 Xiaochun Wang
Xiaochun Wang Kathleen M. Batty
Kathleen M. Batty Philip J. Crowe1,2
Philip J. Crowe1,2 David Goldstein
David Goldstein Jia-Lin Yang
Jia-Lin Yang- 1Sarcoma Nano-Oncology Group, Adult Cancer Program, Lowy Cancer Research Centre, Prince of Wales Clinical School, University of New South Wales (UNSW), Sydney, NSW, Australia
- 2Department of Surgery, Prince of Wales Clinical School, University of New South Wales (UNSW), Sydney, NSW, Australia
- 3Department of Medical Oncology, Prince of Wales Clinical School, University of New South Wales (UNSW), Sydney, NSW, Australia
Purpose: Hyper-activation of the HER (erbB) family receptors, HER 1-4, leads to up-regulation of the three vital signaling pathways: mitogen activated protein kinase, phosphoinositide 3-kinase/AKT, and Janus kinase/signal transducer and activator of transcription pathways. Blocking HER1/EGFR has a limited anticancer effect due to either secondary mutation e.g., T790M or by-pass signaling of other HER members. The emergence of an anti-panHER approach to blockade of these pathways as a cancer treatment may provide a solution to this resistance. This review aimed to provide an overview of the HER signaling pathways and their involvement in tumor progression and examine the current progress in panHER inhibition.
Methods: Recent literature associated with HER signaling pathways and panHER inhibition was reviewed through PubMed and Medline database, followed by critical comparison and analysis.
Results: Pre-clinical studies and clinical trials of panHER inhibitors show promising results, and the potential to improve patient outcomes in solid cancers.
Conclusion: The use of panHER inhibitors in cancers with HER-family hyper-activation, such as other epithelial cancers and sarcoma, is a new direction to research and has potential in clinical cancer therapy in the future.
Introduction
Cancer continues to pose a great problem and burden on society despite new treatment options. In Australia, it is the most common cause of death and the leading cause of burden of disease (19%) (1). In 2007, the incidence of cancer was 485 cases per 100,000 people and the death rate was 176 per 100,000 people. The most common cancers are prostate cancer, bowel cancer, breast cancer, melanoma of skin, and lung cancer. While surgery, radiotherapy, and chemotherapy have led to major improvements in patient prognosis, newer treatments are needed to more effectively manage this disease in its advanced stage.
Over-expression of the human epidermal growth factor receptor (EGFR/HER1) pathway is a feature of many cancers and a potential therapeutic target. Early research suggested that blocking EGFR/HER1 by its specific inhibitor may have activity in some cancers through tyrosine kinase signaling inhibition (2). However, such a blockade can induce secondary mutation (T790M) (3) and, in addition, has no impact on the other tyrosine kinase receptors within the HER-family (HER2/4) (4). This potentially allows by-pass signaling pathways to remain active; a prime example being the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway, which is normally regulated via HER members (5). Other resistance mechanism like c-MET amplification/over-expression may also weaken the effect of HER-family inhibition (6, 7).
Understanding the mechanism of each HER-family member, their signaling pathways and interactions among them will have a great impact on designing treatment approaches to conquer the resistance of EGFR/HER1 targeted therapy. This review has endeavored to provide an overview of the HER signaling pathways and their involvement in tumor progression and to examine the current progress in HER-family inhibition.
HER-Family Members and Their Signaling Pathways
The HER signaling pathways are normally involved in regulation of cell growth and survival as well as adhesion, migration, differentiation, and other cellular responses. An understanding of these pathways is vital in appreciating the action of panHER inhibitors and anti-HER member antibodies. There are four members of the family, including EGFR/HER1, HER2, HER3, and HER4 (also called erbB-1, erbB-2, erbB-3, and erbB-4, respectively).
Hyper-activation of these receptors culminates with downstream up-regulation of the mitogen activated protein kinase (MAPK), phosphoinositide 3-kinase/AKT (PI3K/AKT), and JAK/STAT pathways (8). In cancer, these pathways are linked to many cellular processes including tumor progression, angiogenesis, metastatic spread, and inhibition of apoptosis (9). This hyper-activation may be due to over-expression of HER ligands, receptors or sustained activation of receptors, as summarized in Table 1.
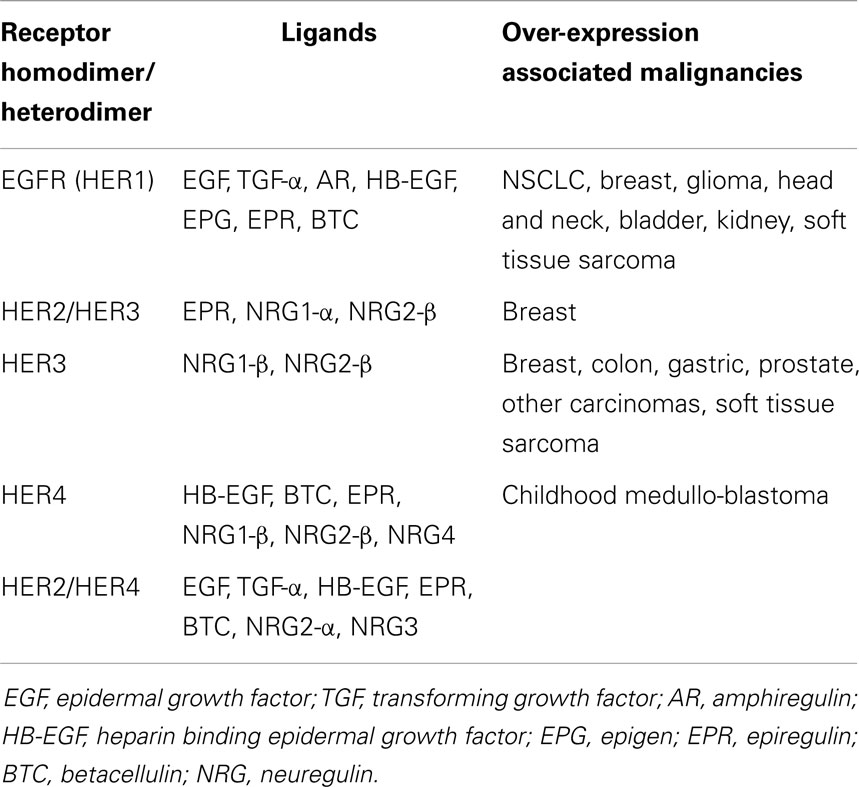
Table 1. HER ligands and receptors.
The HER receptors are composed of a large extra-cellular ligand-binding domain, which has four subdomains (I–IV), followed by a transmembrane domain, a small intracellular juxtamembrane domain preceding the kinase domain, and a C-terminal tail, on which the docking sites for phosphotyrosine-binding effector molecules are found (10). These four receptors form homo-dimers and hetero-dimers, which are associated with instigation of different downstream pathways. Emerging targeted treatment has focused on inhibition of these HER receptors via either external monoclonal antibodies (mAb) or small molecule tyrosine kinase inhibitors (TKIs). Figure 1 presents a simplified summary of the HER signaling pathways.
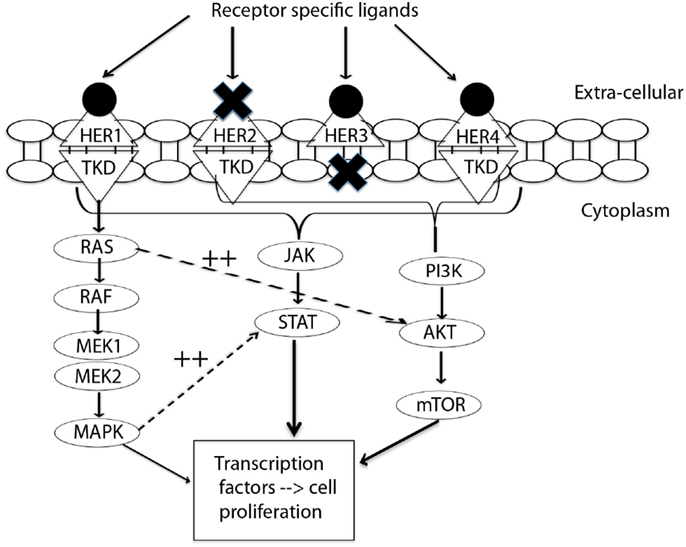
Figure 1. Signal transduction by HER-family. This figure summarizes the interplay between three pathways: MAPK, P13K/AKT, and JAK/STAT. MAPK dramatically enhances transcriptional activation by STAT (11). EGFR/HER1 cannot directly activate the P13K/AKT pathway (12), but it couples to the ras/MAPK pathway as well as to the ras/PI3K/AKT pathway (10). This interplay of pathways forms the source of by-pass resistance to EGFR TKIs. TKD, tyrosine kinase domain; MEK, mitogen activated protein kinase kinase; MAPK, mitogen activated protein kinase; PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin; JAK, janus kinase; STAT, signal transducer and activator of transcription.
EGFR/HER1 Pathways
EGFR/HER1 has been shown to be involved in proliferation and differentiation of epithelial tissues of the skin, lung, pancreas, and gastrointestinal tract (13). Binding of ligands to EGFR leads to auto-phosphorylation of critical tyrosine residues, which serve as attachment sites for various cellular-docking proteins to activate signaling cascades and affect gene transcription (14). Cellular-docking proteins include growth factor receptor bound-2 (GRB2) and Src-homology-2-containing (SHC), both concerned with recruitment of ras and activation of the MAPK cascades (15). Furthermore, MAPK specifically phosphorylates a serine near the C-terminus of most STATs, dramatically enhancing transcriptional activation by STAT (11).
