 Naomi Dicks
Naomi Dicks Karina Gutierrez
Karina Gutierrez Marek Michalak
Marek Michalak Vilceu Bordignon
Vilceu Bordignon Luis B. Agellon
Luis B. Agellon- 1Department of Animal Science, McGill University, Montréal, QC, Canada
- 2Department of Biochemistry, University of Alberta, Edmonton, AB, Canada
- 3School of Dietetics and Human Nutrition, McGill University, Montréal, QC, Canada
Endoplasmic reticulum (ER) stress has been linked to many diseases, including cancer. A large body of work has focused on the activation of the ER stress response in cancer cells to facilitate their survival and tumor growth; however, there are some studies suggesting that the ER stress response can also mitigate cancer progression. Despite these contradictions, it is clear that the ER stress response is closely associated with cancer biology. The ER stress response classically encompasses activation of three separate pathways, which are collectively categorized the unfolded protein response (UPR). The UPR has been extensively studied in various cancers and appears to confer a selective advantage to tumor cells to facilitate their enhanced growth and resistance to anti-cancer agents. It has also been shown that ER stress induces chromatin changes, which can also facilitate cell survival. Chromatin remodeling has been linked with many cancers through repression of tumor suppressor and apoptosis genes. Interplay between the classic UPR and genome damage repair mechanisms may have important implications in the transformation process of normal cells into cancer cells.
Introduction
Cells in the body are continuously exposed to a dynamic environment dictated by the metabolic and nutritional status of the organism. Certain instances, such as exposure of the organism to nutrient excess or deprivation, extremes in temperatures, xenobiotics, and radiation, cause damage to cellular components and disruption of cellular processes. It has long been recognized that cells are adept at compensating for changes in their environment by altering certain cellular processes. The mobilization of such coping mechanisms is designed to maintain or recover proper function, overcome stressful conditions, and increase the chance for survival (Figure 1).
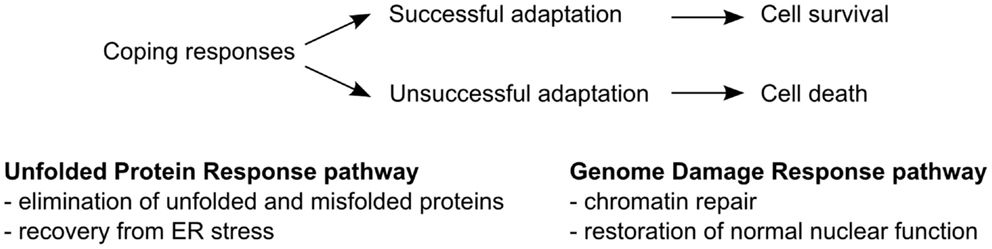
Figure 1. Coping response mechanisms. The unfolded protein response (UPR) pathway operates to restore correct folding of proteins and recovery of the ER from stress. The genome damage response (GDR) pathway enables repair of damaged DNA, histones, and other DNA binding proteins and restores normal nuclear function.
In the past several years, there has been increasing evidence linking endoplasmic reticulum (ER) stress with development of diseases, including certain types of cancers (1–5). In the case of cancers, especially non-inherited cancers that arise from genome damage, the cells capitalize on the ER stress response, which may be adaptive and advantageous at the cellular level, but deleterious to the organism. In this review, we discuss ER stress and genome damage in relation to cancer development. We provide observations supporting a link between different corrective strategies that cells adopt, which may lead to malignancies.
The accumulation of unfolded and misfolded proteins disrupts ER homeostasis and leads to the activation of the classic coping mechanism termed the unfolded protein response (UPR) (Figure 1). The UPR is initiated by the molecular chaperone glucose-regulated protein 78 (GRP78). GRP78 not only binds to the misfolded and unfolded proteins, but also regulates the transmembrane ER stress sensors, namely protein kinase RNA like ER kinase (PERK), inositol-requiring protein 1α (IRE1α), and activating transcription factor 6 (ATF6) (6–8).
Each ER stress sensor activates a separate arm of the UPR to facilitate immediate changes to a set of cellular functions designed to temporarily arrest general protein synthesis, and to produce active transcription factors that ultimately facilitate correct protein folding, degradation of proteins that cannot be properly processed, and regain of ER function. Under extreme conditions, these strategies may not be sufficient to alleviate the ER stress and thus require the removal of the malfunctioning cells. In such cases, cells undergo controlled cell death by activation of the apoptotic pathway. In some situations, certain adaptive strategies provide these cells with a selective growth advantage over other cells (Figure 1). This selective advantage could permit cells to survive and propagate even under chronic ER stress.
ER Stress and Cancer
The high proliferative rates and inadequate vascularization of solid tumors culminate in a very unfavorable microenvironment. The low pH, low oxygen tension, and low nutrient supply result in an accumulation of misfolded proteins and ER stress, which could signal cell death (9, 10). Cancer cells, however, have developed a capacity to survive these extreme conditions, despite the presence of ER stress, through modulation of the UPR response (11–14).
It has been observed that GRP78, a dominant regulator of the ER stress response, is increased in a variety of cancer types including breast, brain, lung, colon, prostate, skin, and some other malignancies (2, 12, 15–20). This chaperone is associated with prolonged cell survival, mainly by preventing ER stress-induced apoptosis and thereby promoting cell malignancy, metastatic development, and resistance to anti-cancer agents (12, 14, 21, 22). High levels of GRP78 are also associated with rapid proliferation and malignancy of tumors (12, 14). In breast cancer cells that express estrogen receptor α [NR3A1], the estrogen-mediated increase in GRP78 abundance confers improved resistance to ER stress and cell proliferation, both of which can be decreased through siRNA-mediated knockdown of estrogen receptor α (12). Similarly, up-regulation of GRP78 has been shown to increase growth of a glioma cell line whereas its down-regulation inhibits tumor development (14). The reduction of GRP78 in glioblastoma cell lines and solid tumors treated with a chemotherapeutic agent increased the expression of CHOP and caspase 7, leading to cell apoptosis and inhibition of tumor formation (11, 14). Moreover, the antitumor agent HKH40A decreases GRP78 not only at the transcriptional level but also at the protein level by directly binding GRP78 to facilitate its degradation (11). Based on these characteristics, GRP78 is considered as a biomarker of cancer progression (21).
