 Kavitha Ramaswamy
Kavitha Ramaswamy Barbara Spitzer
Barbara Spitzer Alex Kentsis
Alex Kentsis- Molecular Pharmacology and Chemistry Program, Department of Pediatrics, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, Weill Medical College of Cornell University, New York, NY, USA
Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that is required for normal cell growth and development. PP2A is a potent tumor suppressor, which is inactivated in cancer cells as a result of genetic deletions and mutations. In myeloid leukemias, genes encoding PP2A subunits are generally intact. Instead, PP2A is functionally inhibited by post-translational modifications of its catalytic C subunit, and interactions with negative regulators by its regulatory B and scaffold A subunits. Here, we review the molecular mechanisms of genetic and functional inactivation of PP2A in human cancers, with a particular focus on human acute myeloid leukemias (AML). By analyzing expression of genes encoding PP2A subunits using transcriptome sequencing, we find that PP2A dysregulation in AML is characterized by silencing and overexpression of distinct A scaffold and B regulatory subunits, respectively. We review the mechanisms of functional PP2A activation by drugs such as fingolimod, forskolin, OP449, and perphenazine. This analysis yields two non-mutually exclusive mechanisms for therapeutic PP2A re-activation: (i) allosteric activation of the phosphatase activity, and (ii) stabilization of active holo-enzyme assembly and displacement of negative regulatory factors from A and B subunits. Future studies should allow the development of specific and potent pharmacologic activators of PP2A, and definition of susceptible disease subsets based on specific mechanisms of PP2A dysregulation.
Introduction
Protein phosphatase 2A (PP2A) is a serine/threonine protein phosphatase that is required for normal cell growth and development and is frequently inactivated in human cancers (1). The functions of PP2A depend on its regulated phosphatase activities toward specific substrates, controlling kinase-dependent signal transduction, protein stability, cell proliferation, and survival (2). The functions of PP2A as a tumor suppressor in human cancers was originally defined as a result of its functional inactivation by the direct binding of the transforming antigens of the SV40 polyoma virus, as well as inactivating deletions and mutations of genes encoding its enzymatic subunits (3). Cells transformed by the polyoma T antigens or mutations of PP2A subunits exhibit reduced phosphorylation of PP2A substrates, including key mediators of mitogenic signaling, apoptosis, and mitosis; reviewed by Sabina et al. (4). Since normal cell growth and development are incompatible with complete inactivation of PP2A (5, 6), cell transformation induced by PP2A inactivation likely involves partial effects on specific substrates.
Specific recruitment of cellular substrates to the phosphatase catalytic subunit of PP2A is mediated by the assembly of its trimeric holo-enzyme (7). Active PP2A holo-enzyme is composed of the regulatory B, catalytic C, and scaffold A subunits, with the AC heterodimer forming the core enzyme (8). There is large diversity in the types of PP2A holo-enzymes formed, which is due to the array of genes and isoforms responsible for PP2A subunits with genetic redundancy of at least two genes encoding each of the different subunits. This diversity leads to heterogeneity in subunit isoforms in terms of genetic alterations, differential tissue expression, and varying types of malignancies. Pharmacological inhibition of PP2A phosphatase activity by toxins or small molecule drugs, including okadaic acid and microcystin, causes cellular transformation (9, 10), raising the possibility that pharmacological re-activation of residual PP2A activity may have anti-tumor efficacy. Here, we review the molecular mechanisms of PP2A in human cancer, with a specific focus on myeloid leukemias, and describe pharmacological strategies for its therapeutic re-activation.
