 Leonel Ampie
Leonel Ampie Eric C. Woolf
Eric C. Woolf Christopher Dardis
Christopher Dardis- Department of Neurology, St. Joseph’s Hospital and Medical Center, Barrow Neurological Institute, Phoenix, AZ, USA
Immunotherapy seeks to improve the body’s immune response to a tumor. Currently, the principal mechanisms employed are: (1) to improve an aspect of the immune response (e.g., T cell activation) and (2) to encourage the targeting of particular antigens. The latter is typically achieved by exposing the immune system to the antigen in question, in vivo, or in vitro followed by re-introduction of the primed cells to the body. The clinical relevance of these approaches has already been demonstrated for solid tumors such as melanoma and prostate cancer. The central nervous system was previously thought to be immune privileged. However, we know now that the immune system is highly active in the brain and interacts with brain tumors. Thus, harnessing and exploiting this interaction represents an important approach for treating malignant brain tumors. We present a summary of progress in this area, focusing particularly on immune-checkpoint inhibition, vaccines, and T cell engineering.
Introduction
Patients with cancer are typically immunosuppressed. This appears to be a survival strategy of the more aggressive tumors and is in excess of that which would be expected by external factors such as chemotherapy, malnutrition and steroid use. When discussing immunotherapy for tumors affecting the nervous system, the prototype remains glioblastoma (GB, grade IV glioma). This is the most common malignant primary central nervous system (CNS) malignancy (1). Aside from developments in the treatment of systemic metastases to the brain, the use of immunotherapy of other CNS tumors is at a relatively less developed stage.
An early observation germane to this field was that tumors may (rarely) resolve following an infection. This phenomenon has been documented, for example, in locally advanced pancreatic cancer (2). Therapeutic applications of this observation began with William Coley in 1891, when he injected inactivated Streptococcus Pyogenes and Serratia Marcescen into patients with sarcoma (3). By inducing systemic immune activation, it was hoped that the immune system would also increase its activity against the tumor. Indeed, the vaccine did cause tumor regression in some patients (4). Another relatively non-specific approach, which has proven to be of clinical value, has been the use of the Bacillus Calmette–Guérin (BCG) vaccine in those affected by bladder cancer (5).
These early, non-specific approaches suffered from unpredictable clinical responses. The use of genetically modified live bacteria remains under active investigation, principally Salmonella (6). In the case of GB, the addition of live bacteria to surgical wounds in the hopes of triggering local inflammation has proved controversial (7).
More tumor-specific therapies have been developed, which do not rely on a generalized immune response. Such approaches have already proven advantageous in highly immunogenic malignancies such as melanoma (4). Tumor-infiltrating lymphocytes are well recognized in GB. Studies to date have yielded conflicting data on the significance of these in relation to patient outcomes (8, 9). Nonetheless, their very presence makes enhancing their activity and specificity an attractive goal.
The gravity of GB has been a motivator for novel approaches. The median survival remains around 15 months and recurrence/progression is almost inevitable (10). Current treatment modalities include surgery, radiation, chemotherapy (temozolomide, bevacizumab, nitrosoureas), and electrical field treatment. This latter, known as NovoTTF-100A®, uses alternating electric fields to inhibit cell growth and has almost no side effects apart from local irritation of skin (11). The use of “targeted” chemotherapy, usually a single-agent specifically aimed at a particular cell-signaling pathway, has thus far been disappointing.
We focus on two emerging methods of harnessing the immune system in the treatment of GB:
• preventing the tumor from evading the immune system.
• exposing the immune system to antigens expressed by the tumor, thus stimulating it to attack the tumor.
To further illustrate these two points, we provide data from recently published clinical trials and from abstracts presented at the 2014 American Society of Clinical Oncology Annual Meeting (ASCO).
CNS Immunology
The CNS was previously considered as a relatively ‘immune-privileged’ site. This was thought to reflect, in part, the protective nature of the blood–brain barrier (BBB). However, we now know that the CNS has an active and tightly regulated immune system (12). The circumventricular organs, which lack a BBB, have the ability to detect infection in the peripheral bloodstream. Areas with high vascularity, such as the leptomeninges and the choroid plexus, may also lead to microglial activation upon detection of pathogen-associated molecular patterns (PAMPs) in the bloodstream (13).
Microglia (phagocytic in function) are part of the evolutionarily older innate immune system. They are concentrated in the brain’s gray matter and are less numerous in white matter (the tracts of which may be used by GB to move to new locations) (14). Aside from the production of pro-inflammatory factors in the presence of infection, microglia are believed to play a role in removing neurotoxic debris (e.g., preventing the amyloid-β accumulation noted in Alzheimer’s disease).
The adaptive arm of the immune system (responsible for immunologic memory) was thought to be limited in the CNS due to the lack of lymphatic channels. Instead, cellular waste from the interstitial fluid is transferred to the CSF for removal via the glymphatic system. Circulating lymphocytes may be found within the CNS in their activated form but naïve T cells are essentially absent (15–17).
