 Galia Ramírez-Toloza
Galia Ramírez-Toloza Lorena Aguilar-Guzmán
Lorena Aguilar-Guzmán Carolina Valck
Carolina Valck Paula Abello
Paula Abello Arturo Ferreira
Arturo Ferreira- 1Faculty of Veterinary Medicine and Livestock Sciences, University of Chile, Santiago, Chile
- 2Program of Immunology, Institute of Biomedical Sciences (ICBM), Faculty of Medicine, University of Chile, Santiago, Chile
The immune system protects against disease, but may aberrantly silence immunity against “altered self,” with consequent development of malignancies. Among the components of the endoplasmic reticulum (ER), important in immunity, is calreticulin (CRT) that, in spite of its residence in the ER, can be translocated to the exterior. Trypanosoma cruzi is the agent of Chagas disease, one of the most important global neglected infections, affecting several hundred thousand people. The syndrome, mainly digestive and circulatory, affects only one-third of those infected. The anti-tumor effects of the infection are known for several decades, but advances in the identification of responsible T. cruzi molecules are scarce. We have shown that T. cruzi CRT (TcCRT) better executes the antiangiogenic and anti-tumor effects of mammal CRT and its N-terminus vasostatin. In this regard, recombinant TcCRT (rTcCRT) and/or its N-terminus inhibit angiogenesis in vitro, ex vivo, and in vivo. TcCRT also inhibits the growth of murine adenocarcinomas and melanomas. Finally, rTcCRT fully reproduces the anti-tumor effect of T. cruzi infection in mice. Thus, we hypothesize that, the long reported anti-tumor effect of T. cruzi infection is mediated at least in part by TcCRT.
Introduction
The immune system protects against disease. However, abnormally silenced protective immunity against “altered self” may lead to the development of malignancies. As such, cancer represents a prominent example of defective immunological surveillance.
Components of the ER play key roles in the development of protective immunity. Among these components, is calreticulin (CRT) that, in spite of its residence in the endoplasmic reticulum (ER), can be translocated to the extracellular milieu, where it displays immune modulating capacities. Work from several laboratories indicates that CRT is an interesting ER candidate to manipulate anti-cancer immunity.
According to the World Health Organization (WHO), Chagas’ disease is endemic in 21 countries, with about 8 million infected people (1). The disease is considered one of the most important neglected tropical infections worldwide, because it causes 15,000 deaths per year and 0.7 million disability adjusted life-years (2). The impact of this parasite on domestic and wild animals (reservoirs) (3) is unknown.
The disease is endemic in Latin America. However, it has now gone global, affecting several hundred thousand people, mainly South American immigrants, in the USA, Canada, Europe, Oceania, and Asia (4), where transmission is independent of the protozoan. In countries without arthropod vectors, transmission is through blood products (5), organ transplants (1, 5), or congenital (6). Infection can also occur per os through parasite-contaminated food (7, 8).
The most frequent treatments for Chagas’ disease have been the administration of Benznidazole or Nifurtimox, with reported efficacy in up to 80% of acute cases after a 60-day course, but with frequent severe side effects and drug resistance (9). Although these drugs reportedly may cure the disease in the acute phase, particularly in children, their efficacy in adults, in the indeterminate or chronic phases, has not been determined.
About 80 years ago, Roskin, Exemplarskaja, and Kliueva, investigators from the former Soviet Union postulated an anti-cancer activity of Trypanosoma cruzi, based on a toxic effect of this parasitic infection, or parasite extracts, over different tumors, both in experimental animals and humans (10, 11). More recently, it was described the parasite capacity to infect preferentially tumor cells as compared to normal host cells (12). In general, these data suggest an antagonism between T. cruzi infection and tumor growth (12). Herein, we will review the available information with regard to possible molecular mechanisms underlying the anti-tumor effects of T. cruzi infection, with emphasis on the experimental rational basis leading to the proposal that the parasite utilizes its calreticulin (TcCRT) to protect its host against neoplastic aggressions. We have provided experimental evidences indicating that TcCRT is an antiangiogenic molecule that inhibits proliferation, migration, and capillary morphogenesis in several in vitro, ex vivo, and in vivo (in ovum) assays (13–15). On the other hand, TcCRT inhibits the growth of a mammary adenocarcinoma and a melanoma in different experimental animal models (13–16).
Is it all That Bad When Living with an Intracellular Protozoan?
The work from the investigators from the former Soviet Union, proposed that T. cruzi infection potential as a biotherapy for cancer treatment (17–19), opened possibilities for several research lines. They produced a “cancerolytic toxin” [Kliueva and Roskin (KR) preparation], from T. cruzi lysed cells. In humans, affected by a variety of tumors, these “toxins,” reduced tumor growth, pain, local inflammation, and bleeding (18). Controversial results followed and the situation, complicated by World War II and “The Cold War,” interrupted or greatly delayed this work (10). Thus, the mechanism and the molecular component responsible for the biotherapy effect have remained largely unknown.
