 Panagiotis J. Vlachostergios1*
Panagiotis J. Vlachostergios1* Christos N. Papandreou2*
Christos N. Papandreou2*- 1Department of Internal Medicine, Lutheran Medical Center, Brooklyn, NY, USA
- 2Department of Medical Oncology, Faculty of Medicine, School of Health Sciences, University of Thessaly, Larissa, Greece
Neuroendocrine prostate carcinoma, either co-present with the local adenocarcinoma disease or as a result of transdifferentiation later in time, was described as one major process of emerging resistance to androgen deprivation therapies, and at the clinical level it is consistent with the development of rapidly progressive visceral disease, often in the absence of elevated serum prostate-specific antigen level. Until present, platinum-based chemotherapy has been the only treatment modality, able to produce a fair amount of responses but of short duration. Recently, several efforts for molecular characterization of this lethal phenotype have resulted in identification of novel signaling factors involved in microenvironment interactions, mitosis, and neural reprograming as potential therapeutic targets. Ongoing clinical testing of specific inhibitors of these targets, for example, Aurora kinase A inhibitors, in carefully selected patients and exploitation of expression changes of the target before and after manipulation is anticipated to increase the existing data and facilitate therapeutic decision making at this late stage of the disease when hormonal manipulations, even with the newest androgen-directed therapies are no longer feasible.
Introduction
For over two decades, several efforts have been made to define and molecularly characterize the frame of neuroendocrine (NE) prostate cancer (NEPC) presenting with distinct clinical features, different from the classic prostatic adenocarcinoma, including frequent visceral metastases, lytic bone involvement, relative low serum prostate-specific antigen (PSA) concentration, resistance to androgen ablation, and high response rate to platinum-based chemotherapy (1). In the era of evolving androgen-directed therapies, better molecular characterization and targeting of the NE phenotype remains of central interest given the inherent or emerging resistance of NEPC cells to current therapies, abrogation of which is warranted in order to improve responses and mortality of prostate cancer (PC) patients.
New Morphological Classification of NEPC
A new histological classification for NEPC has been recently proposed as a refinement of the current WHO morphologic criteria, to better reflect the aspects of NE differentiation in PC (2). According to the new classification, NEPC extends from usual NE differentiation found in usual prostate adenocarcinoma to mixed (small or large cell) NE carcinoma with acinar adenocarcinoma, adenocarcinoma with Paneth cell NE differentiation, carcinoid tumor, and small cell carcinoma. The new classification also satisfies the previously unmet need of characterizing treatment-induced androgen receptor (AR)-independent PC, which is a significant part of the broader category of castration-resistant tumors clinically defined aggressive-variant, previously known as anaplastic PC (2).
In addition, important recommendations regarding the assessment and interpretation methods have emerged. First, it is not recommended to routinely use immunohistochemical (IHC) stains to detect any NE differentiation in an otherwise morphologically typical primary adenocarcinoma of the prostate. In contrast, in cases of metastatic castration-resistant mixed NE-adenocarcinoma, it is recommended that the percentage and grade of the acinar component be provided. Regarding the sampling evaluation, the metastatic site, and/or the histology of the sample, most suspicious for NE differentiation should be evaluated. For confirmation of NE differentiation, markers for NE differentiation include synaptophysin, chromogranin, and CD56. Finally, the IHC expression of AR across the proposed subtypes of NEPC needs to be systematically evaluated with the aim of determining its role in classification of these tumors (2).
Current Treatment and Biomarkers
The majority of patients with NEPC are diagnosed with locally advanced disease, usually associated with nodal or/and distant metastases. Most often, a cisplatin-based combination (e.g., cisplatin plus etoposide) has been used (3). Although the optimal regimen has not been established, most recent evidence from a phase 2 study of first-line carboplatin and docetaxel (CD) and second-line etoposide and cisplatin (EP) in 120 patients with “anaplastic” PC suggests a high response rate of short duration to platinum-containing chemotherapies, similar to small cell PC. Seventy-four of 113 (65.4%) and 24 of 71 (33.8%) were progression free after four cycles of CD and EP, respectively while median overall survival was 16 months. Intriguingly, CEA and LDH emerged as important prognostic indicators in this patient population, in contrast with tissue or serum NE markers including chromogranin A (CgA), synaprophysin, gastric releasing peptide, somatostatin, which did not predict outcome or response to therapy (4).
Plasma anterior gradient 2 (AGR2) has been proposed as a new promising biomarker for characterization, monitoring, and directing therapies for patients with metastatic NEPC. AGR2 is an epithelial marker regulated by androgens through ErbB3 binding protein 1 and Fox A transcription factors and its circulating tumor cell (CTC) mRNA and serum protein levels were found to be significantly elevated in patients with “anaplastic” cancer (5). Likewise, hASH-1 gene transcription is upregulated by androgen deprivation and is associated to the onset of an NE phenotype (6). However, a recent review of studies on the effect of NE differentiation on oncologic outcomes concludes that data are insufficient to recommend the use of NE markers in routine practice, particularly at early PC stage (7).
Emerging Molecular Targets within the Landscape of NEPC Differentiation
A plethora of different signals involving neuropeptides, growth factors, and cytokines, are involved in the several intracellular processes including angiogenesis, cell survival, proliferation, migration, and invasion through paracrine and autocrine pathways. The resulting changes in more or less all of these processes contribute to the induction of NE differentiation in PC. The model, which has been mostly supported among several others proposed recognizes a role of previous treatment of hormone-naïve PC on the natural evolutionary transformation of classic adenocarcinoma with accumulation of new genetic alterations including loss of tumor suppressors and amplification of oncogenes (8). The finding of rearrangement of TMPRSS2–ERG in NEPC supports the origin of NEPC from prostate adenocarcinoma (8). It was thus suggested that there is an evolutionary continuum from conventional prostate adenocarcinoma to hormone naive state and finally to a CRPC/mixed state as the precursor of NEPC. NEPC has been postulated to correspond to the “cell-autonomous” phase of PC progression, which is the latest evolutionary phase following an “androgen-dependent” and a “microenvironment-driven” phase, respectively. The underlying molecular alterations at this point of progression to NEPC include loss of AR and androgen-regulated protein expression, induction of NE and neural programs, loss of tumor suppressors (TP53, RB1, PTEN) and resultant genomic instability, as well as activation of mitotic programs involving Aurora kinase A (AURKA) upregulation and MYCN amplification (5, 8).