However infiltration of lymphocytes, especially T cells, is increased in patients harboring GB as the BBB becomes disrupted, suggesting an important interaction between the immune system and the tumor (18, 19). The tumor responds with a number of strategies to counteract the immune system. These include down regulation of major histocompatibility complex (MHC, responsible for presenting antigens) (20), an increase in cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programed cell death protein 1 (PD-1) (21, 22), IL-10 (23), TGF-β (24), and by damping immune activity by recruiting regulatory T cells (TRegs) (25).
In addition to the BBB, the blood–tumor barrier must be overcome. The formation of new blood vessels by the tumor is often disorganized, with abnormal flow dynamics and immature pericytes, making recruitment of lymphocytes challenging. Experiments in mice and clinical observation support the view that immunotherapy is likely to be much less effective as the vasculature becomes more chaotic (26).
Immune-Checkpoints
Immune-checkpoints prevent excessive immune activation, which may lead to collateral damage in healthy tissue. GB makes use of this apparatus to impair nearby T cell functionality. GB induces a state of chronic antigen exposure, which gradually increases the expression of immune-checkpoint proteins and culminates in lymphocytic exhaustion or anergy (27). By overcoming this habituation, it is hoped that immune-mediated cytotoxicity may be recovered.
While many proteins involved in this process have been identified, we focus here on two for which clinical applications have been developed: CTLA-4 and PD-1. Both are responsible for the down regulation of T cell activity (28). CTLA-4 is located on cytotoxic (CD8+) and the two major subsets of helper (CD4+) T cells. This protein restricts the activity of the T cell (29, 30). The ligand for CTLA-4 is similar to that of the co-stimulatory receptor CD28, (a complex of CD80 and CD86). It is thought to be a competitive agonist at this site (31, 32). T cell activation is inhibited by reducing both the production of IL-2 and the expression of its receptor, as well as arresting lymphocytes in the G1 phase of the cell cycle (33). Additionally, this immune-checkpoint protein has been shown to enhance the suppressive function of TReg cells (34, 35).
Ipilimumab is an antibody, which inactivates CTLA-4. This was the first agent focusing on immune-checkpoint blockade to receive approval from the FDA (36). It is used for patients with melanoma and has proven to be effective for those with brain metastases (37). In GB, a similar approach has been hampered by safety concerns. One review of 10 patients demonstrated that treatment was devoid of significant toxicities in all but 1 patient (38). However, in a subsequent study with five patients, all experienced auto-immune-related adverse effects (39). This typically consisted of a rash with colitis and hypothyroidism; there was also one case each of encephalitis and partial status epilepticus.
PD-1 expression is induced upon activation of a T cell; it serves to limit the potentially deleterious activity of lymphocytes in peripheral tissues. PD-1 has been shown to be expressed by Tregs and activation of its receptor appears to aid in their proliferation (40). PD-1 is also expressed by B cells and NK cells (41).
Nivolumab is a therapeutic antibody against PD-1. Is has proven to be effective when used with ipilimumab in patients with melanoma (42). There is an ongoing phase III trial comparing its efficacy with bevacizumab in patients with recurrent glioblastoma (NCT02017717). Pembrolizumab is another such antibody. Its activity in patients with metastatic melanoma depends on the presence of pre-existing cytotoxic T cells, which are thought to be deactivated by the tumor (43).
PD-1 binds to a ligand, PD-L1. This latter is up-regulated in numerous types of cancer (44). However, the use of PD-L1 as a biomarker for response to therapeutic checkpoint blockade is complicated by its heterogeneous expression in tumors, complex signaling networks, and the normal expression found on lymphocytes and other cells within the tumor microenvironment. In GB, expression of PD-L1 has been linked to the loss of the tumor suppressor PTEN (phosphatase and tensin homolog) and consequently the PI3K–Akt signaling pathway (phosphatidylinositol 3-kinase – protein kinase B a.k.a. Akt) (45). An antibody blocking PD-L1, MPDL3280A, has shown efficacy in the setting of metastatic bladder cancer in a phase I trial (46). This approach appears most effective in those patients in whom pre-existing immunity is suppressed by PD-L1, as evidenced by high levels of PD-L1 and CTLA-4 expression (47).
A more radical approach to recovery of immune function is that of bone-marrow transplant. Autologous progenitor cells have been used in GB to facilitate higher doses of cytotoxic chemotherapy. However, given the mortality with a complete marrow transplant, this has not been the subject of a trial. Experience with other tumor types suggests that this process “resets” the immune system and thus allows for recovery of cytotoxicity (48).
Vaccines
Current approaches to immunotherapy may be classified as active or passive (49). “Passive” refers to antibodies to tumor antigens, or immune-conjugates aimed at targeted drug delivery (50). “Active” vaccines are intended to stimulate the patient’s own immune response. They may be cell-based (e.g., pulsed dendritic cells) or non-cell based (i.e., heat-shock protein-based vaccines).