Of note is the proposal in Science journal in 1948, by the immunologist Theodore S. Hauschka and Margaret Blair Goodwin that tumor-bearing mice, concomitantly infected with the lethal T. cruzi strain died within 8–13 days post infection. They observed that weight loss in tumor-bearing infected animals was important, and that tumor growth was almost completely suppressed. When, in tumor-bearing animals, the infection was treated, the tumors resumed their usual growth rate, and the hosts died of cancer (20). Thus, presence of the parasites was necessary for tumor inhibition. However, the authors’ view (20) that tumor and parasites compete for nutrients with consequent inhibition of the former does not seem now completely satisfactory given the information emerging during the last few years that we review and discuss below.
Earlier this century, experimental data obtained from rats infected with T. cruzi parasites and carcinoma induced by 1,2-dimethylhidrazyne, demonstrated that chronic infection may enhance resistance against tumor growth (21). More recent reports, evaluated the tumor-tropism-parasite capacity to infect host cancer cells rather than normal cells. Normally, the invasiveness (tissue-distribution of parasites of different strains of T. cruzi) in mice, primarily demonstrated a parasite tropism toward heart tissues, since 46% (40–65%) of parasites are found in this organ. The liver and kidney contained 3–4 times less parasites and even less was found in the spinal cord. Finally, only 3–4% were found in the brain, spleen, and lymph nodes. However, the presence of a tumor in the host leads to T. cruzi redistribution between the tissues: the parasites found in the tumor accounted for 18% in the decrease of heart invasion (now down to 28%) and increased invasiveness of spleen and lymph nodes (12). Nevertheless, a relationship between these findings and tumor development was not addressed in these studies.
More recently, a role for T. cruzi infection in controlling tumor growth has been revisited at least in two laboratories, including ours (22, 23). Junqueira et al. (23) reported that the use of a recombinant non-pathogenic T. cruzi clone as vector of a testis tumor antigen (NY-ESO-1) is efficient in generating T cell-immune responses and protection against cancer cells, thus delaying tumor development in mice.
Most recently, we have corroborated that T. cruzi infection greatly reduces the growth of a mammary adenocarcinoma.
Calreticulin
Calreticulin, a 46 kDa pleiotropic protein, participating as a chaperone and in calcium homeostasis (24), has been described in different organisms such as humans (25), insects (26, 27), nematodes (28–31), protozoa (32–35), and plants (36).
Calreticulin, mainly residing in the ER of all nucleated cells (37), contributes in different processes such as the control of glycoprotein folding quality and binding to monoglucosylated glycans with high mannose content. CRT is also present in the cytosol, nucleus, secretory granules, on the plasma membrane and also free in the extracellular environment (37). There, CRT modulates the immune response against apoptotic cancer cells (38–42). The mechanisms involved in CRT translocation and release to the extracellular milieu are still unknown (43). CRT also promotes cutaneous wound healing (44–46), cell adhesion (37), nuclear export of some steroid hormone receptors (47–49), and the stability or translation of a variety of RNAs (50–54). CRT reaches the cytosol and nucleus by a C-terminal domain-dependent retrotranslocation, after ER calcium depletion (55).
Calreticulin has a globular N-terminus (N), a proline-rich (P) domain, and an acidic C-terminus (37). An S-domain (aa 160–289), within N and P, binds complement component C1, a “danger signal detection module” that initiates the classical complement activation pathway (56, 57). The primary CRT sequence starts with a signal peptide and ends with a KDEL-ER retention sequence (58). Human CRT (HuCRT) and its N-terminal fragment binds laminin (59), with antiangiogenic properties in vitro and in vivo (60, 61) and inhibit the growth in several tumor models (62–64). Vasostatin, is a CRT 180 amino acid N-terminal fragment, a potent endogenous inhibitor of angiogenesis and suppressor of tumor growth. Vasostatin inhibits vascular endothelial growth factor (VEGF)-induced endothelial cell proliferation and interactions in matrigel and induces cell apoptosis under limiting oxygen availability (65).
TcCRT and Infectivity
Trypanosoma cruzi, may use its CRT, a putative universal apoptosis cell marker (39, 41, 42), in an “apoptotic mimicry” strategy to generate “eat me” signals (i.e., by capturing C1 in the area of flagellum emergence), thus facilitating the invasion of host cells. C1q bridges the parasite molecule with host cell surface receptors (66), most likely CRT known as cC1qR (67). Thus, host C1, upon binding to the trypomastigote surface, also promotes parasite infectivity (68). The parasite molecule responsible for recruiting this complement component has been identified as TcCRT (69, 70). Increased parasite infectivity is paralleled by significant increases in TcCRT mRNA levels during early (cell contact and penetration) infection stages of a VERO cell line. In spite of its lysine–aspartic acid–glutamic acid–leucine (KDEL)-ER retrieval sequence, TcCRT does translocate from the ER to the parasite area of flagellum emergence. An augmented capacity to recruit C1, an important “eat me” signal for phagocytic cells follows, thus leading to increased infectivity (41, 68–72).
The TcCRT–C1q interaction can be decreased by anti-TcCRT F(ab′)2 antibody fragments (lacking the C1-binding Fc domains) (73). Indeed, passive immunization of mice with these fragments resulted in important decreases in infectivity and improved clinical parameters (69).
Of particular interest and conceivable consequences in pathology is the possibility that, in T. cruzi infected individuals, the parasite molecule may promote autoimmune mechanisms (74).