The components of the UPR pathway have also been implicated in cancer (2, 13, 21, 23). Mutations in IRE1α have been found in some human malignancies (24, 25). Under hypoxia, the effector of the IRE1α pathway, spliced XBP1 (XBP1s), is one of the factors involved in tumor growth and survival. It promotes cancer cell survival under low oxygen conditions by forming a transcriptional complex around hypoxia-inducible factor-1, a major gene regulator under hypoxic conditions (26). This transcription factor is also involved in human breast tumorigenesis as well as in the progression of triple negative breast cancer (26, 27). Similarly, the PERK pathway can contribute to cell survival and growth through ATF4, a transcription factor that induces pro-survival genes (28, 29). ATF4 is overexpressed in solid tumors and is essential for tumor cell survival in various mouse and human cancers whereas elimination of ATF4 in cancer cells induces apoptosis (29). PERK can also facilitate tumor growth by upregulating vascular endothelial growth factor (VEGF) and thereby inducing angiogenesis in tumors (28). Tumors derived from PERK-deficient mouse embryonic fibroblasts are considerably smaller compared to those derived from wildtype embryonic fibroblasts as a result of their impaired ability to stimulate angiogenesis (28).
Despite ample examples suggesting that the activation of the UPR is essential to cancer cell survival and tumor development, there are also indications that ER stress may provide protection against cancer (3, 30, 31). In particular, it has been shown that XBP1 is protective against intestinal tumorigenesis (3). Prostatic cancer cells have been shown to produce high levels of UDP-N-acetylglucosamine pyrophosphorylase 1, which reduces ER stress in these cells and facilitates their growth (30). The flavonoid baicalein has also been shown to induce ER stress in hepatocellular cancer cells, resulting in increased apoptosis (31). Interestingly, in this same study, increased IREα and eIF2α activation provided a survival advantage to theses cancerous cells. This finding highlights the paradoxical role of the UPR in cancer and our incomplete understanding of how signaling pathways may favor cell death or survival under different conditions (32–34). Whichever the outcome produced by ER stress, it is clearly apparent that the UPR plays a critical role in cancer biology.
Genome Damage and Cancer
Genome damage can be caused by a number of endogenous and exogenous genotoxic factors, including reactive oxygen species, altered cell metabolism, xenobiotics, and radiation (35, 36). These factors lead to DNA strand breaks, collapsed DNA replication forks, and damage to histones as well as other DNA-binding proteins (35). In response to chromatin damage, cells can establish a genome damage response (GDR) to repair damage to both DNA and nuclear proteins, adapt to genome damage, and reestablish nuclear function (Figure 1). Adaptation to genome damage can lead to cell survival but also chromatin alterations, which may have severe consequences for tissue function and physiology (36).
The GDR is orchestrated by several factors encompassing sensors, transducers, and effectors proteins (Figure 2), which require post-translational modification and accumulation of proteins to assemble multiprotein foci at the sites of DNA lesions (37–39). In general, activation of GDR involves temporary cell cycle arrest, local inhibition of transcription, and relaxation of chromatin to facilitate repairs. This process requires post-translational modification of proteins including the activation of the kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3 related (ATR), which phosphorylate transducer proteins at the damages sites, including the histone H2A.x (H2AX139ph), which anchors some important effector proteins required for damage repair and cell cycle arrest (37–40). ATM and ATR also activate the serine–threonine checkpoint effector kinases, Chk1 and Chk2, which regulate a number of proteins involved in transcription, cell cycle progression and apoptosis, including the tumor suppressor protein p53 and BRCA1 (41, 42), and the cell cycle regulator proteins Cdc25 and Wee1 (43, 44). Phosphorylation of transcription factors, notably E2F1, NR4A, ATF2, and Sp1, also facilitate DNA repair in a transcription-independent fashion, by direct interaction with damaged DNA, and subsequently the co-localization of other DNA repair proteins (45). In addition to post-translational modifications, genotoxic lesions and DNA damaging agents can also trigger nucleosomal remodeling via eviction of resident histones and reincorporation of new histones into the reassembled nucleosomes after damage repair (37, 46–48).