Peptide Vaccines
Exposing short protein sequences to the immune system is usually done with peptides that are presented by HLA-A2 (human leukocyte antigen). This is the most common of the HLA subtypes but is found in only 50% of Caucasians and 30% of African-Americans. To overcome this limitation, antigens binding other class I HLAs have been developed, bringing population coverage to around 70%. Promising proteins from this line of investigation include: PTPRZ1 (receptor-type tyrosine-protein phosphatase zeta; function unclear but implicated in directional outgrowth of glioma cells), SEC61G (Protein transport protein Sec61 subunit gamma; involved in protein translocation across the endoplasmic reticulum for degradation), TNC (tenascin C; an extracellular glycoprotein typically expressed in development/differentiation and following injury), and EGFR (51).
EGFRvIII is a constitutively active mutant form of the epidermal growth factor receptor, which is present in approximately 33% of GB (52). Its presence is an independent negative prognostic indicator for survival in patients who manage to survive at least 1 year after initial diagnosis (53). A phase II trial was conducted in order to determine the immunogenicity, progression-free survival (PFS), and overall survival (OS) in patients who received a peptide-based vaccine (PEPvIII) targeted at EGFRvIII-expressing GB (54). Eligibility criteria included: gross total resection, Karnofsky performance status (KPS) ≥80%, and no evidence of progression after initial chemo-radiation. Immune reactivity after vaccination was monitored by observation of a delayed-type hypersensitivity (DTH) reaction to intradermal injections of PEPvIII and recall antigens. Eighteen patients were enrolled. Median PFS and OS were 14.2 and 26 months for those vaccinated vs. 6.3 and 15 months for controls. The skin test was performed in 17 patients; all showed no response prior to vaccination and all but 3 after vaccination. Of 14 patients tested, 6 demonstrated a positive humoral response against PEPvIII. The toxicity profile was deemed safe with most adverse reactions consisting of cutaneous reactions at the injection sites. (One patient had a severe allergic reaction). A phase III trial to confirm these results is ongoing.
Heat-Shock Protein Vaccines
Heat-shock proteins (HSP) are molecular chaperones; they provide protein stability by facilitating folding and aid in intra-cellular localization (55). Their activation is induced by adverse environments such as hypoxia, inflammation, and oxidative stress (56). Neoplastic cells are constantly exposed to such stressors; they rely on the HSP for survival.
A vaccine that includes HSP has proved safe and tolerable in a Phase I study of 12 patients with recurrent GB (57). After vaccination, peripheral leukocytes generally showed a response to HSP-96-bound peptides, as demonstrated by IFN-γ production (via real-time PCR). Lymphocytic infiltrates expressing IFN-γ were identified in those undergoing biopsy. Those showing an immune response to the vaccine showed an increase in median OS to 47 weeks vs. 16 in those with no response.
In the subsequent phase II trial, 41 patients with gross total resection of recurrent GB were vaccinated with HSPPC-96 (58). The median PFS of this cohort was 19.1 weeks with a median OS of 42.6 weeks. In both studies, the treatment appeared safe and tolerable.
Autologous Vaccines
These techniques rely on ex vivo modification of the patient’s immune system or of the tumor itself, followed by re-introduction of the altered cells. The immune system, particularly cytotoxic T lymphocytes, may be stimulated with tumor antigens. Neoplastic cells may be irradiated, or altered with viruses, in the hopes of increasing their immunogenicity and lowering their propensity for evasion of the immune system (49, 59).
Newcastle disease virus (NDV) combined with autologous tumor has been used as a vaccine. This virus has been shown to replicate selectively in neoplastic cells and to possess immunogenic properties (60). Twenty-three patients had their tumor surgically resected and incubated with hemagglutinating units of avirulent NDV. Concurrently, a control group was established, which comprised patients receiving standard care with a KPS of ≥60. An improvement in median PFS and OS was seen by comparison with controls: 40 weeks vs. 26 and 100 weeks vs. 49, respectively. Significant DTH skin reactions were noted when vaccinated patients were tested against irradiated tumor cells, both virus-modified and unmodified (61).
Autologous formalin-fixed tumor vaccines (AFTV) use fixed tissue to sensitize T cells to tumor antigens. In a Phase I/IIa trial, 22 newly diagnosed patients with resected GB received AFTV with concomitant fractionated radiotherapy (62–65). Median PFS and OS were promising at 7.6 and 19.8 months. Again, the treatment combination was well tolerated and adverse events were mostly limited to cutaneous reactions induced by the injection (66).
Dendritic-Cell-Based Vaccines
This process involves obtaining dendritic cells from a patient and pulsing them with glioma antigens derived from a resection. A major advantage is that multiple antigens may thus be presented (49, 67). This is of particular relevance to GB, which is known to display high intra-tumoral heterogeneity. Evidence of efficacy has already been established for metastatic prostate cancer with sipuleucel-T, although those with nervous system metastases were excluded from the pivotal trials (68).