Trypanosoma cruzi CRT also binds complement mannan binding lectin (MBL) and Ficolins (75). Together with C1(q,r,s) they are three complement “danger signal” recognition macromolecular modules. Genetically, structurally, and functionally related, they differ in the nature of the recognized danger signals (22). After binding C1(q,r,s), TcCRT or its S and R central domains inhibit the classical pathway of human complement, in a calcium-independent manner (69, 72, 76). More recently, we have also proposed that l-Ficolin binds TcCRT, thus inhibiting the lectin pathway, a likely alternative or concomitant T. cruzi strategy to inhibit the host immune response (75). The roles of MBL and Ficolins in the infectivity process are still under study.
Trypanosoma cruzi Calreticulin, a Molecule with Antiangiogenic and Anti-Tumor Properties
Inhibition of tumor angiogenesis, proposed as a cancer therapy almost 40 years ago (77), is a complex process to form new blood vessels, thus providing the necessary supply of nutrients, oxygen, and ways for waste disposal (78). Antiangiogenesis is currently applicable to a wide variety of tumors, frequently as a supplement to other therapies (79).
Our description of TcCRT provides alternative or concomitant explanations for at least an important part of the anti-tumor effect of this parasite infection. Most likely, TcCRT anti-tumor properties derive from its antiangiogenic properties (13, 73). By direct interaction with endothelial cells, probably through a Scavenger-like receptor, TcCRT acts as a potent angiogenesis inhibitor (13, 14, 71). Antiangiogenic agents may generate a primary stressing challenge to a variety of tumor cells. On the other hand, many tumors have a notorious capacity to produce an array of proangiogenic molecules. Of note are VEGF, the platelet-derived endothelial cell growth factor (PD-ECGF), and the acidic and basic fibroblast growth factors (aFGF and bFGF) (80). Thus, tumor growth and metastasis are indirectly, but importantly promoted by these factors.
Angiogenesis modulators behave differently across species. TcCRT and its N-terminal vasostatin-like domain (N-TcCRT) were studied in mammals, Homo sapiens sapiens included (13). Thus, recombinant TcCRT (rTcCRT) and its N-terminal domain inhibit capillary growth ex vivo in Rattus rattus aortic rings, morphogenesis, proliferation, and chemotaxis in human umbilical cord endothelial cells (HUVECs) (13) and in ovum angiogenesis in the Gallus gallus chorioallantoid membrane (CAM) assay (14). These are valid correlates of important features of angiogenesis in vivo. In most of these assays TcCRT was more effective, in molar terms, than HuCRT (13). Of particular interest is the fact that, in the CAM assay, the antiangiogenic TcCRT effect was fully reverted by polyclonal antibodies against rTcCRT (15). We are currently investigating whether the anti-tumor effect of T. cruzi infection is reverted by F(ab′)2 anti-TcCRT antibody fragments, derived from these immunoglobulins. In such a case, a formal causal link between externalized TcCRT and the anti-tumor effect of T. cruzi infection would be established.
In agreement with the previously described facts, inoculation of rTcCRT inhibits by 60–70% the time-course development of a murine mammary metrotexate multiresistant adenocarcinoma (TA3-MTX-R), with a higher efficiency than the human counterpart (13) (Figure 1).
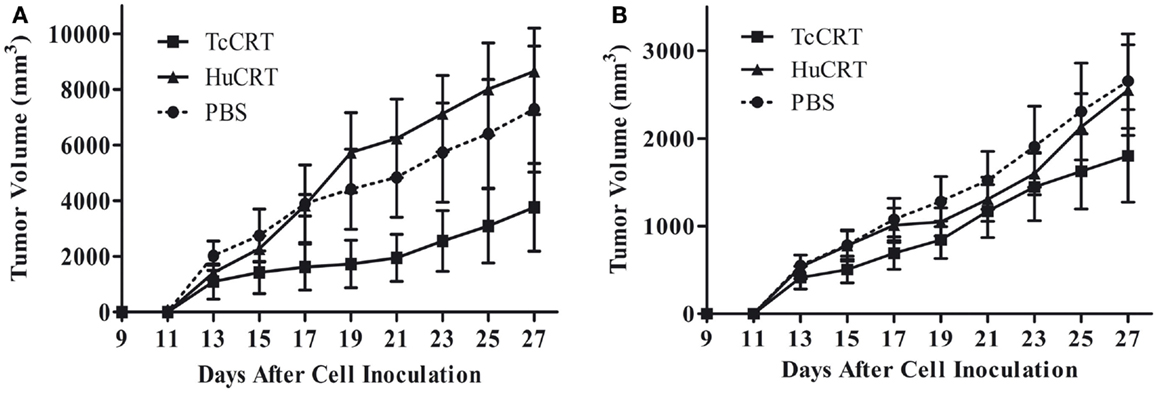
Figure 1. Trypanosoma cruzi CRT-mediated tumor growth inhibition. In both experiments, 5 × 105 murine A/J mammary tumor (TA3 MTXR) cells were inoculated s.c. in A/J female mice, five animals per group. (A,B) Together with tumor cells, and every other day, the animals were inoculated s.c. with 50 μg TcCRT or HuCRT or solvent. While TcCRT had a similar anti-tumor effect in both experiments (p = 0.0078), HuCRT did not show that effect under these conditions (13). In both experiments, the tumor size was determined with a digital caliper (Mitutoyo Corp., Japan), in a double blind procedure. The formula (π/6 × length × width2) was used. Data were statistically validated by Wilcoxon Signed Rank test, GraphPad Prism 4. Reproduced with permission from PLoS Neglected Tropical Diseases (13).
