (184). Bone marrow-derived adherent stromal cell transplantation increased blood levels of intestinal growth factors (R-Spondin1, keratinocyte growth factor, platelet-derived growth factor, fibroblast growth factor-2, and anti-inflammatory cytokines) and induced regeneration of the irradiated host ISCs niche. These findings provided a platform to discover potential radiation mitigators and protectors for ARS and chemoradiation therapy of abdominal malignancies.
Myeloid Progenitor Cells
Cellerant Therapeutics (San Carlos, CA, USA) has developed culture conditions to produce large numbers of mouse myeloid progenitors from hematopoietic stem cells. Myeloid progenitor cells (MPCs) can improve survival against high levels of radiation. In collaboration with Cellerant Therapeutics, one of us (VKS) studied MPCs for use as a bridging therapy for radiation injuries (185). The aim of this study was to elucidate the potential of mouse myeloid progenitor cells (mMPC) to mitigate lethal doses of 60Co γ-radiation and X-rays in various strains of mice. Different cell-doses of pooled allogeneic mMPC generated ex vivo from AKR, C57Bl/6, and FVB mice were transfused iv into haplotype-mismatched recipient BALB/c or CD2F1 mice at various times after irradiation to assess their effect on a 30-day survival. Our results demonstrated that cryopreserved allogeneic mMPC significantly improve survival in both strains of mice irradiated with lethal doses of 60Co γ-radiation (CD2F1, 9.2 Gy) and X-ray exposures (BALB/c, 9 Gy) that are known to cause ARS in hematopoietic tissues (185). The survival benefit was mMPC-dose-dependent and significant even when mMPC administration was delayed up to 7 days post-irradiation. It was further shown that mMPC administration mitigates death from ARS at radiation doses up to 15 Gy (60Co γ-radiation, CD2F1 mice), which are radiation exposure levels that cause mice to succumb to multi-organ failure, and determined that the DRF of 5 million mMPC administered 24 h post-irradiation of CD2F1 mice is 1.73. Even at high doses of up to 14 Gy cobalt-60 gamma-radiation, mMPC administration could be delayed up to 5 days in CD2F1 mice and still provide significant benefit to a 30-day survival. Additional study is needed to monitor mMPC transplanted mice for long term to investigate graft vs. host disease, and to evaluate the histopathology of various organs of transplanted mice. To study the GI tract structural integrity in mice receiving higher doses of radiation exposure causing GI injury and mMPC treatment, intestinal tissues were harvested at different times after irradiation and analyzed for architecture, surviving crypts, villus height, and number. The effect of infused mMPC on bacterial translocation from gut to heart, spleen, and liver in irradiated mice was studied by bacterial tissue cultures and estimated endotoxin levels in serum samples. It was observed that the infusion of mMPC significantly improved survival of mice receiving high doses of radiation, decreased bacterial infection, and lowered endotoxin levels in serum. The histopathology of jejunum from irradiated and mMPC-transfused mice revealed improved gut structural integrity compared to untreated controls. In brief, the results of this study further support our contention that the transfusion of mMPC acts as a bridging therapy, not only for the hematopoietic system, but also for GI system recovery following acute, potentially lethal radiation injury by improving intestinal structural integrity and inhibiting bacterial translocation in the GI tract of lethally irradiated mice.
Mobilized Blood Hematopoietic Stem Cells and Early Progenitors: Tocopherol Succinate-Mobilized Progenitor Cells
It was hypothesized that tocopherol succinate (TS) would stimulate a G-CSF-induced mobilization of bone marrow progenitor cells into the peripheral circulation. This hypothesis was confirmed clearly using several different approaches (186). First, a direct fluorescence flow cytometric approach was used to identify and phenotype the putative, mobilized hematopoietic stem cells in question (186). Second, we evaluated and compared the efficacy of whole blood infusions obtained from TS-treated mice vs. G-CSF-treated mice for survival protection against hematopoietic ARS when transfused into matched groups of acutely irradiated recipient mice. Survival was significantly higher in the group receiving transfused blood from TS-treated animals (187). Further, our results demonstrated that infusions of HSC-enriched, peripheral blood mononuclear cells (PBMC) from TS-injected mice greatly improved survival of lethally irradiated mice (187). Once transfused, these TS-mobilized progenitors acted as a bridging therapy for acutely irradiated, morbidly injured mice and the fostering of time-critical recovery process(es) that principally involve damaged cell-replacement and tissue renewal that aid-and-abet overall restoration of vital organ-system function(s).
Recent studies have yielded a remarkable finding; namely, infusion of whole blood or PBMC from TS- and AMD3100-injected mice significantly improved survival of mice receiving still higher, GI-syndrome-eliciting radiation doses. Histopathology and immunostaining of jejunum from these irradiated and TS- and AMD3100-mobilized PBMC-transfused mice revealed significant protection of GI tissue from radiation injury (188). We also observed that the infusion of PBMC from TS- and AMD3100-injected mice significantly inhibited apoptosis, increased cell proliferation in the analyzed tissues of recipient mice, and inhibited bacterial translocation to various organs compared to mice receiving cells from vehicle-mobilized cells (189). Most recently, we have observed that TS-mobilized progenitors mitigate radiation combined injury (radiation and wound) (190). In aggregate, these rodent-based studies strongly suggest that TS has the capacity to mobilize progenitors from marrow into the blood. This subset of unique, tissue-reparative progenitors not only is therapeutic for a critically injured/failing lymphohematopoietic system but also for the GI system and perhaps other vital organ systems as well. Together these characteristics make TS-mobilized progenitors a suitable candidate as a bridging therapy for acute radiation victims that can be administered in the field with minimal infrastructure requirements.
Conclusion and Perspectives
There are several promising radiation countermeasures under development such as CBLB613 (191), CBLB612 (192), IL-12 (193), epidermal growth factor (194), fibroblast growth factor-2 (195), fibroblast growth factor-peptide (196), insulin-like growth factor-1 (197), tempol (198), TS (13), TPO (thrombopoietin) receptor agonist (ALXN4100TPO) (199), 5-Androstenediol (5-AED)/Neumune® (200), AEOL-10150 (201), cytokines, and growth factors (3, 166, 202), etc. Since it was not possible to discuss all agents under development in this review, we selected some of those agents, which are at advanced stages of the development or are otherwise representative of three general categories of agents: small-molecules, proteins, and cellular therapy. The characteristics of the agents described herein are summarized in Table 1.