Alternatively, adenocarcinoma and small cell carcinoma components of PC may originate from a common tumor clone with stem-like properties, or a cancer stem cell, converted into more differentiated tumor cells that accumulated molecular alterations driving epithelial or NE phenotypes. To date, very limited amounts of data are supportive of normal prostatic NE cell as a cell of origin for NE tumors (9). It is also unclear if NEPC is present at local disease or transdifferentiates later in time. Currently, as the clinical significance remains uncertain, it is not recommended to routinely use IHC stains to detect any NE differentiation in an otherwise morphologically typical primary adenocarcinoma of the prostate (2).
Tumor Suppressor Loss
In both benign prostate and adenocarcinoma, the IL-8–CXCR2–p53 pathway has been shown to be a strong growth inhibitory signal that keeps NE cells quiescent (10). P53 mutation, likely a result of environmental pressure from classical and novel castration strategies inhibiting AR and intratumoral androgen synthesis, inactivates this pathway and leads to hyperproliferation and aggressive behavior of the NE cells, resulting in the development of NEPC. These findings are consistent with a clonal selection model and indicate that clones of NE cells gain a proliferative advantage in an androgen-deprived environment through P53 mutation (11).
Loss of RB1 by deletion is another common event in prostatic small cell carcinoma as Rb protein loss was found by immunohistochemistry in as many as 90% of small cell carcinoma cases (26 of 29) with RB1 allelic loss in 85% of cases (11 of 13). Interestingly, Rb protein loss rarely occurs in high-grade acinar tumors, suggesting that Rb loss is a critical event in the development of small cell carcinomas and may be a useful diagnostic and potential therapeutic target (12).
Activation of Mitosis
The resultant genetic instability leads to additional changes, many of which affect cell-cycle genes, especially those related to M-phase transition, including AURKA and Polo-like kinase 1 (PLK1). PLK1 mediates entry into mitosis as well as centrosome maturation, spindle checkpoint activity, activation of the anaphase-promoting complex, and eventual exit from the M-phase with the initiation of cytokinesis (13). LNCaP androgen-independent cells were found to have upregulation of the mitotic kinase Plk1 and other M-phase cell-cycle proteins, which rendering them highly sensitive to PLK1 inhibition through necroptosis (14).
AURKA regulates entry into mitosis, as well as assembly of the mitotic spindle apparatus, thereby affecting chromosome separation (15). MYCN amplification is frequently associated with AURKA amplification. In addition, AURKA was found to stabilize MYCN via interaction with the Skp1-Cullin-F-box (SCF)-type ubiquitin ligase FBXW7 that ubiquitinates MYCN and counteracts its degradation (16). C-MYC is also involved in NEPC and was shown to cooperate with the Proto-oncogene serine/threonine-protein kinase 1 (PIM1) in a SCID mice NEPC model, supporting the concept of targeting PIM1 (17). In line with the NEPC mitotic reprograming, in SCPC/LCNEC xenograft models, high expression of M-phase genes was found, including Ubiquitin-conjugating enzyme E2 C (UBE2C), coupled with RB and cyclin D1 loss, despite the absence of AR expression (18). A sequence of events was therefore suggested in which loss of RB and/or cyclin D1 precede AR loss and further deregulation of the mitotic apparatus.
Epigenetic Regulation Changes
RE1-silencing transcription factor (REST), also known as neuron-restrictive silencer factor (NRSF) is a transcription factor that represses neuronal differentiation, in NEPC. REST-binding sites were found on 28 of 50 transcriptionally active genes in NEPC and in vivo in a cohort of 218 prostate tumors, in which REST downregulation was observed in 50% of NEPC tumors (19). Gene expression profiling revealed that REST not only acts to repress neuronal genes but also genes involved in cell-cycle progression, including AURKA (20).
Also intriguing was the discovery of an invert correlation between REST and the protocadherin (PCDH) genes PCDH11Y and PCDH11X (9). PCDH-PC overexpression is an early-onset adaptive mechanism following androgen deprivation therapy (ADT) and results in attenuation of the ligand-dependent activity of the AR, enabling certain prostate tumor clones to assume a more NE phenotype and promoting their survival under diverse stress conditions (21) through activation of Wnt signaling and increased nuclear beta-catenin expression (22).
In addition, downregulation of REST level relieves gene silencer REST-mediated transcriptional repression as part of a relay mechanism found in IL-6 induced autophagy through activation AMPK/mTOR pathway (23).
The epigenetic machinery involvement in NE differentiation process is a new field of ongoing research with existing data supporting a role for the inhibition of BET bromodomains in downregulation of MYC expression in PC cell lines and xenografts and more importantly “downstream” of AR (24). The histone deacetylase EZH2 is also highly expressed in NEPC and hypermethylation of key genes within the NEPC genome may be associated with the cellular plasticity seen during transdifferentiation. MYC overexpression leads to EZH2 activation by antagonizing miR-26a and PI3K–AKT-mediated EZH2 inhibition, resulting in suppression of IFNGR1 and downstream JAK–STAT1 signaling with increased cell viability and proliferation (25).
Microenvironment Changes
Acquisition of endogenous IL-6 production and its possible contribution to an autocrine cell growth stimulation may play an important role during androgen-independent progression (26). IL-6 also participates in a feed-forward loop with pigment epithelium-derived factor (PEDF) to induce NE differentiation, in which NFκB induction elicits STAT3 activation and pro-differentiating IL-6 expression causing further expansion of the NE communications (27). Activation of NFκB pathway is sufficient to maintain androgen-independent growth of prostate and PC by regulating AR action (28).