Concluding Remarks
Recombinant TcCRT, and most likely translocated native TcCRT, mediate mechanisms relevant in the host/parasite interplay: (i) through a central S domain (aa 159–281), it interferes with the earliest stages of the complement activation; (ii) C1, bound to the parasite, promotes infectivity (69); and (iii) through an N-terminal domain (20–193), it interacts directly with endothelial cells and inhibits angiogenesis (13). Thus, prolonged host–parasite interactions may be promoted. Several of these features are variably conserved in the HuCRT, but with lower equimolar efficiency. Thus, when the parasite and human chaperones are compared in equimolar terms, the former displays stronger antiangiogenic effects in in vitro, ex vivo, and in vivo (22, 70) and this is reflected in the compared anti-tumor effects.
Perhaps the TcCRT antiangiogenic effects reflect a parasite evolutionary adaptation to protect its host integrity and, as a necessary consequence, its own (71). Concomitantly, by decreasing angiogenesis, access of immunocompetent cells to the sites of parasite locations may be impaired, as well as subsequent inflammatory consequences, both with possible benefits to the aggressor, although the second strategy could also benefit the host from exaggerated immune reactivity.
The ability of TcCRT to delay solid tumor growth may represent an evolutionary adaptation with consequences in host survival and increased possibilities for the parasite to expand its genome. Based on fundamental Darwinian principles, cancer (i.e., mammary, cervix-uterine, prostate, lung, stomach, among others), taken altogether, have prevalence equivalent to an epidemic. These cancers may have exerted a selective pressure on the parasites, to develop molecular mechanisms to protect their hosts. Our experimental evidences indicate that externalized TcCRT, through its antiangiogenic properties may explain, at least in important part, such mechanisms (70).
Trypanosoma cruzi CRT-mediated antiangiogenesis, may provoke a stressful environment in the tumor (decreased nutrients and oxygen supply, accumulation of metabolic waste products, etc.) that induce CRT exteriorization on tumor cells. External CRT captures C1, a signal that increases phagocytosis of tumor cells and consequent immunogenicity (16). These possibilities are summarized in Figure 2. Other stressful agents (i.e., UV, anthracyclins) (39–42, 81) also mediate CRT translocation with similar immune stimulating consequences. The possibility that a concomitant immune boost, mediated by other means, is promoted by the infection (23), is also conceivable.
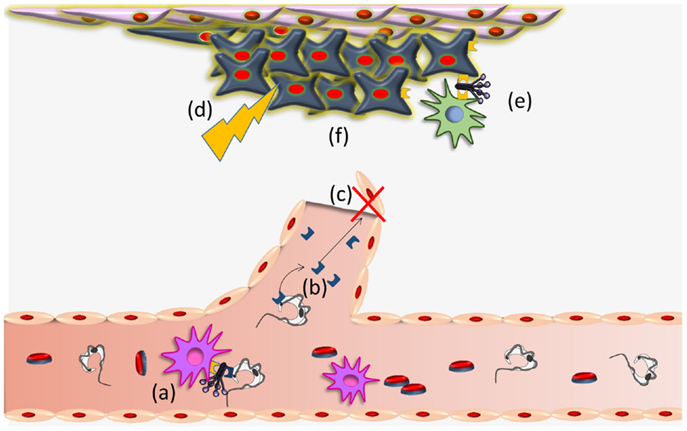
Figure 2. Trypanosoma cruzi CRT participates in infectivity and anti-tumor process. (a) TcCRT, exposed on the parasite surface, binds C1q thus inhibiting the classical pathway of the complement system. TcCRT/C1q interaction participates in the infectivity process binding CRT present on mammalian cells. (b) TcCRT is translocated to the parasite surface and secreted. This TcCRT in the extracellular milieu binds to endothelial cells, (c) inhibiting angiogenesis. (d) This inhibition provokes a stressful environment in the tumor (decreased nutrients and oxygen supply, accumulation of metabolic waste products, etc.) (e) that induces CRT exteriorization on tumor cells. External tumor CRT captures C1, a signal that increases phagocytosis of tumor cells and consequent immunogenicity and (f) reduction of the tumor growth. Whether TcCRT also binds tumor cells in vivo, thus promoting tumor immunogenicity, has not been demonstrated.
Finally, given the current evidences, the old proposal that tumor and parasites compete for nutrients with consequent inhibition of the former (20), now seems less likely.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the support from CONICYT-Chile, through the following projects: PIA ACT 112, ECOS-CONICYT C11S02, and FONDECYT projects: 1130099 (Arturo Ferreira), 11110519 (Carolina Valck), 11110251 (Galia Ramírez-Toloza).
References
1. WHO. Reporte del grupo de trabajo científico sobre la enfermedad de Chagas: 17-20 de Abril de 2005, actualizado en julio de 2007, Buenos Aires, Argentina. Geneva: Word Health Organization (2007).
