 Lloyd Pereira1
Lloyd Pereira1 Amardeep S. Dhillon
Amardeep S. Dhillon- 1Research Division, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
- 2Olivia Newton-John Cancer Research Institute, Austin Hospital, Melbourne, VIC, Australia
- 3Sir Peter MacCallum Department of Oncology, The University of Melbourne, Melbourne, VIC, Australia
- 4Department of Biochemistry and Molecular Biology, The University of Melbourne, Melbourne, VIC, Australia
- 5Department of Pathology, The University of Melbourne, Melbourne, VIC, Australia
Colorectal cancer (CRC) is a genetically heterogeneous disease that develops and progresses through several distinct pathways characterized by genomic instability. In recent years, it has emerged that inherent plasticity in some populations of CRC cells can contribute to heterogeneity in differentiation state, metastatic potential, therapeutic response, and disease relapse. Such plasticity is thought to arise through interactions between aberrant signaling events, including persistent activation of the APC/β-catenin and KRAS/BRAF/ERK pathways, and the tumor microenvironment. Here, we highlight key concepts and evidence relating to the role of epithelial–mesenchymal plasticity as a driver of CRC progression and stratification of the disease into distinct molecular and clinicopathological subsets.
Introduction
Colorectal cancer (CRC) has provided a paradigm for studying tumorigenesis for the past two decades (1, 2). Despite significant advances in understanding how it develops and progresses, CRC remains a major cause of cancer mortality in the developed world, due largely to its propensity to metastasize (3).
Early models of the molecular genetics underlying sporadic and hereditary CRC suggested that it arises via clonal expansion of crypt cells bearing loss-of-function mutations in APC or gain-of-function CTNNB1 mutations. Such mutations result in persistent activation of the Wnt pathway, a central regulator of stem cell compartments and cell fate along the crypt–villus axis. Aberrant Wnt signaling in CRC is characterized by localization of β-catenin to the nucleus, where it interacts with various transcription factor complexes, including TCF/LEF (4) and YAP/Tead (5), and Rel/NFκB (6). These interactions drive growth, proliferation, or stemness programs contributing to formation and progression of adenomas. Subsequent mutations in oncogenes (e.g., KRAS, BRAF) and/or tumor suppressors (e.g., SMAD4, TP53) then drive transition of adenomatous polyps to overt adenocarcinomas and subsequent metastatic disease (1, 2, 7, 8) (Figure 1).
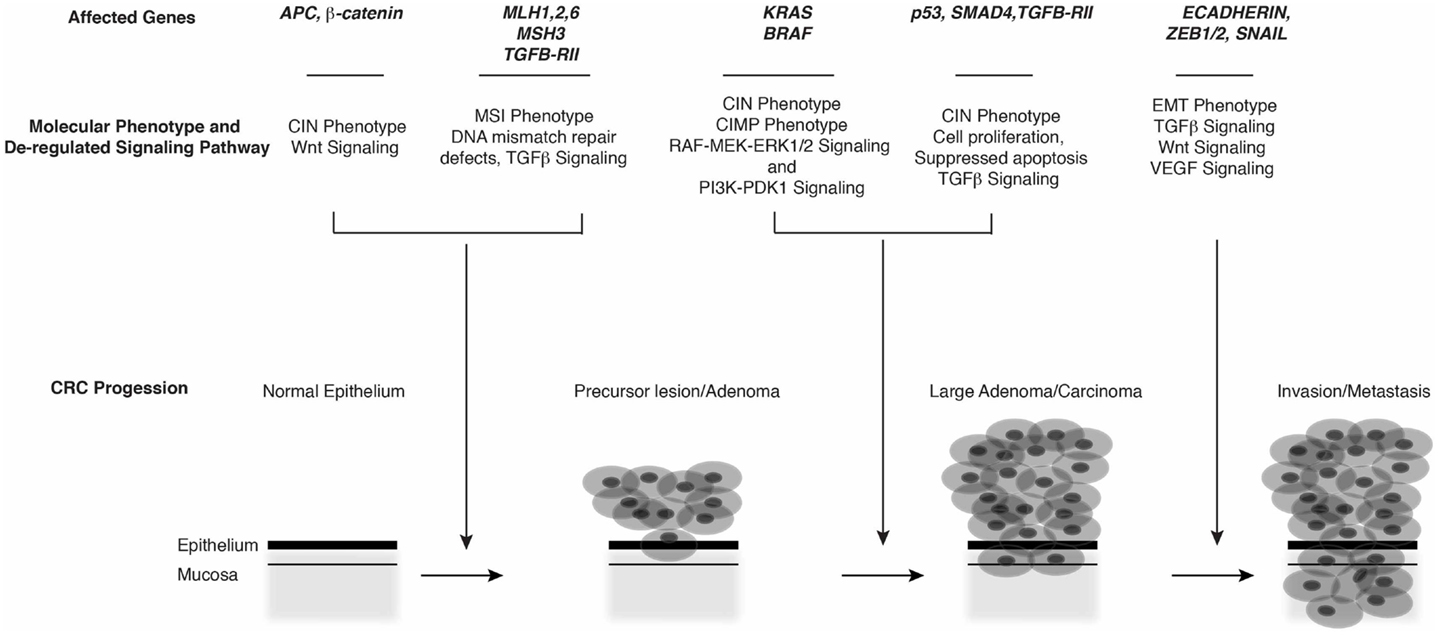
Figure 1. Molecular phenotypes, genetic alterations, and major signaling pathways associated with CRC progression.
The sequential acquisition of mutations within the adenoma-carcinoma axis, coupled with classification of disease stage/grade and histological type has provided an important paradigm to understand the “classic form” of CRC (Table 1). However, it has long been recognized that the disease is often associated with considerable heterogeneity in tumor cell phenotype, therapeutic responses, and prognoses (9–11). Indeed, comprehensive genetic and gene expression analyses have revealed variability in the genetic alterations and pathways that underlie CRC, leading to the view that the disease comprises multiple types and subtypes, which evolve through different routes (12–18). Underlying these classifications are concepts of clonal evolution, cancer stem cells (CSC), and reversible epithelial–mesenchymal transitions (EMT), each with the capacity to drive heterogeneity within CRC (6, 19–22).