DCVax-L® is another such dendritic-cell-based vaccine. In a phase I clinical trial, 23 patients with resected GB had an immunogenic lysate prepared from their tumor plus dendritic-cells derived from peripheral blood mononuclear cells (PBMC). The dendritic cells were supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4 before exposure to the lysate. The treatment was safe, tolerable, and without evidence of dose-limiting toxicity (69). The median PFS and OS were 15.9 and 31.4 months, respectively. A randomized phase III trial is ongoing (NCT00045968).
This approach is also being explored as a way to target glioma stem cells, which represent a radioresistant and chemoresistant subpopulation of cells within a patient’s tumor. In a phase I trial, 17 patients with newly diagnosed GB were given a dendritic-cell-based vaccine with a combination of glioma stem cell antigens. This approach (the ICT-107 vaccine) reported a promising median PFS and OS of 16.9 and 38.4 months, respectively. Interestingly, five patients who underwent a subsequent resection had a decrease or absence of cells positive for CD133, a glioma stem cell marker (70). A phase II trial was initiated with the same vaccine but despite currently unpublished data demonstrating a significant increase in PFS, there was no increase in OS (49). A phase III trial is planned nonetheless. A similar concept has been applied in the production of a vaccine (ICT-121) that targets CD133-positive glioma cells (CD 133 is an enrichment marker for cancer stem cells). A phase I trial involving this vaccine is underway (NCT02049489).
Viral Protein-Based Vaccines
A variety of studies have identified human cytomegalovirus (CMV) proteins and nucleic acids in approximately 90–100% of primary GBs (71–73). Although the role of CMV in the pathogenesis and progression of GB is not fully understood, the prevalence of these antigens in tumor cells and relative absence in normal surrounding tissue provides an important opportunity to develop targeted immunotherapeutics (74). Interestingly, one patient receiving DCVax-L developed a specific anti-CMV (anti-pp65) cytotoxic T cell response (75).
To date, immunotherapeutic targeting of CMV has been tried in a limited number of patients with high-grade gliomas. One case study describes a patient with recurrent GB who received adoptive transfer of CMV-specific T cells concurrently with temozolomide, which resulted in 17 months without disease progression (76). Recently, a trial involving patients with GB demonstrated that the transfer of expanded CMV-specific T cells lead to a median OS of 403 days (vs. historical median OS of 180 days) and 4/10 patients who completed the treatment remained progression-free during the study period (77). Ongoing trials are assessing the use of CMV-specific dendritic-cell vaccines (NCT00639639) and CMV-specific T cells following drug-induced lymphopenia in GB (NCT00693095). Direct targeting of CMV with valganciclovir has been the subject of some controversy and is not currently recommended outside the context of a clinical trial (78).
T Cell Engineering
Adoptive cell transfer using genetically engineered T cells represents another attractive immunotherapeutic approach to treating GB. T cells that recognize specific tumor-associated antigens (TAAs) can be generated by fusing an extracellular binding domain (usually derived from a TAA-specific monoclonal antibody) to the intra-cellular signaling domain of the T cell receptor (TCR) to form a chimeric antigen receptor (CAR) (79). CAR T cell activation is MHC-independent and therefore circumvents issues involving down regulation of HLA class I molecules and defects in antigen processing that tumors use to evade T cell recognition (80). These engineered cells are also potentially more useful than antibody-based immunotherapies because they have the ability to migrate through blood vessel walls, penetrate solid tumor, and recruit addition components of the immune response (81). CARs have been developed for glioma-specific antigens, including HER2, IL-13Rα2, and EGFRvIII, and have demonstrated potent antitumor activity with in vivo models (81, 82).
Interestingly, the CARs generated against HER2 in GB patients, also recognized the CD133+ stem cell populations, that are thought to contribute to tumor recurrence (80). Mounting evidence that this has led to a number of clinical trials exploring the safety and effectiveness of CARs against HER2 (NCT01109095), IL-13Rα2 (NCT00730613; NCT01082926), and EGFRvIII (NCT01454596).
What have We Learned?
Although immunotherapy has been with us for over a century, we are still in the preliminary stages of refining this therapeutic approach. Thus far, immune-based treatments have proven to be relatively safe with minimal toxicities, especially by comparison with traditional cytotoxic chemotherapy. Currently, it is estimated that <20% of patients with GB enroll in clinical trials, so increasing participation would appear to be a clear priority. Given the variety of methods receiving attention, much of the field is anticipated to be in phase I and II trials for some years (Table 1). Hence, the usual caveats apply regarding lack of power, lack of randomization, and the use of historical controls. In spite of this, the preliminary survival data have, on the whole, been encouraging.