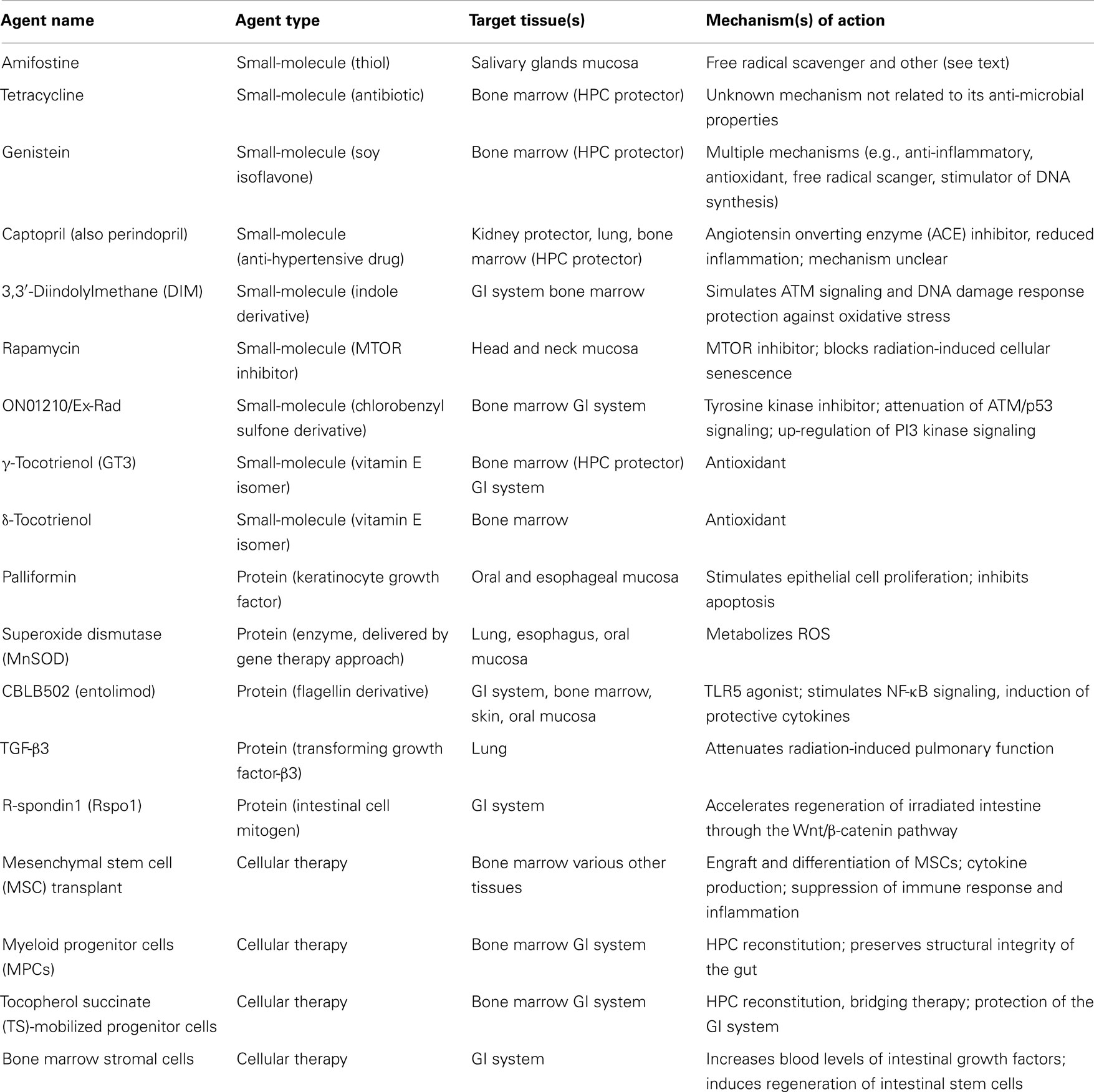
Table 1. Summary of radioprotector/mitigator agents and their characteristics.
We have reviewed some of the principles of radiation protection and mitigation and discussed some of the agents under development. The agents described represent various molecule types, including gene therapy, small-molecule drugs, and drug-like compounds (e.g., captopril, Ex-RAD), phytochemicals (plant-derived agents) (e.g., DIM, genistein), vitamins (e.g., vitamin E derivatives: gamma and delta tocotrienol), protein (e.g., truncated flagellin, CBLB502), and cell-based agents. The wide variety of agents that can function as protectors or mitigators is consistent with the complexity of the responses of different cell types and tissues to radiation. Examples of agents that protect normal tissues but not tumors have been provided. Some such agents may exhibit antitumor activity, particularly at higher concentrations, for example DIM and genistein, which are proposed cancer prevention agents. Many questions remain, such as why some compounds are strong protectants but weak mitigators (e.g., vitamin E derivatives) and why protectants often selectively target normal tissues and not tumors.
Compounds being considered as radioprotectors and/or mitigators are typically tested in rodents using a 30-day survival as the major end-point and in non-human primates (monkeys) using a 60-day survival end-point. These time intervals were chosen to reflect the ability of the compound to protect against or mitigate ARS following whole body exposure to nuclear radiation (e.g., 60Co or 137Cs). Later effects of whole body, near whole body, or partial body exposures, including survivors of ARS are a relatively understudied area in the field. These may be important because there are other radiosensitive tissues than bone marrow and intestine, including skin, esophagus, lung, and kidney. The performance of radioprotectors/mitigators in the setting of exposure to particulate radiation – for example – neutrons, protons, and heavier ions – is another unexplored area, as most studies utilize gamma-radiation or X-rays. It is also of interest whether a potential radioprotector/mitigator can block radiation-induced mutagenesis and, thus, carcinogenesis, since certain medical procedures (e.g., computerized axial tomography) are associated with exposure to low doses of radiation. The use of combinations of protective agents has not been extensively tested. Thus, combinations of agents with differing mechanisms of action and/or different toxicities may be superior to single agents in the same manner as combination cancer chemotherapy is often superior to individual agents.
Relative to radiation mitigators, it is an open question as to how long after exposure to radiation the mitigator should still work. Obviously, an agent that works within the first 24 h or longer would be more valuable than an agent that only works within a few hours after exposure, since it may take time to deliver the compound to the site of a nuclear disaster. This would not be a consideration for usage of a protector/mitigator in the radiotherapy clinic, since here the agents can conveniently be given within a few hours of each radiation treatment. An additional consideration applicable to use of a mitigator in civilian or military populations exposed to radiation is that the agent in question should have a convenient mode of administration, e.g., by oral route or by intramuscular or subcutaneous injection. Ideally, the compound should be easily self-administered since access to medical care may be delayed or limited.
Finally, it would be interesting to know if there are other FDA-approved drugs (see section on captopril) or food additives (see sections on DIM and genistein) that exert radioprotective or mitigative activity and could be “repurposed” for these indications.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The opinions or assertions contained herein are the private views of the authors and are not necessarily those of the Armed Forces Radiobiology Research Institute, the Uniformed Services University of the Health Sciences, or the Department of Defense. Dr. Rosen has been supported in part by the United States Public Health Service (1R01CA184168-01) and by a Dean’s Pilot Research Award at Georgetown University.
References
1. Anno GH, Baum SJ, Withers HR, Young RW. Symptomatology of acute radiation effects in humans after exposure to doses of 0.5-30 Gy. Health Phys (1989) 56(6):821–38. doi:10.1097/00004032-198906000-00001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Dainiak N. Hematologic consequences of exposure to ionizing radiation. Exp Hematol (2002) 30(6):513–28. doi:10.1016/S0301-472X(02)00802-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Waselenko JK, MacVittie TJ, Blakely WF, Pesik N, Wiley AL, Dickerson WE, et al. Medical management of the acute radiation syndrome: recommendations of the Strategic National Stockpile Radiation Working Group. Ann Intern Med (2004) 140(12):1037–51. doi:10.7326/0003-4819-140-12-200406150-00015
4. Williams JP, Brown SL, Georges GE, Hauer-Jensen M, Hill RP, Huser AK, et al. Animal models for medical countermeasures to radiation exposure. Radiat Res (2010) 173(4):557–78. doi:10.1667/RR1880.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Drouet M, Herodin F. Radiation victim management and the haematologist in the future: time to revisit therapeutic guidelines? Int J Radiat Biol (2010) 86(8):636–48. doi:10.3109/09553001003789604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Booth C, Tudor G, Tudor J, Katz BP, MacVittie TJ. Acute gastrointestinal syndrome in high-dose irradiated mice. Health Phys (2012) 103(4):383–99. doi:10.1097/HP.0b013e318266ee13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Hosseinimehr SJ. Trends in the development of radioprotective agents. Drug Discov Today (2007) 12(19–20):794–805. doi:10.1016/j.drudis.2007.07.017
8. Na Nakorn T, Traver D, Weissman IL, Akashi K. Myeloerythroid-restricted progenitors are sufficient to confer radioprotection and provide the majority of day 8 CFU-S. J Clin Invest (2002) 109(12):1579–85. doi:10.1172/JCI200215272
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Meijne EI, van der Winden-van Groenewegen RJ, Ploemacher RE, Vos O, David JA, Huiskamp R. The effects of x-irradiation on hematopoietic stem cell compartments in the mouse. Exp Hematol (1991) 19(7):617–23.
10. Takahashi K, Monzen S, Hayashi N, Kashiwakura I. Correlations of cell surface antigens with individual differences in radiosensitivity in human hematopoietic stem/progenitor cells. Radiat Res (2010) 173(2):184–90. doi:10.1667/RR1839.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Qiu W, Carson-Walter EB, Liu H, Epperly M, Greenberger JS, Zambetti GP, et al. PUMA regulates intestinal progenitor cell radiosensitivity and gastrointestinal syndrome. Cell Stem Cell (2008) 2(6):576–83. doi:10.1016/j.stem.2008.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Qiu W, Leibowitz B, Zhang L, Yu J. Growth factors protect intestinal stem cells from radiation-induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene (2010) 29(11):1622–32. doi:10.1038/onc.2009.451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Singh PK, Wise SY, Ducey EJ, Fatanmi OO, Elliott TB, Singh VK. Alpha-tocopherol succinate protects mice against radiation-induced gastrointestinal injury. Radiat Res (2012) 177(2):133–45. doi:10.1667/RR2627.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Calveley VL, Khan MA, Yeung IW, Vandyk J, Hill RP. Partial volume rat lung irradiation: temporal fluctuations of in-field and out-of-field DNA damage and inflammatory cytokines following irradiation. Int J Radiat Biol (2005) 81(12):887–99. doi:10.1080/09553000600568002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Augustine AD, Gondre-Lewis T, McBride W, Miller L, Pellmar TC, Rockwell S. Animal models for radiation injury, protection and therapy. Radiat Res (2005) 164(1):100–9. doi:10.1667/RR3388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Down JD. The nature and relevance of late lung pathology following localised irradiation of the thorax in mice and rats. Br J Cancer Suppl (1986) 7:330–2.