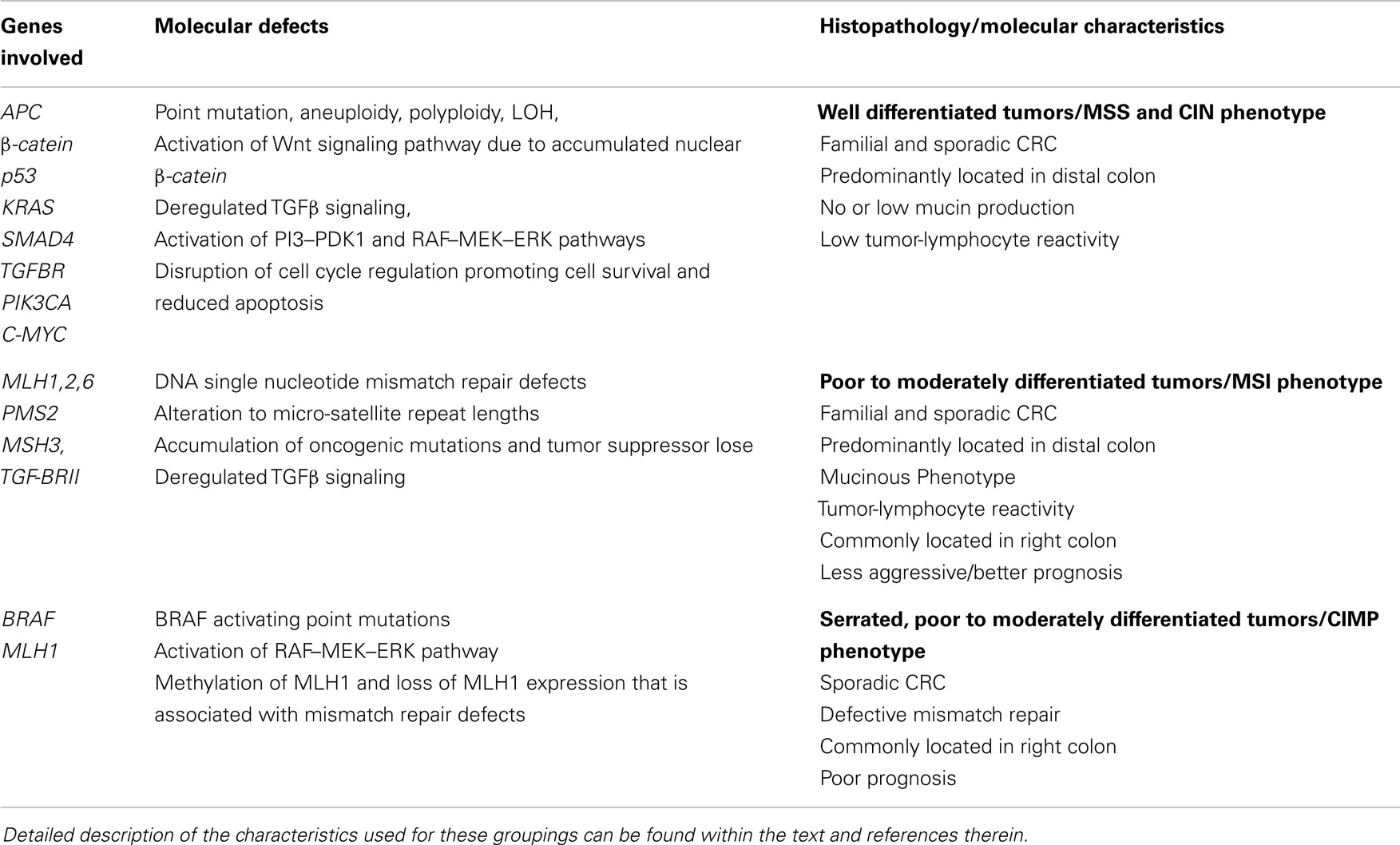
Table 1. Classification of CRC on the basis of the occurrence of genetic lesions, genomic stability, and histopathology.
EMT and Tumor Cell Plasticity During CRC Progression
That tumor heterogeneity arises through selection and expansion of different cancer cell clones bearing perturbations (e.g., mutations, epigenetic changes) conferring survival and proliferative fitness is widely accepted (1, 2, 8, 12). Heterogeneity can also arise from plasticity in tumor cell behavior, via reversible phenotypic changes driven by micro-environmental, morphogenetic, or therapeutic factors (21). These observations have in part been linked to the cancer stem cell idea, according to which a small but highly tumorigenic population of CSC having the potential to form metastases regenerates itself and progeny exhibiting a cellular hierarchy resembling normal tissue (6, 19–22).
An important source of plasticity in CRC and some other solid cancers is the EMT, which together with its reverse process, a mesenchymal–epithelial transition (MET), is essential for tissue remodeling during embryogenesis and in some pathological contexts (23, 24). Importantly, EMT–MET events also provide a framework through which solid cancers can disseminate and colonize distant sites (21, 25–31). During EMT, hallmarks of differentiated epithelia such as apico-basal polarity and cell–cell adhesions are replaced with mesenchymal traits, including rear-to-front polarity, capacity for individual cell migration, and invasion of basal lamina and blood vessels.
In addition to providing a mechanism for tumor dissemination, recent studies have identified a further pathological manifestation of EMT – endowing cancer cells with stem-like potential (32, 33) that appears critical for tumor initiation, metastasis, and relapse in CRC (6, 34, 35). The coexistence of mesenchymal and stem-like traits in cancer cells that have undergone EMT has led to the idea that they constitute “migrating CSC” from which metastases are derived (21, 36). Such cells acquire the capacity to both disseminate and successfully colonize new sites, where they are thought to redifferentiate via an MET and regain the organization of cells present in the primary tumor. This model thus provides a mechanism to explain the observation that CRC metastases often retain a similar degree of differentiation as the primary tumor.