Increased paracrine release of the pro-inflammatory cytokine macrophage migration inhibitory factor (MIF) during NE differentiation in PC may facilitate cancer progression or recurrence, especially following androgen deprivation, through stimulation of AKT and ERK1/2 signaling pathways. Thus, MIF could represent an attractive target for NEPC therapy (29).
Continued focal adhesion kinase (FAK) expression (and activity) emerged as an essential factor for the androgen-independent formation of NE carcinoma in the TRAMP model (30). Targeting FAK might be an appropriate strategy in the context of arising NE phenotype in the microenvironment phase of NEPC differentiation.
Recent studies indicate the importance of the ubiquitin ligase Siah2 in control of NEPC and prostate adenocarcinoma harboring NE lesions. Siah2-dependent expression and activity of HIF-1α regulate its availability to form a transcriptional complex with FoxA2, resulting in expression of specific target genes, including Hes6, Sox9, and Jmjd1a, whose co-expression is sufficient for formation of NE tumors and NE lesions in PC. Siah is likely the best candidate, since its loss abolishes formation of TRAMP NE tumors and restoring HIF expression in such tumor cells only partially (30%) rescues formation of NE tumors. Menadione is a Siah2 inhibitor. Menadione treatment inhibited HIF levels in cultured cells, increased expression of direct Siah2 targets, and inhibited formation of melanoma xenografts. Several inhibitors directed against HIF have been recently developed. It is of importance to assess their effects in prostate tumor models (31). Hypoxia itself was shown to induce NE differentiation of LNCaP cells in vitro, which seems to be driven by the inhibition of Notch signaling with subsequent downregulation of hairy and enhancer of split-1 (Hes1) transcription (32).
MMP-9 produced by mast cells within the tumor microenvironment enables well-differentiated adenocarcinoma outgrowth by favoring angiogenesis and invasion to the surrounding tissue in TRAMP mice. Upon tumor progression, well-differentiated adenocarcinoma undergoes epithelial-mesenchymal transition (EMT) and foci of poorly differentiated adenocarcinoma are originated. Poorly differentiated adenocarcinoma produces MMP-9 autocrinously, thus becoming independent from mast cells. In the absence of functional mast cells (i.e., when mast cells are inhibited and/or genetically ablated), stem cell factor (SCF) in the prostatic environment is no longer sequestered and becomes available for binding to c-Kit receptor on prostate stem cells. Upon enhanced c-Kit signaling, prostate stem cells continue to proliferate, undergo NE differentiation, and progress to NE tumors, which grow fast and quickly invade the surrounding tissue. The common expression of c-Kit by mast cells and NE clones suggests a possible competition for the ligand SCF and offers the chance of curing early-stage disease while preventing NE tumors using c-Kit–targeted therapy (33, 34).
The implication of EMT in NE transdifferentiation can also occur through the effect of Snail. LNCaP PC cells transfected with Snail displayed increase in the NE markers, neuron-specific enolase (NSE) and CgA, while LNCaP C-33 cells that have been previously reported as a NE differentiation model exhibited increased expression levels of Snail protein as compared with LNCaP parental cells. Functionally, Snail-mediated NE differentiation was associated with increased paracrine cell proliferation. The novel proteasome inhibitor NPI-0052 (salinosporamide A) can inhibit Snail mRNA and protein and thereby promote sensitivity to cisplatin- and TRAIL-mediated apoptosis in DU145 PC cells. Natural products including flavonoids and parthenolide have also shown some promise toward targeting Snail signaling in PC (35, 36). Likewise, adrenomedullin, an autocrine/paracrine factor induced by androgen withdrawal, stimulates the NE phenotype in LNCaP prostate tumor cells (37).
Src kinase is another important player in the EMT process as it is activated by several neuropeptides, including CgA, NSE, serotonin, neurophysin, bombesin, and synaptophysin. Through direct physical interaction with the AR, Src is able to phosphorylate the AR and thereby induce ligand-independent AR activation (one of the key mechanisms of castration-resistant PC) (38). Neuropeptides, notably bombesin were also found to enhance early growth response 1 (Egr-1) expression leading to increased human protease-activated receptor 1 (PAR1) expression and function directly correlating with invasiveness and the degree of PC malignancy (39). Src is also a mediator for the NE-derived parathyroid hormone-related protein (PTHrP), which induces tyrosine phosphorylation and subsequent reduced AR ubiquitination thus increased accumulation of AR, enhancing growth of PC cells at low levels of androgen (40).
In addition, in co-cultures of macrophages with LNCaP and TRAMP-C2 PC cells, a feedback loop between bone morphogenetic protein-6 (BMP-6) derived from PC cells and IL-6 produced by macrophages, resulted in IL-6-induced NE differentiation in PC cells (41).
Receptor activator of nuclear factor kappa-B ligand (RANKL) either derived from the tumor or from the host plays a key role in cancer bone metastasis. A small population of RANKL-expressing cells was observed to initiate and promote cancer bone and soft tissue metastases by recruiting bystander cells to form tumors in bone. The mechanism underlying this recruitment appears to involve a feed-forward mechanism in which RANKL, RANK, and c-Met expression is increased and AR is downregulated. RANKL alters a large transcriptional program that appears to govern formation of the premetastatic niche as well as emergence of osteomimetic, EMT, and stem and NE differentiation (42). When the anti-tumor effects of the bisphosphonate zolendronic acid and somatostatin analogs (SMS) were tested on NE carcinoma models, zolendronic acid, but not SMS induced apoptosis and inhibition of proliferation and migration through impaired prenylation of Ras, thus offering the possibility of therapeutic use in the early phase for controlling NE cells (43).
The concept of “epithelial immune cell-like transition” (EIT), similar to NE-like transdifferentiation of prostate adenocarcinoma cells has been proposed to describe the acquisition of immune properties from cancer cells, which enable them to “communicate” with immune cells, leading to suppression of anti-cancer immune activity in their microenvironment and facilitation of the expansion and malignant progression of the disease (44).