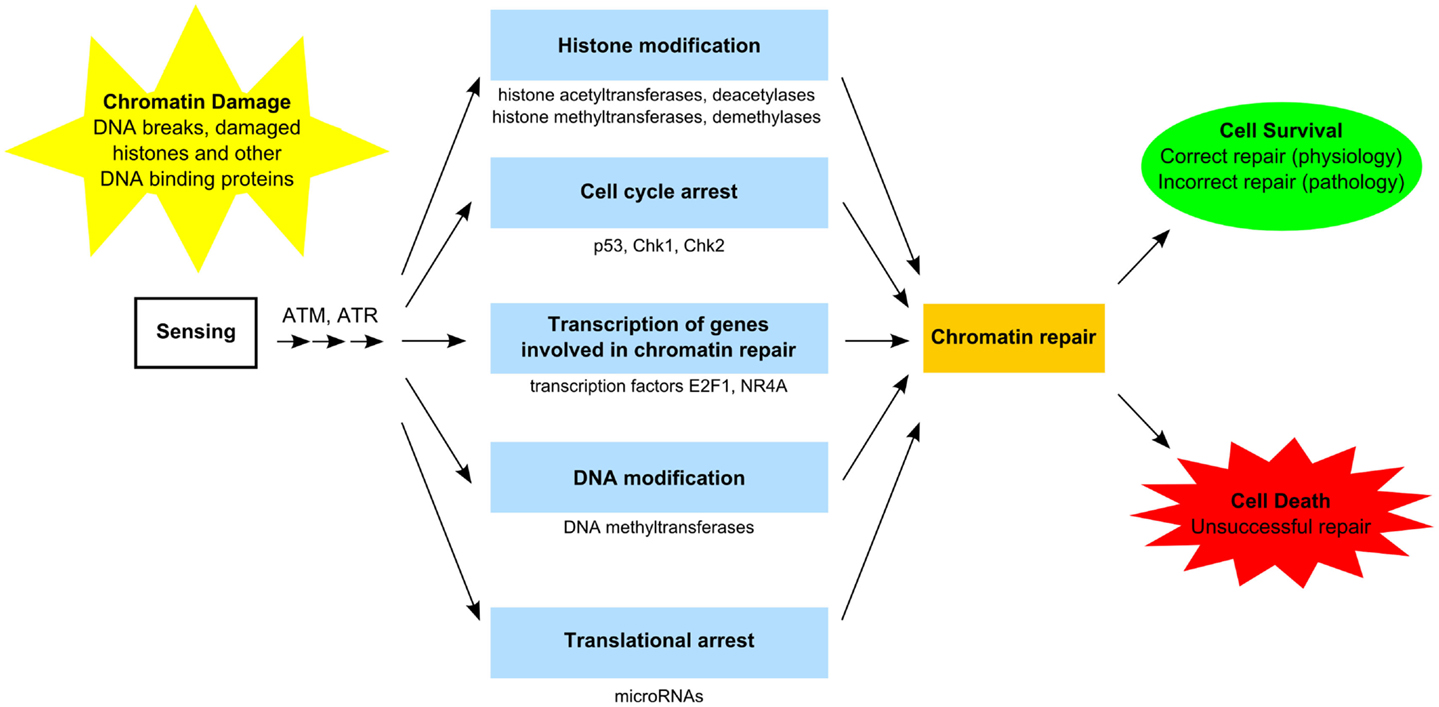
Figure 2. Functional components of the GDR pathway. Sensing of damaged DNA, histones, and other DNA-binding proteins results in activation of specific kinases [e.g., ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) proteins] (37–40). Access to damaged chromatin is facilitated by histone modification involving histone acetyltransferases/deacetylases and histone methyltransferase/demethylases as well as by DNA modification involving DNA methyltransferase (37, 38, 48–50). Certain transcription factors (e.g., E2F1, NR4A) stimulate genes involved in chromatin repair (45). Translational arrest is facilitated by microRNAs (e.g., mIR-155, miR-18a) (51, 52). Cell cycle arrest (e.g., via p53, Chk1, Chk2) may be required in order to complete chromatin repair (41–44). Unsuccessful chromatin repair due to extensive damage commits the cell to die. Successful chromatin repair enables cells to survive and restore normal function. In certain cases, incorrect repair escapes quality control surveillance and leads to altered cell function, which may provide the cell with a survival advantage, but manifest as pathology at the organismal level.
Local transcriptional arrest associated with GDR may be transient or stable (49, 53). Linked to this arrest are numerous histone modifications, including hypoacetylation of histone H4, increased histone H3K9me3 and H3K27me3, and decreased histone H3K4me3 (49, 50). Histone chaperones, which transfer histones to the nucleosomes, are required for transcriptional reinitiation following DNA damage (49, 54). GDR also involves the participation of ATP-dependent chromatin remodeling complexes, including the switch/sucrose non-fermentable (SWI2/SNF2), imitation switch (ISWI), inositol requiring 80 (INO80), and chromodomain helicase DNA-binding protein, which mediate nucleosome sliding and histone displacement promoting access for DNA repair proteins (37, 38, 48).
Non-coding RNAs are also known to affect DNA repair and genome instability. Indeed, DNA damage responsive microRNAs have been shown to be misexpressed in cancer cells and to affect chemotherapy sensitivity (55–57). It has also been shown that down-regulation of Dicer and Ago2, two essential microRNA processing components, reduced cell survival and checkpoint response after UV-induced DNA damage (58). Moreover, the miR-18a was shown to downregulate ATM expression, reduce DNA damage repair, and sensitize breast cancer cells to γ-irradiation treatment (51). Also, up-regulation of the miR-24 decreases H2AX and renders cells more vulnerable to DNA damage induced by γ-irradiation and genotoxic drugs (59). Another microRNA, miR-155, was shown to reduce the levels of RAD51, a recombinase required to repair double strand breaks by DNA homologous recombination, and consequently decreased DNA repair and enhanced sensitivity to ionizing radiation in human breast cancer cells (52).
UPR and GDR Crosstalk
There is accumulating evidence suggesting that ER stress and GDRs are intertwined. Indeed, ER stress induced with tunicamycin treatment or glucose deprivation decreases genomic DNA damage repair by stimulating proteasomal degradation of Rad51 (60). On the other hand, down-regulation of PERK enhances DNA damage repair in irradiated cancer cells (61). Interestingly, induction of ER stress recruits the histone acetyltransferase p300 to the GRP78 promoter and this correlates with increased histone H4 acetylation and GRP78 gene expression (62). Increased GRP78 transcription is associated with the recruitment of arginine histone methyltransferase, PRMT1 (62). It was suggested that arginine methylation of MRE11 by PRMT1 regulates the activity of MRN complex, which is required for proper DNA damage checkpoint control (63). Therefore, it appears that increased GRP78 transcription from ER stress can also facilitate DNA damage repair. These contradictory effects further demonstrate our incomplete understanding of the stress signaling pathways and how they interact to determine cell fate. However, it also illustrates how ER stress can cause chromatin remodeling and affect the GDR pathway. If GDRs are impaired by alterations in the UPR, this can affect DNA integrity and subsequently increase risks of carcinogenesis.