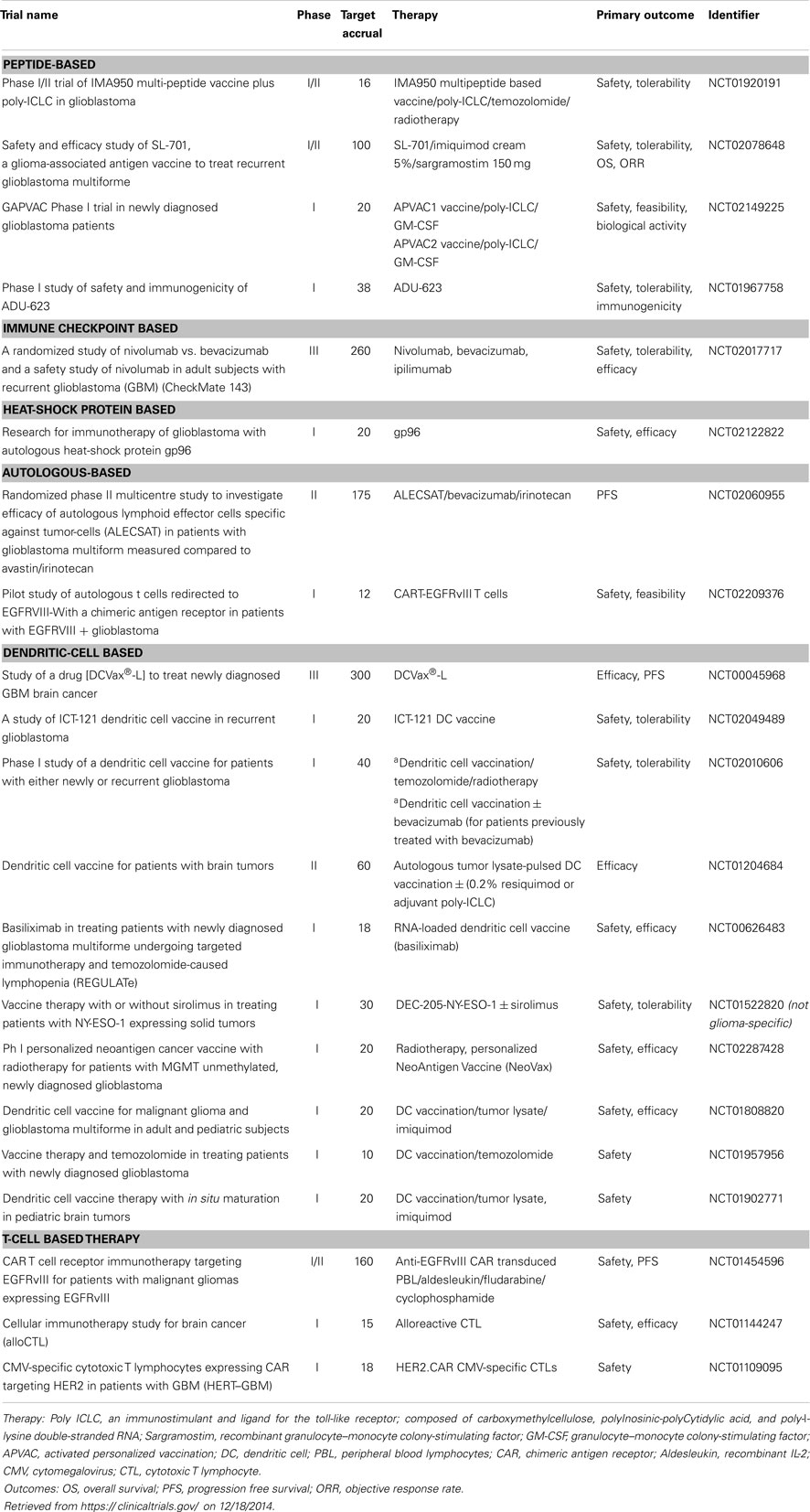
Table 1. Immunotherapy-based clinical trials for glioblastoma, which are currently recruiting.
Using peripheral immune reactivity as a surrogate marker for disease activity (and thus outcomes) is attractive, in that it may allow for more rapid development of active agents. In practice, it has thus far led to mixed results. While some trials link immune reactivity with a better prognosis, others show no such association (83). It is hoped that greater standardization and more refined methods will overcome these difficulties.
Trials to date have studied the effects of immune-checkpoint inhibitors and vaccines separately. As our knowledge of these treatments increases, we can begin to consider combining both. Such an approach has already been shown to be efficacious in a murine model of glioma (84).
Approaches targeting specifically just one antigen have the drawback that evolution of resistance appears almost inevitable in those with GB. Such difficulties are well recognized in solid tumors to which “targeted” approaches have been applied: at least two such agents are thought to be necessary (to inhibit tumor growth) and preferably three (85). Those which aim to simulate the immune system or expose it to a broad range of antigens thus hold greater promise. As data on the safety of single-agent approaches accrues and as patents expire, rational multi-agent combinations are likely to become the norm for most patients.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors declare that there are no conflicts of interest. One author received grant support from the Howard Hughes Medical Institute (Leonel Ampie).
References
1. Field KM, Jordan JT, Wen PY, Rosenthal MA, Reardon DA. Bevacizumab and glioblastoma: scientific review, newly reported updates, and ongoing controversies. Cancer (2014). doi:10.1002/cncr.28935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Hoption Cann SF, Gunn HD, van Netten JP. Spontaneous regression of pancreatic cancer. Case Rep Clin Pract Rev (2004) 5:293–6.
3. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J (2006) 26:154–8.
4. Lebbe C, Weber JS, Maio M, Neyns B, Harmankaya K, Hamid O, et al. Survival follow-up and ipilimumab retreatment for patients with advanced melanoma who received ipilimumab in prior phase II studies. Ann Oncol (2014) 25(11):2277–84. doi:10.1093/annonc/mdu441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Chakrabarty AM. Microorganisms and cancer: quest for a therapy. J Bacteriol (2003) 185:2683–6. doi:10.1128/JB.185.9.2683-2686.2003
6. Chorobik P, Czaplicki D, Ossysek K, Bereta J. Salmonella and cancer: from pathogens to therapeutics. Acta Biochim Pol (2013) 60:285–97.
7. Lundstrom M. 2 UC Davis Neurosurgeons Accused of Experimental Surgery are Banned from Human Research. California, CA: Sacramento Bee (2012).
8. Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol (2013) 264:71–83. doi:10.1016/j.jneuroim.2013.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Yue Q, Zhang X, Ye HX, Wang Y, Du ZG, Yao Y, et al. The prognostic value of Foxp3+ tumor-infiltrating lymphocytes in patients with glioblastoma. J Neurooncol (2014) 116:251–9. doi:10.1007/s11060-013-1314-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352:987–96. doi:10.1056/NEJMoa043330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Fonkem E, Wong ET. NovoTTF-100A: a new treatment modality for recurrent glioblastoma. Expert Rev Neurother (2012) 12:895–9. doi:10.1586/ern.12.80
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12:623–35. doi:10.1038/nri3265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol (2009) 9:429–39. doi:10.1038/nri2565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Torres-Platas SG, Comeau S, Rachalski A, Bo GD, Cruceanu C, Turecki G, et al. Morphometric characterization of microglial phenotypes in human cerebral cortex. J Neuroinflammation (2014) 11:12. doi:10.1186/1742-2094-11-12
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res (1991) 28:254–60. doi:10.1002/jnr.490280213
16. Heimberger AB, Sampson JH. Immunotherapy coming of age: what will it take to make it standard of care for glioblastoma? Neuro Oncol (2011) 13:3–13. doi:10.1093/neuonc/noq169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Thomas AA, Ernstoff MS, Fadul CE. Immunotherapy for the treatment of glioblastoma. Cancer J (2012) 18:59–68. doi:10.1097/PPO.0b013e3182431a73
18. Vauleon E, Avril T, Collet B, Mosser J, Quillien V. Overview of cellular immunotherapy for patients with glioblastoma. Clin Dev Immunol (2010) 2010:689171. doi:10.1155/2010/689171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest (2010) 120:1368–79. doi:10.1172/JCI41911
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Mehling M, Simon P, Mittelbronn M, Meyermann R, Ferrone S, Weller M, et al. WHO grade associated downregulation of MHC class I antigen-processing machinery components in human astrocytomas: does it reflect a potential immune escape mechanism? Acta Neuropathol (2007) 114:111–9. doi:10.1007/s00401-007-0231-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. El Andaloussi A, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol (2006) 8:234–43. doi:10.1215/15228517-2006-006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Jacobs JF, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, et al. Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol (2009) 11:394–402. doi:10.1215/15228517-2008-104
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Zisakis A, Piperi C, Themistocleous MS, Korkolopoulou P, Boviatsis EI, Sakas DE, et al. Comparative analysis of peripheral and localised cytokine secretion in glioblastoma patients. Cytokine (2007) 39:99–105. doi:10.1016/j.cyto.2007.05.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Joseph JV, Balasubramaniyan V, Walenkamp A, Kruyt FA. TGF-beta as a therapeutic target in high grade gliomas – promises and challenges. Biochem Pharmacol (2013) 85:478–85. doi:10.1016/j.bcp.2012.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Learn CA, Fecci PE, Schmittling RJ, Xie W, Karikari I, Mitchell DA, et al. Profiling of CD4+, CD8+, and CD4+CD25+CD45RO+FoxP3+ T cells in patients with malignant glioma reveals differential expression of the immunologic transcriptome compared with T cells from healthy volunteers. Clin Cancer Res (2006) 12:7306–15. doi:10.1158/1078-0432.CCR-06-1727
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Manzur M, Hamzah J, Ganss R. Modulation of the “blood-tumor” barrier improves immunotherapy. Cell Cycle (2008) 7:2452–5. doi:10.4161/cc.7.16.6451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Ngiow SF, Teng MW, Smyth MJ. Prospects for TIM3-targeted antitumor immunotherapy. Cancer Res (2011) 71:6567–71. doi:10.1158/0008-5472.CAN-11-1487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi:10.1038/nrc3239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell (1992) 71:1065–8. doi:10.1016/S0092-8674(05)80055-8
30. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev (2009) 229:12–26. doi:10.1111/j.1600-065X.2009.00770.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174:561–9. doi:10.1084/jem.174.3.561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Freeman GJ, Gribben JG, Boussiotis VA, Ng JW, Restivo VA Jr, Lombard LA, et al. Cloning of B7-2: a CTLA-4 counter-receptor that costimulates human T cell proliferation. Science (1993) 262:909–11. doi:10.1126/science.7694363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol (2001) 1:220–8. doi:10.1038/35105024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi:10.1126/science.1160062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206:1717–25. doi:10.1084/jem.20082492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol (2013) 94:41–53. doi:10.1189/jlb.1212631
37. Knisely JP, Yu JB, Flanigan J, Sznol M, Kluger HM, Chiang VL. Radiosurgery for melanoma brain metastases in the ipilimumab era and the possibility of longer survival. J Neurosurg (2012) 117:227–33. doi:10.3171/2012.5.JNS111929
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Schaff LR, Lassman AB, Goldlust SA, Cloughesy TF, Singer S, Schwartz GK, et al. Ipilimumab for recurrent glioblastoma (GB). J Clin Oncol (2014) 32:e13026.