Genetic Mechanisms of PP2A Inactivation
Consistent with the requirement of PP2A activity for normal cell growth and development, genetic inactivation of PP2A appears to involve deletions and mutations of A or B subunits, but not the catalytic C subunit. The PP2A holo-enzyme is assembled by the A scaffold subunit, as structured by its HEAT repeat domain (11). In this way, the HEAT domain serves to juxtapose the regulatory B subunits that recruit specific substrates with the catalytic C subunit, as a result of protein–protein interactions of the N-terminal HEAT repeats 1–10 and C-terminal repeats 11–15 of the scaffold A subunit, respectively (12). The scaffold subunit A is expressed as constitutive and tissue-specific isoforms and encoded by different genes, PR65α (PPP2R1A) and PR65β (PPP2R1B), respectively with the PPP2R1B isoform responsible for 5–10% of the scaffold subunit (13). For example, breast and lung carcinomas inactivate PP2A by point mutations of the PPP2R1A gene and exon 9 deletion in the PPP2R1B genes, allowing for inactivation of PP2A phosphatase activity (14). Breast carcinomas also produce defective A subunits due to the deletions of amino acids 171–589. Similarly, deletion of E344–E388 that spans HEAT repeats 9 and 10 causes defective binding of specific regulatory B subunits in breast carcinomas. Mutations of the A scaffold PR65α also include mis-sense mutations E64G in breast carcinomas and R418W in melanomas, leading to the impaired recruitment of the B regulatory and C catalytic subunits, respectively (15). The PR65β A scaffold isoform appears to be more frequently affected by mis-sense mutations, including G8R, P65S, G90D, K343E, D504G, and V545A (16). Mutants of the PR65α isoform of subunit A have impaired binding to the B56γ B subunit, induce functional haploinsufficiency and contribute to cell transformation and near complete suppression of this Aα isoform leads to growth arrest (17).
In addition to the mutations of the A scaffold subunit, human cancers also frequently exhibit altered gene expression of the regulatory B subunits. For example, the PPP2R2A gene, which codes for the regulatory subunit B55α, is deleted in breast and prostate carcinomas and myelomas (18–20). In melanomas, there is reduced gene expression of the B56γ subunit encoded by PPP2R5C (21). In acute myeloid leukemias (AML), genes encoding PP2A subunits are generally intact, without apparent somatic mutations or deletions identified to date. One exception is the haploinsufficiency of the PPP2CA gene encoding the catalytic C subunit in AML cases with deletions of chromosome 5, including del(5q) specifically (contribution in this issue by Sallman et al.). The 5q commonly deleted region also includes several additional tumor suppressor genes (22), suggesting that haploinsufficiency of PPP2CA may cooperate with other gene deletions in AML.
Analysis of PP2A subunits and genetic mutations have until now focused at the level of genetic alteration and post-translational modifications. The correlation of these mutations with protein levels in AML cells of abnormal PP2A components has yet to be determined. In order to understand the expression of PP2A subunits in human AML, we analyzed transcriptome data collected as part of the Cancer Genome Atlas (TCGA) from 138 patients with AML and 5 healthy controls (23). Raw sequences were aligned to the hg19 human reference genome using STAR (24) and differential expression analysis was determined using DESeq2 Bioconductor package (25). As can be seen in Figure 1, most genes encoding PP2A subunits do not have statistically significantly altered expression in AML cells relative to normal CD34+ bone marrow precursor cells. We find that PPP2R1B, encoding the Aβ scaffold subunit, is significantly down-regulated in AML cells (Benjamini Hochberg-adjusted p = 0.036). This analysis also reveals the relative overexpression of genes encoding two regulatory B subunits in AML cells, PPP2R2B and PPP2R5B (Benjamini Hochberg-adjusted p = 0.0025 and 0.00021, respectively). Genes encoding regulatory B subunits PPP2R2C and PPP2R3B were found to be minimally expressed in AML cells in this analysis. In addition, PPP2CA encoding one of the catalytic C subunits is distinctly down-regulated in patients with TP53 mutant AML, consistent with cytogenetically complex karyotypes in this disease subset, and with prior observations of its loss in del(5q) AML (26). We did not find any other apparent associations between PP2A subunit gene expression and molecular subtypes of AML in this analysis. Combined with the analysis of the expression of PP2A subunits in normal hematopoietic cells (contribution in this issue by Haesen et al.), these findings indicate that AML cells express a distinct repertoire of PP2A enzymes, characterized by down-regulation of scaffold A subunits and up-regulation of specific regulatory B subunits.