Induction of EMT requires extensive reprograming of gene expression in response to activation of various signaling pathways. Among the best studied are the Wnt, MAPK, TGFβ, and NFκB pathways, which converge on one or more transcription factors (TFs) driving EMT in the embryo, including members of the zinc finger (SNAIL1, SNAIL2/SLUG, ZEB1, ZEB2/SIP1), bHLH (TWIST1, TWIST2), forkhead (FOXC2), or homeobox (Goosecoid, SIX1, PRRX1, PREP1) families (37–39). In CRC, multiple TFs were reported as being aberrantly expressed based on immunohistochemical and transcriptome studies, including ZEB1, ZEB2/SIP1, SNAIL1, SNAIL2/SLUG TWIST, and FOXC2 (21, 40–48). Although these TFs typically function as repressors of epithelial genes, and/or genes required for cell cycle progression, they also activate transcription in some contexts, including that of stemness-promoting genes and cell cycle inhibitors (21, 49, 50).
The effects of EMT-driven TF activation can be antagonized by several species of micro-RNA (miRNA) that in addition to repressing expression of TFs, are themselves repressed by these TFs. Such reciprocal inhibition creates self-enforcing double-negative feedback loops that dictate the epithelial–mesenchymal balance. Two such loops have been well documented to operate in colorectal and other cancer cells – ZEB/miR-200 and SNAIL/mir-34 loops (51–53). In addition to repressing EMT-TFs, the miRNAs also directly target other genes involved in regulating EMT (e.g., cytoskeletal components, Wnt pathway components) and stemness (e.g., BMI1, KLF4, SOX2), underscoring their critical functions in regulating cellular plasticity during cancer progression (26, 51, 54–57). Notably, both miR-200 family members and miR-34 are induced by the tumor suppressor p53 (58–60), whose induction of miR-34 expression was found to reduce levels of several Wnt pathway components, including LEF-1, β-catenin, WNT1, WNT3, LPR6, and AXIN2 (60–62). Reduction in Axin2 via this mechanism was also reported to promote nuclear accumulation of GSK3β, where it can phosphorylate to destabilize SNAIL1 (63).
Association of EMT with CRC Pathology
The majority of CRCs appear moderately differentiated, with smaller subsets being well or poorly differentiated. The latter cancers are characterized by highly irregular glandular structure, aggressiveness, poor prognosis, and resistance to treatment. However, moderately differentiated tumors can also contain regions of poor differentiation, typically observed at the invasive front (21, 27, 36). Often, these cancers exhibit budding phenotype, in which individual or clusters of tumor cells detach from the tumor mass and invade into the adjacent stroma. This feature is adversely prognostic and linked with enhanced probability of metastasis to the lymph nodes, liver, or lung (36, 64, 65).
Budding tumor cells are thought to have undergone an EMT-like event, losing expression of epithelial differentiation markers while gaining the capacity to express mesenchymal and stemness markers (36, 66). In contrast to central regions of the tumor, budding cells at the invasive front also typically strongly express nuclear β-catenin, which is critical for induction of EMT programs characterized by expression of ZEB1 (42) and altered basement membrane components (67). This intra-tumoral heterogeneity in β-catenin expression is likely to arise from a range of factors, including micro-environmental signals, altered cell–cell and cell–matrix adhesion, and through cross-talk with other signaling pathways such as the ERK module (27, 36, 68, 69).
While EMT–MET events provide a framework for how differentiated CRCs may metastasize, a different model was proposed by Brabletz to account for progression of poorly differentiated cancers (21). Rather than exhibiting high plasticity, these tumors retain a poorly differentiated mesenchymal phenotype that is driven primarily by mutational events. Such cancers may have arisen prior to differentiation of stem or progenitor cells in the crypt, or from cells that have evolved from differentiated tumors but selected for mutations that render them in a stable mesenchymal-like state. A further mechanism through which selection may occur is as a result of therapies, where the relapsing tumors often displaying a mesenchymal, stem-like phenotype (21). Finally, it was suggested that the highly aggressive nature of poorly differentiated tumors may result form their propensity to metastasize through “parallel progression” routes (70), in which tumors and metastasis develop and progress concurrently.
Association of EMT with CRC Subtypes
An important question is whether models of tumor cell plasticity involving EMT–MET events and CSC can be incorporated into current approaches for CRC subtyping. Collectively, this approach may help better define the heterogeneity observed in CRC and progress the development of targeted therapies.
CIN, MSS/MSI, CIMP Subtyping
Conventional approaches to classify colorectal tumors have centered primarily on molecular [chromosomal instability (CIN); micro-satellite stability/instability (MSS/MSI); CpG island methylator phenotype (CIMP)], and pathological (TNM grade, degree of differentiation, immunohistological markers) characteristics of the tumor (9, 71) (Table 1). These classifications recognize the various forms of global genomic and epigenetic alterations that occur during tumorigenesis (Table 1). CIN is the most common form of genomic instability in CRC that underlies the sequential deregulation of classical tumor suppressor and oncogenes including APC, KRAS, and TP53. In the MSI classification, genomic instability arises from the mutation or methylation-mediated silencing of genes required for DNA mismatch repair (hMLH1, hMSH2, hMSH6, and hPMS2) and based on the level of MSI, CRCs can be classified as MSI-high (MSI-H), MSI-low, or MSS. MSI tumors have a lower frequency of mutations in KRAS and TP53 compared to MSS cancers, and a higher frequency of mutations in genes harboring repetitive elements in their coding sequence such as TGFBR2 (72). Recent work indicates that as a result of this loss of TGFβRII function, MSI tumor cells lines fail to undergo EMT in response to TGFβ, which may contribute to their better prognosis (73). In the CIMP classification, tumors harbor aberrant DNA methylation patterns that result in the global epigenetic silencing of genes. Each of these pathways serves as an important classifier of disease progression and response to therapy (Table 1).