EGFR/HER1 has highly specific recognition sites, and is unable to recruit PI3K (12). As such, EGFR/HER1 cannot directly activate the PI3K/AKT/protein kinase B (PKB) pathway, but it converges with the ras/MAPK pathway as well as via ras to the ras/PI3K/AKT/PKB pathway (10).
HER2 Pathways
HER2 only functions when partnered with either one of the other three type I receptor tyrosine kinase family members (HER1, HER3, or HER4) or type II receptor tyrosine kinase family members (IR or IGF-IR) (16–20). It is a current understanding that HER2 as an orphan receptor cannot directly bind any ligands. Rather, it dimerizes with ligand-bound receptors. However, when expressed as a heterodimer, the combination-receptor shows higher affinity and broader specificity for ligands than other heterodimer couples (21). These advantages are due to slower growth factor dissociation, as well as slow endocytosis of HER2 containing hetero-dimers. Hence, HER2-associated hetero-dimers show a strong proliferative potential as there is simultaneous and prolonged recruitment of multiple signaling pathways including the JAK/STAT pathway (22). Several types of cancer are associated with HER2 over-expression, being most thoroughly studied in breast cancer where there is gene amplification in 15–30% of invasive ductal carcinomas (23).
Trastuzumab is a humanized mAb directed against the extra-cellular domain of the HER2 receptor. It has been shown to have major clinical benefits when used in HER2 overexpressing breast cancer, which otherwise carries a very poor prognosis (24). The mechanism of action is complex, ultimately providing antibody-dependent cellular cytotoxicity (ADCC) against cells overexpressing HER2 as well as down-regulation of signaling pathways including MAPK and AKT (18).
In malignant tumors, constitutively activated HER2 and EGFR/HER1 not surprisingly stimulate many of the same intracellular signaling proteins and pathways as wild-type receptors, such as the MAPK, PI3K/AKT/mTOR pathway, Src kinase, and STAT transcription factors (25). However, differences arise due to impairment of the usual negative regulatory loops functioning in normal cells (26). Without such negative feedback, cells are able to proliferate in an uncontrolled manner.
HER3 Pathways
HER3 is another non-autonomous receptor, which forms functional hetero-dimers with the other members of the HER-family. It has defective kinase activity and only acquires signaling potential, that is, tyrosine phosphorylation, when dimerized with another receptor. Upon heterodimerization, the cytoplasmic domain of HER3 undergoes aforementioned tyrosine phosphorylation allowing recruitment of PI3K as well as SHC, although not GRB2 (27). Additionally, upon heterodimerization, HER3 strongly promotes PI3K activation; especially when binding to its’ preferred partner HER2 (28). Activation of PI3K pathway is closely associated with survival signals allowing avoidance of apoptosis (29).
HER3 is expressed in many cancers, although there is no evidence to suggest gene amplification and over-expression (30). In malignancies such as ovarian cancer where HER3 is frequently expressed (53.4%), expression has been associated with a shorter survival time (3.31 vs. 1.8 years median survival time: low HER3 expression and HER3 over-expression, respectively) (31).
HER4 Pathways
HER4 has many characteristics likening it to EGFR/HER1 including recruitment of GRB2, SHC, and STAT5. One isoform of HER4 is able to activate the PI3K/AKT pathway (32). HER4 tends to have low expression in breast and prostate cancers, yet over-expression is frequently seen in other cancers (e.g., >50% childhood medullo-blastomas) (33). In such cases, heterodimerization with HER2 is correlated with malignancy, suggesting increased autocrine or paracrine loop signaling involving Neuregulin 1 (NRG1) (34).
HER Pathway Synergy
As referred to above, co-operation between HER receptors has been observed in oncogenic transformation, both in vitro in cultured cells and in primary human tumors. For example, HER3 expression increases HER2-mediated transformation and tumorigenic growth in NIH3T3 cells (35). Additionally, co-expression of EGFR/HER1 or HER2 is necessary for NRG-1-induced transformation of fibroblasts by HER4 (8). The enhanced proliferative activity of cells expressing multiple HER receptors are presumably due to the interaction and strength of signaling in HER receptor combinations (36). HER pathway synergy results in additional autocrine loops driven by co-expression of multiple HER proteins. Such pathways are closely linked with tumor progression (8). This synergy of multiple pathways also provides an “escape route” whereby blocking specific member of the HER-family results in functional compensation by parallel HER pathways. It follows that panHER inhibition may overcome this shortcoming of single targeted therapies. Table 2 summarizes the regulation mechanisms of the HER signaling pathways.

Table 2. HER signaling regulation.
Single HER Inhibitors
Single molecular tyrosine kinase inhibitors (TKIs) are a class of extensively investigated and developed therapeutics that are effective alone and in combination with conventional chemotherapeutics in treating a variety of cancer subtypes. Their mechanism of action is via inhibition of the single members of the HER-family culminating in inhibition of downstream signaling (37, 38). More specifically, TKIs such as erlotinib take effect via reversible binding to the kinase catalytic domain of EGFR/HER1 preventing auto-phosphorylation and downstream activation (39). Alternatively, mAbs, such as trastuzumab (40) and cetuximab (41), bind to the extra-cellular segment of HER-family members leading to down-regulation.
The success of reversible TKIs has been most marked in patients harboring an EGFR/HER1 gene mutation that increases TKI sensitivity. Analysis of non-small cell lung cancer (NSCLC) patients has shown that 70–80% of EGFR/HER1 gene-mutated patients are responsive to TKIs compared with 10–20% of EGFR/HER1 wild-type patients (42). In contrast, mAbs mode of action varies from those of small molecule inhibitors. Accordingly their inhibitory effects are less dependent upon activation-inducing mutations in the receptor and even downstream changes. This may explain the increased therapeutic impact compared to TKIs on patients with breast and colon cancers (41).
Shortcomings of single HER inhibitor treatment and strategies to overcome resistance
Despite successful drug development targeting single HER receptors, patient response has been less than expected, and development of resistance has been a major issue. The primary efficacy of erlotinib and gefitinib in malignancies harboring an EGFR/HER1 activating mutation is well established. However, this efficacy has shown to be short lived with patients developing secondary mutations conferring drug resistance. These mutations occur at a median time of 12 months from commencement of treatment (42). The EGFR/HER1 T790M resistant mutation involves substitution of threonine 790 with methionine, conferring drug resistance by increasing ATP affinity. Inhibition is initially possible due to the compromised ATP affinity resulting from EGFR/HER1 mutations, and as such, the T790M mutation culminates in restoration of wild-type level ATP affinity and a decreased affinity of TKIs targeting HER receptors. The T790M mutation is believed to account for 50% of the resistance to the aforementioned drugs (43). Other secondary mutations specific to EGFR/HER1 resistance development include D761Y, L747S, and T854A (44). However, these have only been reported with low frequency and are not the focus of treatments to overcome resistance. Recently, several third-generation, irreversible, selective EGFR inhibitors such as AZD9291 and CO1686 have shown promise in pre-clinical studies and provided hope for patients with advanced lung cancers that have become resistant to gefitinib or erlotinib. AZD9291 and CO1686 inhibited both the activating (L858R) and resistant (T790M) EGFR mutations in cell culture and in animal models. Conversely, these drugs did not inhibit the wild-type EGFR that is present in normal skin and gut cells, thereby reducing the side effects encountered with existing reversible EGFR inhibitors (45–48). Both compounds are undergoing clinical trials and the preliminary data have shown partial responses with good tolerability, according to RECIST (response evaluation criteria in solid tumors), which is a set of published rules that define when cancer patients improve (“respond”), stay the same (“stable”), or worse (“progression”) during treatment (49, 50).
In addition to EGFR mutations, MET amplification/over-expression was also reported as another common EGFR TKI resistance mechanism in lung cancer. MET amplification was observed in 21% of patients with acquired resistance to EGFR TKI (in only 3% of untreated patients) and developed resistance to gefitinib by driving HER3-dependent activation of PI3K in a gefitinib-sensitive lung cancer cell line (HCC827) (6, 7), as well as by activating STAT3 transcription factor directly, through an SH2 domain (51). Targeting MET can enhance EGFR inhibition. EGFR inhibitors combined with MET tyrosine kinase inhibitors such as SU11274 and PHA665752 or MET antibody such as DN-30 synergistically inhibited cell proliferation and promoted apoptosis in NSCLC (52, 53). A number of MET inhibitors are currently in late phase clinical trials (54, 55).
Trastuzumab is a monoclonal antibody against HER2 and was approved for the treatment of HER2-overexpressed metastatic breast cancer in 1998 (16). However, the majority of patients who initially responded to trastuzumab showed partial or complete resistance and disease progression in <1 year post treatment (18). Multiple mechanisms of trastuzumab resistance have been reported (56) including constitutive activation of downstream PI3K/AKT signaling through PTEN down-regulation or PIK3CA hyper-activating mutations, lack of an effective ADCC immune response as well as increased expression or compensatory signaling through other receptor tyrosine kinases (IGF-IR, EGFR, or HER3). IGF-IR signaling (17, 57) was demonstrated to contribute the molecular resistance mechanism to HER2-targeted therapy via cross-talk between IGFR-IR and HER2, which activated the PI3K/AKT and Ras/Raf/MAPK signaling cascades. In addition, trastuzumab only blocks HER2-mediated signaling, but it may not inhibit signaling mediated from other HER receptors (such as EGFR/HER3 or EGFR/EGFR) (18). Therefore, inhibiting multiple HER-family receptors concurrently may be more effective than trastuzumab-based therapy alone.