2. Coura JR, Vinas PA. Chagas disease: a new worldwide challenge. Nature (2010) 465(7301):S6–7. doi: 10.1038/nature09221
3. Coura JR, Dias JC. Epidemiology, control and surveillance of Chagas disease: 100 years after its discovery. Mem Inst Oswaldo Cruz (2009) 104(Suppl 1):31–40. doi:10.1590/S0074-02762009000900006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Tanowitz HB, Weiss LM, Montgomery SP. Chagas disease has now gone global. PLoS Negl Trop Dis (2011) 5(4):e1136. doi:10.1371/journal.pntd.0001136
5. Schmunis GA. Epidemiology of Chagas disease in non-endemic countries: the role of international migration. Mem Inst Oswaldo Cruz (2007) 102(Suppl 1):75–85. doi:10.1590/S0074-02762007005000093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Castillo C, Ramirez G, Valck C, Aguilar L, Maldonado I, Rosas C, et al. The interaction of classical complement component C1 with parasite and host calreticulin mediates Trypanosoma cruzi infection of human placenta. PLoS Negl Trop Dis (2013) 7(8):e2376. doi:10.1371/journal.pntd.0002376
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Pereira KS, Schmidt FL, Guaraldo AM, Franco RM, Dias VL, Passos LA. Chagas’ disease as a foodborne illness. J Food Prot (2009) 72(2):441–6.
8. Yoshida N. Trypanosoma cruzi infection by oral route: how the interplay between parasite and host components modulates infectivity. Parasitol Int (2008) 57(2):105–9. doi:10.1016/j.parint.2007.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Clayton J. Chagas disease: pushing through the pipeline. Nature (2010) 465(7301):S12–5. doi:10.1038/nature09224
10. Krementsov N. The Cure: A Story of Cancer and Politics from the Annals of the Cold War. Chicago, IL: University of Chicago Press (2002). 261 p.
11. Klyueva NG, Roskin G. Cancerolytic substance of Schizotrypanum cruzi. Am Rev Sov Med (1946) 4(2):127–9.
12. Kallinikova VD, Matekin PV, Ogloblina TA, Leikina MI, Kononenko AF, Sokolova NM, et al. [Anticancer properties of flagellate protozoan Trypanosoma cruzi Chagas, 1909]. Izv Akad Nauk Ser Biol (2001) (3):299–311.
13. Lopez NC, Valck C, Ramirez G, Rodriguez M, Ribeiro C, Orellana J, et al. Antiangiogenic and antitumor effects of Trypanosoma cruzi calreticulin. PLoS Negl Trop Dis (2010) 4(7):e730. doi:10.1371/journal.pntd.0000730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Molina MC, Ferreira V, Valck C, Aguilar L, Orellana J, Rojas A, et al. An in vivo role for Trypanosoma cruzi calreticulin in antiangiogenesis. Mol Biochem Parasitol (2005) 140(2):133–40. doi:10.1016/j.molbiopara.2004.12.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Toledo V, Ramirez G, Valck C, Lopez N, Ribeiro CH, Maldonado I, et al. Comparative in vivo antiangiogenic effects of calreticulin from Trypanosoma cruzi and Homo sapiens sapiens. Biol Res (2010) 43(3):287–9. doi:10.4067/S0716-97602010000300004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Aguilar-Guzman L, Lobos-Gonzalez L, Rosas C, Vallejos G, Falcon C, Sosoniuk E, et al. Human survivin and Trypanosoma cruzi calreticulin act in synergy against a murine melanoma in vivo. PLoS One (2014) 9(4):e95457. doi:10.1371/journal.pone.0095457
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Roskin G. Toxin therapy of experimental cancer; the influence of protozoan infections upon transplanted cancer. Cancer Res (1946) 6:363–5.
18. Kliueva NG, Roskin GI. Biotherapy of Malignant Tumours. New York, NY: Pergamon Press (1963). 315 p.
19. Roskin GI. EE. Protozoen infektion und experimentellen Krebs. Z Krebsforsch (1931) 31:628–45. doi:10.1007/BF01625403
20. Hauschka TS, Goodwin MB. Trypanosoma cruzi endotoxin (KR) in the treatment of malignant mouse tumors. Science (1948) 107(2788):600–2. doi:10.1126/science.107.2788.600
21. Oliveira EC, Leite MS, Miranda JA, Andrade AL, Garcia SB, Luquetti AO, et al. Chronic Trypanosoma cruzi infection associated with low incidence of 1,2-dimethylhydrazine-induced colon cancer in rats. Carcinogenesis (2001) 22(5):737–40. doi:10.1093/carcin/22.5.737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Ramirez G, Valck C, Aguilar L, Kemmerling U, Lopez-Munoz R, Cabrera G, et al. Roles of Trypanosoma cruzi calreticulin in parasite-host interactions and in tumor growth. Mol Immunol (2012) 52(3–4):133–40. doi:10.1016/j.molimm.2012.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Junqueira C, Santos LI, Galvao-Filho B, Teixeira SM, Rodrigues FG, DaRocha WD, et al. Trypanosoma cruzi as an effective cancer antigen delivery vector. Proc Natl Acad Sci U S A (2011) 108(49):19695–700. doi:10.1073/pnas.1110030108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Zamanian M, Veerakumarasivam A, Abdullah S, Rosli R. Calreticulin and cancer. Pathol Oncol Res (2013) 19(2):149–54. doi:10.1007/s12253-012-9600-2
25. McCauliffe DP, Zappi E, Lieu TS, Michalak M, Sontheimer RD, Capra JD. A human Ro/SS-A autoantigen is the homologue of calreticulin and is highly homologous with onchocercal RAL-1 antigen and an aplysia “memory molecule”. J Clin Invest (1990) 86(1):332–5. doi:10.1172/JCI114704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Ferreira CA, Da Silva Vaz I, da Silva SS, Haag KL, Valenzuela JG, Masuda A. Cloning and partial characterization of a Boophilus microplus (Acari: Ixodidae) calreticulin. Exp Parasitol (2002) 101(1):25–34. doi:10.1016/S0014-4894(02)00032-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Smith MJ. Nucleotide sequence of a Drosophila melanogaster gene encoding a calreticulin homologue. DNA Seq (1992) 3(4):247–50.