Within this context, a dendritic cell vaccine sipuleucel-T was developed from peripheral blood mononuclear cells obtained by leukapheresis. In randomized trials, sipuleucel-T prolonged overall survival compared with placebo in men with minimally symptomatic, metastatic castrate-resistant PC (CRPC). However, sipuleucel-T did not affect the serum PSA, restricting this approach to patients with slowly progressive disease where a relatively rapid response to treatment is not required (45). In addition, treatment is contraindicated in patients who are on steroids or opioids for cancer-related pain, and should be used with caution in patients with liver metastases. Thus, with so far available data, assessing the impact of immunotherapy on an individual patient with NEPC can be difficult or impossible.
Upcoming Targeted Approaches and Predictive Tools in NEPC
In the “cell-autonomous” phase of the disease, inhibitors that affect mitotic function may be efficacious, as opposed to earlier stages when AR signaling affects more “classic” AR-mediated pathways. Currently, first-line treatment for this phase is chemotherapy, but patients become rapidly resistant to this approach. As the molecular basis for NEPC becomes better understood, individualized therapy may be possible.
The AURKA inhibitor danusertib (PHA-739358) was tested in a phase II clinical trial but failed to achieve the primary endpoint of PSA response (46). However, PSA as an endpoint is unlikely to be suitable for tumors that are in the “tumor cell-autonomous” phase. In addition, therapeutic treatment in this trial was not directed specifically to patients with amplified AURKA; hence, it is not certain whether better response would have been achieved by focusing on NEPC patients with amplified AURKA (46). Also, given the enzymatic activity of AURKA depends not only on the amount of protein present but also on the activity of several cofactors (such as TPX2, BORA, and Ajuba), and has numerous substrates (including p53, BRCA1, and even AR), it is likely that the effects of AURKA (and thus the impact of its inhibition) are dependent, at least in part, on the activity of the cofactors and the role of its substrates in a given cell (47). A clinical trial evaluating the AURKA inhibitor alisertib for patients with NEPC is under way, and AURKA and MYCN co-amplification are being explored as potential predictive biomarkers and may be used to select NEPC and patients with high-risk PC for early intervention with AURKA-targeted therapy (48).
Dasatinib is a Src family/abl inhibitor with preclinical activity in PC and encouraging results in phase II studies (49). However, there was no increase in overall survival when dasatinib was given in combination with docetaxel plus prednisone compared with chemotherapy alone. In the phase III READY trial, 1522 men with metastatic CRPC were randomly assigned to either dasatinib with docetaxel plus prednisone or docetaxel plus prednisone alone. With a median follow-up of 19 months, the median survival was approximately 21 months on both treatment arms. This failure can be explained by several factors including inadequate study design, potential pharmacokinetic interactions between dasatinib and docetaxel, a stronger effect on stromal cells than on epithelial cells (despite association with the epithelial-targeted docetaxel), and the too broad specificity of inhibitory effect of dasatinib for numerous receptor and non-receptor tyrosine kinases (50).
PLK1 inhibitors have recently entered clinical trials for solid tumors. BI 2536 is a PLK1 selective inhibitor that reached phase II trial in several solid tumors, but not PC, with little efficacy (51). Volasertib (BI 6727) is a potent and relatively selective inhibitor for PLK1. A phase I study in patients with advanced disease showed a favorable pharmacokinetic profile and limited toxicities in patients with advanced solid tumors (52). Phase II studies are ongoing. On the basis of the three-phase model, PLK1 inhibitors might be effective in “cell-autonomous” tumors where PLK1 is overexpressed (8).
The role of the tumor microenvironment and data supporting MET as a potential targetable driver of NEPC was also presented as an area of active investigation (53). Cabozantinib (XL184) is an inhibitor of MET, VEGFR2, and RET. A phase II trial in patients with progressive, metastatic PC provided preliminary evidence of activity in men with bone metastases (54). Based upon these results, two phase III trials were initiated in patients who had progressed on docetaxel and either abiraterone or enzalutamide as treatment for CRPC. In the COMET-1 trial, patients were randomly assigned to either cabozantinib or prednisone; the primary endpoint was overall survival. In the COMET-2 trial, patients were randomly assigned to cabozanitinib or mitoxantrone plus prednisone and the primary endpoint was pain control. Preliminary results from the COMET-1 trial released by the corporate sponsor indicated that cabozantinib did not achieve a statistically significant increase in overall survival (55). Enrollment in the COMET-2 trial was discontinued based upon these results.
MK-2206 is an oral AKT inhibitor that was tested in a phase 1 trial using a QOD, QW, or Q3W dosing schedule in combination with carboplatin and paclitaxel, docetaxel, or erlotinib, and was well-tolerated at doses that inhibit AKT signaling. Within a diverse population of 72 patients including breast, melanoma, pancreas, prostate, colon, esophageal, small cell lung cancer, a partial response with a PFS of 6 months was shown in a patient with NEPC and minor responses were demonstrated in two patients with NE pancreatic cancers (56). Randomized phase 2 studies in specific cancer types and more homogenous cohorts are expected before being able to draw any conclusions about the clinical effects of MK-2206 with other standard cytotoxic or targeted treatment options.
Two oral endothelin receptor antagonists, atrasentan and zibotentan, have been extensively studied, to target the supporting environment for metastatic growth. Multiple phase II and subsequent phase III trials were conducted with both atrasentan and zibotentan; however, none was able to show significant benefit compared with placebo (57–61).
Given the critical roles of CTCs and EMT in PC tumorigenesis and the current immunotherapeutic strategies targeting prostate tumor antigens, such as sipuleucel-T, there may be a need to design new immunotherapies targeting cancer stem cells and cells involved in EMT.