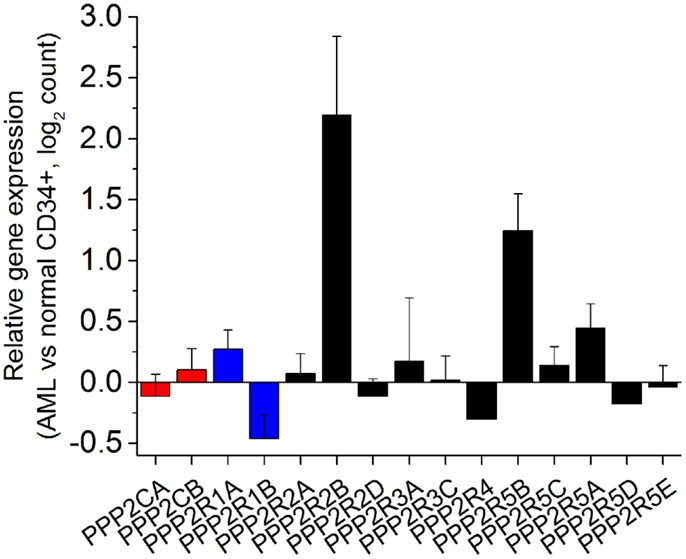
Figure 1. Human acute myeloid leukemia cells exhibit distinct alterations in the expression of specific genes encoding PP2A subunits. Relative expression of genes encoding PP2A subunits (catalytic C subunits in red, scaffold A subunits in blue, and regulatory B subunits in black) measured as a ratio of expression in AML cells relative to normal CD34+ bone marrow hematopoietic progenitor cells in normalized read counts. PPP2R2C and PPP2R3B are not significantly expressed and thus not shown, whereas PPP2R1B, PPP2R2B, and PPP2R5B are statistically significantly altered in their expression in AML cells relative to normal CD34+ cells (Benjamini Hochberg-adjusted p < 0.05).
Functional PP2A Inactivation
In addition to the apparent alterations in expression of PP2A enzymatic subunits, PP2A activity can be dysregulated through functional mechanisms, including post-translational modifications of the enzymatic C subunit and effects of regulatory proteins that interact with the A and B subunits. The PP2A catalytic C subunit is known to undergo phosphorylation and carboxymethylation, both of which have been found to regulate its enzymatic activity in the context of active holo-enzyme. Several oncogenic kinases, including BCR–ABL1 in myeloid leukemias, phosphorylate T304 and T307 of the C subunit, leading to reduced PP2A phosphatase activity (27, 28). Carboxymethylation of L309 of the C subunit by the methylesterases LCMT1 and PME-1 leads to altered recruitment of regulatory B55 subunits (29). Finally, oxidative nitration of Y289 of the B56δ subunit was recently described to interfere with the assembly of the PP2A holo-enzyme, leading to dysregulation of BCL-2 phosphorylation and aberrant cell survival in lymphoblastic leukemia and lymphoma cells (30). In summary, PP2A activity appears to be controlled by two non-mutually exclusive mechanisms: (i) allosteric regulation of the phosphatase activity, and (ii) regulation of holo-enzyme assembly and recruitment of specific substrates by the A and B subunits. These two mechanisms inform possible pharmacological strategies for therapeutic re-activation of PP2A, as discussed below.
In addition to regulatory post-translational modifications of PP2A, several endogenous cellular factors have been found to inhibit PP2A activity, including CIP2A and SET. Overexpression of these cellular inhibitors of PP2A leads to functional inactivation of the PP2A phosphatase activity, and contributes to cellular transformation. For example, CIP2A appears to transform cells by direct interaction with the c-Myc transcription factor and interfering with substrate recruitment by PP2A B subunits. In colon and head and neck squamous cell carcinomas, overexpression of CIP2A leads to the potentiation of MYC S62 phosphorylation, the PP2A target, thereby inhibiting proteolysis and resulting in increased oncogenic MYC protein stability (31, 32). Overexpression of CIP2A is also associated with increased phosphorylation of other PP2A substrates, including AKT1, MEK1, and MDM2, presumably due to interference with the assembly of additional PP2A holo-enzyme complexes targeting a variety of substrates (33).