Signaling from both ER stress and DNA damage also appear to result in similar chromatin remodeling changes to respond to cellular insults. Increased H3K14 acetylation as a consequence of ER stress has been observed, and this can activate the expression of other target ER stress response genes (64). Similarly, GDR results in increased H3K14ac, which promotes the binding of BRG1, an ATPase component of SWI2/SNF2 complex, to H2AXph139 at the sites of DNA damage enabling chromatin remodeling for DNA repair (65, 66). Phosphorylation of the histone H2Ax also enables recruitment of other chromatin remodeling complexes including INO80 and SWR1, and the histone acetyltransferase complex NuA4 to facilitate DNA repair (67–70). Therefore, H3K14ac and H2AXph139 seem to be important in connecting ER stress and GDR.
Chromatin remodeling has also been shown to occur as a result of hypoxia and heat stress, two common causes of ER stress that also have effects on GDR (71). Hypoxia-induced ER stress leads to global deacetylation and methylation of histones in the proximity of genes involved in the hypoxia-inducible factor-1-mediated response (72–74). This facilitates transcription of the genes needed for an adaptive response to hypoxia (75, 76). Yet, there is evidence confirming that hypoxia can lead to defective DNA repair, genomic instability, and consequently, to cellular transformation (76). In addition, it has been well documented that chromatin remodeling in response to heat stress results in increased transcription of heat shock proteins (77). These proteins have been shown to reduce accumulation of H2AXph139, decrease DNA damage repair, and increase radiation sensitivity and genome instability (78, 79).
While there is evidence demonstrating crosstalk between the UPR and GDR, it is not well understood at this time. Increased reactive oxygen species appears to be a common by-product of most cellular insults, ER stress, and DNA damage included (80). Oxidative stress can modulate multiple signaling pathways through activation of common transducers and transcription factors (81).
Role of UPR and GDR in Carcinogenesis
Classically, the development of cancer is largely associated with inherited or acquired mutations of specific genes that regulate cell cycle, proliferation, and apoptosis (82, 83). However, similar effects can be seen with epigenetic changes, which, alone or associated with genetic mutations, can alter the expression of tumor suppressor genes (84–87). There are many examples of chromatin changes that lead to cancers. Hypermethylation of the DNA repair gene BRCA1 has been associated with both breast and ovarian cancer (88–90). Aberrant promoter methylation of the Kelch-like-ECH-associated protein 1 gene, which codes for an adaptor protein involved in degradation of cell survival and anti-apoptosis gene products, has been linked to a poorer prognosis and increased carcinogenesis in breast cancer patients (91). Hypermethylation of tumor suppressor genes has been observed in renal carcinomas and hematopoietic cancers (92–94). Hypermethylation of the cell cycle regulation gene RB1 and cyclin-dependent kinase inhibitor genes, CDKN2B and CDKN2A, which are, respectively, associated with the ocular tumor, retinoblastoma (95), and various leukemias and lymphomas (93). Histone deacetylation has been associated with a more aggressive form of acute myeloid leukemia (AML) through its repressive effect on the tumor suppressor gene death-associated protein kinase 1 (96). AML has also been associated with changes in histone methylation patterns (97). Finally, chromatin remodeling agents, including inhibitors of histone deacetylases, histone lysine demethylases, and DNA methyltransferases, have been tested for the treatment of various cancers (94, 98–102).
Since both ER stress and GDR coping mechanisms affect chromatin remodeling and DNA repair, adaptations based on these mechanisms could lead to emergence of malignant cells with self-renewal properties due to both genomic and epigenomic alterations. For example, hypermethylation of promoter regions around ER stress response genes have been implicated in the development of alcohol-induced liver cancer (103). GRP78-deficient mice fed large quantities of alcohol throughout their lives show high incidence of hepatic tumors, and correlate with hypermethylation of ATF6, which upregulates genes involved in ER-associated degradation to deal with the accumulation of misfolded proteins (103). Also, increased GRP78 stimulates the VEGF receptor 2 and subsequently VEGF-induced endothelial cell proliferation, which facilitates angiogenesis and tumor survival and growth (104–106). The apparent contradictory effect on neoplasticity as both inhibition and promotion of cancer progression, predicted by GRP78 abundance, suggests that the nature and context of coping response activation are important determinants of the outcome.
Acetylation of H3K14 has also been implicated in cell survival and carcinogenesis, both with respect to the UPR and GDR. Increase in H3K14 acetylation in response to ER stress results in stimulation of transcription, promoting cell survival (64). Increased H3K14ac during GDR enhances access of BRG1 to the sites of DNA damage to promote chromatin remodeling required for DNA repair (65, 66). However, in addition to promoting DNA repair, BRG1 has been associated with cancer development. For example, BRG1 was shown to impair the recruitment of BRCA1 to DNA damage sites, which is important in DNA damage repair and in the maintenance of genomic stability (107); to activate the melanoma inhibitor of apoptosis gene (108); and to support oncogenic transcriptional program, including Myc (109), for the survival of leukemic cells (110). Finally, chromatin changes in response to genotoxic conditions have been shown to alter the regulation of the Hedgehog–Gli signaling pathway, which has been implicated in genome instability and in several types of cancers (111–113).