17. Day RM, Barshishat-Kupper M, Mog SR, McCart EA, Prasanna PG, Davis TA, et al. Genistein protects against biomarkers of delayed lung sequelae in mice surviving high-dose total body irradiation. J Radiat Res (2008) 49(4):361–72. doi:10.1269/jrr.07121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Bloomer WD, Hellman S. Normal tissue responses to radiation therapy. N Engl J Med (1975) 293(2):80–3. doi:10.1056/NEJM197507102930206
19. Hellman S. Cell kinetics, models, and cancer treatment – some principles for the radiation oncologist. Radiology (1975) 114(1):219–23. doi:10.1148/114.1.219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Monson JM, Stark P, Reilly JJ, Sugarbaker DJ, Strauss GM, Swanson SJ, et al. Clinical radiation pneumonitis and radiographic changes after thoracic radiation therapy for lung carcinoma. Cancer (1998) 82(5):842–50. doi:10.1002/(SICI)1097-0142(19980301)82:5<842::AID-CNCR7>3.3.CO;2-O
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Johansson S, Bjermer L, Franzen L, Henriksson R. Effects of ongoing smoking on the development of radiation-induced pneumonitis in breast cancer and oesophagus cancer patients. Radiother Oncol (1998) 49(1):41–7. doi:10.1016/S0167-8140(98)00064-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Bradley J, Movsas B. Radiation pneumonitis and oesophagitis in thoracic irradiation. In: Small W, Woloschak GE, editors. Radiation Toxicity: A Practical Guide. New York, NY: Springer Science and Media Business, Inc. (2006). p. 43–52.
23. Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer (2006) 6(9):702–13. doi:10.1038/nrc1950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Hopewell JW, Millar WT, Ang KK. Toward improving the therapeutic ratio in stereotactic radiosurgery: selective modulation of the radiation responses of both normal tissues and tumor. J Neurosurg (2007) 107(1):84–93. doi:10.3171/JNS-07/07/0084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Berkey FJ. Managing the adverse effects of radiation therapy. Am Fam Physician (2010) 82(4):381–8, 394.
26. Petersen C, Wurschmidt F. Late toxicity of radiotherapy: a problem or a challenge for the radiation oncologist? Breast Care (Basel) (2011) 6(5):369–74. doi:10.1159/000334220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Werner-Wasik M, Pequignot E, Leeper D, Hauck W, Curran W. Predictors of severe esophagitis include use of concurrent chemotherapy, but not the length of irradiated esophagus: a multivariate analysis of patients with lung cancer treated with nonoperative therapy. Int J Radiat Oncol Biol Phys (2000) 48(3):689–96. doi:10.1016/S0360-3016(00)00699-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Nguyen NP, Sallah S, Karlsson U, Antoine JE. Combined chemotherapy and radiation therapy for head and neck malignancies: quality of life issues. Cancer (2002) 94(4):1131–41. doi:10.1002/cncr.10257
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene (2007) 26(56):7741–8. doi:10.1038/sj.onc.1210872
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Guida C, Maione P, Rossi A, Bareschino M, Schettino C, Barzaghi D, et al. Combined chemo-radiotherapy for locally advanced non-small cell lung cancer: current status and future development. Crit Rev Oncol Hematol (2008) 68(3):222–32. doi:10.1016/j.critrevonc.2008.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Marur S, Forastiere AA. Challenges of integrating chemotherapy and targeted therapy with radiation in locally advanced head and neck squamous cell cancer. Curr Opin Oncol (2010) 22(3):206–11. doi:10.1097/CCO.0b013e328338475c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Ring KL, Young JL, Dunlap NE, Andersen WA, Schneider BF. Extended-field radiation therapy with whole pelvis radiotherapy and cisplatin chemosensitization in the treatment of IB2-IIIB cervical carcinoma: a retrospective review. Am J Obstet Gynecol (2009) 201(1):109.e1–6. doi:10.1016/j.ajog.2009.03.022
33. Merchant TE, Pollack IF, Loeffler JS. Brain tumors across the age spectrum: biology, therapy, and late effects. Semin Radiat Oncol (2010) 20(1):58–66. doi:10.1016/j.semradonc.2009.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Duffner PK. Risk factors for cognitive decline in children treated for brain tumors. Eur J Paediatr Neurol (2010) 14:106–15. doi:10.1016/j.ejpn.2009.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Boerma M, Hauer-Jensen M. Potential targets for intervention in radiation-induced heart disease. Curr Drug Targets (2010) 11(11):1405–12. doi:10.2174/1389450111009011405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Andratschke N, Maurer J, Molls M, Trott KR. Late radiation-induced heart disease after radiotherapy. Clinical importance, radiobiological mechanisms and strategies of prevention. Radiother Oncol (2011) 100(2):160–6. doi:10.1016/j.radonc.2010.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Campen CJ, Kranick SM, Kasner SE, Kessler SK, Zimmerman RA, Lustig R, et al. Cranial irradiation increases risk of stroke in pediatric brain tumor survivors. Stroke (2012) 43(11):3035–40. doi:10.1161/STROKEAHA.112.661561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Adams GE, Jameson DG. Time effects in molecular radiation biology. Radiat Environ Biophys (1980) 17(2):95–113. doi:10.1007/BF02027847
39. Tallentire A. Oxygen effect, hydrogen peroxide yields, and time scale of interaction of potentially damaging species in electron pulse irradiated bacterial spores. Adv Space Res (1983) 3(8):105–12. doi:10.1016/0273-1177(83)90180-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Wardman P, Ross AB. “Radiation chemistry” literature compilations: their wider value in free radical research. Free Radic Biol Med (1991) 10(3–4):243–7. doi:10.1016/0891-5849(91)90081-D
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann N Y Acad Sci (2000) 899:136–47. doi:10.1111/j.1749-6632.2000.tb06182.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Revesz L, Palcic B. Radiation dose dependence of the sensitization by oxygen and oxygen mimic sensitizers. Acta Radiol Oncol (1985) 24(3):209–17. doi:10.3109/02841868509134389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Guichard M. Hypoxia as a determinant factor in radiotherapy? Where are we? BJR Suppl (1992) 24:142–5.