Resistance to monoclonal antibody (e.g., cetuximab) or small molecule receptor inhibitor (gefitinib or dacomitinib) therapy is most frequently through mutations that result in downstream activation of the pathway eliminating the opportunity for upstream blockade of external signaling; for example, cetuximab blockade to EGFR/HER1. Karapetis et al. (58) in a randomized trial of single agent cetuximab vs. best supportive care showed that a k-ras mutation was a predictor for resistance to cetuximab in patients. Drugs that target molecules downstream of k-ras (e.g., b-raf or MEK inhibitors) are needed to effectively manage these patients.
Ciardello et al. (59) have shown that HER2 gene amplification is another mechanism of resistance to EGFR/HER1 targeted therapy. Their study revealed that resistance to cetuximab in metastatic colorectal cancer “xenopatients” (patient-derived xenografts) was frequently coupled with HER2 over-expression. When treated with EGFR and HER2 inhibition, long lasting tumor regression suggested that panHER inhibition will have clinical benefit in such patients. In addition, blocking of EGFR/HER1 has no impact on the HER2, HER4, and JAK/STAT pathways (by-pass signaling pathways), in which activation of STAT3 is associated with tumor growth and malignancy (5, 60). HER2 acts as a STAT3 co-activator for cyclin D1 promoter activation to promote tumor proliferation (61).
This gives context to the potential benefit of panHER inhibitors. These irreversible TKIs overcome resistance by binding covalently to HER receptors, which inhibits tumor growth in cancers, which are driven by HER heterodimerization and co-expression (62).
panHER Inhibition Approach
In response to the clinical limitations of EGFR/HER1 TKIs, the role of panHER inhibitors is gaining increased attention. In cancers responsive to HER pathway stimulation, as discussed above, panHER inhibition could provide a means of blocking signaling pathways that are not associated with EGFR/HER1 activating mutations or gene amplification (hence lower response rates to reversible TKIs). While panHER inhibitor therapy seems to be most effective in patients harboring an activating EGFR mutation, it also shows therapeutic benefit in patients who do not have a HER activating mutation. Pre-clinical studies have shown inhibition of wild-type EGFR/HER1 as well as wild-type HER2 using novel panHER inhibitors (63). Late stage cancers such as lung, breast, head and neck, gastric, and colorectal, which are associated with over-activity of the HER pathways, may all benefit from this new class of drugs (9).
Effects of a range of panHER inhibitors targeting EGFR/HER1, HER2, and HER4 have been explored including AC-480 (formerly known as BMS 599626), HM781-36B, Canertinib (CI-1033), Nertinib (HKI272), afatinib (BIBW2992), and dacomitinib (PF299804). Multi-target TKIs such as lapatinib (an inhibitor of HER1 and HER2) are already approved and in regular use in clinical settings, however, resistance has become apparent via escape route signaling (64). A panHER approach demonstrated potential to prevent escape route resistance via the JAK/STAT pathway. AC-480 and HM 781-36B have only just reached or completed phase I testing, respectively (65, 66). Canertinib and Nertinib have already entered phase II (67, 68), while dacomitinib and afatinib have been extensively tested in a range of cancer types and are currently undergoing Phase III studies (69). Accordingly, dacomitinib and afatinib will be introduced in the sessions that follow and Tables 3 and 4 include examples of targeting panHER for cancer therapy. Both compounds have two attractive features including irreversible binding to the targeted receptors to extend the efficacy of an agent and overcome resistance and, in addition, broad inhibition of all cancer-relevant HER-family homo-dimers and hetero-dimers to potentially improve efficacy and limit alternative signaling from receptor cross-talk.
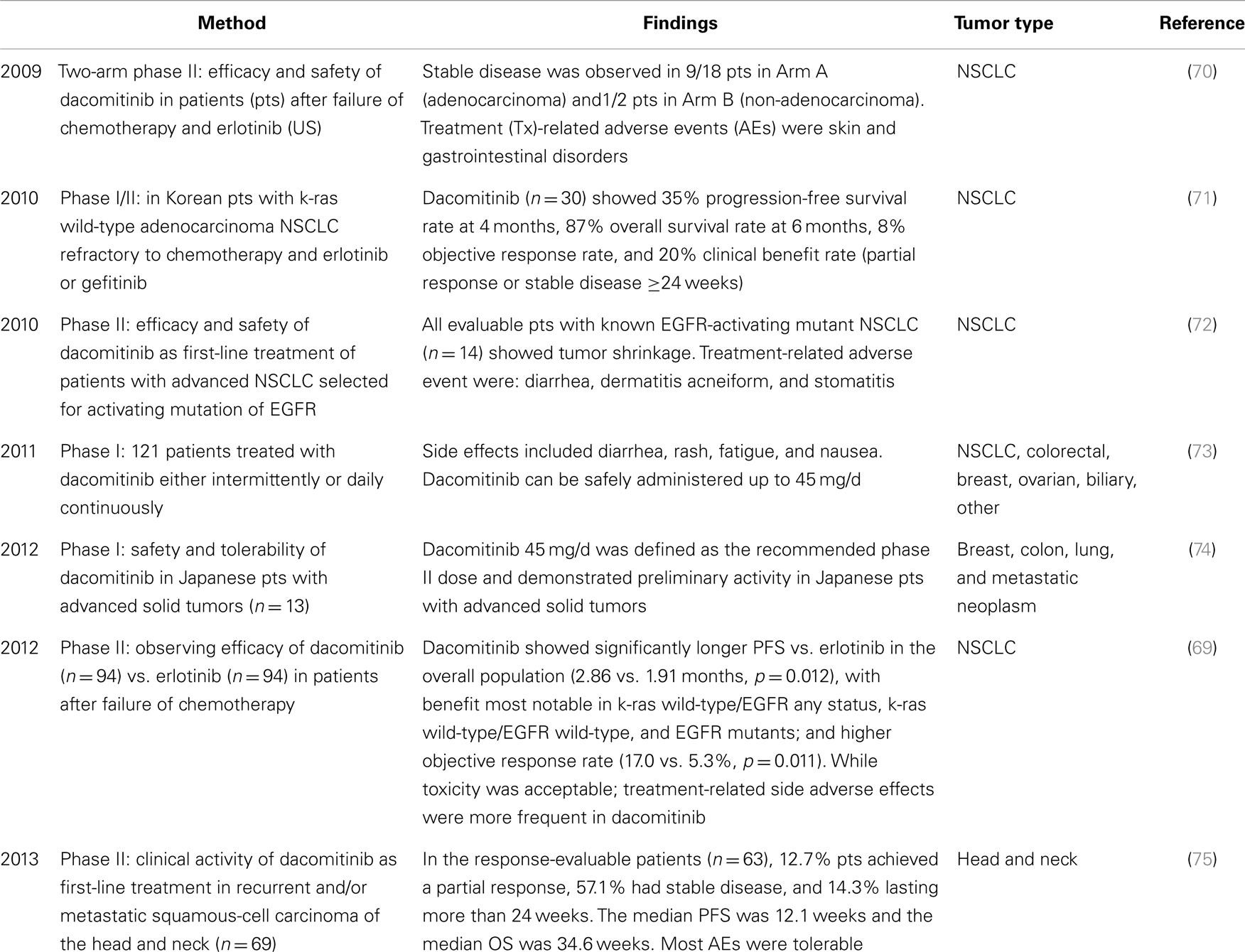
Table 3. Clinical trials with dacomitinib.
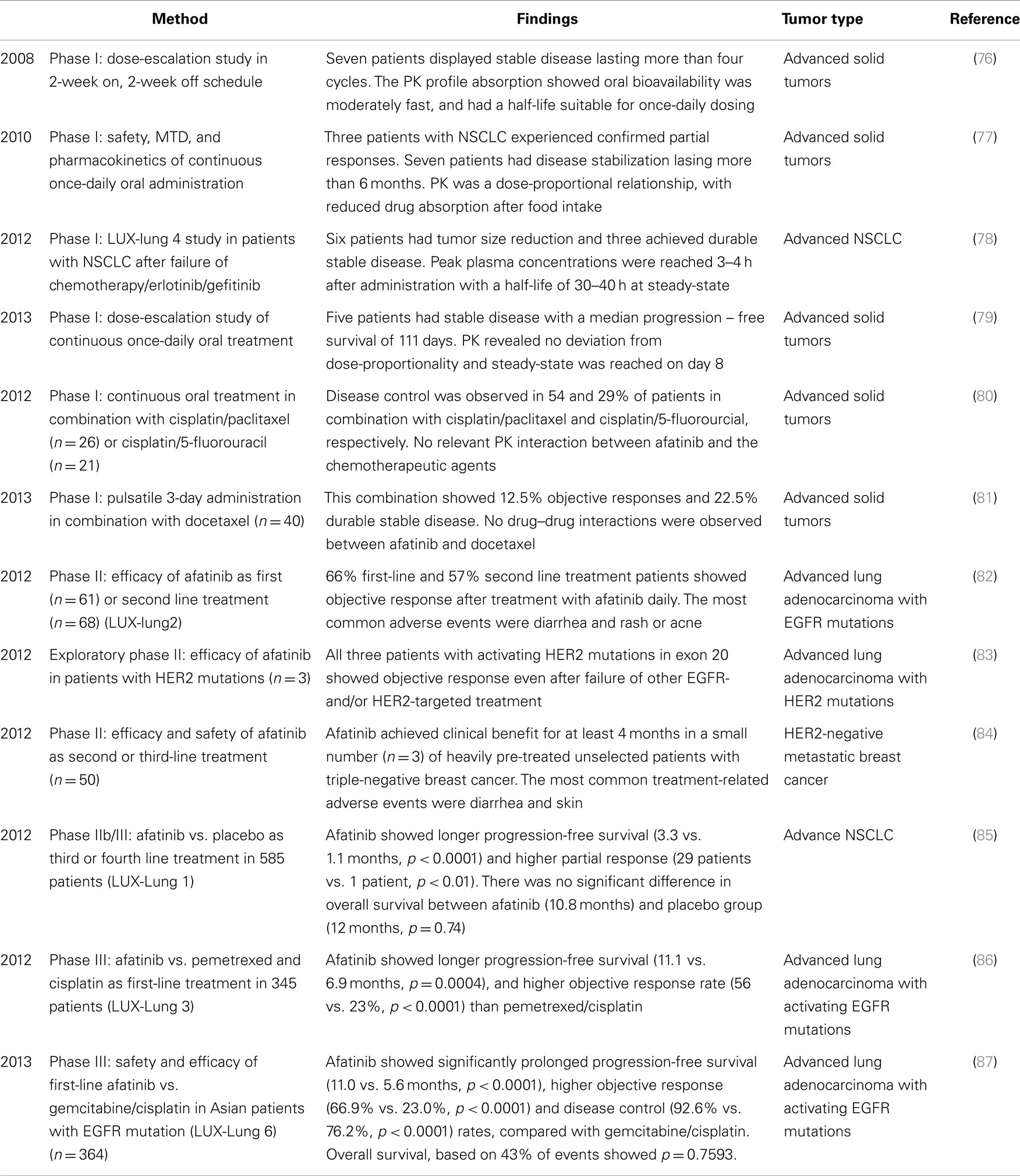
Table 4. Clinical trials with afatinib.