Summary
Coping mechanisms are designed to correct, minimize, or overcome damage caused by harsh environments, and promote cell survival. The UPR pathway is mobilized in response to the accumulation of unfolded proteins and to ultimately regain ER homeostasis. Similarly, the GDR pathway operates in response to chromatin damage and to restore normal nuclear function. Some adaptive strategies allow cells to overcome defects in cellular function through metabolic adaptation and gain a survival advantage, such as in certain types of malignancies. A better understanding of the interplay between UPR and GDR pathways may provide new insights into the pathogenesis of cancers, which could give rise to more effective anti-cancer therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Research in our laboratories is funded by grants from the Canadian Institutes of Health Research (to Luis B. Agellon and to Marek Michalak) and the Natural Sciences and Engineering Research Council of Canada (to Luis B. Agellon and to Vilceu Bordignon). Naomi Dicks is supported by an Alexander Graham Bell Canada Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada. Karina Gutierrez is supported by a scholarship from the Science without Borders Program of the Brazilian Coordination for the Improvement of Higher Education Personnel (CAPES).
References
1. Wu X, Xin Z, Zhang W, Zheng S, Wu J, Chen K, et al. A missense polymorphism in ATF6 gene is associated with susceptibility to hepatocellular carcinoma probably by altering ATF6 level. Int J Cancer (2014) 135:61–8. doi: 10.1002/ijc.28649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer (2014) 14:581–97. doi:10.1038/nrc3800
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Niederreiter L, Fritz TM, Adolph TE, Krismer AM, Offner FA, Tschurtschenthaler M, et al. ER stress transcription factor Xbp1 suppresses intestinal tumorigenesis and directs intestinal stem cells. J Exp Med (2013) 210:2041–56. doi:10.1084/jem.20122341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Yoshida H. ER stress and diseases. FEBS J (2007) 274:630–58. doi:10.1111/j.1742-4658.2007.05639.x
5. Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev (2007) 87:1377–408. doi:10.1152/physrev.00050.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell (2001) 107:881–91. doi:10.1016/S0092-8674(01)00611-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell (2002) 3:99–111. doi:10.1016/S1534-5807(02)00203-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol (2000) 2:326–32. doi:10.1038/35014014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Saito S, Furuno A, Sakurai J, Sakamoto A, Park HR, Shin-Ya K, et al. Chemical genomics identifies the unfolded protein response as a target for selective cancer cell killing during glucose deprivation. Cancer Res (2009) 69:4225–34. doi:10.1158/0008-5472.CAN-08-2689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer (2004) 4:966–77. doi:10.1038/nrc1505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Kosakowska-Cholody T, Lin J, Srideshikan SM, Scheffer L, Tarasova NI, Acharya JK. HKH40A downregulates GRP78/BiP expression in cancer cells. Cell Death Dis (2014) 5:e1240. doi:10.1038/cddis.2014.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Andruska N, Zheng X, Yang X, Helferich WG, Shapiro DJ. Anticipatory estrogen activation of the unfolded protein response is linked to cell proliferation and poor survival in estrogen receptor α-positive breast cancer. Oncogene (2014). doi:10.1038/onc.2014.292
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta (2014) 1843:2143–9. doi:10.1016/j.bbamcr.2014.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Pyrko P, Schonthal AH, Hofman FM, Chen TC, Lee AS. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res (2007) 67(20):9809–16. doi:10.1158/0008-5472.CAN-07-0625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res (2006) 66(16):7849–53. doi:10.1158/0008-5472.CAN-06-1660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Wang Q, He Z, Zhang J, Wang Y, Wang T, Tong S, et al. Overexpression of endoplasmic reticulum molecular chaperone GRP94 and GRP78 in human lung cancer tissues and its significance. Cancer Detect Prev (2005) 29:544–51. doi:10.1016/j.cdp.2005.09.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Lee HK, Xiang C, Cazacu S, Finniss S, Kazimirsky G, Lemke N, et al. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol (2008) 10:236–43. doi:10.1215/15228517-2008-006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Xing X, Lai M, Wang Y, Xu E, Huang Q. Overexpression of glucose-regulated protein 78 in colon cancer. Clin Chim Acta (2006) 364:308–15. doi:10.1016/j.cca.2005.07.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Pootrakul L, Datar RH, Shi SR, Cai J, Hawes D, Groshen SG, et al. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin Cancer Res (2006) 12:5987–93. doi:10.1158/1078-0432.CCR-06-0133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Zhuang L, Scolyer RA, Lee CS, McCarthy SW, Cooper WA, Zhang XD, et al. Expression of glucose-regulated stress protein GRP78 is related to progression of melanoma. Histopathology (2009) 54:462–70. doi:10.1111/j.1365-2559.2009.03242.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res (2007) 67:3496–9. doi:10.1158/0008-5472.CAN-07-0325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Visioli F, Wang Y, Alam GN, Ning Y, Rados PV, Nor JE, et al. Glucose-regulated protein 78 (Grp78) confers chemoresistance to tumor endothelial cells under acidic stress. PLoS One (2014) 9:e101053. doi:10.1371/journal.pone.0101053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Manie SN, Lebeau J, Chevet E. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. Orchestrating the unfolded protein response in oncogenesis: an update. Am J Physiol Cell Physiol (2014) 307:c901–7. doi:10.1152/ajpcell.00292.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet (2012) 44:694–8. doi:10.1038/ng.2256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, et al. Patterns of somatic mutation in human cancer genomes. Nature (2007) 446:153–8. doi:10.1038/nature05610
26. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature (2014) 508:103–7. doi:10.1038/nature13119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Fujimoto T, Onda M, Nagai H, Nagahata T, Ogawa K, Emi M. Upregulation and overexpression of human X-box binding protein 1 (hXBP-1) gene in primary breast cancers. Breast Cancer (2003) 10:301–6. doi:10.1007/BF02967649
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol (2006) 26:9517–32. doi:10.1128/MCB.01145-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J (2010) 29:2082–96. doi:10.1038/emboj.2010.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Itkonen HM, Engedal N, Babaie E, Luhr M, Guldvik IJ, Minner S, et al. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene (2014). doi:10.1038/onc.2014.307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Wang Z, Jiang C, Chen W, Zhang G, Luo D, Cao Y, et al. Baicalein induces apoptosis and autophagy via endoplasmic reticulum stress in hepatocellular carcinoma cells. Biomed Res Int (2014) 2014:732516. doi:10.1155/2014/732516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Vandewynckel YP, Laukens D, Geerts A, Bogaerts E, Paridaens A, Verhelst X, et al. The paradox of the unfolded protein response in cancer. Anticancer Res (2013) 33:4683–94.
33. Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell (2014) 25:563–73. doi:10.1016/j.ccr.2014.03.015
34. Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer Lett (2013) 332:249–64. doi:10.1016/j.canlet.2010.07.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet (2001) 27:247–54. doi:10.1038/85798
36. Furgason JM, Bahassi el M. Targeting DNA repair mechanisms in cancer. Pharmacol Ther (2013) 137:298–308. doi:10.1016/j.pharmthera.2012.10.009
37. Luijsterburg MS, van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol Oncol (2011) 5:349–67. doi:10.1016/j.molonc.2011.06.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Soria G, Polo SE, Almouzni G. Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell (2012) 46:722–34. doi:10.1016/j.molcel.2012.06.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Roos WP, Kaina B. DNA damage-induced cell death: from specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett (2013) 332:237–48. doi:10.1016/j.canlet.2012.01.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol (2011) 13:1161–9. doi:10.1038/ncb2344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J (2005) 24:3411–22. doi:10.1038/sj.emboj.7600812
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Lee JS, Collins KM, Brown AL, Lee CH, Chung JH. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature (2000) 404:201–4. doi:10.1038/35004614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet (2002) 30:290–4. doi:10.1038/ng845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell (2001) 12:551–63. doi:10.1091/mbc.12.3.551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Malewicz M, Perlmann T. Function of transcription factors at DNA lesions in DNA repair. Exp Cell Res (2014) 329:94–100. doi:10.1016/j.yexcr.2014.08.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Ikura T, Tashiro S, Kakino A, Shima H, Jacob N, Amunugama R, et al. DNA damage-dependent acetylation and ubiquitination of H2AX enhances chromatin dynamics. Mol Cell Biol (2007) 27:7028–40. doi:10.1128/MCB.00579-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Dinant C, Ampatziadis-Michailidis G, Lans H, Tresini M, Lagarou A, Grosbart M, et al. Enhanced chromatin dynamics by FACT promotes transcriptional restart after UV-induced DNA damage. Mol Cell (2013) 51:469–79. doi:10.1016/j.molcel.2013.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Polo SE. Reshaping chromatin after DNA damage: the choreography of histone proteins. J Mol Biol (2014). doi:10.1016/j.jmb.2014.05.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Adam S, Polo SE. Blurring the line between the DNA damage response and transcription: the importance of chromatin dynamics. Exp Cell Res (2014) 329:148–53. doi:10.1016/j.yexcr.2014.07.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Seiler DM, Rouquette J, Schmid VJ, Strickfaden H, Ottmann C, Drexler GA, et al. Double-strand break-induced transcriptional silencing is associated with loss of tri-methylation at H3K4. Chromosome Res (2011) 19:883–99. doi:10.1007/s10577-011-9244-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Song L, Lin C, Wu Z, Gong H, Zeng Y, Wu J, et al. miR-18a impairs DNA damage response through downregulation of ataxia telangiectasia mutated (ATM) kinase. PLoS One (2011) 6:e25454. doi:10.1371/journal.pone.0025454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Gasparini P, Lovat F, Fassan M, Casadei L, Cascione L, Jacob NK, et al. Protective role of miR-155 in breast cancer through RAD51 targeting impairs homologous recombination after irradiation. Proc Natl Acad Sci U S A (2014) 111:4536–41. doi:10.1073/pnas.1402604111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Svejstrup JQ. The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem Sci (2010) 35:333–8. doi:10.1016/j.tibs.2010.02.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Hamiche A, Shuaib M. Chaperoning the histone H3 family. Biochim Biophys Acta (2013) 1819:230–7. doi:10.1016/j.bbagrm.2011.08.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Wang Y, Taniguchi T. MicroRNAs and DNA damage response: implications for cancer therapy. Cell Cycle (2013) 12:32–42. doi:10.4161/cc.23051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. van Jaarsveld MT, Wouters MD, Boersma AW, Smid M, van Ijcken WF, Mathijssen RH, et al. DNA damage responsive microRNAs misexpressed in human cancer modulate therapy sensitivity. Mol Oncol (2014) 8:458–68. doi:10.1016/j.molonc.2013.12.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Wan G, Liu Y, Han C, Zhang X, Lu X. Noncoding RNAs in DNA repair and genome integrity. Antioxid Redox Signal (2014) 20:655–77. doi:10.1089/ars.2013.5514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Pothof J, Verkaik NS, van IW, Wiemer EA, Ta VT, van der Horst GT, et al. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J (2009) 28:2090–9. doi:10.1038/emboj.2009.156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Lal A, Pan Y, Navarro F, Dykxhoorn DM, Moreau L, Meire E, et al. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat Struct Mol Biol (2009) 16:492–8. doi:10.1038/nsmb.1589