Intrinsic EMT-Associated CRC Subtypes
While the CIN, MSI, and CIMP are important disease sub-classifiers, it is now well-established that tumors defined by these groupings can be additionally stratified into molecularly defined subtypes. Over the past decade, genomic and expression analyses involving large patient cohorts have provided insight into the diversity within CRC. Combined with existing mutational, clinical, and pathological classifiers, these studies have identified several distinct molecularly defined CRC subtypes (e.g., stem-like, mesenchymal, immune, and epithelial/differentiated), each driven by unique and/or overlapping biological pathways and exhibiting differing prognostic and/or therapeutic response (11, 13, 15–17, 41, 74, 75) (Figure 2). A unifying feature from each of these studies was the identification of a CRC subtype significantly enriched for genes associated with a poorly differentiated, mesenchymal/invasive phenotype, and that were often co-enriched with genes indicative of a stem-like state (Figure 2).
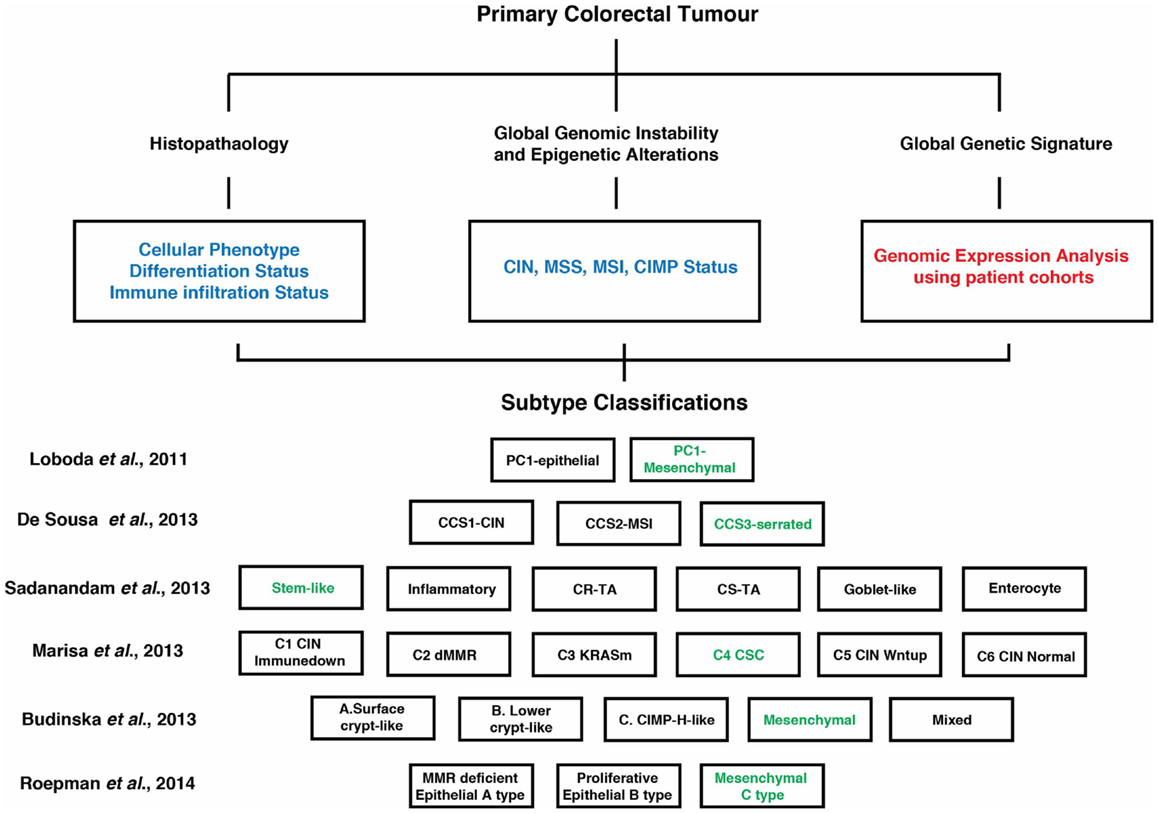
Figure 2. Overview of suggested contemporary subtype classification of CRC. Genomic and expression analyses involving large patient cohorts (highlighted in red) combined with existing mutational, clinical, and pathological classifiers (highlighted in blue) have identified several distinct molecularly defined CRC subtypes as indicated by the various studies. Each of these subtypes is driven by unique and/or overlapping signaling pathways (see Figure 1) and exhibit different prognostic and therapeutic responses. A unifying feature is a CRC subtype enriched for genes associated with a poorly differentiated, mesenchymal/invasive phenotype, and often co-enriched for genes indicative of a stem-like state (highlighted in green). A more detailed description of these subtypes and their clinical/therapeutic response can be found within the text (13, 15–17, 41, 74).
Loboda et al. (41) defined two subsets, epithelial and mesenchymal, where the latter was linked to TGFβ signaling and low expression levels of anti-EMT miRNAs. Examination of the heterogeneity within CRC gene expression profiles also revealed a strong association between EMT gene signatures and subtyping (13). Marisa et al. (17) identified six molecular subtypes (C1–C6) from stage I–IV CRC patients, with two subtypes (C4 and C6) showing a distinct down-regulation of proliferative and upregulation of EMT/motility pathways. Subtype C4 was also characterized by a stem cell-like phenotype. Furthermore, both subtypes were distinct with regard to harboring a serrated tumor signature. Roepman et al. (74) identified three subtypes (A–C) within stages II and III CRC, with C-type tumors featuring an EMT phenotype and low proliferative activity. Two additional studies (15, 16) examined large patient-derived CRC gene expression datasets and defined CRC subtypes characterized by a mesenchymal gene signature. In the study by Sadanandam et al. (16), six subtypes were described on the basis of gene expression signatures associated with their cell of origin within the colon crypt. In this context, a stem cell subgroup was associated with expression of mesenchymal genes. De Sousa et al. (15) described three CRC subtypes (CCS1–3) and in the CCS3 grouping EMT and genes involved with migration, invasion, and TGFβ signaling were elevated. Subsequent analysis suggests that the EMT subgroups identified in both studies show strong overlap (76). Importantly, several of the above studies demonstrated that EMT signature defined tumors consistently display a worse prognosis and were least sensitive to conventional chemotherapy regimes. Thus, a mesenchymal/invasive poor differentiation signature is a defining feature of CRC subtyping and clinical response.