44. Yoshimura M, Itasaka S, Harada H, Hiraoka M. Microenvironment and radiation therapy. Biomed Res Int (2013) 2013:685308. doi:10.1155/2013/685308
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Skov KA. The contribution of hydroxyl radical to radiosensitization: a study of DNA damage. Radiat Res (1984) 99(3):502–10. doi:10.2307/3576326
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Roots R, Chatterjee A, Chang P, Lommel L, Blakely EA. Characterization of hydroxyl radical-induced damage after sparsely and densely ionizing irradiation. Int J Radiat Biol Relat Stud Phys Chem Med (1985) 47(2):157–66. doi:10.1080/09553008514550231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Corre I, Niaudet C, Paris F. Plasma membrane signaling induced by ionizing radiation. Mutat Res (2010) 704(1–3):61–7. doi:10.1016/j.mrrev.2010.01.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Chatgilialoglu C, Ferreri C, Torreggiani A, Salzano AM, Renzone G, Scaloni A. Radiation-induced reductive modifications of sulfur-containing amino acids within peptides and proteins. J Proteomics (2011) 74(11):2264–73. doi:10.1016/j.jprot.2011.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Breen AP, Murphy JA. Reactions of oxyl radicals with DNA. Free Radic Biol Med (1995) 18(6):1033–77. doi:10.1016/0891-5849(94)00209-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Epe B. DNA damage profiles induced by oxidizing agents. Rev Physiol Biochem Pharmacol (1996) 127:223–49. doi:10.1007/BFb0048268
51. Wallace SS. Enzymatic processing of radiation-induced free radical damage in DNA. Radiat Res (1998) 150(5 Suppl):S60–79. doi:10.2307/3579809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res (2003) 531(1–2):231–51. doi:10.1016/j.mrfmmm.2003.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science (2005) 308(5721):551–4. doi:10.1126/science.1108297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle (2007) 6(8):931–42. doi:10.4161/cc.6.8.4180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol (2010) 11(3):196–207. doi:10.1038/nrm2851
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:181–211. doi:10.1146/annurev.biochem.052308.093131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Stracker TH, Roig I, Knobel PA, Marjanovic M. The ATM signaling network in development and disease. Front Genet (2013) 4:37. doi:10.3389/fgene.2013.00037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKgamma by SUMO-1 and ubiquitin mediates NF-kappaB activation by genotoxic stress. Cell (2003) 115(5):565–76. doi:10.1016/S0092-8674(03)00895-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Sakamoto K, Hikiba Y, Nakagawa H, Hirata Y, Hayakawa Y, Kinoshita H, et al. Promotion of DNA repair by nuclear IKKbeta phosphorylation of ATM in response to genotoxic stimuli. Oncogene (2013) 32(14):1854–62. doi:10.1038/onc.2012.192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Little MP, Heidenreich WF, Moolgavkar SH, Schollnberger H, Thomas DC. Systems biological and mechanistic modelling of radiation-induced cancer. Radiat Environ Biophys (2008) 47(1):39–47. doi:10.1007/s00411-007-0150-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Newhauser WD, Durante M. Assessing the risk of second malignancies after modern radiotherapy. Nat Rev Cancer (2011) 11(6):438–48. doi:10.1038/nrc3069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Williams JP, McBride WH. After the bomb drops: a new look at radiation-induced multiple organ dysfunction syndrome (MODS). Int J Radiat Biol (2011) 87(8):851–68. doi:10.3109/09553002.2011.560996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Hong JH, Chiang CS, Tsao CY, Lin PY, Wu CJ, McBride WH. Can short-term administration of dexamethasone abrogate radiation-induced acute cytokine gene response in lung and modify subsequent molecular responses? Int J Radiat Oncol Biol Phys (2001) 51(2):296–303. doi:10.1016/S0360-3016(01)01702-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Hong JH, Jung SM, Tsao TC, Wu CJ, Lee CY, Chen FH, et al. Bronchoalveolar lavage and interstitial cells have different roles in radiation-induced lung injury. Int J Radiat Biol (2003) 79(3):159–67. doi:10.1080/0955300031000076894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol (2010) 97(1):149–61. doi:10.1016/j.radonc.2010.09.002
66. Kouvaris JR, Kouloulias VE, Vlahos LJ. Amifostine: the first selective-target and broad-spectrum radioprotector. Oncologist (2007) 12(6):738–47. doi:10.1634/theoncologist.12-6-738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Schuchter LM. Exploration of platinum-based dose-intensive chemotherapy strategies with amifostine (Ethyol). Eur J Cancer (1996) 32A(Suppl 4):S40–2. doi:10.1016/S0959-8049(96)00316-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Ali BH, Al Moundhri MS. Agents ameliorating or augmenting the nephrotoxicity of cisplatin and other platinum compounds: a review of some recent research. Food Chem Toxicol (2006) 44(8):1173–83. doi:10.1016/j.fct.2006.01.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Chirino YI, Pedraza-Chaverri J. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp Toxicol Pathol (2009) 61(3):223–42. doi:10.1016/j.etp.2008.09.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. van der Vijgh WJ, Peters GJ. Protection of normal tissues from the cytotoxic effects of chemotherapy and radiation by amifostine (Ethyol): preclinical aspects. Semin Oncol (1994) 21(5 Suppl 11):2–7.
71. Bourhis J, Rosine D. Radioprotective effect of amifostine in patients with head and neck squamous cell carcinoma. Semin Oncol (2002) 29(6 Suppl 19):61–2. doi:10.1053/sonc.2002.37349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Karacetin D, Yucel B, Leblebicioglu B, Aksakal O, Maral O, Incekara O. A randomized trial of amifostine as radioprotector in the radiotherapy of head and neck cancer. J BUON (2004) 9(1):23–6.
73. Bourhis J, Blanchard P, Maillard E, Brizel DM, Movsas B, Buentzel J, et al. Effect of amifostine on survival among patients treated with radiotherapy: a meta-analysis of individual patient data. J Clin Oncol (2011) 29(18):2590–7. doi:10.1200/JCO.2010.33.1454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Nicolatou-Galitis O, Sarri T, Bowen J, Di Palma M, Kouloulias VE, Niscola P, et al. Systematic review of amifostine for the management of oral mucositis in cancer patients. Support Care Cancer (2013) 21(1):357–64. doi:10.1007/s00520-012-1613-6
75. Duval M, Daniel SJ. Meta-analysis of the efficacy of amifostine in the prevention of cisplatin ototoxicity. J Otolaryngol Head Neck Surg (2012) 41(5):309–15.
76. Koukourakis MI, Panteliadou M, Abatzoglou IM, Sismanidou K, Sivridis E, Giatromanolaki A. Postmastectomy hypofractionated and accelerated radiation therapy with (and without) subcutaneous amifostine cytoprotection. Int J Radiat Oncol Biol Phys (2013) 85(1):e7–13. doi:10.1016/j.ijrobp.2012.08.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Praetorius NP, Mandal TK. Alternate delivery route for amifostine as a radio-/chemo-protecting agent. J Pharm Pharmacol (2008) 60(7):809–15. doi:10.1211/jpp.60.7.0001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Seed TM, Inal CE, Singh VK. Radioprotection of hematopoietic progenitors by low dose amifostine prophylaxis. Int J Radiat Biol (2014) 90(7):594–604. doi:10.3109/09553002.2014.899450
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Finch PW, Mark Cross LJ, McAuley DF, Farrell CL. Palifermin for the protection and regeneration of epithelial tissues following injury: new findings in basic research and pre-clinical models. J Cell Mol Med (2013) 17(9):1065–87. doi:10.1111/jcmm.12091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Vadhan-Raj S, Goldberg JD, Perales MA, Berger DP, van den Brink MR. Clinical applications of palifermin: amelioration of oral mucositis and other potential indications. J Cell Mol Med (2013) 17(11):1371–84. doi:10.1111/jcmm.12169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Lauritano D, Petruzzi M, Di Stasio D, Lucchese A. Clinical effectiveness of palifermin in prevention and treatment of oral mucositis in children with acute lymphoblastic leukaemia: a case-control study. Int J Oral Sci (2014) 6(1):27–30. doi:10.1038/ijos.2013.93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Epperly MW, Bray JA, Krager S, Berry LM, Gooding W, Engelhardt JF, et al. Intratracheal injection of adenovirus containing the human MnSOD transgene protects athymic nude mice from irradiation-induced organizing alveolitis. Int J Radiat Oncol Biol Phys (1999) 43(1):169–81. doi:10.1016/S0360-3016(98)00355-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Epperly MW, Defilippi S, Sikora C, Gretton J, Kalend A, Greenberger JS. Intratracheal injection of manganese superoxide dismutase (MnSOD) plasmid/liposomes protects normal lung but not orthotopic tumors from irradiation. Gene Ther (2000) 7(12):1011–8. doi:10.1038/sj.gt.3301207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Epperly MW, Gretton JA, DeFilippi SJ, Greenberger JS, Sikora CA, Liggitt D, et al. Modulation of radiation-induced cytokine elevation associated with esophagitis and esophageal stricture by manganese superoxide dismutase-plasmid/liposome (SOD2-PL) gene therapy. Radiat Res (2001) 155(1 Pt 1):2–14. doi:10.1667/0033-7587(2001)155[0002:MORICE]2.0.CO;2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Epperly MW, Kagan VE, Sikora CA, Gretton JE, Defilippi SJ, Bar-Sagi D, et al. Manganese superoxide dismutase-plasmid/liposome (MnSOD-PL) administration protects mice from esophagitis associated with fractionated radiation. Int J Cancer (2001) 96(4):221–31. doi:10.1002/ijc.1023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Epperly MW, Sikora CA, DeFilippi SJ, Gretton JA, Zhan Q, Kufe DW, et al. Manganese superoxide dismutase (SOD2) inhibits radiation-induced apoptosis by stabilization of the mitochondrial membrane. Radiat Res (2002) 157(5):568–77. doi:10.1667/0033-7587(2002)157[0568:MSDSIR]2.0.CO;2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Guo H, Seixas-Silva JA Jr, Epperly MW, Gretton JE, Shin DM, Bar-Sagi D, et al. Prevention of radiation-induced oral cavity mucositis by plasmid/liposome delivery of the human manganese superoxide dismutase (SOD2) transgene. Radiat Res (2003) 159(3):361–70. doi:10.1667/0033-7587(2003)159[0361:PORIOC]2.0.CO;2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Epperly MW, Gretton JE, Sikora CA, Jefferson M, Bernarding M, Nie S, et al. Mitochondrial localization of superoxide dismutase is required for decreasing radiation-induced cellular damage. Radiat Res (2003) 160(5):568–78. doi:10.1667/RR3081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Greenberger JS. Gene therapy approaches for stem cell protection. Gene Ther (2008) 15(2):100–8. doi:10.1038/sj.gt.3303004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Epperly MW, Chaillet JR, Kalash R, Shaffer B, Goff J, Franicola D, et al. Conditional radioresistance of Tet-inducible manganese superoxide dismutase bone marrow stromal cell lines. Radiat Res (2013) 180(2):189–204. doi:10.1667/RR3177.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Epperly MW, Wang H, Jones JA, Dixon T, Montesinos CA, Greenberger JS. Antioxidant-chemoprevention diet ameliorates late effects of total-body irradiation and supplements radioprotection by MnSOD-plasmid liposome administration. Radiat Res (2011) 175(6):759–65. doi:10.1667/RR2398.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Epperly MW, Smith T, Zhang X, Goff JP, Franicola D, Greenberger B, et al. Modulation of in utero total body irradiation induced newborn mouse growth retardation by maternal manganese superoxide dismutase-plasmid liposome (MnSOD-PL) gene therapy. Gene Ther (2011) 18(6):579–83. doi:10.1038/gt.2010.178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Epperly MW, Franicola D, Shields D, Rwigema JC, Stone B, Zhang X, et al. Screening of antimicrobial agents for in vitro radiation protection and mitigation capacity, including those used in supportive care regimens for bone marrow transplant recipients. In vivo (2010) 24(1):9–19.