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Yamamori T, Meike S, Nagane M, Yasui H, Inanami O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett (2013) 587:3348–53. doi:10.1016/j.febslet.2013.08.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Oommen D, Prise KM. Down-regulation of PERK enhances resistance to ionizing radiation. Biochem Biophys Res Commun (2013) 441:31–5. doi:10.1016/j.bbrc.2013.09.129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Baumeister P, Luo S, Skarnes WC, Sui G, Seto E, Shi Y, et al. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol Cell Biol (2005) 25:4529–40. doi:10.1128/MCB.25.11.4529-4540.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Boisvert FM, Dery U, Masson JY, Richard S. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev (2005) 19:671–6. doi:10.1101/gad.1279805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Schram AW, Baas R, Jansen PW, Riss A, Tora L, Vermeulen M, et al. A dual role for SAGA-associated factor 29 (SGF29) in ER stress survival by coordination of both histone H3 acetylation and histone H3 lysine-4 trimethylation. PLoS One (2013) 8:e70035. doi:10.1371/journal.pone.0070035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. A cooperative activation loop among SWI/SNF, γ-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J (2010) 29:1434–45. doi:10.1038/emboj.2010.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, et al. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene (2011) 30:2135–46. doi:10.1038/onc.2010.592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Bird AW, Yu DY, Pray-Grant MG, Qiu Q, Harmon KE, Megee PC, et al. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature (2002) 419:411–5. doi:10.1038/nature01035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, et al. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell (2004) 16:979–90. doi:10.1016/j.molcel.2004.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, et al. INO80 and γ-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell (2004) 119:767–75. doi:10.1016/j.cell.2004.11.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell (2004) 119:777–88. doi:10.1016/j.cell.2004.11.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol (2013) 75:49–67. doi:10.1146/annurev-physiol-030212-183707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, et al. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J (2005) 24:3846–58. doi:10.1038/sj.emboj.7600846
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J (2005) 19:1296–8. doi:10.1096/fj.04-3099fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Islam KN, Mendelson CR. Permissive effects of oxygen on cyclic AMP and interleukin-1 stimulation of surfactant protein A gene expression are mediated by epigenetic mechanisms. Mol Cell Biol (2006) 26:2901–12. doi:10.1128/MCB.26.8.2901-2912.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Johnson AB, Barton MC. Hypoxia-induced and stress-specific changes in chromatin structure and function. Mutat Res (2007) 618:149–62. doi:10.1016/j.mrfmmm.2006.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat Rev Cancer (2008) 8:180–92. doi:10.1038/nrc2344
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Sullivan EK, Weirich CS, Guyon JR, Sif S, Kingston RE. Transcriptional activation domains of human heat shock factor 1 recruit human SWI/SNF. Mol Cell Biol (2001) 21:5826–37. doi:10.1128/MCB.21.17.5826-5837.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Laszlo A, Fleischer I. Heat-induced perturbations of DNA damage signaling pathways are modulated by molecular chaperones. Cancer Res (2009) 69:2042–9. doi:10.1158/0008-5472.CAN-08-1639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Gabai VL, Sherman MY, Yaglom JA. HSP72 depletion suppresses γH2AX activation by genotoxic stresses via p53/p21 signaling. Oncogene (2010) 29:1952–62. doi:10.1038/onc.2009.480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Jiang F, Zhang Y, Dusting GJ. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev (2011) 63:218–42. doi:10.1124/pr.110.002980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Simmons SO, Fan CY, Ramabhadran R. Cellular stress response pathway system as a sentinel ensemble in toxicological screening. Toxicol Sci (2009) 111:202–25. doi:10.1093/toxsci/kfp140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature (2009) 458:719–24. doi:10.1038/nature07943
83. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (2013) 339:1546–58. doi:10.1126/science.1235122
84. Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer (2011) 11:726–34. doi:10.1038/nrc3130
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell (2012) 22:9–20. doi:10.1016/j.ccr.2012.06.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell (2013) 153:38–55. doi:10.1016/j.cell.2013.03.008
87. Easwaran H, Tsai HC, Baylin SB. Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol Cell (2014) 54:716–27. doi:10.1016/j.molcel.2014.05.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Birgisdottir V, Stefansson OA, Bodvarsdottir SK, Hilmarsdottir H, Jonasson JG, Eyfjord JE. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res (2006) 8:R38. doi:10.1186/bcr1522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst (2000) 92:564–9. doi:10.1093/jnci/92.7.564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Ibanez de Caceres I, Battagli C, Esteller M, Herman JG, Dulaimi E, Edelson MI, et al. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res (2004) 64:6476–81. doi:10.1158/0008-5472.CAN-04-1529
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Barbano R, Muscarella LA, Pasculli B, Valori VM, Fontana A, Coco M, et al. Aberrant Keap1 methylation in breast cancer and association with clinicopathological features. Epigenetics (2013) 8:105–12. doi:10.4161/epi.23319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A (1994) 91:9700–4. doi:10.1073/pnas.91.21.9700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Herman JG, Civin CI, Issa JP, Collector MI, Sharkis SJ, Baylin SB. Distinct patterns of inactivation of p15INK4B and p16INK4A characterize the major types of hematological malignancies. Cancer Res (1997) 57:837–41.