An important issue to emerge from the above publications is the extent to which activation of mesenchymal and stem-like programs are linked in CRC subtypes. Consistent with the role that Wnt signaling plays in regulating the fate of stem cells at the base of the crypt (8), Sadanandam et al. (16) found elevated activation of this pathway in stem-like tumors and cell lines, which co-expressed markers of intestinal and colorectal stem cells and EMT genes (34). However, whether Wnt signaling alone is sufficient to drive stem-like/mesenchymal programs expression requires further clarification as Zhu et al. (75) suggest that the pathway is not only active in mesenchymal-type tumors but also in those exhibiting differentiated or proliferative expression signatures. Instead, they found that the context of Wnt activation differed between these cancers, with migratory/EMT subsets also enriched for VEGF signaling, whereas Wnt and Notch were active in differentiated/epithelial-type tumors. Only the proliferative group (enriched for genes involved in early colon development) showed Wnt activation alone. The notion that VEGF signaling may be important for activating EMT/migration programs in the context of Wnt signaling is also supported by the finding that genes associated with sprouting angiogenesis, a process regulated by the VEGF pathway were co-enriched in mesenchymal-type tumors identified by Marisa et al. (17).
A second pathway that appears to be critical for activation of EMT programs in mesenchymal tumors is the TGFβ pathway (77, 78). Transcriptional outputs of this pathway were significantly enriched in several studies and associated with the mesenchymal phenotype (15, 17, 41, 74). Interestingly, in one study (15), TGFβ and EMT programs appeared to be active in the absence of Wnt transcriptional signatures or activation of stem cell programs. One implication of this observation is that Wnt signaling is required for stemness programs but not necessarily required for EMT in poorly differentiated cancers. Interestingly, the CCS3 group (15) enriched for sessile-serrated adenoma (SSA) tumors comprised both differentiated and poorly differentiated tumors, suggesting that further stratification based on differentiation status may be possible.
Sessile-Serrated Adenoma Pathway
A distinct feature of CRC that has emerged from recent studies is that groups harboring an EMT gene expression signature may display a pathology related to serrated adenoma (13, 15, 17, 76). As such, the CRC subtype displaying a serrated pathology provides an important context to examine the role of EMT events in driving CRC progression.
In the classical adenoma-carcinoma sequence, tumors are often located in the distal colon or rectum and genetically are defined by CIN. In contrast, the serrated adenoma represents an alternative pathway to tumorigenesis. Typically, the serrated adenoma is located in the proximal or right colon and is characterized by the sawtooth appearance of the crypt epithelium (79). Traditionally viewed to have limited potential to progress to a neoplastic lesion, it is now established that precursor “serrated polyp” can be subdivided into hyperplastic polyp (HP), SSA, and traditional serrated adenoma (TSA) with both the SSA and TSA having significant potential to develop into neoplastic lesions (80, 81).
It has been suggested that up to 30–35% of CRCs evolve through a serrated pathway (82–84). In addition to their distinct morphology, serrated CRCs are also distinct in the genetic background that drives their development. For example, serrated colon tumors predominately display mutations in BRAF and KRAS rather than APC. With respect to the MSI and CIN classification, serrated tumors usually lack CIN but are often MSI-H and CIMP-H (71, 85, 86). Thus, serrated tumors have been classified in three subtypes: CIMP-low/MSS/MSI-low/KRAS mutant; CIMP-H/MSI-H/BRAF mutant; CIMP-low/MSS/MSI-low/BRAF mutant (9, 87). In the context of EMT-driven cellular plasticity, it is important to note that clinically CIMP-low/MSS/MSI-low/BRAF mutant tumors confer a poor prognosis and display high tumor budding. This observation is consistent with the increased EMT potential associated with wild-type TGFβRII and active TGFβ signaling and MSI-low status. In contrast CIMP-H/MSI-H/BRAF mutant tumors have a more favorable prognosis (86, 88, 89). Here, EMT potential is reduced due to the increased incidence of mutated TGFβRII (72, 73).
Clinical Implications and Concluding Comments
The CRC classifications outlined above may provide new opportunities for the more targeted therapeutic/clinical management of CRC disease progression. This possibility is illustrated in the studies by Sadanandam et al. (16), De Sousa et al. (15), and Roepman et al. (74). Each of these studies revealed subtype-specific responses to therapy that could potentially contribute to more effective manage of disease. In case of the study by De Sousa et al., the CCS3-serrated subtype was reported to be resistant to cetuximab therapy, suggesting that new targeted therapies would be required for this subtype (15). The identification of CCS3 specific elevated TGFβ signaling suggested that this pathway may be an avenue for targeted therapy (15). The six CRC subtypes identified in the study by Sadanandam et al. (16) also displayed subtype-specific responses to therapy. Here, three subtypes, CR-TA, CS-TA, and Goblet were suggested to not respond to FOLFIRI chemotherapy treatment and patients with this form of disease may better spared this therapy in the context of local disease. However, in the context of metastatic disease, the CR-TA and CS-TA subtypes were suggested to respond to cetuximab therapy (16). In contrast, stem cell-like-subtypes and inflammatory subtypes may respond best to FOLFIRI treatment. The specific treatment of a stem cell-like subtype is an important consideration given that such populations of cells are key drivers of the moderately differentiated phenotype that are seen in most CRCs and which due to their stem-like behavior (e.g., low proliferative index) have thus far proved highly resilient to current therapies. Collectively, these studies strongly support the idea that distinct, clinically relevant CRC subtypes can be used as a guide for subtype-specific therapy.