28. Smith MJ. A C. elegans gene encodes a protein homologous to mammalian calreticulin. DNA Seq (1992) 2(4):235–40.
29. Kasper G, Brown A, Eberl M, Vallar L, Kieffer N, Berry C, et al. A calreticulin-like molecule from the human hookworm Necator americanus interacts with C1q and the cytoplasmic signalling domains of some integrins. Parasite Immunol (2001) 23(3):141–52. doi:10.1046/j.1365-3024.2001.00366.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Khalife J, Pierce RJ, Godin C, Capron A. Cloning and sequencing of the gene encoding Schistosoma mansoni calreticulin. Mol Biochem Parasitol (1993) 62(2):313–5. doi:10.1016/0166-6851(93)90120-M
31. Rokeach LA, Zimmerman PA, Unnasch TR. Epitopes of the Onchocerca volvulus RAL1 antigen, a member of the calreticulin family of proteins, recognized by sera from patients with onchocerciasis. Infect Immun (1994) 62(9):3696–704.
32. Aguillon JC, Ferreira L, Perez C, Colombo A, Molina MC, Wallace A, et al. Tc45, a dimorphic Trypanosoma cruzi immunogen with variable chromosomal localization, is calreticulin. Am J Trop Med Hyg (2000) 63(5–6):306–12.
33. Joshi M, Pogue GP, Duncan RC, Lee NS, Singh NK, Atreya CD, et al. Isolation and characterization of Leishmania donovani calreticulin gene and its conservation of the RNA binding activity. Mol Biochem Parasitol (1996) 81(1):53–64. doi:10.1016/0166-6851(96)02676-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Labriola C, Cazzulo JJ, Parodi AJ. Trypanosoma cruzi calreticulin is a lectin that binds monoglucosylated oligosaccharides but not protein moieties of glycoproteins. Mol Biol Cell (1999) 10(5):1381–94. doi:10.1091/mbc.10.5.1381
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Oladiran A, Belosevic M. Trypanosoma carassii calreticulin binds host complement component C1q and inhibits classical complement pathway-mediated lysis. Dev Comp Immunol (2010) 34(4):396–405. doi:10.1016/j.dci.2009.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Denecke J, Carlsson LE, Vidal S, Hoglund AS, Ek B, van Zeijl MJ, et al. The tobacco homolog of mammalian calreticulin is present in protein complexes in vivo. Plant Cell (1995) 7(4):391–406. doi:10.1105/tpc.7.4.391
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Michalak M, Groenendyk J, Szabo E, Gold LI, Opas M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem J (2009) 417(3):651–66. doi:10.1042/BJ20081847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Tesniere A, Apetoh L, Ghiringhelli F, Joza N, Panaretakis T, Kepp O, et al. Immunogenic cancer cell death: a key-lock paradigm. Curr Opin Immunol (2008) 20(5):504–11. doi:10.1016/j.coi.2008.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ (2007) 14(10):1848–50. doi:10.1038/sj.cdd.4402201
40. Obeid M, Panaretakis T, Tesniere A, Joza N, Tufi R, Apetoh L, et al. Leveraging the immune system during chemotherapy: moving calreticulin to the cell surface converts apoptotic death from “silent” to immunogenic. Cancer Res (2007) 67(17):7941–4. doi:10.1158/0008-5472.CAN-07-1622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi:10.1038/nm1523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Obeid M, Tesniere A, Panaretakis T, Tufi R, Joza N, van Endert P, et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol Rev (2007) 220:22–34. doi:10.1111/j.1600-065X.2007.00567.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Gold L, Eggleton P, Sweetwyne M, Van Duyn L, Greives M, Naylor S, et al. Calreticulin: non-endoplasmic reticulum functions in physiology and disease. FASEB J (2010) 24(3):665–83. doi:10.1096/fj.09-145482
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Gold LI, Rahman M, Blechman KM, Greives MR, Churgin S, Michaels J, et al. Overview of the role for calreticulin in the enhancement of wound healing through multiple biological effects. J Investig Dermatol Symp Proc (2006) 11(1):57–65. doi:10.1038/sj.jidsymp.5650011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Nanney LB, Woodrell CD, Greives MR, Cardwell NL, Pollins AC, Bancroft TA, et al. Calreticulin enhances porcine wound repair by diverse biological effects. Am J Pathol (2008) 173(3):610–30. doi:10.2353/ajpath.2008.071027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Arias I, Sepulveda C, Bravo P, Hamilton-West C, Maldonado I, Ferreira A. Comparative effect of human and Trypanosoma cruzi calreticulin in wound healing. J Tissue Eng Regen Med (2012). doi:10.1002/term.1613
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Holaska JM, Black BE, Love DC, Hanover JA, Leszyk J, Paschal BM. Calreticulin Is a receptor for nuclear export. J Cell Biol (2001) 152(1):127–40. doi:10.1083/jcb.152.1.127