39. Hu JL, Sharma P, Yu J, Black KL, Rudnick JD. Ipilimumab for recurrent glioblastoma: a single-institution case series. J Clin Oncol (2014) 32:e13010.
40. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi:10.1084/jem.20090847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature (2009) 458:206–10. doi:10.1038/nature07662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369:122–33. doi:10.1056/NEJMoa1302369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature (2014) 515:568–71. doi:10.1038/nature13954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8:793–800. doi:10.1038/nm730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13:84–8. doi:10.1038/nm1517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) 515:558–62. doi:10.1038/nature13904
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature (2014) 515:563–7. doi:10.1038/nature14011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Petrosiute A, Auletta JJ, Lazarus HM. Achieving graft-versus-tumor effect in brain tumor patients: from autologous progenitor cell transplant to active immunotherapy. Immunotherapy (2012) 4:1139–51. doi:10.2217/imt.12.96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Sayegh ET, Oh T, Fakurnejad S, Bloch O, Parsa AT. Vaccine therapies for patients with glioblastoma. J Neurooncol (2014) 119:531–46. doi:10.1007/s11060-014-1502-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Smaglo BG, Aldeghaither D, Weiner LM. The development of immunoconjugates for targeted cancer therapy. Nat Rev Clin Oncol (2014) 11(11):637–48. doi:10.1038/nrclinonc.2014.159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Neidert MC, Schoor O, Trautwein C, Trautwein N, Christ L, Melms A, et al. Natural HLA class I ligands from glioblastoma: extending the options for immunotherapy. J Neurooncol (2013) 111:285–94. doi:10.1007/s11060-012-1028-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A (1992) 89:2965–9. doi:10.1073/pnas.89.7.2965
53. Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res (2005) 11:1462–6. doi:10.1158/1078-0432.CCR-04-1737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol (2010) 28:4722–9. doi:10.1200/JCO.2010.28.6963
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, Workman P. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle (2010) 9:1542–50. doi:10.4161/cc.9.8.11204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol (2004) 5:781–91. doi:10.1038/nrm1492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Crane CA, Han SJ, Ahn B, Oehlke J, Kivett V, Fedoroff A, et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin Cancer Res (2013) 19:205–14. doi:10.1158/1078-0432.CCR-11-3358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Bloch O, Crane CA, Fuks Y, Kaur R, Aghi MK, Berger MS, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro Oncol (2014) 16:274–9. doi:10.1093/neuonc/not203
59. Jackson C, Ruzevick J, Brem H, Lim M. Vaccine strategies for glioblastoma: progress and future directions. Immunotherapy (2013) 5:155–67. doi:10.2217/imt.12.155
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Schirrmacher V, Haas C, Bonifer R, Ahlert T, Gerhards R, Ertel C. Human tumor cell modification by virus infection: an efficient and safe way to produce cancer vaccine with pleiotropic immune stimulatory properties when using Newcastle disease virus. Gene Ther (1999) 6:63–73. doi:10.1038/sj.gt.3300787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Steiner HH, Bonsanto MM, Beckhove P, Brysch M, Geletneky K, Ahmadi R, et al. Antitumor vaccination of patients with glioblastoma multiforme: a pilot study to assess feasibility, safety, and clinical benefit. J Clin Oncol (2004) 22:4272–81. doi:10.1200/JCO.2004.09.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Liu SQ, Saijo K, Todoroki T, Ohno T. Induction of human autologous cytotoxic T lymphocytes on formalin-fixed and paraffin-embedded tumour sections. Nat Med (1995) 1:267–71. doi:10.1038/nm0395-267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Liu SQ, Shiraiwa H, Kawai K, Hayashi H, Akaza H, Kim BS, et al. Tumor-specific autologous cytotoxic T lymphocytes from tissue sections. Nat Med (1996) 2:1283. doi:10.1038/nm1296-1283
64. Ohno T. Autologous formalin-fixed tumor vaccine. Curr Pharm Des (2005) 11:1181–8. doi:10.2174/1381612053507620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Ohno T. Autologous cancer vaccine: a novel formulation. Microbiol Immunol (2003) 47:255–63. doi:10.1111/j.1348-0421.2003.tb03393.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Muragaki Y, Maruyama T, Iseki H, Tanaka M, Shinohara C, Takakura K, et al. Phase I/IIa trial of autologous formalin-fixed tumor vaccine concomitant with fractionated radiotherapy for newly diagnosed glioblastoma. J Neurosurg (2011) 115:248–55. doi:10.3171/2011.4.JNS10377
67. Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J. Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol (2013) 10:14–26. doi:10.1038/nrclinonc.2012.204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Tse BW, Jovanovic L, Nelson CC, De Souza P, Power CA, Russell PJ. From bench to bedside: immunotherapy for prostate cancer. Biomed Res Int (2014) 2014:981434. doi:10.1155/2014/981434
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res (2011) 17:1603–15. doi:10.1158/1078-0432.CCR-10-2563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol Immunother (2013) 62:125–35. doi:10.1007/s00262-012-1319-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res (2002) 62:3347–50.