Cumulative data on currently established and potential future targets of NEPC therapies within corresponding pathways are presented in Table 1. Most evidence for NEPC targets and corresponding targeted agents is derived from preclinical studies or in vivo mouse models (62–68). At the clinical level, there is no direct evidence and all data are extrapolated from studies in CRPC (50, 69–75) (Table 1). Thus, there is an urgent need for exploitation of emerging targets through design and implementation of studies in this particular subpopulation of PC patients with NE differentiated PC.
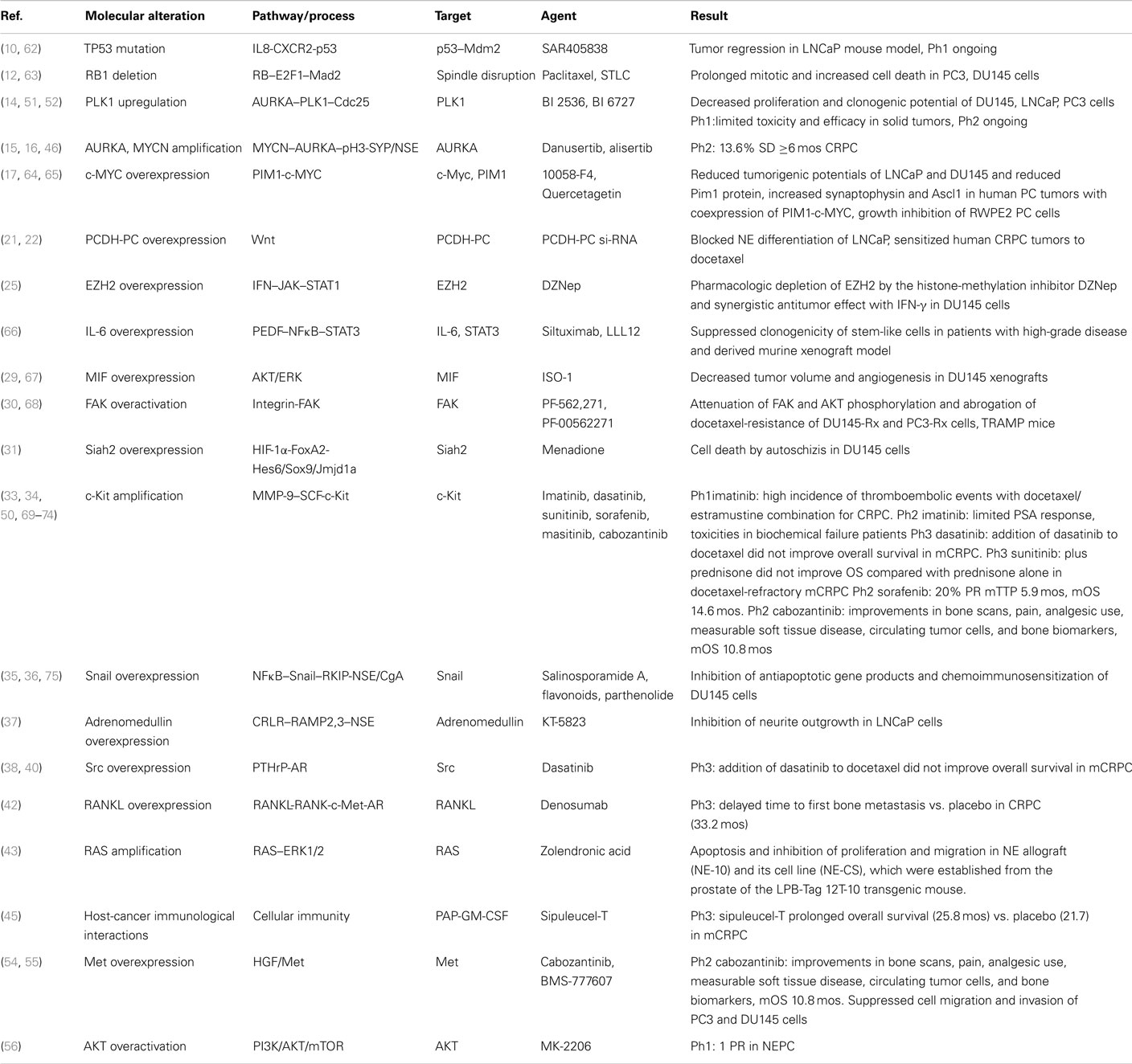
Table 1. Targeted therapies in NEPC.