94. Mustata G, Li M, Zevola N, Bakan A, Zhang L, Epperly M, et al. Development of small-molecule PUMA inhibitors for mitigating radiation-induced cell death. Curr Top Med Chem (2011) 11(3):281–90. doi:10.2174/156802611794072641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Greenberger JS, Clump D, Kagan V, Bayir H, Lazo JS, Wipf P, et al. Strategies for discovery of small molecule radiation protectors and radiation mitigators. Front Oncol (2011) 1:59. doi:10.3389/fonc.2011.00059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Kim H, Bernard ME, Epperly MW, Shen H, Amoscato A, Dixon TM, et al. Amelioration of radiation esophagitis by orally administered p53/Mdm2/Mdm4 inhibitor (BEB55) or GS-nitroxide. In vivo (2011) 25(6):841–8.
97. Kruk I, Aboul-Enein HY, Michalska T, Lichszteld K, Kladna A. Scavenging of reactive oxygen species by the plant phenols genistein and oleuropein. Luminescence (2005) 20(2):81–9. doi:10.1002/bio.808
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, et al. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem (1987) 262(12):5592–5.
99. Valachovicova T, Slivova V, Sliva D. Cellular and physiological effects of soy flavonoids. Mini Rev Med Chem (2004) 4(8):881–7. doi:10.2174/1389557043403387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Wu HJ, Chan WH. Genistein protects methylglyoxal-induced oxidative DNA damage and cell injury in human mononuclear cells. Toxicol In vitro (2007) 21(3):335–42. doi:10.1016/j.tiv.2006.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Hong H, Landauer MR, Foriska MA, Ledney GD. Antibacterial activity of the soy isoflavone genistein. J Basic Microbiol (2006) 46(4):329–35. doi:10.1002/jobm.200510073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Verdrengh M, Jonsson IM, Holmdahl R, Tarkowski A. Genistein as an anti-inflammatory agent. Inflamm Res (2003) 52(8):341–6. doi:10.1007/s00011-003-1182-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Kang JL, Lee HW, Lee HS, Pack IS, Chong Y, Castranova V, et al. Genistein prevents nuclear factor-kappa B activation and acute lung injury induced by lipopolysaccharide. Am J Respir Crit Care Med (2001) 164(12):2206–12. doi:10.1164/ajrccm.164.12.2104017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Lian F, Li Y, Bhuiyan M, Sarkar FH. p53-independent apoptosis induced by genistein in lung cancer cells. Nutr Cancer (1999) 33(2):125–31. doi:10.1207/S15327914NC330202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Comalada M, Ballester I, Bailon E, Sierra S, Xaus J, Galvez J, et al. Inhibition of pro-inflammatory markers in primary bone marrow-derived mouse macrophages by naturally occurring flavonoids: analysis of the structure-activity relationship. Biochem Pharmacol (2006) 72(8):1010–21. doi:10.1016/j.bcp.2006.07.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Tacyildiz N, Ozyoruk D, Yavuz G, Unal E, Dincaslan H, Dogu F, et al. Soy isoflavones ameliorate the adverse effects of chemotherapy in children. Nutr Cancer (2010) 62(7):1001–5. doi:10.1080/01635581.2010.509841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Ahmad IU, Forman JD, Sarkar FH, Hillman GG, Heath E, Vaishampayan U, et al. Soy isoflavones in conjunction with radiation therapy in patients with prostate cancer. Nutr Cancer (2010) 62(7):996–1000. doi:10.1080/01635581.2010.509839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Landauer MR, Srinivasan V, Seed TM. Genistein treatment protects mice from ionizing radiation injury. J Appl Toxicol (2003) 23(6):379–85. doi:10.1002/jat.904
109. Davis TA, Clarke TK, Mog SR, Landauer MR. Subcutaneous administration of genistein prior to lethal irradiation supports multilineage, hematopoietic progenitor cell recovery and survival. Int J Radiat Biol (2007) 83(3):141–51. doi:10.1080/09553000601132642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Davis TA, Mungunsukh O, Zins S, Day RM, Landauer MR. Genistein induces radioprotection by hematopoietic stem cell quiescence. Int J Radiat Biol (2008) 84(9):713–26. doi:10.1080/09553000802317778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Para AE, Bezjak A, Yeung IW, Van Dyk J, Hill RP. Effects of genistein following fractionated lung irradiation in mice. Radiother Oncol (2009) 92(3):500–10. doi:10.1016/j.radonc.2009.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Day RM, Davis TA, Barshishat-Kupper M, McCart EA, Tipton AJ, Landauer MR. Enhanced hematopoietic protection from radiation by the combination of genistein and captopril. Int Immunopharmacol (2013) 15(2):348–56. doi:10.1016/j.intimp.2012.12.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Mahmood J, Jelveh S, Calveley V, Zaidi A, Doctrow SR, Hill RP. Mitigation of radiation-induced lung injury by genistein and EUK-207. Int J Radiat Biol (2011) 87(8):889–901. doi:10.3109/09553002.2011.583315
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Grace MB, Blakely WF, Landauer MR. Genistein-induced alterations of radiation-responsive gene expression. Radiat Meas (2007) 42:1152–7. doi:10.1016/j.radmeas.2007.05.011
115. Oki T, Sowa Y, Hirose T, Takagaki N, Horinaka M, Nakanishi R, et al. Genistein induces Gadd45 gene and G2/M cell cycle arrest in the DU145 human prostate cancer cell line. FEBS Lett (2004) 577(1–2):55–9. doi:10.1016/j.febslet.2004.09.085
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Caetano B, Le Corre L, Chalabi N, Delort L, Bignon YJ, Bernard-Gallon DJ. Soya phytonutrients act on a panel of genes implicated with BRCA1 and BRCA2 oncosuppressors in human breast cell lines. Br J Nutr (2006) 95(2):406–13. doi:10.1079/BJN20051640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Tamulevicius P, Wang M, Iliakis G. Homology-directed repair is required for the development of radioresistance during S phase: interplay between double-strand break repair and checkpoint response. Radiat Res (2007) 167(1):1–11. doi:10.1667/RR0751.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Ha CT, Li XH, Fu D, Xiao M, Landauer MR. Genistein nanoparticles protect mouse hematopoietic system and prevent proinflammatory factors after gamma irradiation. Radiat Res (2013) 180(3):316–25. doi:10.1667/RR3326.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Kim H, Peterson TG, Barnes S. Mechanisms of action of the soy isoflavone genistein: emerging role for its effects via transforming growth factor β signaling pathways. Am J Clin Nutr (1998) 68S:1418S–25S.