72. Scheurer ME, Bondy ML, Aldape KD, Albrecht T, El-Zein R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol (2008) 116:79–86. doi:10.1007/s00401-008-0359-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Dziurzynski K, Chang SM, Heimberger AB, Kalejta RF, McGregor Dallas SR, Smit M, et al. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol (2012) 14:246–55. doi:10.1093/neuonc/nor227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Landi D, Hegde M, Ahmed N. Human cytomegalovirus antigens in malignant gliomas as targets for adoptive cellular therapy. Front Oncol (2014) 4:338. doi:10.3389/fonc.2014.00338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Prins RM, Cloughesy TF, Liau LM. Cytomegalovirus immunity after vaccination with autologous glioblastoma lysate. N Engl J Med (2008) 359:539–41. doi:10.1056/NEJMc0804818
76. Crough T, Beagley L, Smith C, Jones L, Walker DG, Khanna R. Ex vivo functional analysis, expansion and adoptive transfer of cytomegalovirus-specific T-cells in patients with glioblastoma multiforme. Immunol Cell Biol (2012) 90:872–80. doi:10.1038/icb.2012.19
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Schuessler A, Smith C, Beagley L, Boyle GM, Rehan S, Matthews K, et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res (2014) 74:3466–76. doi:10.1158/0008-5472.CAN-14-0296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Weller M, Soffietti R, Brada M. The legend of cytomegalovirus and glioblastoma lives on. Neuro Oncol (2014) 16:166. doi:10.1093/neuonc/not204
79. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A (1993) 90:720–4. doi:10.1073/pnas.90.2.720
80. Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res (2010) 16:474–85. doi:10.1158/1078-0432.CCR-09-1322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Miao H, Choi BD, Suryadevara CM, Sanchez-Perez L, Yang S, De Leon G, et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PLoS One (2014) 9(4):e94281. doi:10.1371/journal.pone.0094281
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Bullain SS, Sahin A, Szentirmai O, Sanchez C, Lin N, Baratta E, et al. Genetically engineered T cells to target EGFRvIII expressing glioblastoma. J Neurooncol (2009) 94:373–82. doi:10.1007/s11060-009-9889-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Maki RG, Livingston PO, Lewis JJ, Janetzki S, Klimstra D, Desantis D, et al. A phase I pilot study of autologous heat shock protein vaccine HSPPC-96 in patients with resected pancreatic adenocarcinoma. Dig Dis Sci (2007) 52:1964–72. doi:10.1007/s10620-006-9205-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT Jr. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother (2012) 35:385–9. doi:10.1097/CJI.0b013e3182562d59
85. Bozic I, Reiter JG, Allen B, Antal T, Chatterjee K, Shah P, et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife (2013) 25(2):e00747. doi:10.7554/eLife.00747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: immunotherapy, glioblastoma, vaccines, antibodies, monoclonal, checkpoint modulators, T cell engineering
Citation: Ampie L, Woolf EC and Dardis C (2015) Immunotherapeutic advancements for glioblastoma. Front. Oncol. 5:12. doi: 10.3389/fonc.2015.00012
Received: 12 November 2014; Accepted: 12 January 2015;
Published online: 29 January 2015.
Edited by:
Lois A. Lampson, Harvard Medical School, USAReviewed by:
Justin Lathia, Cleveland Clinic, USARajiv Khanna, QIMR Berghofer Medical Research Institute, Australia
Vinesh Puliyappadamba, University of Alabama, USA
Stephen Gottschalk, Baylor College of Medicine, USA
Copyright: © 2015 Ampie, Woolf and Dardis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher Dardis, Department of Neurology, Barrow Neurological Institute, Suite 300, 500 W. Thomas Road, Phoenix, AZ 85013, USA e-mail:Y2hyaXN0b3BoZXJkYXJkaXNAZ21haWwuY29t