Representative panHER Inhibitor: Dacomitinib
Dacomitinib (PF299804) is a second-generation small molecule tyrosine kinase irreversible panHER inhibitor, which is currently involved in Phase III trials for NSCLC and Phase II for Head and Neck cancer (published clinical trials involving dacomitinib to date are summarized in Table 3). It comprises a structurally related quinazoline based core scaffold, as well as an electrophilic motif that covalently binds Cys-797 of EGFR (88). Dacomitinib is potent and highly selective for the EGFR/HER1, HER2, and HER4 members of the HER (erbB) signaling pathway (73). It is believed to irreversibly inhibit HER tyrosine kinase activity through binding at the ATP site and covalent modification of nucleophilic cysteine residues in the catalytic domains of HER-family members.
Pre-clinical studies
Dacomitinib is a highly effective inhibitor of EGFR activating and EGFR T790M acquired resistant mutations as well as the wild-type HER2, gefitinib-resistant oncogenic HER2 mutation, and HER2 amplification both in vitro and in vivo in a broad range of human cancer cell lines including lung cancer, gastric cancer, biliary tract cancer, breast cancer, head and neck cancer, ovarian carcinoma, and squamous-cell carcinoma (63, 89–93). Treatment with dacomitinib reduced phosphorylation of HER-family members (EGFR, HER2, and HER4) and downstream AKT and Erk pathways, as well as induced apoptosis and caused G0/G1 arrest (91, 92). Furthermore, excellent pharmacodynamic effects (such as high bioavailability, long half-life, and large volume of distribution) with this compound were observed across species including rats, monkeys, and dogs (93). Despite apparent success in treating tumors with an EGFR/HER1 T790M resistant mutation, pre-clinical studies (88) using cell lines with EGFR T790M showed that dacomitinib resistance can develop both in vitro and using a xenograft model in vivo by EGFR/HER1 T790M amplification. The activation of IL-6R/JAK1/STAT3 signaling has been identified as a mechanism of de novo resistance to irreversible panHER inhibitors in NSCLC with T790M resistant mutation (94).
Phase I studies
Three Phase I studies in patients with advanced malignant solid tumors in United States, Japan, and South Korea indicated that dacomitinib has attractive pharmacokinetics and metabolism including great bioavailability, long half-life (59–85 h with dosing ranging from 30 to 60 mg), large volume of distribution (2,610 L with the dosing of 45 mg), and low clearance (23.7–32 L/h across the dosing of 15–45 mg) (71, 73, 74). These studies suggested the maximum tolerated dose (MTD) was 45 mg/d, and both continuous and intermittent treatment schedules were well-tolerated. However, this has revealed that concentrations, which are clinically achievable may not allow maximum inhibition of cells harboring the T790M resistant mutation. Treatment-related adverse effects (diarrhea, acne, and rash) associated with EGFR/HER1 blocking TKIs were more common in dacomitinib compared with erlotinib (95).
Phase II studies
In two subsequent phase II trials in NSCLC as a third-line treatment, dacomitinib was well-tolerated and showed encouraging activity in patients after failure of prior chemotherapy and erlotinib or gefitinib treatment (70, 71). A randomized Phase II trial of 188 patients with advanced NSCLC demonstrated a significant improvement of progression-free survival (PFS) in patients receiving dacomitinib (2.86 months) compared to erlotinib (1.91 months) (69). In two Phase II studies of dacomitinib as a first-line therapy, dacomitinib demonstrated clinical benefits in patients with recurrent and/or metastatic squamous-cell carcinoma of the head and neck (RM-SCCHN) and advanced NSCLC (72, 75).
Dacomitinib, currently under phase III studies (JBR-26 and NCT01360554), appears to be an example of how the panHER approach to targeted therapy may hold promise for improving targeted treatment outcomes. Activity demonstrated in NSCLC may also have an application within other cancer types with positive HER-family expression such as other epithelial cancers (96) and sarcoma (97).
Representative panHER Inhibitor: Afatinib
Afatinib, an aniline–quinazoline derivative with a functional Michael acceptor group, is designed to covalently bind the catalytic domains of EGFR (Cys-797), HER2 (Cys805), and HER4 (Cys803) and irreversibly block enzymatically active HER-family members (98).
Pre-clinical studies
In cell-free in vitro kinase assays, afatinib potently inhibited wild-type EGFR (EC50 = 0.5 nM), HER2 (14 nM), and HER4 (1 nM), as well as oncogenic L858R activating mutant EGFR (0.4 nM) and L858R-T790M resistant mutant EGFR (10 nM) (98, 99). Afatinib inhibited auto-phosphorylation of HER members and proliferation in cancer cell lines representing different mutational status of EGFR and HER2, including wild-type EGFR (A431), activating mutant EGFR (PC-9 and H3255, EC50 = 0.4 and 0.5 nM, respectively), L858R-T790M mutant EGFR (NCI-H1975), L858R-T854A mutant EGFR (transfected 293T), and HER2 amplification (BT474) with EC50 below 100 nM, whereas NCI-H1975, T854A transfected 293T, and BT474 were resistant to erlotinib and/or gefitinib (98, 100). Furthermore, afatinib induced tumor regression in a broad spectrum of xenograft models carrying epidermoid carcinoma A431 with wild-type EGFR, gastric cancer NCI-N87 with HER2 over-expression, lung cancer NCI-H1975 with L858R-T790M mutant EGFR, and adenosquamous lung tumor with HER2YVMA mutation, with superior activity over erlotinib (99, 101). In a panel of seven human pancreatic tumor cell lines, afatinib indicated greater efficacy in antiproliferation and signaling blockage (phosphorylation of EGFR, MAPK, and AKT) (102). In vivo, daily administration of 15 mg/kg afatinib showed potent antitumor activity in the BxPC-3 human pancreatic xenograft model (102).
Clinical trials using Afatinib are listed in Table 4.
Phase I studies
Several Phase I dose-escalation studies with afatinib indicated that afatinib is well-tolerated (76–79, 103). The recommended dose in 2-week on, 2-week off schedule is 70 mg/d (76), while in once-daily oral treatment is 40–50 mg/d in advanced solid tumors and in advanced NSCLC after failure of chemotherapy/erlotinib/gefitinib (LUX-Lung4) (77–79). The most frequent treatment-related adverse events were diarrhea, mucosal inflammation, and skin rash. A pharmacokinetic study in healthy male volunteers with afatinib showed the main route of excretion is via feces, and it undergoes minimal metabolism (103).
Phase II studies
LUX-Lung 2, a single-arm phase II study in 129 patients with lung adenocarcinoma containing activating EGFR mutations within exons 18-21, showed antitumor activity after treatment as first or second line with afatinib daily 40 or 50 mg (82). Afatinib was more effective (objective response: 66%) in patients with the common EGFR mutations (deletion 19 and L858R) compared to patients with less common mutations (39%) (82). In addition, afatinib indicated therapeutic activity in three patients with lung adenocarcinoma and a non-smoking history, whose tumors exhibited activating HER2 mutation in exon 20 (83). However, afatinib had limited activity in HER2-negative breast cancer (84). None of the fifty patients achieved an objective response, while three patients with triple-negative metastatic breast cancer had stable disease for more than four treatment courses. This indicated that further examination of afatinib in patients should select population with EGFR/HER2 mutations or HER receptor/ligand over-expression or activation to increase the likelihood of a meaningful clinical benefit.
Phase III studies
LUX-Lung 1 was a randomized, double-blind phase IIb/III study comparing afatinib with placebo in 585 NSCLC patients after failure of chemotherapy/erlotinib/gefitinib (85). Afatinib failed to show a difference between groups at its primary endpoint and overall survival, although the PFS findings were promising. One of the potential reasons is that the number of patients with EGFR mutations was unknown, since EGFR mutation status was not required for study entry. Accordingly, LUX-Lung 3 examined the efficacy of afatinib compared with pemetrexed and cisplatin as first-line treatment for patients with advanced lung adenocarcinoma harboring EGFR-activating mutations. Encouragingly, afatinib led to a prolonged PFS, more significantly with common Del19 and L858R mutations (86, 87).
In July 2013, afatinib was approved for the first-line treatment of patients with metastatic NSCLC with EGFR exon 19 deletions or exon 21 (L858R) substitution mutations by the US Food and Drug Administration (104).
panHER Combination Therapy
Combination therapy involving panHER inhibitors is also under investigation. This varies from combinations of targeted therapies with either radiation or chemotherapy or combinations of targeted therapies directed against different aspects of the EGFR pathway.