94. Schafer E, Irizarry R, Negi S, McIntyre E, Small D, Figueroa ME, et al. Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: biology and therapeutic targeting. Blood (2010) 115:4798–809. doi:10.1182/blood-2009-09-243634
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Greger V, Debus N, Lohmann D, Hopping W, Passarge E, Horsthemke B. Frequency and parental origin of hypermethylated RB1 alleles in retinoblastoma. Hum Genet (1994) 94:491–6. doi:10.1007/BF00211013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Shanmugam R, Gade P, Wilson-Weekes A, Sayar H, Suvannasankha A, Goswami C, et al. A noncanonical Flt3ITD/NF-kappaB signaling pathway represses DAPK1 in acute myeloid leukemia. Clin Cancer Res (2012) 18:360–9. doi:10.1158/1078-0432.CCR-10-3022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Yamazaki J, Estecio MR, Lu Y, Long H, Malouf GG, Graber D, et al. The epigenome of AML stem and progenitor cells. Epigenetics (2013) 8:92–104. doi:10.4161/epi.23243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Zhou J, Bi C, Cheong LL, Mahara S, Liu SC, Tay KG, et al. The histone methyltransferase inhibitor, DZNep, up-regulates TXNIP, increases ROS production, and targets leukemia cells in AML. Blood (2011) 118:2830–9. doi:10.1182/blood-2010-07-294827
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours – lessons from the past. Nat Rev Clin Oncol (2013) 10:256–66. doi:10.1038/nrclinonc.2013.42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Yang PM, Lin YT, Shun CT, Lin SH, Wei TT, Chuang SH, et al. Zebularine inhibits tumorigenesis and stemness of colorectal cancer via p53-dependent endoplasmic reticulum stress. Sci Rep (2013) 3:3219. doi:10.1038/srep03219
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Bojang P Jr, Ramos KS. The promise and failures of epigenetic therapies for cancer treatment. Cancer Treat Rev (2014) 40:153–69. doi:10.1016/j.ctrv.2013.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Thinnes CC, England KS, Kawamura A, Chowdhury R, Schofield CJ, Hopkinson RJ. Targeting histone lysine demethylases – progress, challenges, and the future. Biochim Biophys Acta (2014) 1839:1416–32. doi:10.1016/j.bbagrm.2014.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Han H, Hu J, Lau MY, Feng M, Petrovic LM, Ji C. Altered methylation and expression of ER-associated degradation factors in long-term alcohol and constitutive ER stress-induced murine hepatic tumors. Front Genet (2013) 4:224. doi:10.3389/fgene.2013.00224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res (2008) 68:498–505. doi:10.1158/0008-5472.CAN-07-2950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Virrey JJ, Dong D, Stiles C, Patterson JB, Pen L, Ni M, et al. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol Cancer Res (2008) 6:1268–75. doi:10.1158/1541-7786.MCR-08-0060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Katanasaka Y, Ishii T, Asai T, Naitou H, Maeda N, Koizumi F, et al. Cancer antineovascular therapy with liposome drug delivery systems targeted to BiP/GRP78. Int J Cancer (2010) 127:2685–98. doi:10.1002/ijc.25276
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Zhang L, Chen H, Gong M, Gong F. The chromatin remodeling protein BRG1 modulates BRCA1 response to UV irradiation by regulating ATR/ATM activation. Front Oncol (2013) 3:7. doi:10.3389/fonc.2013.00007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Saladi SV, Wong PG, Trivedi AR, Marathe HG, Keenen B, Aras S, et al. BRG1 promotes survival of UV-irradiated melanoma cells by cooperating with MITF to activate the melanoma inhibitor of apoptosis gene. Pigment Cell Melanoma Res (2013) 26:377–91. doi:10.1111/pcmr.12088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev (2013) 27:2648–62. doi:10.1101/gad.232710.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Buscarlet M, Krasteva V, Ho L, Simon C, Hebert J, Wilhelm B, et al. Essential role of BRG, the ATPase subunit of BAF chromatin remodeling complexes, in leukemia maintenance. Blood (2014) 123:1720–8. doi:10.1182/blood-2013-02-483495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Mazza D, Infante P, Colicchia V, Greco A, Alfonsi R, Siler M, et al. PCAF ubiquitin ligase activity inhibits Hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ (2013) 20:1688–97. doi:10.1038/cdd.2013.120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Malatesta M, Steinhauer C, Mohammad F, Pandey DP, Squatrito M, Helin K. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res (2013) 73:6323–33. doi:10.1158/0008-5472.CAN-12-4660
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Carrillo AM, Bouska A, Arrate MP, Eischen CM. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene (2014). doi:10.1038/onc.2014.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: carcinogenesis, cell death, chromatin damage, coping responses, DNA breaks, endoplasmic reticulum, unfolded proteins
Citation: Dicks N, Gutierrez K, Michalak M, Bordignon V and Agellon LB (2015) Endoplasmic reticulum stress, genome damage, and cancer. Front. Oncol. 5:11. doi: 10.3389/fonc.2015.00011
Received: 01 November 2014; Paper pending published: 28 November 2014;
Accepted: 12 January 2015; Published online: 03 February 2015.
Edited by:
Paul Eggleton, Exeter University Medical School, UKReviewed by:
Aleksandra Fucic, Institute for Medical Research and Occupational Health, CroatiaSushma S. Iyengar, University of Southern California, USA
Copyright: © 2015 Dicks, Gutierrez, Michalak, Bordignon and Agellon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis B. Agellon, School of Dietetics and Human Nutrition, McGill University, 21111 Lakeshore Road, Ste. Anne de Bellevue, Montréal, QC H9X3V9, Canada e-mail:bHVpcy5hZ2VsbG9uQG1jZ2lsbC5jYQ==;
Vilceu Bordignon, Department of Animal Science, McGill University, 21111 Lakeshore Road, Ste. Anne de Bellevue, Montréal, QC H9X3V9, Canada e-mail:dmlsY2V1LmJvcmRpZ25vbkBtY2dpbGwuY2E=