Conclusive Remarks
The molecular characterization of NEPC is a challenging area of ongoing research with encouraging new findings on potential new targeted therapeutic approaches as well as emerging surrogate biological markers for early identification of treatment responses and failures. However, the determination of appropriate target at the right timepoint within the evolving genotype and phenotype of the disease requires constant reassessment of the underlying molecular changes. Collection and analysis of CTCs offers a great opportunity of repetitively studying these changes as a non-interventional approach compared to tissue biopsy. Ultimately, identification of appropriate targets within the signaling networks that “drive” the evolution of NEPC may not only guide development of newer biologic treatments but may also enable a more appropriate, in terms of sequencing, utilization of existing therapies to correspond to the underlying molecular biology, which dictates NEPC differentiation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Papandreou CN, Daliani DD, Thall PF, Tu SM, Wang X, Reyes A, et al. Results of a phase II study with doxorubicin, etoposide, and cisplatin in patients with fully characterized small-cell carcinoma of the prostate. J Clin Oncol (2002) 20:3072–80. doi:10.1200/JCO.2002.12.065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol (2014) 38:756–67. doi:10.1097/PAS.0000000000000208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Culine S, El Demery M, Lamy PJ, Iborra F, Avancès C, Pinguet F. Docetaxel and cisplatin in patients with metastatic androgen independent prostate cancer and circulating neuroendocrine markers. J Urol (2007) 178:844. doi:10.1016/j.juro.2007.05.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, Tu SM, et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res (2013) 19:3621–30. doi:10.1158/1078-0432.CCR-12-3791
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Kani K, Malihi PD, Jiang Y, Wang H, Wang Y, Ruderman DL, et al. Anterior gradient 2 (AGR2): blood-based biomarker elevated in metastatic prostate cancer associated with the neuroendocrine phenotype. Prostate (2013) 73:306–15. doi:10.1002/pros.22569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Rapa I, Volante M, Migliore C, Farsetti A, Berruti A, Vittorio Scagliotti G, et al. Human ASH-1 promotes neuroendocrine differentiation in androgen deprivation conditions and interferes with androgen responsiveness in prostate cancer cells. Prostate (2013) 73:1241–9. doi:10.1002/pros.22679
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Surcel CI, van Oort IM, Sooriakumaran P, Briganti A, De Visschere PJ, Fütterer JJ, et al. Prognostic effect of neuroendocrine differentiation in prostate cancer: a critical review. Urol Oncol (2014). doi:10.1016/j.urolonc.2014.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Logothetis CJ, Gallick GE, Maity SN, Kim J, Aparicio A, Efstathiou E, et al. Molecular classification of prostate cancer progression: foundation for marker-driven treatment of prostate cancer. Cancer Discov (2013) 3:849–61. doi:10.1158/2159-8290.CD-12-0460
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Terry S, Beltran H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front Oncol (2014) 4:60. doi:10.3389/fonc.2014.00060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Chen H, Sun Y, Wu C, Magyar CE, Li X, Cheng L, et al. Pathogenesis of prostatic small cell carcinoma involves the inactivation of the P53 pathway. Endocr Relat Cancer (2012) 19:321–31. doi:10.1530/ERC-11-0368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Li Z, Chen CJ, Wang JK, Hsia E, Li W, Squires J, et al. Neuroendocrine differentiation of prostate cancer. Asian J Androl (2013) 15:328–32. doi:10.1038/aja.2013.7
12. Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res (2014) 20:890–903. doi:10.1158/1078-0432.CCR-13-1982
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat Rev Cancer (2010) 10:825–41. doi:10.1038/nrc2964
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Deeraksa A, Pan J, Sha Y, Liu XD, Eissa NT, Lin SH, et al. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene (2013) 32:2973–83. doi:10.1038/onc.2012.309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Mosquera JM, Beltran H, Park K, MacDonald TY, Robinson BD, Tagawa ST, et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia (2013) 15:1–10. doi:10.1593/neo.121550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell (2009) 15:67–78. doi:10.1016/j.ccr.2008.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Wang J, Kim J, Roh M, Franco OE, Hayward SW, Wills ML, et al. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene (2010) 29:2477–87. doi:10.1038/onc.2010.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Tzelepi V, Zhang J, Lu JF, Kleb B, Wu G, Wan X, et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin Cancer Res (2012) 18:666–77. doi:10.1158/1078-0432.CCR-11-1867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lapuk AV, Wu C, Wyatt AW, McPherson A, McConeghy BJ, Brahmbhatt S, et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol (2012) 227:286–97. doi:10.1002/path.4047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Svensson C, Ceder J, Iglesias-Gato D, Chuan YC, Pang ST, Bjartell A, et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res (2014) 42:999–1015. doi:10.1093/nar/gkt921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Terry S, Maillé P, Baaddi H, Kheuang L, Soyeux P, Nicolaiew N, et al. Cross modulation between the androgen receptor axis and protocadherin-PC in mediating neuroendocrine transdifferentiation and therapeutic resistance of prostate cancer. Neoplasia (2013) 15:761–72. doi:10.1593/neo.122070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Yang X, Chen MW, Terry S, Vacherot F, Chopin DK, Bemis DL, et al. A human- and male-specific protocadherin that acts through the wnt signaling pathway to induce neuroendocrine transdifferentiation of prostate cancer cells. Cancer Res (2005) 65:5263–71. doi:10.1158/0008-5472.CAN-05-0162
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Chang PC, Wang TY, Chang YT, Chu CY, Lee CL, Hsu HW, et al. Autophagy pathway is required for IL-6 induced neuroendocrine differentiation and chemoresistance of prostate cancer LNCaP cells. PLoS One (2014) 9:e88556. doi:10.1371/journal.pone.0088556
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature (2014) 510:278–82. doi:10.1038/nature13229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Wee ZN, Li Z, Lee PL, Lee ST, Lim YP, Yu Q. EZH2-mediated inactivation of IFN-γ-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep (2014) 8:204–16. doi:10.1016/j.celrep.2014.05.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Lee SO, Chun JY, Nadiminty N, Lou W, Gao AC. Interleukin-6 undergoes transition from growth inhibitor associated with neuroendocrine differentiation to stimulator accompanied by androgen receptor activation during LNCaP prostate cancer cell progression. Prostate (2007) 67:764–73. doi:10.1002/pros.20553
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Smith ND, Schulze-Hoepfner FT, Veliceasa D, Filleur S, Shareef S, Huang L, et al. Pigment epithelium-derived factor and interleukin-6 control prostate neuroendocrine differentiation via feed-forward mechanism. J Urol (2008) 179:2427–34. doi:10.1016/j.juro.2008.01.081