Following radiotherapy, tissue recovery is associated with increased generation of HER ligands to promote the survival and cell proliferation that HER signaling provides. One potential outcome of this tissue repair event is tumor recurrence in patients with high HER expression. It follows biologically that blocking HER pathways simultaneously may impair this process and thus enhance radiation effectiveness (105). Torres et al. (106) showed that panHER inhibitor AC-480 increased tumor growth delay (enhancement factor = 1.94) when used in combination with radiotherapy in head and neck squamous-cell carcinoma in mice.
panHER inhibitors in combination with chemotherapy have successfully showed synergistic results in both pre-clinical studies and clinical trials. Combination therapy in NSCLC cells harboring a T790M mutation both in vitro and in vivo showed (107) that gefitinib (EGFR/HER1 TKI) used in combination with antimetabolites, either fluorouracil (5FU) or pemetrexed, had an antagonistic growth inhibitory effect in vitro. In contrast, afatinib both in vitro and in vivo was synergistic with these drugs. In two phase I studies of afatinib combination with chemotherapy, the MTD for afatinib was 20 mg with cisplatin/paclitaxel, 30 mg with cisplatin/5-fluorouracil, and 90 mg for 3 days after docetaxel in advanced solid tumors (80, 81). Both studies showed antitumor activities with a manageable adverse-event profile. CI-1033 is another panHER inhibitor with promising lab data showing it synergizes with cisplatin in vitro (108). This is believed to be because cisplatin inhibits key genes in cell survival when the EGFR pathway is blocked simultaneously. CI-1033 has undergone a phase I trial in combination with docetaxel in advanced solid tumor patients. It appeared to be safe with acceptable side effects following an intermittent administration schedule (109). CI-1033 has also been examined in combination with paclitaxel and carboplatin in advanced NSCLC patients (110). Phase I studies showed it was well-tolerated as well as having possible synergistic antitumor trends, which await confirmation in phase II studies. Lapatinib (HER1/HER2 TKI) has been approved by the FDA for routine clinical use in combination with capecitabine (prodrug of 5FU). This is on the basis of phase III studies showing improved PFS in HER2 positive breast cancer (12.0 weeks in combination therapy vs. 8.4 weeks in lapatinib alone) (111).
Another approach to target the HER-family is through concurrent treatment with new bispecific antibodies such as MM-111 and an HER2 antibody or small molecular inhibitor. MM-111 targets the HER2/HER3 heterodimer with specificity and avidity by docking onto HER2 and subsequently binding to HER3 and blocking Heregulin-induced activation of HER3, therefore, it is more effective at inhibiting HER3 activation than a HER2 or HER3 monoclonal antibody (trastuzumab and pertuzumab) and TKI (lapatinib) (112). In addition, combining MM-111 with other HER2-targeted agents (trastuzumab and/or lapatinib) and chemotherapy (such as paclitaxel) synergistically inhibit tumor cell growth and prevent the development of HER3-driven drug resistance in gastric and breast cancer cells and xenografts (113–115). Currently, a Phase II clinical trial combining MM-111 with paclitaxel and trastuzumab is being investigate (116).
The development of bispecific antibodies, along with the third-generation irreversible small molecular inhibitors, have improved the efficacy to target HER-family, even though they clearly differ in their mode of action at target level: for example, MM-111, consisting of fully human anti-HER2 and anti-HER3 single chain antibody moieties linked by modified human serum albumin, targets the extra-cellular domain of HER2/HER3 heterodimer and promotes the formation of inactive trimetric complexes. In comparison, the third-generation EGFR TKIs (AZD9291 and CO1686) act on the intracellular ATP binding domain and irreversibly inhibit both the activating and resistant EGFR mutations (45, 48, 112). Based on our current knowledge, there is no apparent distinction between antibodies and TKIs regarding HER-family targeting effects. However, their differences could be exploited in certain clinical situations. For example, patients with activating and/or resistant EGFR mutations should be considered for the use of third-generation EGFR TKIs, whereas patients with over-expression of HER2/HER3 should be treated by bispecific antibody MM-111. Generally, TKIs as small molecules are able to penetrate the blood-brain barrier, indicating that these agents may be therapeutic in patients with primary or metastatic central nervous system disease (117). By contrast more clarity about the potential for ADCC to have a role in antibody-mediated antitumor activity may encourage development of this as the favored therapeutic approach. Especially FcγR genotype may be a predictor of efficacy (118). Indeed rather than focusing on identifying which approach is optimal, it is possible that inhibition of multiple sites of HER receptor activation using both antibody and TKI will prove to be the best way forward. A recent example showed an enhanced antitumor activity when HER-antibody trastuzumab was combined with an EGFR TKI gefitinib or erlotinib (117).
Soft Tissue Sarcomas as a Novel Target
EGFR/HER1 is frequently overexpressed in soft tissue sarcoma, with 78% of patient tissue samples showing positive EGFR/HER1 expression in a study by Yang et al. (119). Early data suggest that blocking EGFR/HER1 by its specific inhibitor may have activity in sarcomas through tyrosine kinase signaling inhibition (14). However, such blockade has no impact on the HER2, HER4, and JAK/STAT pathways, in which activation of STAT3 is associated with tumor growth (120). This problem with single receptor blockage is also confirmed in clinical studies (121), showing that erlotinib was not active in malignant peripheral nerve sheath tumors (MPNST), which are relatively resistant to chemotherapy. Similarly, cetuximab has minimal activity in EGFR positive advanced sarcoma (122).
Recently, a phase II trial in osteosarcoma patients with HER2 over-expression showed that blocking HER2 using trastuzumab in combination with cytotoxic chemotherapy had no clinical benefit (123). The promising data discussed earlier regarding panHER combination therapy could justify a trial in such disease groups to overcome the failure of single targeting.
Conclusion
Targeted therapy against the HER-family individual members or panHER-family has shown potential to improve prognosis in sensitive patients in some tumor types. One mechanism of resistance to EGFR/HER1 therapies is HER2, 3, 4, and JAK/STAT by-pass signaling. Pre-clinical studies and clinical trials of panHER inhibitors show promising results, and the potential to improve patient outcomes in NSCLC and head and neck cancers. In some cancer patients, the panHER inhibition approach met resistance from HER-independent JAK/STAT by-pass signaling. The use of STAT3 blockage in combination with panHER inhibition in cancers including epithelial cancers and sarcoma, with HER-family hyper-activation and resistance to panHER inhibitor, is a new direction to explore and has potential in clinical cancer therapy in the future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
EGFR, the epidermal growth factor receptor; GRB2, growth-factor-receptor bound-2; HER, the human epidermal growth factor receptor; JAK/STAT, the Janus kinas/signal transducer and activator of transcription; MAPK, the mitogen activated protein kinase; MEK, mitogen activated protein kinase kinase; MTD, maximum tolerated dose; mTOR, mammalian target of rapamycin; NSCLC, non-small cell lung cancer; PI3K/AKT, the phosphoinositide 3-kinase/AKT; PKB, protein kinase B; SHC, Src-homology-2-containing; TKD, tyrosine kinase domain; TKIs, tyrosine kinase inhibitors.
References
1. Begg S, Vos T, Barker B, Stevenson C, Stanley L, Lopez AD. The Burden of Disease and Injury in Australia 2003. Australian Institute of Health and Welfare (2007). Available from: http://www.aihw.gov.au/publication-detail/?id=6442467990
2. Normanno N, Maiello MR, De Luca A. Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs): simple drugs with a complex mechanism of action? J Cell Physiol (2003) 194(1):13–9. doi:10.1002/jcp.10194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Ogino A, Kitao H, Hirano S, Uchida A, Ishiai M, Kozuki T, et al. Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res (2007) 67(16):7807–14. doi:10.1158/0008-5472.CAN-07-0681
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Kong A, Calleja V, Leboucher P, Harris A, Parker PJ, Larijani B. HER2 oncogenic function escapes EGFR tyrosine kinase inhibitors via activation of alternative HER receptors in breast cancer cells. PLoS One (2008) 3(8):e2881. doi:10.1371/journal.pone.0002881
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Wang X, Crowe PJ, Goldstein D, Yang J-L. STAT3 inhibition, a novel approach to enhancing targeted therapy in human cancers (review). Int J Oncol (2012) 41(4):1181–91. doi:10.3892/ijo.2012.1568
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316(5827):1039–43. doi:10.2307/20036288
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Bean J, Brennan C, Shih J-Y, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A (2007) 104(52):20932–7. doi:10.1073/pnas.0710370104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Zaczek A, Brandt B, Bielawski KP. The diverse signaling network of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol Histopathol (2005) 20(3):1005–15.
9. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol (2001) 2(2):127–37. doi:10.1038/35052073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol (2006) 7(7):505–16. doi:10.1038/nrm1962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Rawlings JS, Rosler KM, Harrison DA. The JAK/STAT signaling pathway. Cell Sci Glance (2004) 117(8):1281–3. doi:10.1242/jcs.00963
12. Oliviera-Cunha M, Newman WG, Siriwardena AK. Epidermal growth factor receptor in pancreatic cancer. Cancers (Basel) (2011) 3(2):1513–26. doi:10.3390/cancers3021513
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Yano S, Kondo K, Yamaguchi M, Richmond G, Hutchison M, Wakeling A, et al. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res (2003) 23(5A):3639–50.