120. Robbins ME, Diz DI. Pathogenic role of the renin-angiotensin system in modulating radiation-induced late effects. Int J Radiat Oncol Biol Phys (2006) 64(1):6–12. doi:10.1016/j.ijrobp.2005.08.033
121. Robbins ME, Hopewell JW. Physiological factors effecting renal radiation tolerance: a guide to the treatment of late effects. Br J Cancer Suppl (1986) 7:265–7.
122. Davis TA, Landauer MR, Mog SR, Barshishat-Kupper M, Zins SR, Amare MF, et al. Timing of captopril administration determines radiation protection or radiation sensitization in a murine model of total body irradiation. Exp Hematol (2010) 38(4):270–81. doi:10.1016/j.exphem.2010.01.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Moulder JE, Cohen EP, Fish BL. Captopril and losartan for mitigation of renal injury caused by single-dose total-body irradiation. Radiat Res (2011) 175(1):29–36. doi:10.1667/RR2400.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Kma L, Gao F, Fish BL, Moulder JE, Jacobs ER, Medhora M. Angiotensin converting enzyme inhibitors mitigate collagen synthesis induced by a single dose of radiation to the whole thorax. J Radiat Res (2012) 53(1):10–7. doi:10.1269/jrr.11035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Medhora M, Gao F, Jacobs ER, Moulder JE. Radiation damage to the lung: mitigation by angiotensin-converting enzyme (ACE) inhibitors. Respirology (2012) 17(1):66–71. doi:10.1111/j.1440-1843.2011.02092.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Ghosh SN, Zhang R, Fish BL, Semenenko VA, Li XA, Moulder JE, et al. Renin-angiotensin system suppression mitigates experimental radiation pneumonitis. Int J Radiat Oncol Biol Phys (2009) 75(5):1528–36. doi:10.1016/j.ijrobp.2009.07.1743
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Moulder JE, Cohen EP. Future strategies for mitigation and treatment of chronic radiation-induced normal tissue injury. Semin Radiat Oncol (2007) 17(2):141–8. doi:10.1016/j.semradonc.2006.11.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Ward WF, Kim YT, Molteni A, Solliday NH. Radiation-induced pulmonary endothelial dysfunction in rats: modification by an inhibitor of angiotensin converting enzyme. Int J Radiat Oncol Biol Phys (1988) 15(1):135–40. doi:10.1016/0360-3016(88)90357-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Ward WF, Molteni A, Ts’ao C, Hinz JM. The effect of captopril on benign and malignant reactions in irradiated rat skin. Br J Radiol (1990) 63(749):349–54. doi:10.1259/0007-1285-63-749-349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Cohen EP, Fish BL, Moulder JE. Treatment of radiation nephropathy with captopril. Radiat Res (1992) 132(3):346–50. doi:10.2307/3578243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Moulder JE, Cohen EP, Fish BL, Hill P. Prophylaxis of bone marrow transplant nephropathy with captopril, an inhibitor of angiotensin-converting enzyme. Radiat Res (1993) 136(3):404–7. doi:10.2307/3578554
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Cohen EP, Irving AA, Drobyski WR, Klein JP, Passweg J, Talano JA, et al. Captopril to mitigate chronic renal failure after hematopoietic stem cell transplantation: a randomized controlled trial. Int J Radiat Oncol Biol Phys (2008) 70(5):1546–51. doi:10.1016/j.ijrobp.2007.08.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Charrier S, Michaud A, Badaoui S, Giroux S, Ezan E, Sainteny F, et al. Inhibition of angiotensin I-converting enzyme induces radioprotection by preserving murine hematopoietic short-term reconstituting cells. Blood (2004) 104(4):978–85. doi:10.1182/blood-2003-11-3828
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Zakheim RM, Mattioli L, Molteni A, Mullis KB, Bartley J. Prevention of pulmonary vascular changes of chronic alveolar hypoxia by inhibition of angiotensin I-converting enzyme in the rat. Lab Invest (1975) 33(1):57–61.
135. Chisi JE, Briscoe CV, Ezan E, Genet R, Riches AC, Wdzieczak-Bakala J. Captopril inhibits in vitro and in vivo the proliferation of primitive haematopoietic cells induced into cell cycle by cytotoxic drug administration or irradiation but has no effect on myeloid leukaemia cell proliferation. Br J Haematol (2000) 109(3):563–70. doi:10.1046/j.1365-2141.2000.02073.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Aggarwal BB, Ichikawa H. Molecular targets and anticancer potential of indole-3-carbinol and its derivatives. Cell Cycle (2005) 4(9):1201–15. doi:10.4161/cc.4.9.1993
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Reed GA, Arneson DW, Putnam WC, Smith HJ, Gray JC, Sullivan DK, et al. Single-dose and multiple-dose administration of indole-3-carbinol to women: pharmacokinetics based on 3,3’-diindolylmethane. Cancer Epidemiol Biomarkers Prev (2006) 15(12):2477–81. doi:10.1158/1055-9965.EPI-06-0396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Reed GA, Sunega JM, Sullivan DK, Gray JC, Mayo MS, Crowell JA, et al. Single-dose pharmacokinetics and tolerability of absorption-enhanced 3,3’-diindolylmethane in healthy subjects. Cancer Epidemiol Biomarkers Prev (2008) 17(10):2619–24. doi:10.1158/1055-9965.EPI-08-0520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Heath EI, Heilbrun LK, Li J, Vaishampayan U, Harper F, Pemberton P, et al. A phase I dose-escalation study of oral BR-DIM (BioResponse 3,3’-diindolylmethane) in castrate-resistant, non-metastatic prostate cancer. Am J Transl Res (2010) 2(4):402–11.
140. Del Priore G, Gudipudi DK, Montemarano N, Restivo AM, Malanowska-Stega J, Arslan AA. Oral diindolylmethane (DIM): pilot evaluation of a nonsurgical treatment for cervical dysplasia. Gynecol Oncol (2010) 116(3):464–7. doi:10.1016/j.ygyno.2009.10.060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Fan S, Meng Q, Xu J, Jiao Y, Zhao L, Zhang X, et al. DIM (3,3’-diindolylmethane) confers protection against ionizing radiation by a unique mechanism. Proc Natl Acad Sci U S A (2013) 110(46):18650–5. doi:10.1073/pnas.1308206110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Yao Z, Hu W, Yin S, Huang Z, Zhu Q, Chen J, et al. 3,3’-Diindolymethane ameliorates adriamycin-induced cardiac fibrosis via activation of a BRCA1-dependent anti-oxidant pathway. Pharmacol Res (2013) 70(1):139–46. doi:10.1016/j.phrs.2013.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Zong J, Deng W, Zhou H, Bian ZY, Dai J, Yuan Y, et al. 3,3’-Diindolylmethane protects against cardiac hypertrophy via 5’-adenosine monophosphate-activated protein kinase-alpha2. PLoS One (2013) 8(1):e53427. doi:10.1371/journal.pone.0053427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Panganiban RA, Mungunsukh O, Day RM. X-irradiation induces ER stress, apoptosis, and senescence in pulmonary artery endothelial cells. Int J Radiat Biol (2012) 89(8):656–67. doi:10.3109/09553002.2012.711502
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, et al. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell (2012) 11(3):401–14. doi:10.1016/j.stem.2012.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Igarashi K, Sakimoto I, Kataoka K, Ohta K, Miura M. Radiation-induced senescence-like phenotype in proliferating and plateau-phase vascular endothelial cells. Exp Cell Res (2007) 313(15):3326–36. doi:10.1016/j.yexcr.2007.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Oh CW, Bump EA, Kim JS, Janigro D, Mayberg MR. Induction of a senescence-like phenotype in bovine aortic endothelial cells by ionizing radiation. Radiat Res (2001) 156(3):232–40. doi:10.1667/0033-7587(2001)156[0232:IOASLP]2.0.CO;2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal (2009) 11(1):59–98. doi:10.1089/ars.2008.2104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Chen J, Goligorsky MS. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am J Physiol Heart Circ Physiol (2006) 290(5):H1729–39. doi:10.1152/ajpheart.01103.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Cmielova J, Havelek R, Soukup T, Jiroutova A, Visek B, Suchanek J, et al. Gamma radiation induces senescence in human adult mesenchymal stem cells from bone marrow and periodontal ligaments. Int J Radiat Biol (2012) 88(5):393–404. doi:10.3109/09553002.2012.666001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Lewis DA, Yi Q, Travers JB, Spandau DF. UVB-induced senescence in human keratinocytes requires a functional insulin-like growth factor-1 receptor and p53. Mol Biol Cell (2008) 19(4):1346–53. doi:10.1091/mbc.E07-10-1041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature (2003) 421(6919):182–7. doi:10.1038/nature01298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Handayaningsih AE, Takahashi M, Fukuoka H, Iguchi G, Nishizawa H, Yamamoto M, et al. IGF-I enhances cellular senescence via the reactive oxygen species-p53 pathway. Biochem Biophys Res Commun (2012) 425(2):478–84. doi:10.1016/j.bbrc.2012.07.140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Tran D. IGF-1 Induces Premature Cellular Senescence Through Inhibition of Sirt1 and Activation Ofp53 [Dissertation]. Boston, MA: Division of Graduate Medical Sciences, Boston University (2008).