Another well-characterized cellular inhibitor of PP2A is SET, and its regulator SETBP1. Although the exact mechanism is not well understood, SET is thought to inhibit PP2A by direct binding to the PP2A catalytic subunit (34). SET is overexpressed in breast carcinoma and acute leukemia cells, and can directly bind to the PP2A catalytic C subunit, causing reduced dephosphorylation of PP2A substrates (35). In AML, SET activity is regulated by proteolysis, mediated by its binding partner SETBP1, leading to the formation of an inhibitory SETBP1–SET–PP2A complex (36). When SETBP1 is overexpressed, SET is stabilized and protected from protease cleavage and facilitates PP2A inhibition, potentially explaining the relatively poor prognosis in patients with AML with altered expression of these regulatory proteins (37). In addition, some patients with AML exhibit SETBP1 overexpression due to the t(12;18)(p13;q12) chromosomal translocation (36), and patients with atypical CML can have recurrent G870S mutations of SETBP1 (38).
Therapeutic Strategies to Reactivate PP2A
Absence of genetic mutations that cause complete loss-of-function of PP2A phosphatase activity, such as homozygous deletions of the C or A subunits and their genetic redundancy with two genes encoding these subunits, makes it possible to potentially reactivate PP2A for anti-cancer therapy. Such therapeutic PP2A re-activation strategies can be classified in two non-mutually exclusive mechanisms: (i) allosteric activation of the phosphatase activity, and (ii) stabilization of active holo-enzyme assembly and displacement of negative regulatory factors from A and B subunits. The C phosphatase subunit is known to be regulated by post-translational modifications of its C-terminal TPDYFL tail, including nitration of Y289, phosphorylation of T304 and Y307, and carboxymethylation of L309, which can potentially regulate the allosteric relay of the structurally juxtaposed 120–126 helix and 183–195 loop switches regulating the phosphatase catalytic site (39). Indeed, inhibition of PP2A by carcinogenic antibiotics such as okadaic acid and microcystin, involves changes in the allosteric regulation of the PP2A catalytic subunit (40). This raises the possibility that either natural products or small molecules can be used to pharmacologically modulate the PP2A C subunit to allosterically activate its phosphatase activity in the context of an active holo-enzyme (mechanism I). Indeed, forskolin, a diterpene antibiotic, can reduce the phosphorylation of the C-terminal tail PP2A C subunit Y307, leading to PP2A activation (41, 42). This effect appears to be independent of forskolin’s activation of adenylate cyclase, as evidenced by the apparently equal anti-leukemic efficacy of 1,9-dideoxy-forskolin, which does not alter cyclic AMP levels. Consistent with this, forskolin treatment was found to be synergistic with cytotoxic AML chemotherapeutic agents, idarubicin and cytarabine (41).
Fingolimod or FTY720, a derivative of the antibiotic myriocin, activates PP2A in part by inhibition of the negative PP2A regulator SET (mechanism II). Fingolimod can bind SET directly, causing its displacement from and assembly of active PP2A holo-enzymes (43). Treatment of AML cells with fingolimod leads to dephosphorylation of PP2A substrates, such as the KIT receptor, AKT1, and STAT5, causing cell death (44, 45). Because fingolimod also inhibits sphingosine kinase 1, current efforts are focused on using fingolimod derivatives that lack this activity, such as (R)-FTY720-OMe and OSU-2S to specifically activate PP2A (46). Similarly, the cell-penetrating peptide OP449 or COG449 is an apolipoprotein E-mimetic that directly binds SET, relieving SET-mediated PP2A phosphatase inhibition (47, 48).
Recently, perphenazine and related phenothiazines were found to activate PP2A, causing dephosphorylation of PP2A substrates and leukemia cell death (49). Perphenazine can bind directly to the PP2A Aα scaffold subunit (49). This can cause PP2A activation either due to the displacement of a negative A subunit regulator, such as CIP2A (31), and/or allosteric effects of the C subunit activity when bound to the A scaffold HEAT domains 11–15. This mechanism may also explain the pervasive inhibition of serine/threonine kinase signaling by the chemically related trifluoperazine drugs (50). Elucidation of the structural basis and effects on PP2A holo-enzyme assembly and phosphatase activity should lead to the development of more potent and specific phenothiazine-like PP2A activators.