Tumor heterogeneity has posed a major obstacle for the successful treatment of metastatic forms of CRC and several other common cancers. The studies highlighted here have provided a substantial insight into CRC heterogeneity. The identification of various degrees of epithelial–mesenchymal plasticity, acting in concert with clonal evolution and the concept of CSC, have helped dissect the heterogeneity underlying CRC and resulted in a more detailed classification of CRC into distinct molecularly defined subtypes. These classifications will provide new opportunities for understanding CRC and the key oncogenic pathways and mechanisms required for disease progression. This new information may also be invaluable for re-focusing basic and translational/pre-clinical studies on identifying and targeting key pathways required for the malignant growth of the most aggressive subtypes.
Author Contributions
Lloyd Pereira and Amardeep Singh Dhillon conceived and drafted the manuscript. John M. Mariadason and Ross D. Hannan provided critical intellectual input and assisted with revision of the text. All authors approved the final version to be published and agree to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to colleagues whose work was not cited due to space constraints. This work was supported by grants from the National Health and Medical Research Council of Australia (to Amardeep Singh Dhillon).
References
1. Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol (2011) 6:479–507. doi: 10.1146/annurev-pathol-011110-130235
2. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell (1990) 61:759–67. doi:10.1016/0092-8674(90)90186-I
3. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet (2014) 383:1490–502. doi:10.1016/S0140-6736(13)61649-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol (2012) 4(11):a007906. doi:10.1101/cshperspect.a007906
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. Beta-catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell (2012) 151:1457–73. doi:10.1016/j.cell.2012.11.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell (2013) 152:25–38. doi:10.1016/j.cell.2012.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Ong BA, Vega KJ, Houchen CW. Intestinal stem cells and the colorectal cancer microenvironment. World J Gastroenterol (2014) 20:1898–909. doi:10.3748/wjg.v20.i8.1898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature (2009) 457:608–11. doi:10.1038/nature07602
9. Kanthan R, Senger JL, Kanthan SC. Molecular events in primary and metastatic colorectal carcinoma: a review. Pathol Res Int (2012) 2012:597497. doi:10.1155/2012/597497
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Troiani T, Martinelli E, Napolitano S, Morgillo F, Belli G, Cioffi L, et al. Molecular aspects of resistance to biological and non-biological drugs and strategies to overcome resistance in colorectal cancer. Curr Med Chem (2014) 21:1639–53. doi:10.2174/09298673113209990224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Schlicker A, Beran G, Chresta CM, McWalter G, Pritchard A, Weston S, et al. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genomics (2012) 5:66. doi:10.1186/1755-8794-5-66
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Markowitz SD, Bertagnolli MM. Molecular origins of cancer: molecular basis of colorectal cancer. N Engl J Med (2009) 361:2449–60. doi:10.1056/NEJMra0804588
13. Budinska E, Popovici V, Tejpar S, D’Ario G, Lapique N, Sikora KO, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol (2013) 231:63–76. doi:10.1002/path.4212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Perez-Villamil B, Romera-Lopez A, Hernandez-Prieto S, Lopez-Campos G, Calles A, Lopez-Asenjo JA, et al. Colon cancer molecular subtypes identified by expression profiling and associated to stroma, mucinous type and different clinical behavior. BMC Cancer (2012) 12:260. doi:10.1186/1471-2407-12-260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. De Sousa EMF, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LP, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med (2013) 19:614–8. doi:10.1038/nm.3174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med (2013) 19:619–25. doi:10.1038/nm.3175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med (2013) 10:e1001453. doi:10.1371/journal.pmed.1001453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487:330–7. doi:10.1038/nature11252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Vermeulen L, Snippert HJ. Stem cell dynamics in homeostasis and cancer of the intestine. Nat Rev Cancer (2014) 14:468–80. doi:10.1038/nrc3744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Fanali C, Lucchetti D, Farina M, Corbi M, Cufino V, Cittadini A, et al. Cancer stem cells in colorectal cancer from pathogenesis to therapy: controversies and perspectives. World J Gastroenterol (2014) 20:923–42. doi:10.3748/wjg.v20.i4.923
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Brabletz T. To differentiate or not – routes towards metastasis. Nat Rev Cancer (2012) 12:425–36. doi:10.1038/nrc3265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature (2013) 501:328–37. doi:10.1038/nature12624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139:871–90. doi:10.1016/j.cell.2009.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer (2009) 9:265–73. doi:10.1038/nrc2620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science (2013) 342:1234850. doi:10.1126/science.1234850
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Korpal M, Ell BJ, Buffa FM, Ibrahim T, Blanco MA, Celia-Terrassa T, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med (2011) 17:1101–8. doi:10.1038/nm.2401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A (2001) 98:10356–61. doi:10.1073/pnas.171610498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell (2012) 148:349–61. doi:10.1016/j.cell.2011.11.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell (2012) 22:725–36. doi:10.1016/j.ccr.2012.09.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell (2012) 22:709–24. doi:10.1016/j.ccr.2012.10.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Aokage K, Ishii G, Ohtaki Y, Yamaguchi Y, Hishida T, Yoshida J, et al. Dynamic molecular changes associated with epithelial-mesenchymal transition and subsequent mesenchymal-epithelial transition in the early phase of metastatic tumor formation. Int J Cancer (2011) 128:1585–95. doi:10.1002/ijc.25500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One (2008) 3:e2888. doi:10.1371/journal.pone.0002888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Merlos-Suarez A, Barriga FM, Jung P, Iglesias M, Cespedes MV, Rossell D, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell (2011) 8:511–24. doi:10.1016/j.stem.2011.02.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell (2012) 22:571–84. doi:10.1016/j.ccr.2012.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells – an integrated concept of malignant tumour progression. Nat Rev Cancer (2005) 5:744–9. doi:10.1038/nrc1694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene (2014) 33:1755–63. doi:10.1038/onc.2013.128