14. Zahorowska B, Crowe PJ, Yang JL. Combined therapies for cancer: a review of EGFR-targeted monotherapy and combination treatment with other drugs. J Cancer Res Clin Oncol (2009) 135(9):1137–48. doi:10.1007/s00432-009-0622-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Mor A, Philips MR. Compartmentalized Ras/MAPK signaling. Annu Rev Immunol (2006) 24:771–800. doi:10.1146/annurev.immunol.24.021605.090723
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Nahta R. New developments in the treatment of HER2-positive breast cancer. Breast Cancer Targets Ther (2012) 4:53–64. doi:10.2147/BCTT.S24976
17. Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res (2005) 65(23):11118–28. doi:10.1158/0008-5472.CAN-04-3841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol (2006) 3(5):269–80. doi:10.1038/ncponc0509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene (2007) 26(25):3637–43. doi:10.1038/sj.onc.1210379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Browne BC, Crown J, Venkatesan N, Duffy MJ, Clynes M, Slamon D, et al. Inhibition of IGF1R activity enhances response to trastuzumab in HER-2-positive breast cancer cells. Ann Oncol (2011) 22(1):68–73. doi:10.1093/annonc/mdq349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Rubin I, Yarden Y. The basic biology of HER2. Ann Oncol (2001) 12(Suppl 1):S3–8. doi:10.1023/A:1013026527758
22. Liu J, Kern JA. Neuregulin-1 activates the JAK-STAT pathway and regulates lung epithelial cell proliferation. Am J Respir Cell Mol Biol (2002) 27(3):306–13. doi:10.1165/rcmb.4850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Tanner M, Jarvinen P, Isola J. Amplification of HER-2/neu and topoisomerase IIalpha in primary and metastatic breast cancer. Cancer Res (2001) 61(14):5345–8.
24. Hudis CA. Trastuzumab – mechanism of action and use in clinical practice. N Engl J Med (2007) 357(1):39–51. doi:10.1056/NEJMra043186
25. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst (2004) 96(12):926–35. doi:10.1093/jnci/djh166
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol (2009) 21(2):177–84. doi:10.1016/j.ceb.2008.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Way TD, Lin JK. Role of HER2/HER3 co-receptor in breast carcinogenesis. Future Oncol (2005) 1(6):841–9. doi:10.2217/14796694.1.6.841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Hsieh AC, Moasser MM. Targeting HER proteins in cancer therapy and the role of the non-target HER3. Br J Cancer (2007) 97(4):453–7. doi:10.1038/sj.bjc.6603910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Fresno Vara JA, Casado E, de Castro J, Cejas P, Belda-Iniesta C, Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev (2004) 30(2):193–204. doi:10.1016/j.ctrv.2003.07.007
30. Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res (2010) 16(5):1373–83. doi:10.1158/1078-0432.CCR-09-1218
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Tanner B, Hasenclever D, Stern K, Schormann W, Bezler M, Hermes M, et al. ErbB-3 predicts survival in ovarian cancer. J Clin Oncol (2006) 24(26):4317–23. doi:10.1200/JCO.2005.04.8397
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Karamouzis MV, Badra FA, Papavassiliou AG. Breast cancer: the upgraded role of HER-3 and HER-4. Int J Biochem Cell Biol (2007) 39(5):851–6. doi:10.1016/j.biocel.2006.11.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Gilbertson RJ, Perry RH, Kelly PJ, Pearson AD, Lunec J. Prognostic significance of HER2 and HER4 coexpression in childhood medulloblastoma. Cancer Res (1997) 57(15):3272–80.
34. Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J (2000) 19(13):3159–67. doi:10.1093/emboj/19.13.3159
35. Zhang K, Sun J, Liu N, Wen D, Chang D, Thomason A, et al. Transformation of NIH 3T3 cells by HER3 or HER4 receptors requires the presence of HER1 or HER2. J Biol Chem (1996) 271(7):3884–90. doi:10.1074/jbc.271.7.3884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Holbro T, Hynes NE. ErbB receptors: directing key signaling networks throughout life. Annu Rev Pharmacol Toxicol (2004) 44:195–217. doi:10.1146/annurev.pharmtox.44.101802.121440
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res (2014) 79:34–74. doi:10.1016/j.phrs.2013.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Shor AC, Agresta SV, D’Amato GZ, Sondak VK. Therapeutic potential of directed tyrosine kinase inhibitor therapy in sarcomas. Cancer Control (2008) 15(1):47–54.
39. Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther (2005) 315(3):971–9. doi:10.1124/jpet.105.084145
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol (2007) 18(6):977–84. doi:10.1093/annonc/mdl475
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Lenz HJ. Anti-EGFR mechanism of action: antitumor effect and underlying cause of adverse events. Oncology (2006) 20(5 Suppl 2):5–13.
42. Kosaka T, Yamaki E, Mogi A, Kuwano H. Mechanisms of resistance to EGFR TKIs and development of a new generation of drugs in non-small-cell lung cancer. J Biomed Biotechnol (2011) 2011:165214. doi:10.1155/2011/165214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A (2008) 105(6):2070–5. doi:10.1073/pnas.0709662105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Nguyen KS, Kobayashi S, Costa DB. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway. Clin Lung Cancer (2009) 10(4):281–9. doi:10.3816/CLC.2009.n.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Cross D, Ashton S, Nebhan C, Eberlein C, Finlay MRV, Hughes G, et al. eds. AZD9291: an irreversible, potent and selective third generation tyrosine kinase inhibitor (TKI) targeting EGFR activating (EGFRm+) and resistance (T790M) mutations in advanced lung adenocarcinoma. Mol Cancer Ther (2013) 12(11 Suppl):A109. doi:10.1158/1535-7163.TARG-13-A109
46. Ballard P, Ashton S, Cross D, Dimelow R, Yates J, eds. Integrating the pre-clinical pharmacokinetic, pharmacodynamics, and efficacy data for AZD9291, an oral, irreversible inhibitor of EGFR activating (EGFRm+) and resistant (EGFRm+/T790M) mutations and an active metabolite to predict the human pharmacokinetics and potential efficacious dose in patients. Mol Cancer Ther (2013) 12(11 Suppl):B212.
47. Finlay MRV, Anderson M, Ashton S, Ballard PG, Bradbury RH, Butterworth S, et al. eds. Discovery of and first disclosure of the clinical candidate AZD9291, a potent and selective third-generation EGFR inhibitor of both activating and T790M resistant mutations that spares the wild type form of the receptor. Mol Cancer Ther (2013) 12(11 Suppl):B94.
48. Carmi C, Mor M, Petronini PG, Alfieri RR. Clinical perspectives for irreversible tyrosine kinase inhibitors in cancer. Biochem Pharmacol (2012) 84(11):1388–99. doi:10.1016/j.bcp.2012.07.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Soria J-C, Sequist LV, Gadgeel S, Goldman J, Wakelee H, Varga A, et al. eds. First-in-human evaluation of CO-1686, an irreversible, highly selective tyrosine kinase inhibitor of mutations of EGFR (activating and T790M). J Thorac Oncol (2013) 8(2 Suppl):S141.
50. Ranson M, Pao W, Kim D-W, Kim S-W, Ohe Y, Felip E, et al. eds. AZD9291: an irreversible, potent and selective tyrosine kinase inhibitor (TKI) of activating (EGFRm+) and resistance (T790M) mutations in advanced NSCLC. J Thorac Oncol (2013) 8(2 Suppl):S389.
51. Boccaccio C, Ando M, Tamagnone L, Bardelli A, Michieli P, Battistini C, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature (1998) 391(6664):285–8. doi:10.1038/34657
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Puri N, Salgia R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog (2008) 7:9. doi:10.4103/1477-3163.44372
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Zucali PA, Ruiz MG, Giovannetti E, Destro A, Varella-Garcia M, Floor K, et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Ann Oncol (2008) 19(9):1605–12. doi:10.1093/annonc/mdn240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev (2013) 39(7):793–801. doi:10.1016/j.ctrv.2013.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Robinson KW, Sandler AB. The role of MET receptor tyrosine kinase in non-small cell lung cancer and clinical development of targeted anti-MET agents. Oncologist (2013) 18(2):115–22. doi:10.1634/theoncologist.2012-0262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Sanabria-Figueroa E, Donnelly S, Foy K, Buss M, Castellino RC, Paplomata E, et al. Insulin-like growth factor-1 receptor signaling increases the invasive potential of Human Epidermal Growth Factor Receptor 2-overexpressing breast cancer cells via Src-Focal Adhesion Kinase and Foxhead Box Protein M1. Mol Pharmacol (2015) 87(2):150–61. doi:10.1124/mol.114.095380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Lu Y, Zi X, Zhao Y, Mascarenhas D, Pollak M. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin). J Natl Cancer Inst (2001) 93(24):1852–7. doi:10.1093/jnci/93.24.1852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med (2008) 359(17):1757–65. doi:10.1056/NEJMoa0804385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Ciardello F, Normanno N. HER2 signaling and resistance to the anti-EGFR monoclonal antibody cetuximab: a furthermore step toward personalised medicine for patients with colorectal cancer. Cancer Discov (2011) 1(6):472–4. doi:10.1158/2159-8290.CD-11-0261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Corvinus FM, Orth C, Moriggl R, Tsareva SA, Wagner S, Pfitzner EB, et al. Persistent STAT3 activation in colon cancer is associated with enhanced cell proliferation and tumor growth. Neoplasia (2005) 7(6):545–55. doi:10.1593/neo.04571
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Béguelin W, Flaqué MCD, Proietti CJ, Cayrol F, Rivas MA, Tkach M, et al. Progesterone receptor induces ErbB-2 nuclear translocation to promote breast cancer growth via a novel transcriptional effect: ErbB-2 Function as a coactivator of Stat3. Mol Cell Biol (2010) 30(23):5456–72. doi:10.1128/MCB.00012-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Wong TW, Lee FY, Yu C, Luo FR, Oppenheimer S, Zhang H, et al. Preclinical antitumor activity of BMS-599626, a pan-HER kinase inhibitor that inhibits HER1/HER2 homodimer and heterodimer signaling. Clin Cancer Res (2006) 12(20):6186–93. doi:10.1158/1078-0432.CCR-06-0642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Engelman JA, Zejnullahu K, Gale C-M, Lifshits E, Gonzales AJ, Shimamura T, et al. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res (2007) 67(24):11924–32. doi:10.1158/0008-5472.can-07-1885
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Liu L, Greger J, Shi H, Liu Y, Greshock J, Annan R, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res (2009) 69(17):6871–8. doi:10.1158/0008-5472.CAN-08-4490
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Soria JC, Cortes J, Massard C, Armand JP, De Andreis D, Ropert S, et al. Phase I safety, pharmacokinetic and pharmacodynamic trial of BMS-599626 (AC480), an oral pan-HER receptor tyrosine kinase inhibitor, in patients with advanced solid tumors. Ann Oncol (2012) 23(2):463–71. doi:10.1093/annonc/mdr137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Kim T, Lee K-W, Oh D-Y. A phase I study of HM781-36B, a novel pan-HER inhibitor, in patients (pts) with advanced solid tumors. J Clin Oncol (2012) 30:3076.