Future Therapeutic Strategies
Cellular PP2A comprises an ensemble of distinct holo-enzymes associated with specific regulatory subunits. Cancer cells with non-genetic mechanisms of PP2A inactivation, such as altered expression of genes encoding PP2A subunits or aberrant expression of PP2A negative regulators, may therefore be candidates for pharmacological functional re-activation of PP2A for anti-cancer therapy. In AML specifically, PP2A phosphatase activity appears to be dysregulated as a result of reduced expression of specific A scaffold subunits, and altered expression of regulatory B subunits. Consequently, leukemogenic effects of PP2A dysregulation are mediated by specific phospho-serine and phospho-threonine substrates. Likewise, pharmacological strategies for therapeutic PP2A re-activation will depend on the specific details of PP2A dysregulation. For example, pharmacologic inhibitors of PP2A negative regulator SET, such as fingolimod, OP449 and its analogs, will likely be ineffective in leukemias without SET overexpression or activation. In contrast, pharmacological PP2A activators that modulate PP2A phosphatase activity through allosteric regulation of phosphatase activity, such as forskolin and possibly perphenazine and related drugs, may have broader efficacy. Finally, detailed understanding of the molecular mediators of carcinogenic PP2A dysregulation, including specific substrates and signaling pathways, will be necessary to define not only biomarkers that can be used to prospectively identify susceptible tumors but also to determine effective combination therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the Charles Trobman Scholarship to Kavitha Ramaswamy, National Institutes of Health grant K08CA160660, Gabrielle’s Angel Foundation, Alex’s Lemonade Stand Foundation, Burroughs Wellcome Fund, and Josie Robertson Foundation Alex Kentsis.
References
1. Sontag E. Protein phosphatase 2A: the Trojan horse of cellular signaling. Cell Signal (2001) 13(1):7–16. doi:10.1016/S0898-6568(00)00123-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J (2001) 353(Pt 3):417–39. doi:10.1042/0264-6021:3530417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene (2005) 24(52):7746–55. doi:10.1038/sj.onc.1209038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Sablina AA, Hahn WC. SV40 small T antigen and PP2A phosphatase in cell transformation. Cancer Metastasis Rev (2008) 27(2):137–46. doi:10.1007/s10555-008-9116-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Götz J, Probst A, Ehler E, Hemmings B, Kues W. Delayed embryonic lethality in mice lacking protein phosphatase 2A catalytic subunit Cα. Proc Natl Acad Sci U S A (1998) 95(21):12370–5. doi:10.1073/pnas.95.21.12370
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Götz J, Schild A. Transgenic and knockout models of PP2A. In: Klumpp S, Krieglstein J, editors. Methods in Enzymology. (Vol. 366), New York: Academic Press (2003). p. 390–403.
7. Mumby MC, Walter G. Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol Rev (1993) 73(4):673–99.
8. Xu Y, Xing Y, Chen Y, Chao Y, Lin Z, Fan E, et al. Structure of the protein phosphatase 2A holoenzyme. Cell (2006) 127(6):1239–51. doi:10.1016/j.cell.2006.11.033
9. Suganuma M, Fujiki H. [Tumor promotion by inhibitors of protein phosphatase 1 and 2A]. Tanpakushitsu Kakusan Koso (1998) 43(8 Suppl):1102–10.