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Jin RJ, Lho Y, Connelly L, Wang Y, Yu X, Saint Jean L, et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res (2008) 68:6762–9. doi:10.1158/0008-5472.CAN-08-0107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Tawadros T, Alonso F, Jichlinski P, Clarke N, Calandra T, Haefliger JA, et al. Release of macrophage migration inhibitory factor by neuroendocrine-differentiated LNCaP cells sustains the proliferation and survival of prostate cancer cells. Endocr Relat Cancer (2013) 20:137–49. doi:10.1530/ERC-12-0286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Slack-Davis JK, Hershey ED, Theodorescu D, Frierson HF, Parsons JT. Differential requirement for focal adhesion kinase signaling in cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol Cancer Ther (2009) 8:2470–7. doi:10.1158/1535-7163.MCT-09-0262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell (2010) 18:23–38. doi:10.1016/j.ccr.2010.05.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Danza G, Di Serio C, Rosati F, Lonetto G, Sturli N, Kacer D, et al. Notch signaling modulates hypoxia-induced neuroendocrine differentiation of human prostate cancer cells. Mol Cancer Res (2012) 10:230–8. doi:10.1158/1541-7786.MCR-11-0296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Pittoni P, Tripodo C, Piconese S, Mauri G, Parenza M, Rigoni A, et al. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res (2011) 71:5987–97. doi:10.1158/0008-5472.CAN-11-1637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Pittoni P, Colombo MP. The dark side of mast cell-targeted therapy in prostate cancer. Cancer Res (2012) 72:831–5. doi:10.1158/0008-5472.CAN-11-3110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. McKeithen D, Graham T, Chung LW, Odero-Marah V. Snail transcription factor regulates neuroendocrine differentiation in LNCaP prostate cancer cells. Prostate (2010) 70:982–92. doi:10.1002/pros.21132
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Smith BN, Odero-Marah VA. The role of Snail in prostate cancer. Cell Adh Migr (2012) 6:433–41. doi:10.4161/cam.21687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Berenguer C, Boudouresque F, Dussert C, Daniel L, Muracciole X, Grino M, et al. Adrenomedullin, an autocrine/paracrine factor induced by androgen withdrawal, stimulates ‘neuroendocrine phenotype’ in LNCaP prostate tumor cells. Oncogene (2008) 27:506–18. doi:10.1038/sj.onc.1210656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Vlaeminck-Guillem V, Gillet G, Rimokh R. SRC: marker or actor in prostate cancer aggressiveness. Front Oncol (2014) 4:222. doi:10.3389/fonc.2014.00222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Salah Z, Maoz M, Pizov G, Bar-Shavit R. Transcriptional regulation of human protease-activated receptor 1: a role for the early growth response-1 protein in prostate cancer. Cancer Res (2007) 67:9835–43. doi:10.1158/0008-5472.CAN-07-1886
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. DaSilva J, Gioeli D, Weber MJ, Parsons SJ. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res (2009) 69:7402–11. doi:10.1158/0008-5472.CAN-08-4687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Lee GT, Kwon SJ, Lee JH, Jeon SS, Jang KT, Choi HY, et al. Macrophages induce neuroendocrine differentiation of prostate cancer cells via BMP6-IL6 loop. Prostate (2011) 71:1525–37. doi:10.1002/pros.21369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Chu GC, Zhau HE, Wang R, Rogatko A, Feng X, Zayzafoon M, et al. RANK- and c-Met-mediated signal network promotes prostate cancer metastatic colonization. Endocr Relat Cancer (2014) 21:311–26. doi:10.1530/ERC-13-0548
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Hashimoto K, Masumori N, Tanaka T, Maeda T, Kobayashi K, Kitamura H, et al. Zoledronic acid but not somatostatin analogs exerts anti-tumor effects in a model of murine prostatic neuroendocrine carcinoma of the development of castration-resistant prostate cancer. Prostate (2013) 73:500–11. doi:10.1002/pros.22590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Choi SY, Gout PW, Collins CC, Wang Y. Epithelial immune cell-like transition (EIT): a proposed transdifferentiation process underlying immune-suppressive activity of epithelial cancers. Differentiation (2012) 83:293–8. doi:10.1016/j.diff.2012.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med (2010) 363:411–22. doi:10.1056/NEJMoa1001294
46. Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, et al. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int (2013) 111:44–52. doi:10.1111/j.1464-410X.2012.11404.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Aparicio A, Logothetis CJ, Maity SN. Understanding the lethal variant of prostate cancer: power of examining extremes. Cancer Discov (2011) 1:466–8. doi:10.1158/2159-8290.CD-11-0259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Beltran H, Rubin MA. New strategies in prostate cancer: translating genomics into the clinic. Clin Cancer Res (2013) 19:517–23. doi:10.1158/1078-0432.CCR-12-1452
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Yu EY, Wilding G, Posadas E, Gross M, Culine S, Massard C, et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res (2009) 15:7421–8. doi:10.1158/1078-0432.CCR-09-1691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Araujo JC, Trudel GC, Saad F, Armstrong AJ, Yu EY, Bellmunt J, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol (2013) 14:1307–16. doi:10.1016/S1470-2045(13)70479-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Schöffski P, Blay JY, De Greve J, Brain E, Machiels JP, Soria JC, et al. Multicentric parallel phase II trial of the polo-like kinase 1 inhibitor BI 2536 in patients with advanced head and neck cancer, breast cancer, ovarian cancer, soft tissue sarcoma and melanoma. The first protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network of Core Institutes (NOCI). Eur J Cancer (2010) 46:2206–15. doi:10.1016/j.ejca.2010.03.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Schöffski P, Awada A, Dumez H, Gil T, Bartholomeus S, Wolter P, et al. A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur J Cancer (2012) 48:179–86. doi:10.1016/j.ejca.2011.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res (2014) 20:2846–50. doi:10.1158/1078-0432.CCR-13-3309
54. Smith DC, Smith MR, Sweeney C, Elfiky AA, Logothetis C, Corn PG, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol (2013) 31:412–9. doi:10.1200/JCO.2012.45.0494
55. Available from: http://www.exelixis.com/investors-media/press-releases
56. Molife LR, Yan L, Vitfell-Rasmussen J, Zernhelt AM, Sullivan DM, Cassier PA, et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol (2014) 7:1. doi:10.1186/1756-8722-7-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Carducci MA, Saad F, Abrahamsson PA, Dearneley DP, Schulman CC, North SA, et al. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer (2007) 110:1959–66. doi:10.1002/cncr.22996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Nelson JB, Love W, Chin JL, Saad F, Schulman CC, Sleep DJ, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer (2008) 113:2478–87. doi:10.1002/cncr.23864
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Quinn DI, Tangen CM, Hussain M, Lara PN Jr, Goldkorn A, Moinpour CM, et al. Docetaxel and atrasentan versus docetaxel and placebo for men with advanced castration-resistant prostate cancer (SWOG S0421): a randomised phase 3 trial. Lancet Oncol (2013) 14:893–900. doi:10.1016/S1470-2045(13)70294-8
60. Nelson JB, Fizazi K, Miller K, Higano C, Moul JW, Akaza H, et al. Phase 3, randomized, placebo-controlled study of zibotentan (ZD4054) in patients with castration-resistant prostate cancer metastatic to bone. Cancer (2012) 118:5709–18. doi:10.1002/cncr.27674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Fizazi KS, Higano CS, Nelson JB, Gleave M, Miller K, Morris T, et al. Phase III, randomized, placebo-controlled study of docetaxel in combination with zibotentan in patients with metastatic castration-resistant prostate cancer. J Clin Oncol (2013) 31:1740–7. doi:10.1200/JCO.2012.46.4149
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barrière C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res (2014) 74:5855–65. doi:10.1158/0008-5472.CAN-14-0799
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Zhao J, Zhang Z, Liao Y, Du W. Mutation of the retinoblastoma tumor suppressor gene sensitizes cancers to mitotic inhibitor induced cell death. Am J Cancer Res (2014) 4:42–52.