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol (2014) 16:488–94. doi:10.1038/ncb2976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Risolino M, Mandia N, Iavarone F, Dardaei L, Longobardi E, Fernandez S, et al. Transcription factor PREP1 induces EMT and metastasis by controlling the TGF-beta-SMAD3 pathway in non-small cell lung adenocarcinoma. Proc Natl Acad Sci U S A (2014) 111:E3775–84. doi:10.1073/pnas.1407074111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kouso H, Yano T, Maruyama R, Shikada Y, Okamoto T, Haro A, et al. Differences in the expression of epithelial-mesenchymal transition related molecules between primary tumors and pulmonary metastatic tumors in colorectal cancer. Surg Today (2013) 43:73–80. doi:10.1007/s00595-012-0344-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Loboda A, Nebozhyn MV, Watters JW, Buser CA, Shaw PM, Huang PS, et al. EMT is the dominant program in human colon cancer. BMC Med Genomics (2011) 4:9. doi:10.1186/1755-8794-4-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Sanchez-Tillo E, de Barrios O, Siles L, Cuatrecasas M, Castells A, Postigo A. Beta-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc Natl Acad Sci U S A (2011) 108:19204–9. doi:10.1073/pnas.1108977108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Kim YH, Kim G, Kwon CI, Kim JW, Park PW, Hahm KB. TWIST1 and SNAI1 as markers of poor prognosis in human colorectal cancer are associated with the expression of ALDH1 and TGF-beta1. Oncol Rep (2014) 31:1380–8. doi:10.3892/or.2014.2970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Larriba MJ, Martin-Villar E, Garcia JM, Pereira F, Pena C, de Herreros AG, et al. Snail2 cooperates with Snail1 in the repression of vitamin D receptor in colon cancer. Carcinogenesis (2009) 30:1459–68. doi:10.1093/carcin/bgp140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Pena C, Garcia JM, Larriba MJ, Barderas R, Gomez I, Herrera M, et al. SNAI1 expression in colon cancer related with CDH1 and VDR downregulation in normal adjacent tissue. Oncogene (2009) 28:4375–85. doi:10.1038/onc.2009.285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Pena C, Garcia JM, Silva J, Garcia V, Rodriguez R, Alonso I, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet (2005) 14:3361–70. doi:10.1093/hmg/ddi366
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Toiyama Y, Yasuda H, Saigusa S, Tanaka K, Inoue Y, Goel A, et al. Increased expression of Slug and Vimentin as novel predictive biomarkers for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis (2013) 34:2548–57. doi:10.1093/carcin/bgt282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Diesch J, Sanij E, Gilan O, Love C, Tran H, Fleming NI, et al. Widespread FRA1-dependent control of mesenchymal transdifferentiation programs in colorectal cancer cells. PLoS One (2014) 9:e88950. doi:10.1371/journal.pone.0088950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Mejlvang J, Kriajevska M, Vandewalle C, Chernova T, Sayan AE, Berx G, et al. Direct repression of cyclin D1 by SIP1 attenuates cell cycle progression in cells undergoing an epithelial mesenchymal transition. Mol Biol Cell (2007) 18:4615–24. doi:10.1091/mbc.E07-05-0406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol (2010) 12:982–92. doi:10.1038/ncb2099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Siemens H, Jackstadt R, Hunten S, Kaller M, Menssen A, Gotz U, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle (2011) 10:4256–71. doi:10.4161/cc.10.24.18552
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol (2008) 10:593–601. doi:10.1038/ncb1722
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Paterson EL, Kazenwadel J, Bert AG, Khew-Goodall Y, Ruszkiewicz A, Goodall GJ. Down-regulation of the miRNA-200 family at the invasive front of colorectal cancers with degraded basement membrane indicates EMT is involved in cancer progression. Neoplasia (2013) 15:180–91.
54. Li X, Roslan S, Johnstone CN, Wright JA, Bracken CP, Anderson M, et al. MiR-200 can repress breast cancer metastasis through ZEB1-independent but moesin-dependent pathways. Oncogene (2014) 33:4077–88. doi:10.1038/onc.2013.370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell (2009) 138:592–603. doi:10.1016/j.cell.2009.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol (2009) 11:1487–95. doi:10.1038/ncb1998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Ahn YH, Gibbons DL, Chakravarti D, Creighton CJ, Rizvi ZH, Adams HP, et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating miR-34a expression. J Clin Invest (2012) 122:3170–83. doi:10.1172/JCI63608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med (2011) 208:875–83. doi:10.1084/jem.20110235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Chang CJ, Chao CH, Xia W, Yang JY, Xiong Y, Li CW, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol (2011) 13:317–23. doi:10.1038/ncb2173
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Cha YH, Kim NH, Park C, Lee I, Kim HS, Yook JI. MiRNA-34 intrinsically links p53 tumor suppressor and Wnt signaling. Cell Cycle (2012) 11:1273–81. doi:10.4161/cc.19618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Kim NH, Kim HS, Li XY, Lee I, Choi HS, Kang SE, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol (2011) 195:417–33. doi:10.1083/jcb.201103097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Kim NH, Kim HS, Kim NG, Lee I, Choi HS, Li XY, et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci Signal (2011) 4:ra71. doi:10.1126/scisignal.2001744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol (2006) 8:1398–406. doi:10.1038/ncb1508
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Zlobec I, Lugli A. Epithelial mesenchymal transition and tumor budding in aggressive colorectal cancer: tumor budding as oncotarget. Oncotarget (2010) 1:651–61.