48. Burns K, Duggan B, Atkinson EA, Famulski KS, Nemer M, Bleackley RC, et al. Modulation of gene expression by calreticulin binding to the glucocorticoid receptor. Nature (1994) 367(6462):476–80. doi:10.1038/367476a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Dedhar S, Rennie PS, Shago M, Hagesteijn CY, Yang H, Filmus J, et al. Inhibition of nuclear hormone receptor activity by calreticulin. Nature (1994) 367(6462):480–3. doi:10.1038/367480a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Nickenig G, Michaelsen F, Muller C, Berger A, Vogel T, Sachinidis A, et al. Destabilization of AT(1) receptor mRNA by calreticulin. Circ Res (2002) 90(1):53–8. doi:10.1161/hh0102.102503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Singh NK, Atreya CD, Nakhasi HL. Identification of calreticulin as a rubella virus RNA binding protein. Proc Natl Acad Sci U S A (1994) 91(26):12770–4. doi:10.1073/pnas.91.26.12770
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Timchenko LT, Iakova P, Welm AL, Cai ZJ, Timchenko NA. Calreticulin interacts with C/EBPalpha and C/EBPbeta mRNAs and represses translation of C/EBP proteins. Mol Cell Biol (2002) 22(20):7242–57. doi:10.1128/MCB.22.20.7242-7257.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Iakova P, Wang GL, Timchenko L, Michalak M, Pereira-Smith OM, Smith JR, et al. Competition of CUGBP1 and calreticulin for the regulation of p21 translation determines cell fate. EMBO J (2004) 23(2):406–17. doi:10.1038/sj.emboj.7600052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Totary-Jain H, Naveh-Many T, Riahi Y, Kaiser N, Eckel J, Sasson S. Calreticulin destabilizes glucose transporter-1 mRNA in vascular endothelial and smooth muscle cells under high-glucose conditions. Circ Res (2005) 97(10):1001–8. doi:10.1161/01.RES.0000189260.46084.e5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Labriola C, Conte I, López Medus M, Parodi A, Caramelo J. Endoplasmic reticulum calcium regulates the retrotranslocation of Trypanosoma cruzi calreticulin to the cytosol. PLoS One (2010) 5(10):e13141. doi:10.1371/journal.pone.0013141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Stuart GR, Lynch NJ, Day AJ, Schwaeble WJ, Sim RB. The C1q and collectin binding site within C1q receptor (cell surface calreticulin). Immunopharmacology (1997) 38(1–2):73–80. doi:10.1016/S0162-3109(97)00076-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Stuart GR, Lynch NJ, Lu J, Geick A, Moffatt BE, Sim RB, et al. Localisation of the C1q binding site within C1q receptor/calreticulin. FEBS Lett (1996) 397(2–3):245–9. doi:10.1016/S0014-5793(96)01156-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Michalak M, Corbett EF, Mesaeli N, Nakamura K, Opas M. Calreticulin: one protein, one gene, many functions. Biochem J (1999) 344(Pt 2):281–92. doi:10.1042/0264-6021:3440281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Yao L, Pike SE, Tosato G. Laminin binding to the calreticulin fragment vasostatin regulates endothelial cell function. J Leukoc Biol (2002) 71(1):47–53.
60. Pike SE, Yao L, Jones KD, Cherney B, Appella E, Sakaguchi K, et al. Vasostatin, a calreticulin fragment, inhibits angiogenesis and suppresses tumor growth. J Exp Med (1998) 188(12):2349–56. doi:10.1084/jem.188.12.2349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Pike SE, Yao L, Setsuda J, Jones KD, Cherney B, Appella E, et al. Calreticulin and calreticulin fragments are endothelial cell inhibitors that suppress tumor growth. Blood (1999) 94(7):2461–8.
62. Cai KX, Tse LY, Leung C, Tam PK, Xu R, Sham MH. Suppression of lung tumor growth and metastasis in mice by adeno-associated virus-mediated expression of vasostatin. Clin Cancer Res (2008) 14(3):939–49. doi:10.1158/1078-0432.CCR-07-1930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Jazowiecka-Rakus J, Jarosz M, Kozlowska D, Sochanik A, Szala S. Combination of vasostatin and cyclophosphamide in the therapy of murine melanoma tumors. Acta Biochim Pol (2007) 54(1):125–33.
64. Yao L, Pike SE, Pittaluga S, Cherney B, Gupta G, Jaffe ES, et al. Anti-tumor activities of the angiogenesis inhibitors interferon-inducible protein-10 and the calreticulin fragment vasostatin. Cancer Immunol Immunother (2002) 51(7):358–66. doi:10.1007/s00262-002-0294-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Shu Q, Li W, Li H, Sun G. Vasostatin inhibits VEGF-induced endothelial cell proliferation, tube formation and induces cell apoptosis under oxygen deprivation. Int J Mol Sci (2014) 15(4):6019–30. doi:10.3390/ijms15046019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Malhotra R. Collectin receptor (C1q receptor): structure and function. Behring Inst Mitt (1993) (93):254–61.