67. Campos S, Hamid O, Seiden MV, Oza A, Plante M, Potkul RK, et al. Multicenter, randomized phase II trial of oral CI-1033 for previously treated advanced ovarian cancer. J Clin Oncol (2005) 23(24):5597–604. doi:10.1200/jco.2005.08.091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Wong K-K. HKI-272 in non-small cell lung cancer. Clin Cancer Res (2007) 13(15):4593s–6s. doi:10.1158/1078-0432.ccr-07-0369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Ramalingam SS, Blackhall F, Krzakowski M, Barrios CH, Park K, Bover I, et al. Randomized phase II study of dacomitinib (PF-00299804), an irreversible pan-human epidermal growth factor receptor inhibitor, versus erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol (2012) 30(27):3337–44. doi:10.1200/jco.2011.40.9433
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Janne PA, Reckamp K, Koczywas M, Engelman JA, Camidge DR, Rajan A, et al. eds. Efficacy and safety of PF-00299804 (PF299) in patients (pt) with advanced NSCLC after failure of at least one prior chemotherapy regimen and prior treatment with erlotinib (E): a two-arm, phase II trial. J Clin Oncol (2009) 27(15 Suppl):8063.
71. Park K, Heo DS, Cho B, Kim D, Ahn M, Lee S, et al. eds. PF-00299804 (PF299) in Asian patients (pts) with non-small cell lung cancer (NSCLC) refractory to chemotherapy (CT) and erlotinib (E) or gefitinib (G): a phase (P) I/II study. J Clin Oncol (2010) 28(15 Suppl):7599.
72. Mok T, Spigel DR, Park K, Socinski MA, Tung SY, Kim D, et al. eds. Efficacy and safety of PF-299804 (PF299), an oral, irreversible, pan-human epidermal growth factor receptor (pan-HER) tyrosine kinase inhibitor (TKI), as first-line treatment (tx) of selected patients (pts) with advanced (adv) non-small cell lung cancer (NSCLC). J Clin Oncol (2010) 28(15 Suppl):7537.
73. Jänne PA, Boss DS, Camidge DR, Britten CD, Engelman JA, Garon EB, et al. Phase I dose-escalation study of the pan-HER inhibitor, PF299804, in patients with advanced malignant solid tumors. Clin Cancer Res (2011) 17(5):1131–9. doi:10.1158/1078-0432.ccr-10-1220
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Takahashi T, Boku N, Murakami H, Naito T, Tsuya A, Nakamura Y, et al. Phase I and pharmacokinetic study of dacomitinib (PF-00299804), an oral irreversible, small molecule inhibitor of human epidermal growth factor receptor-1, -2, and -4 tyrosine kinases, in Japanese patients with advanced solid tumors. Invest New Drugs (2012) 30(6):2352–63. doi:10.1007/s10637-011-9789-z
75. Abdul Razak AR, Soulières D, Laurie SA, Hotte SJ, Singh S, Winquist E, et al. A phase II trial of dacomitinib, an oral pan-human EGF receptor (HER) inhibitor, as first-line treatment in recurrent and/or metastatic squamous-cell carcinoma of the head and neck. Ann Oncol (2013) 24(3):761–9. doi:10.1093/annonc/mds503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Eskens FALM, Mom CH, Planting AST, Gietema JA, Amelsberg A, Huisman H, et al. A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br J Cancer (2008) 98(1):80–5. doi:10.1038/sj.bjc.6604108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Yap TA, Vidal L, Adam J, Stephens P, Spicer J, Shaw H, et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol (2010) 28(25):3965–72. doi:10.1200/jco.2009.26.7278
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Murakami H, Tamura T, Takahashi T, Nokihara H, Naito T, Nakamura Y, et al. Phase I study of continuous afatinib (BIBW 2992) in patients with advanced non-small cell lung cancer after prior chemotherapy/erlotinib/gefitinib (LUX-Lung 4). Cancer Chemother Pharmacol (2012) 69(4):891–9. doi:10.1007/s00280-011-1738-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Gordon M, Mendelson D, Gross M, Uttenreuther-Fischer M, Ould-Kaci M, Zhao Y, et al. A Phase I, open-label, dose-escalation study of continuous once-daily oral treatment with afatinib in patients with advanced solid tumors. Invest New Drugs (2013) 31(2):409–16. doi:10.1007/s10637-012-9904-9
80. Vermorken JB, Rottey S, Ehrnrooth E, Pelling K, Lahogue A, Wind S, et al. A phase Ib, open-label study to assess the safety of continuous oral treatment with afatinib in combination with two chemotherapy regimens: cisplatin plus paclitaxel and cisplatin plus 5-fluorouracil, in patients with advanced solid tumors. Ann Oncol (2013) 24(5):1392–400. doi:10.1093/annonc/mds633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Awada AH, Dumez H, Hendlisz A, Wolter P, Besse-Hammer T, Uttenreuther-Fischer M, et al. Phase I study of pulsatile 3-day administration of afatinib (BIBW 2992) in combination with docetaxel in advanced solid tumors. Invest New Drugs (2013) 31(3):734–41. doi:10.1007/s10637-012-9880-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Yang JC-H, Shih J-Y, Su W-C, Hsia T-C, Tsai C-M, Ou S-HI, et al. Afatinib for patients with lung adenocarcinoma and epidermal growth factor receptor mutations (LUX-Lung 2): a phase 2 trial. Lancet Oncol (2012) 13(5):539–48. doi:10.1016/S1470-2045(12)70086-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. De Grève J, Teugels E, Geers C, Decoster L, Galdermans D, De Mey J, et al. Clinical activity of afatinib (BIBW 2992) in patients with lung adenocarcinoma with mutations in the kinase domain of HER2/neu. Lung Cancer (2012) 76(1):123–7. doi:10.1016/j.lungcan.2012.01.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Schuler M, Awada A, Harter P, Canon J, Possinger K, Schmidt M, et al. A phase II trial to assess efficacy and safety of afatinib in extensively pretreated patients with HER2-negative metastatic breast cancer. Breast Cancer Res Treat (2012) 134(3):1149–59. doi:10.1007/s10549-012-2126-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Miller VA, Hirsh V, Cadranel J, Chen Y-M, Park K, Kim S-W, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol (2012) 13(5):528–38. doi:10.1016/S1470-2045(12)70087-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Yang JC-H, Schuler MH, Yamamoto N, O’Byrne KJ, Hirsh V, Mok T, et al. LUX-Lung 3: a randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol (2012) 30(18_suppl):LBA7500.
87. Wu YL, Zhou C, Hu C-P, Feng JF, Lu S, Huang Y, et al. LUX-Lung 6: a randomized, open-label, phase III study of afatinib (A) versus gemcitabine/cisplatin (GC) as first-line treatment for Asian patients (pts) with EGFR mutation-positive (EGFR M1) advanced adenocarcinoma of the lung. J Clin Oncol (2013) 31(15S):8016.
88. Ercan D, Zejnullahu K, Yonesaka K, Xiao Y, Capelletti M, Rogers A, et al. Amplification of EGFR T790M causes resistance to an irreversible EGFR inhibitor. Oncogene (2010) 29(16):2346–56. doi:10.1038/onc.2009.526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Nam H-J, Ching KA, Kan J, Kim H-P, Han S-W, Im S-A, et al. Evaluation of the antitumor effects and mechanisms of PF00299804, a pan-HER inhibitor, alone or in combination with chemotherapy or targeted agents in gastric cancer. Mol Cancer Ther (2012) 11(2):439–51. doi:10.1158/1535-7163.mct-11-0494
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Nam H-J, Kim H-P, Yoon Y-K, Song S-H, Min A-R, Han S-W, et al. The irreversible pan-HER inhibitor PF00299804 alone or combined with gemcitabine has an antitumor effect in biliary tract cancer cell lines. Invest New Drugs (2012) 30(6):2148–60. doi:10.1007/s10637-011-9782-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Kalous O, Conklin D, Desai AJ, O’Brien NA, Ginther C, Anderson L, et al. Dacomitinib (PF-00299804), an irreversible pan-HER inhibitor, inhibits proliferation of HER2-amplified breast cancer cell lines resistant to trastuzumab and lapatinib. Mol Cancer Ther (2012) 11(9):1978–87. doi:10.1158/1535-7163.mct-11-0730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Ather F, Hamidi H, Fejzo MS, Letrent S, Finn RS, Kabbinavar F, et al. Dacomitinib, an irreversible pan-ErbB inhibitor significantly abrogates growth in head and neck cancer models that exhibit low response to cetuximab. PLoS One (2013) 8(2):e56112. doi:10.1371/journal.pone.0056112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Gonzales AJ, Hook KE, Althaus IW, Ellis PA, Trachet E, Delaney AM, et al. Antitumor activity and pharmacokinetic properties of PF-00299804, a second-generation irreversible pan-erbB receptor tyrosine kinase inhibitor. Mol Cancer Ther (2008) 7(7):1880–9. doi:10.1158/1535-7163.mct-07-2232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Kim SM, Kwon O-J, Hong YK, Kim JH, Solca F, Ha S-J, et al. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther (2012) 11(10):2254–64. doi:10.1158/1535-7163.mct-12-0311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Boyer MJ, Blackhall FH, Park K, Barrios CH, Krzakowski MJ, Taylor I, et al. Efficacy and safety of PF299804 versus erlotinib (E): a global, randomized phase II trial in patients (pts) with advanced non-small cell lung cancer (NSCLC) after failure of chemotherapy (CT). J Clin Oncol (2010) 28(18 Suppl):LBA7523.