155. Costantini C, Lorenzetto E, Cellini B, Buffelli M, Rossi F, Della-Bianca V. Astrocytes regulate the expression of insulin-like growth factor 1 receptor (IGF1-R) in primary cortical neurons during in vitro senescence. J Mol Neurosci (2010) 40(3):342–52. doi:10.1007/s12031-009-9305-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Panganiban RA, Day RM. Inhibition of IGF-1R prevents ionizing radiation-induced primary endothelial cell senescence. PLoS One (2013) 8:e78589. doi:10.1371/journal.pone.0078589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Vijay-Kumar M, Aitken JD, Sanders CJ, Frias A, Sloane VM, Xu J, et al. Flagellin treatment protects against chemicals, bacteria, viruses, and radiation. J Immunol (2008) 180(12):8280–5. doi:10.4049/jimmunol.180.12.8280
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Burdelya LG, Krivokrysenko VI, Tallant TC, Strom E, Gleiberman AS, Gupta D, et al. An agonist of toll-like receptor 5 has radioprotective activity in mouse and primate models. Science (2008) 320(5873):226–30. doi:10.1126/science.1154986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Krivokrysenko VI, Shakhov AN, Singh VK, Bone F, Kononov Y, Shyshynova I, et al. Identification of granulocyte colony-stimulating factor and interleukin-6 as candidate biomarkers of CBLB502 efficacy as a medical radiation countermeasure. J Pharmacol Exp Ther (2012) 343(2):497–508. doi:10.1124/jpet.112.196071
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Burdelya LG, Gleiberman AS, Toshkov I, Aygun-Sunar S, Bapardekar M, Manderscheid-Kern P, et al. Toll-like receptor 5 agonist protects mice from dermatitis and oral mucositis caused by local radiation: implications for head-and-neck cancer radiotherapy. Int J Radiat Oncol Biol Phys (2012) 83(1):228–34. doi:10.1016/j.ijrobp.2011.05.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Ghosh SP, Perkins MW, Hieber K, Kulkarni S, Kao TC, Reddy EP, et al. Radiation protection by a new chemical entity, Ex-Rad: efficacy and mechanisms. Radiat Res (2009) 171(2):173–9. doi:10.1667/RR1367.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Suman S, Datta K, Doiron K, Ren C, Kumar R, Taft DR, et al. Radioprotective effects of ON 01210.Na upon oral administration. J Radiat Res (2012) 53(3):368–76.
163. Ghosh SP, Kulkarni S, Perkins MW, Hieber K, Pessu RL, Gambles K, et al. Amelioration of radiation-induced hematopoietic and gastrointestinal damage by Ex-RAD(R) in mice. J Radiat Res (2012) 53(4):526–36. doi:10.1093/jrr/rrs001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Suman S, Maniar M, Fornace AJ Jr, Datta K. Administration of ON 01210.Na after exposure to ionizing radiation protects bone marrow cells by attenuating DNA damage response. Radiat Oncol (2012) 7:6. doi:10.1186/1748-717X-7-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Kang AD, Cosenza SC, Bonagura M, Manair M, Reddy MV, Reddy EP. ON01210.Na (Ex-RAD(R)) mitigates radiation damage through activation of the AKT pathway. PLoS One (2013) 8(3):e58355. doi:10.1371/journal.pone.0058355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Singh VK, Newman VL, Romaine PL, Wise SY, Seed TM. Radiation countermeasure agents: an update (2011-2014). Expert Opin Ther Pat (2014) 24(11):1229–55. doi:10.1517/13543776.2014.964684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Ghosh SP, Kulkarni S, Hieber K, Toles R, Romanyukha L, Kao TC, et al. Gamma-tocotrienol, a tocol antioxidant as a potent radioprotector. Int J Radiat Biol (2009) 85(7):598–606. doi:10.1080/09553000902985128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Kulkarni S, Ghosh SP, Satyamitra M, Mog S, Hieber K, Romanyukha L, et al. Gamma-tocotrienol protects hematopoietic stem and progenitor cells in mice after total-body irradiation. Radiat Res (2010) 173(6):738–47. doi:10.1667/RR1824.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Suman S, Datta K, Chakraborty K, Kulkarni SS, Doiron K, Fornace AJ Jr, et al. Gamma tocotrienol, a potent radioprotector, preferentially upregulates expression of anti-apoptotic genes to promote intestinal cell survival. Food Chem Toxicol (2013) 60:488–96. doi:10.1016/j.fct.2013.08.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Kulkarni S, Singh PK, Ghosh SP, Posarac A, Singh VK. Granulocyte colony-stimulating factor antibody abrogates radioprotective efficacy of gamma-tocotrienol, a promising radiation countermeasure. Cytokine (2013) 62(2):278–85. doi:10.1016/j.cyto.2013.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Satyamitra M, Kulkarni S, Ghosh SP, Mullaney C, Condliffe D, Srinivasan V. Hematopoietic recovey and amelioration of radiation-induced lethality by the vitamin E isoform, delta-tocotrienol. Radiat Res (2011) 175:736–45. doi:10.1667/RR2460.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Li XH, Fu D, Latif NH, Mullaney CP, Ney PH, Mog SR, et al. Delta-tocotrienol protects mouse and human hematopoietic progenitors from gamma-irradiation through extracellular signal-regulated kinase/mammalian target of rapamycin signaling. Haematologica (2010) 95(12):1996–2004. doi:10.3324/haematol.2010.026492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Singh VK, Wise SY, Scott JR, Romaine LP, Newman VL, Fatanmi OO. Radioprotective efficacy of delta-tocotrienol, a vitamin E isoform, is mediated through granulocyte colony-stimulating factor. Life Sci (2014) 98:113–22. doi:10.1016/j.lfs.2014.01.065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Singh VK, Romaine PL, Newman VL, Seed TM. Tocols induce G-CSF and mobilise progenitors that mitigate radiation injury. Radiat Prot Dosimetry (2014) 162(1–2):83–7. doi:10.1093/rpd/ncu223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Satyamitra M, Ney P, Graves J III, Mullaney C, Srinivasan V. Mechanism of radioprotection by delta-tocotrienol: pharmacokinetics, pharmacodynamics and modulation of signalling pathways. Br J Radiol (2012) 85(1019):e1093–103. doi:10.1259/bjr/63355844
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Bhanja P, Saha S, Kabarriti R, Liu L, Roy-Chowdhury N, Roy-Chowdhury J, et al. Protective role of R-spondin1, an intestinal stem cell growth factor, against radiation-induced gastrointestinal syndrome in mice. PLoS One (2009) 4(11):e8014. doi:10.1371/journal.pone.0008014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Xu L, Xiong S, Guo R, Yang Z, Wang Q, Xiao F, et al. Transforming growth factor beta3 attenuates the development of radiation-induced pulmonary fibrosis in mice by decreasing fibrocyte recruitment and regulating IFN-gamma/IL-4 balance. Immunol Lett (2014) 162(1 Pt A):27–33. doi:10.1016/j.imlet.2014.06.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Prockop DJ. Further proof for an unpopular concept: a single cell from bone marrow can serve as a stem cell for both hematopoiesis and osteogenesis. Mol Ther (2013) 21(6):1116–7. doi:10.1038/mt.2013.103