65. Bhangu A, Wood G, Mirnezami A, Darzi A, Tekkis P, Goldin R. Epithelial mesenchymal transition in colorectal cancer: seminal role in promoting disease progression and resistance to neoadjuvant therapy. Surg Oncol (2012) 21:316–23. doi:10.1016/j.suronc.2012.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Yusra , Semba S, Yokozaki H. Biological significance of tumor budding at the invasive front of human colorectal carcinoma cells. Int J Oncol (2012) 41:201–10. doi:10.3892/ijo.2012.1459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Spaderna S, Schmalhofer O, Hlubek F, Berx G, Eger A, Merkel S, et al. A transient, EMT-linked loss of basement membranes indicates metastasis and poor survival in colorectal cancer. Gastroenterology (2006) 131:830–40. doi:10.1053/j.gastro.2006.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol (2007) 19:150–8. doi:10.1016/j.ceb.2007.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Horst D, Chen J, Morikawa T, Ogino S, Kirchner T, Shivdasani RA. Differential WNT activity in colorectal cancer confers limited tumorigenic potential and is regulated by MAPK signaling. Cancer Res (2012) 72:1547–56. doi:10.1158/0008-5472.CAN-11-3222
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer (2009) 9:302–12. doi:10.1038/nrc2627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Pancione M, Remo A, Colantuoni V. Genetic and epigenetic events generate multiple pathways in colorectal cancer progression. Pathol Res Int (2012) 2012:509348. doi:10.1155/2012/509348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science (1995) 268:1336–8. doi:10.1126/science.7761852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Pino MS, Kikuchi H, Zeng M, Herraiz MT, Sperduti I, Berger D, et al. Epithelial to mesenchymal transition is impaired in colon cancer cells with microsatellite instability. Gastroenterology (2010) 138:1406–17. doi:10.1053/j.gastro.2009.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Roepman P, Schlicker A, Tabernero J, Majewski I, Tian S, Moreno V, et al. Colorectal cancer intrinsic subtypes predict chemotherapy benefit, deficient mismatch repair and epithelial-to-mesenchymal transition. Int J Cancer (2014) 134:552–62. doi:10.1002/ijc.28387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Zhu J, Wang J, Shi Z, Franklin JL, Deane NG, Coffey RJ, et al. Deciphering genomic alterations in colorectal cancer through transcriptional subtype-based network analysis. PLoS One (2013) 8:e79282. doi:10.1371/journal.pone.0079282
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Sadanandam A, Wang X, de Sousa EMF, Gray JW, Vermeulen L, Hanahan D, et al. Reconciliation of classification systems defining molecular subtypes of colorectal cancer: interrelationships and clinical implications. Cell Cycle (2014) 13:353–7. doi:10.4161/cc.27769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Drabsch Y, ten Dijke P. TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev (2012) 31:553–68. doi:10.1007/s10555-012-9375-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res (2009) 19:156–72. doi:10.1038/cr.2009.5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology (2010) 138:2088–100. doi:10.1053/j.gastro.2009.12.066
80. Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, et al. A genetic progression model of Braf(V600E)-induced intestinal tumorigenesis reveals targets for therapeutic intervention. Cancer Cell (2013) 24:15–29. doi:10.1016/j.ccr.2013.05.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Patai AV, Molnar B, Tulassay Z, Sipos F. Serrated pathway: alternative route to colorectal cancer. World J Gastroenterol (2013) 19:607–15. doi:10.3748/wjg.v19.i5.607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Kahi CJ, Hewett DG, Norton DL, Eckert GJ, Rex DK. Prevalence and variable detection of proximal colon serrated polyps during screening colonoscopy. Clin Gastroenterol Hepatol (2011) 9:42–6. doi:10.1016/j.cgh.2010.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Rex DK, Ahnen DJ, Baron JA, Batts KP, Burke CA, Burt RW, et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol (2012) 107:1315–29. doi:10.1038/ajg.2012.161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol (2011) 42:1–10. doi:10.1016/j.humpath.2010.06.002
85. Bond CE, Umapathy A, Buttenshaw RL, Wockner L, Leggett BA, Whitehall VL. Chromosomal instability in BRAF mutant, microsatellite stable colorectal cancers. PLoS One (2012) 7:e47483. doi:10.1371/journal.pone.0047483
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Pai RK, Jayachandran P, Koong AC, Chang DT, Kwok S, Ma L, et al. BRAF-mutated, microsatellite-stable adenocarcinoma of the proximal colon: an aggressive adenocarcinoma with poor survival, mucinous differentiation, and adverse morphologic features. Am J Surg Pathol (2012) 36:744–52. doi:10.1097/PAS.0b013e31824430d7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Jass JR. Molecular heterogeneity of colorectal cancer: implications for cancer control. Surg Oncol (2007) 16(Suppl 1):S7–9. doi:10.1016/j.suronc.2007.10.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, et al. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res (2005) 65:6063–9. doi:10.1158/0008-5472.CAN-05-0404
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Patil DT, Shadrach BL, Rybicki LA, Leach BH, Pai RK. Proximal colon cancers and the serrated pathway: a systematic analysis of precursor histology and BRAF mutation status. Mod Pathol (2012) 25:1423–31. doi:10.1038/modpathol.2012.98
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: CRC, epithelial–mesenchymal transition, cancer stem cell, tumor progression, subtypes, serrated
Citation: Pereira L, Mariadason JM, Hannan RD and Dhillon AS (2015) Implications of epithelial–mesenchymal plasticity for heterogeneity in colorectal cancer. Front. Oncol. 5:13. doi: 10.3389/fonc.2015.00013
Received: 12 November 2014; Accepted: 12 January 2015;
Published online: 02 February 2015.
Edited by:
Scott Bultman, University of North Carolina, USAReviewed by:
Pier Giorgio Petronini, University of Parma, ItalyValerio Donato, New York University Medical Center, USA
Copyright: © 2015 Pereira, Mariadason, Hannan and Dhillon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amardeep S. Dhillon, Research Division, Peter MacCallum Cancer Centre, St. Andrews Place, Melbourne, VIC 3002, Australia e-mail:YW1hcmRlZXAuZGhpbGxvbkBwZXRlcm1hYy5vcmc=