96. Press MF, Cordon-Cardo C, Slamon DJ. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene (1990) 5(7):953–62.
97. Scotlandi K, Manara MC, Hattinger CM, Benini S, Perdichizzi S, Pasello M, et al. Prognostic and therapeutic relevance of HER2 expression in osteosarcoma and Ewing’s sarcoma. Eur J Cancer (2005) 41(9):1349–61. doi:10.1016/j.ejca.2005.03.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther (2012) 343(2):342–50. doi:10.1124/jpet.112.197756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene (2008) 27(34):4702–11. doi:10.1038/onc.2008.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Bean J, Riely GJ, Balak M, Marks JL, Ladanyi M, Miller VA, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res (2008) 14(22):7519–25. doi:10.1158/1078-0432.ccr-08-0151
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L, et al. HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci U S A (2009) 106(2):474–9. doi:10.1073/pnas.0808930106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Ioannou N, Dalgleish AG, Seddon AM, Mackintosh D, Guertler U, Solca F, et al. Anti-tumour activity of afatinib, an irreversible ErbB family blocker, in human pancreatic tumour cells. Br J Cancer (2011) 105(10):1554–62. doi:10.1038/bjc.2011.396
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Stopfer P, Marzin K, Narjes H, Gansser D, Shahidi M, Uttereuther-Fischer M, et al. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother Pharmacol (2012) 69(4):1051–61. doi:10.1007/s00280-011-1803-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Afatinib US. Food and Drug Administration. (2013). Available from: http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm360574.htm
105. Huang Z, Brdlik C, Jin P, Shepard HM. A pan-HER approach for cancer therapy: background, current status and future development. Expert Opin Biol Ther (2009) 9(1):97–110. doi:10.1517/14712590802630427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Torres MA, Raju U, Molkentine D, Riesterer O, Milas L, Ang KK. AC480, formerly BMS-599626, a pan Her inhibitor, enhances radiosensitivity and radioresponse of head and neck squamous cell carcinoma cells in vitro and in vivo. Invest New Drugs (2011) 29(4):554–61. doi:10.1007/s10637-010-9389-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Takezawa K, Okamoto I, Tanizaki J, Kuwata K, Yamaguchi H, Fukuoka M, et al. Enhanced anticancer effect of the combination of BIBW2992 and thymidylate synthase-targeted agents in non-small cell lung cancer with the T790M mutation of epidermal growth factor receptor. Mol Cancer Ther (2010) 9(6):1647–56. doi:10.1158/1535-7163.mct-09-1009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Gieseg MA, de Bock C, Ferguson LR, Denny WA. Evidence for epidermal growth factor receptor-enhanced chemosensitivity in combinations of cisplatin and the new irreversible tyrosine kinase inhibitor CI-1033. Anticancer Drugs (2001) 12(8):683–90. doi:10.1097/00001813-200109000-00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Garland LL, Hidalgo M, Mendelson DS, Ryan DP, Arun BK, Lovalvo JL, et al. A phase I clinical and pharmacokinetic study of oral CI-1033 in combination with docetaxel in patients with advanced solid tumors. Clin Cancer Res (2006) 12(14 Pt 1):4274–82. doi:10.1158/1078-0432.CCR-05-2507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Chiappori AA, Ellis PM, Hamm JT, Bitran JD, Eiseman I, Lovalvo J, et al. A phase I evaluation of oral CI-1033 in combination with paclitaxel and carboplatin as first-line chemotherapy in patients with advanced non-small cell lung cancer. J Thorac Oncol (2006) 1(9):1010–9. doi:10.1097/01243894-200611000-00015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Storniolo AM, Pegram MD, Overmoyer B, Silverman P, Peacock NW, Jones SF, et al. Phase I dose escalation and pharmacokinetic study of lapatinib in combination with trastuzumab in patients with advanced ErbB2-positive breast cancer. J Clin Oncol (2008) 26(20):3317–23. doi:10.1200/JCO.2007.13.5202
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. McDonagh CF, Huhalov A, Harms BD, Adams S, Paragas V, Oyama S, et al. Antitumor activity of a novel bispecific antibody that targets the ErbB2/ErbB3 oncogenic unit and inhibits heregulin-induced activation of ErbB3. Mol Cancer Ther (2012) 11(3):582–93. doi:10.1158/1535-7163.mct-11-0820
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Zhang B, Lahdenranta J, Du J, Kirouac D, Nguyen S, Overland R, et al. eds. MM-111, a bispecific HER2 and HER3 antibody, synergistically combines with trastuzumab and paclitaxel in preclincal models of gastric cancer. Cancer Res (2013) 73(8 Suppl):4633. doi:10.1158/1538-7445.AM2013-4633
114. Lahdenranta J, Paragas V, Kudla AJ, Overland R, Moyo VM, Nielsen UB, et al. eds. Preclinical activity of MM-111, a bispecific ErbB2/ErbB3 antibody in previously treated ErbB-2-positive gastric and gastrophageal junction cancer. J Clin Oncol (2013) 31(4 Suppl):48.
115. Kirouac DC, Du JY, Lahdenranta J, Overland R, Yarar D, Paragas V, et al. Computational modeling of ERBB2-amplified breast cancer identifies combined ErbB2/3 blockade as superior to the combination of MEK and AKT inhibitors. Sci Signal (2013) 6(288):ra68. doi:10.1126/scisignal.2004008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Denlinger CS, Sym S, Bendell J, Alsina M, Watkins D, Chao Y, et al. Randomized open-label phase 2 study of MM-111 and paclitaxel (PTX) with trastuzumab (TRAS) in patients with HER2-expressing carcinomas of the distal esophagus, gastroesophageal (GE) junction, and stomach who have failed front-line metastatic or locally advanced therapy. J Clin Oncol (2014) 32(5s):abstrTS4148.
117. Britten CD. Targeting ErbB receptor signaling: a pan-ErbB approach to cancer. Mol Cancer Ther (2004) 3(10):1335–42.
118. Dassonville O, Bozec A, Fischel JL, Milano G. EGFR targeting therapies: monoclonal antibodies versus tyrosine kinase inhibitors: similarities and differences. Crit Rev Oncol Hematol (2007) 62(1):53–61. doi:10.1016/j.critrevonc.2006.12.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Yang JL, Hannan MT, Russell PJ, Crowe PJ. Expression of HER1/EGFR protein in human soft tissue sarcomas. Eur J Surg Oncol (2006) 32(4):466–8. doi:10.1016/j.ejso.2006.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Wang L, Zhang Q, Zhang J, Sun S, Guo H, Jia Z, et al. PI3K pathway activation results in low efficacy of both trastuzumab and lapatinib. BMC Cancer (2011) 11:248. doi:10.1186/1471-2407-11-248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Albritton KH, Rankin C, Coffin CM, Ratner N, Budd GT, Schuetze SM, et al. Phase II study of erlotinib in metastatic or unresectable malignant peripheral nerve sheath tumors (MPNST). J Clin Oncol (2006) 24(18S):9518.
122. Ha HT, Griffith KA, Zalupski MM, Schuetze SM, Thomas DG, Lucas DR, et al. Phase II trial of cetuximab in patients with metastatic or locally advanced soft tissue or bone sarcoma. Am J Clin Oncol (2013) 36(1):77–82. doi:10.1097/COC.0b013e31823a4970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Ebb D, Meyers P, Grier H, Bernstein M, Gorlick R, Lipshultz SE, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. J Clin Oncol (2012) 30(20):2545–51. doi:10.1200/JCO.2011.37.4546
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: targeted therapy, panHER inhibitors, drug resistance, HER signaling pathways, EGFR
Citation: Wang X, Batty KM, Crowe PJ, Goldstein D and Yang J-L (2015) The potential of panHER inhibition in cancer. Front. Oncol. 5:2. doi: 10.3389/fonc.2015.00002
Received: 10 September 2014; Accepted: 07 January 2015;
Published online: 28 January 2015.
Edited by:
Francois X. Claret, University of Texas, USAReviewed by:
Ruth Keri, Case Western Reserve University, USANune Markosyan, University of Pennsylvania, USA
Anastasia Pazaiti, emBIO Medical Center, Greece
Copyright: © 2015 Wang, Batty, Crowe, Goldstein and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia-Lin Yang, Adult Cancer Program, Lowy Cancer Research Centre, University of New South Wales (UNSW), Room 209, Building C25, Sydney, NSW 2052, Australia e-mail:ai55YW5nQHVuc3cuZWR1LmF1