64. Kim J, Roh M, Abdulkadir SA. Pim1 promotes human prostate cancer cell tumorigenicity and c-MYC transcriptional activity. BMC Cancer (2010) 10:248. doi:10.1186/1471-2407-10-248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Holder S, Zemskova M, Zhang C, Tabrizizad M, Bremer R, Neidigh JW, et al. Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase. Mol Cancer Ther (2007) 6:163–72. doi:10.1158/1535-7163.MCT-06-0397
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Kroon P, Berry PA, Stower MJ, Rodrigues G, Mann VM, Simms M, et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res (2013) 73:5288–98. doi:10.1158/0008-5472.CAN-13-0874
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Meyer-Siegler KL, Iczkowski KA, Leng L, Bucala R, Vera PL. Inhibition of macrophage migration inhibitory factor or its receptor (CD74) attenuates growth and invasion of DU-145 prostate cancer cells. J Immunol (2006) 177:8730–9. doi:10.4049/jimmunol.177.12.8730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Lee BY, Hochgräfe F, Lin HM, Castillo L, Wu J, Raftery MJ, et al. Phosphoproteomic profiling identifies focal adhesion kinase as a mediator of docetaxel resistance in castrate-resistant prostate cancer. Mol Cancer Ther (2014) 13:190–201. doi:10.1158/1535-7163.MCT-13-0225-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Lin AM, Rini BI, Derynck MK, Weinberg V, Park M, Ryan CJ, et al. A phase I trial of docetaxel/estramustine/imatinib in patients with hormone-refractory prostate cancer. Clin Genitourin Cancer (2007) 5:323–8. doi:10.3816/CGC.2007.n.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Bajaj GK, Zhang Z, Garrett-Mayer E, Drew R, Sinibaldi V, Pili R, et al. Phase II study of imatinib mesylate in patients with prostate cancer with evidence of biochemical relapse after definitive radical retropubic prostatectomy or radiotherapy. Urology (2007) 69:526–31. doi:10.1016/j.urology.2006.12.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Lin AM, Rini BI, Weinberg V, Fong K, Ryan CJ, Rosenberg JE, et al. A phase II trial of imatinib mesylate in patients with biochemical relapse of prostate cancer after definitive local therapy. BJU Int (2006) 98:763–9. doi:10.1111/j.1464-410X.2006.06396.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Michaelson MD, Oudard S, Ou YC, Sengeløv L, Saad F, Houede N, et al. Randomized, placebo-controlled, phase III trial of sunitinib plus prednisone versus prednisone alone in progressive, metastatic, castration-resistant prostate cancer. J Clin Oncol (2014) 32:76–82. doi:10.1200/JCO.2012.48.5268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Safarinejad MR. Safety and efficacy of sorafenib in patients with castrate resistant prostate cancer: a phase II study. Urol Oncol (2010) 28:21–7. doi:10.1016/j.urolonc.2008.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Soria JC, Massard C, Magné N, Bader T, Mansfield CD, Blay JY, et al. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. Eur J Cancer (2009) 45:2333–41. doi:10.1016/j.ejca.2009.05.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Baritaki S, Yeung K, Palladino M, Berenson J, Bonavida B. Pivotal roles of Snail inhibition and RKIP induction by the proteasome inhibitor NPI-0052 in tumor cell chemoimmunosensitization. Cancer Res (2009) 69:8376–85. doi:10.1158/0008-5472.CAN-09-1069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: neuroendocrine prostate cancer, small cell prostate carcinoma, targeted therapy, androgen-independent, castration-resistant
Citation: Vlachostergios PJ and Papandreou CN (2015) Targeting neuroendocrine prostate cancer: molecular and clinical perspectives. Front. Oncol. 5:6. doi: 10.3389/fonc.2015.00006
Received: 22 October 2014; Accepted: 09 January 2015;
Published online: 02 February 2015.
Edited by:
Mercedes Salido, University of Cadiz, SpainReviewed by:
Vitaly Margulis, The University of Texas Southwestern Medical Center at Dallas, USARobert B. Den, Jefferson Medical College of Thomas Jefferson University, USA
Copyright: © 2015 Vlachostergios and Papandreou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiotis J. Vlachostergios, Department of Internal Medicine, Lutheran Medical Center, 150 55th Street, Brooklyn, NY 11220, USA e-mail:cHZsYWNob3N0ZXJnaW9zQGxtY21jLmNvbQ==;
Christos N. Papandreou, Department of Medical Oncology, Faculty of Medicine, School of Health Sciences, University of Thessaly, Biopolis, Larissa 41110, Greece e-mail:Y3BhcGFuZHJlb3VAbWVkLnV0aC5ncg==