179. Prockop DJ, Gregory CA, Spees JL. One strategy for cell and gene therapy: harnessing the power of adult stem cells to repair tissues. Proc Natl Acad Sci U S A (2003) 100(Suppl 1):11917–23. doi:10.1073/pnas.1834138100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Semont A, Francois S, Mouiseddine M, Francois A, Sache A, Frick J, et al. Mesenchymal stem cells increase self-renewal of small intestinal epithelium and accelerate structural recovery after radiation injury. Adv Exp Med Biol (2006) 585:19–30. doi:10.1007/978-0-387-34133-0_2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Prockop DJ, Brenner M, Fibbe WE, Horwitz E, Le Blanc K, Phinney DG, et al. Defining the risks of mesenchymal stromal cell therapy. Cytotherapy (2010) 12(5):576–8. doi:10.3109/14653249.2010.507330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Nadir Y, Brenner B. Thrombotic complications associated with stem cell transplantation. Blood Rev (2012) 26(5):183–7. doi:10.1016/j.blre.2012.05.001
183. Abdel-Mageed AS, Senagore AJ, Pietryga DW, Connors RH, Giambernardi TA, Hay RV, et al. Intravenous administration of mesenchymal stem cells genetically modified with extracellular superoxide dismutase improves survival in irradiated mice. Blood (2009) 113(5):1201–3. doi:10.1182/blood-2008-07-170936
184. Saha S, Bhanja P, Kabarriti R, Liu L, Alfieri AA, Guha C. Bone marrow stromal cell transplantation mitigates radiation-induced gastrointestinal syndrome in mice. PLoS One (2011) 6(9):e24072. doi:10.1371/journal.pone.0024072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Singh VK, Christensen J, Fatanmi OO, Gille D, Ducey EJ, Wise SY, et al. Myeloid progenitors: a radiation countermeasure that is effective when initiated days after irradiation. Radiat Res (2012) 177:781–91. doi:10.1667/RR2894.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Singh VK, Brown DS, Kao TC. Alpha-tocopherol succinate protects mice from gamma-radiation by induction of granulocyte-colony stimulating factor. Int J Radiat Biol (2010) 86(1):12–21. doi:10.3109/09553000903264515
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Singh VK, Brown DS, Kao TC, Seed TM. Preclinical development of a bridging therapy for radiation casualties. Exp Hematol (2010) 38(1):61–70. doi:10.1016/j.exphem.2009.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Singh VK, Wise SY, Singh PK, Ducey EJ, Fatanmi OO, Seed TM. Alpha-tocopherol succinate- and AMD3100-mobilized progenitors mitigate radiation-induced gastrointestinal injury in mice. Exp Hematol (2012) 40(5):407–17. doi:10.1016/j.exphem.2012.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Singh VK, Wise SY, Singh PK, Posarac A, Fatanmi OO, Ducey EJ, et al. Alpha-tocopherol succinate-mobilized progenitors improve intestinal integrity after whole body irradiation. Int J Radiat Biol (2013) 89(5):334–45. doi:10.3109/09553002.2013.762137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Singh VK, Wise SY, Fatanmi OO, Beattie LA, Ducey EJ, Seed TM. Alpha-tocopherol succinate- and AMD3100-mobilized progenitors mitigate radiation combined injury in mice. J Radiat Res (2014) 55(1):41–53. doi:10.1093/jrr/rrt088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Singh VK, Ducey EJ, Fatanmi OO, Singh PK, Brown DS, Purmal A, et al. CBLB613: a TLR 2/6 agonist, natural lipopeptide of Mycoplasma arginini, as a novel radiation countermeasure. Radiat Res (2012) 177(5):628–42. doi:10.1667/RR2657.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Shakhov AN, Singh VK, Bone F, Cheney A, Kononov Y, Krasnov P, et al. Prevention and mitigation of acute radiation syndrome in mice by synthetic lipopeptide agonists of toll-like receptor 2 (TLR2). PLoS One (2012) 7(3):e33044. doi:10.1371/journal.pone.0033044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Gluzman-Poltorak Z, Mendonca SR, Vainstein V, Kha H, Basile LA. Randomized comparison of single dose of recombinant human IL-12 versus placebo for restoration of hematopoiesis and improved survival in rhesus monkeys exposed to lethal radiation. J Hematol Oncol (2014) 7:31. doi:10.1186/1756-8722-7-31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Doan PL, Himburg HA, Helms K, Russell JL, Fixsen E, Quarmyne M, et al. Epidermal growth factor regulates hematopoietic regeneration after radiation injury. Nat Med (2013) 19(3):295–304. doi:10.1038/nm.3070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Zhang L, Sun W, Wang J, Zhang M, Yang S, Tian Y, et al. Mitigation effect of an FGF-2 peptide on acute gastrointestinal syndrome after high-dose ionizing radiation. Int J Radiat Oncol Biol Phys (2010) 77(1):261–8. doi:10.1016/j.ijrobp.2009.11.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Ma J, Hou Y, Han D, Zhang M, Chen C, Zhang B, et al. Fibroblast growth factor-peptide promotes bone marrow recovery after irradiation. Adv Exp Med Biol (2013) 765:155–61. doi:10.1007/978-1-4614-4989-8_22
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Zhou D, Deoliveira D, Kang Y, Choi SS, Li Z, Chao NJ, et al. Insulin-like growth factor 1 mitigates hematopoietic toxicity after lethal total body irradiation. Int J Radiat Oncol Biol Phys (2013) 85(4):1141–8. doi:10.1016/j.ijrobp.2012.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
198. Mitchell JB, Anver MR, Sowers AL, Rosenberg PS, Figueroa M, Thetford A, et al. The antioxidant tempol reduces carcinogenesis and enhances survival in mice when administered after nonlethal total body radiation. Cancer Res (2012) 72(18):4846–55. doi:10.1158/0008-5472.CAN-12-1879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Satyamitra M, Lombardini E, Graves IIIJ, Mullaney C, Ney P, Hunter A, et al. A TPO receptor agonist, ALXN4100TPO, mitigates radiation-induced lethality and stimulates hematopoiesis in CD2F1 mice. Radiat Res (2011) 175(6):746–58. doi:10.1667/RR2462.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Singh VK, Ducey EJ, Brown DS, Whitnall MH. A review of radiation countermeasure work ongoing at the Armed Forces Radiobiology Research Institute. Int J Radiat Biol (2012) 88(4):296–310. doi:10.3109/09553002.2012.652726
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Garofalo MC, Ward AA, Farese AM, Bennett A, Taylor-Howell C, Cui W, et al. A pilot study in rhesus macaques to assess the treatment efficacy of a small molecular weight catalytic metalloporphyrin antioxidant (AEOL 10150) in mitigating radiation-induced lung damage. Health Phys (2014) 106(1):73–83. doi:10.1097/HP.0b013e3182a4d967
202. Singh VK, Newman VL, Seed TM. Colony-stimulating factors for the treatment of the hematopoietic component of the acute radiation syndrome (H-ARS): a review. Cytokine (2015) 71(1):22–37. doi:10.1016/j.cyto.2014.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: radiation protection, radioprotectors, mitigators, cancer treatment, irradiation
Citation: Rosen EM, Day R and Singh VK (2015) New approaches to radiation protection. Front. Oncol. 4:381. doi: 10.3389/fonc.2014.00381
Received: 24 July 2014; Accepted: 19 December 2014;
Published online: 20 January 2015.
Edited by:
Adam Paul Dicker, Thomas Jefferson University, USAReviewed by:
Chandan Guha, Albert Einstein College of Medicine, USAMichael Wayne Epperly, University of Pittsburgh Cancer Institute, USA
Copyright: © 2015 Rosen, Day and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eliot M. Rosen, Department of Oncology, Georgetown University School of Medicine, Preclinical Sciences Building, Room GM12B, 3970 Reservoir Road, NW, Washington, DC 20057, USA e-mail:ZW1yMzZAZ2VvcmdldG93bi5lZHU=