67. Eggleton P, Tenner AJ, Reid KB. C1q receptors. Clin Exp Immunol (2000) 120(3):406–12. doi:10.1046/j.1365-2249.2000.01218.x
68. Rimoldi MT, Tenner AJ, Bobak DA, Joiner KA. Complement component C1q enhances invasion of human mononuclear phagocytes and fibroblasts by Trypanosoma cruzi trypomastigotes. J Clin Invest (1989) 84(6):1982–9. doi:10.1172/JCI114388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Ramirez G, Valck C, Molina MC, Ribeiro CH, Lopez N, Sanchez G, et al. Trypanosoma cruzi calreticulin: a novel virulence factor that binds complement C1 on the parasite surface and promotes infectivity. Immunobiology (2011) 216(1–2):265–73. doi:10.1016/j.imbio.2010.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Ramirez G, Valck C, Ferreira VP, Lopez N, Ferreira A. Extracellular Trypanosoma cruzi calreticulin in the host-parasite interplay. Trends Parasitol (2011) 27(3):115–22. doi:10.1016/j.pt.2010.12.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Ferreira V, Molina MC, Schwaeble W, Lemus D, Ferreira A. Does Trypanosoma cruzi calreticulin modulate the complement system and angiogenesis? Trends Parasitol (2005) 21(4):169–74. doi:10.1016/j.pt.2005.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Ferreira V, Valck C, Sanchez G, Gingras A, Tzima S, Molina MC, et al. The classical activation pathway of the human complement system is specifically inhibited by calreticulin from Trypanosoma cruzi. J Immunol (2004) 172(5):3042–50. doi:10.4049/jimmunol.172.5.3042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Aguilar L, Ramirez G, Valck C, Molina MC, Rojas A, Schwaeble W, et al. F(ab’)2 antibody fragments against Trypanosoma cruzi calreticulin inhibit its interaction with the first component of human complement. Biol Res (2005) 38(2–3):187–95. doi:10.4067/S0716-97602005000200008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Ribeiro CH, Lopez NC, Ramirez GA, Valck CE, Molina MC, Aguilar L, et al. Trypanosoma cruzi calreticulin: a possible role in Chagas’ disease autoimmunity. Mol Immunol (2009) 46(6):1092–9. doi:10.1016/j.molimm.2008.10.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Sosoniuk E, Vallejos G, Kenawy H, Gaboriaud C, Thielens N, Fujita T, et al. Trypanosoma cruzi calreticulin inhibits the complement lectin pathway activation by direct interaction with L-Ficolin. Mol Immunol (2014) 60(1):80–5. doi:10.1016/j.molimm.2014.03.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Valck C, Ramirez G, Lopez N, Ribeiro CH, Maldonado I, Sanchez G, et al. Molecular mechanisms involved in the inactivation of the first component of human complement by Trypanosoma cruzi calreticulin. Mol Immunol (2010) 47(7–8):1516–21. doi:10.1016/j.molimm.2010.01.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Baillie CT, Winslet MC, Bradley NJ. Tumour vasculature – a potential therapeutic target. Br J Cancer (1995) 72(2):257–67. doi:10.1038/bjc.1995.323
78. Folkman J. Fundamental concepts of the angiogenic process. Curr Mol Med (2003) 3(7):643–51. doi:10.2174/1566524033479465
79. Griffioen AW, Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacol Rev (2000) 52(2):237–68.
80. Zhu L, Niu G, Fang X, Chen X. Preclinical molecular imaging of tumor angiogenesis. Q J Nucl Med Mol Imaging (2010) 54(3):291–308.
81. Aguilera R, Saffie C, Tittarelli A, Gonzalez FE, Ramirez M, Reyes D, et al. Heat-shock induction of tumor-derived danger signals mediates rapid monocyte differentiation into clinically effective dendritic cells. Clin Cancer Res (2011) 17(8):2474–83. doi:10.1158/1078-0432.CCR-10-2384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: Trypanosoma cruzi calreticulin, angiogenesis, cancer, infectivity, c1q
Citation: Ramírez-Toloza G, Aguilar-Guzmán L, Valck C, Abello P and Ferreira A (2015) Is it all that bad when living with an intracellular protozoan? The role of Trypanosoma cruzi calreticulin in angiogenesis and tumor growth. Front. Oncol. 4:382. doi: 10.3389/fonc.2014.00382
Received: 24 October 2014; Paper pending published: 25 November 2014;
Accepted: 19 December 2014; Published online: 13 January 2015.
Edited by:
Paul Eggleton, Exeter University Medical School, UKReviewed by:
Fabian Benencia, Ohio University Heritage College of Osteopathic Medicine, USAMoisés Santillán, Centro de Investigación y Estudios Avanzados del IPN, Mexico
Copyright: © 2015 Ramírez-Toloza, Aguilar-Guzmán, Valck, Abello and Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arturo Ferreira, Program of Immunology, Institute of Biomedical Sciences (ICBM), Faculty of Medicine, University of Chile, Av. Independencia 1027, Santiago 8380453, Chile e-mail:YWZlcnJlaXJhX3VjaGlsZUB5YWhvby5jb20=
†Galia Ramírez-Toloza and Lorena Aguilar-Guzmán have contributed equally to this work.