10. Fujiki H, Suganuma M. Tumor promotion by inhibitors of protein phosphatases 1 and 2A: the okadaic acid class of compounds. Adv Cancer Res (1993) 61:143–94. doi:10.1016/S0065-230X(08)60958-6
11. Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature (2007) 445(7123):53–7. doi:10.1038/nature05351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell (1999) 96(1):99–110. doi:10.1016/S0092-8674(00)80963-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Hemmings BA, Adams-Pearson C, Maurer F, Muller P, Goris J, Merlevede W, et al. Alpha- and beta-forms of the 65-kDa subunit of protein phosphatase 2A have a similar 39 amino acid repeating structure. Biochemistry (1990) 29(13):3166–73. doi:10.1021/bi00465a002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Calin GA, di Iasio MG, Caprini E, Vorechovsky I, Natali PG, Sozzi G, et al. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene (2000) 19(9):1191–5. doi:10.1038/sj.onc.1203389
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Ruediger R, Pham HT, Walter G. Alterations in protein phosphatase 2A subunit interaction in human carcinomas of the lung and colon with mutations in the A beta subunit gene. Oncogene (2001) 20(15):1892–9. doi:10.1038/sj.onc.1204279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Wang DH, Ishii K, Seno E, Yane S, Horike T, Yamamoto H, et al. Reduced serum levels of ALT and GGT and high carbohydrate intake among workers exposed to toluene below the threshold limit values. Ind Health (1998) 36(1):14–9. doi:10.2486/indhealth.36.14
17. Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res (2005) 65(18):8183–92. doi:10.1158/0008-5472.CAN-05-1103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature (2012) 486(7403):346–52. doi:10.1038/nature10983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Liu R, Zhou XW, Tanila H, Bjorkdahl C, Wang JZ, Guan ZZ, et al. Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology. J Cell Mol Med (2008) 12(1):241–57. doi:10.1111/j.1582-4934.2008.00249.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Mosca L, Musto P, Todoerti K, Barbieri M, Agnelli L, Fabris S, et al. Genome-wide analysis of primary plasma cell leukemia identifies recurrent imbalances associated with changes in transcriptional profiles. Am J Hematol (2013) 88(1):16–23. doi:10.1002/ajh.23339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Deichmann M, Polychronidis M, Wacker J, Thome M, Naher H. The protein phosphatase 2A subunit Bgamma gene is identified to be differentially expressed in malignant melanomas by subtractive suppression hybridization. Melanoma Res (2001) 11(6):577–85. doi:10.1097/00008390-200112000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Wei S, Chen X, Rocha K, Epling-Burnette PK, Djeu JY, Liu Q, et al. A critical role for phosphatase haplodeficiency in the selective suppression of deletion 5q MDS by lenalidomide. Proc Natl Acad Sci U S A (2009) 106(31):12974–9. doi:10.1073/pnas.0811267106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368(22):2059–74. doi:10.1056/NEJMoa1301689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (2013) 29(1):15–21. doi:10.1093/bioinformatics/bts635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol (2010) 11(10):R106. doi:10.1186/gb-2010-11-10-r106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Sallman D, Wei S, List A. PP2A: the Achilles heal in MDS with 5q deletion. Frontiers in Oncology (2014) 4:264.
27. Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science (1992) 257(5074):1261–4. doi:10.1126/science.1325671
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell (2005) 8(5):355–68. doi:10.1016/j.ccr.2005.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Favre B, Zolnierowicz S, Turowski P, Hemmings BA. The catalytic subunit of protein phosphatase 2A is carboxyl-methylated in vivo. J Biol Chem (1994) 269(23):16311–7.
30. Low ICC, Loh T, Huang Y, Virshup DM, Pervaiz S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56δ stabilizes its antiapoptotic activity. Blood (2014) 24(14):2223–34. doi:10.1182/blood-2014-03-563296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell (2007) 130(1):51–62. doi:10.1016/j.cell.2007.04.044
32. Dong QZ, Wang Y, Dong XJ, Li ZX, Tang ZP, Cui QZ, et al. CIP2A is overexpressed in non-small cell lung cancer and correlates with poor prognosis. Ann Surg Oncol (2011) 18(3):857–65. doi:10.1245/s10434-010-1313-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. De P, Carlson J, Leyland-Jones B, Dey N. Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A (CIP2A): an oncoprotein with many hands. Oncotarget (2014) 5(13):4581–602. doi:10.1126/scisignal.2004088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem (1996) 271(19):11059–62. doi:10.1074/jbc.271.19.11059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Arnaud L, Chen S, Liu F, Li B, Khatoon S, Grundke-Iqbal I, et al. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I(2)(PP2A)/SET. FEBS Lett (2011) 585(17):2653–9. doi:10.1016/j.febslet.2011.07.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Cristóbal I, Blanco FJ, Garcia-Orti L, Marcotegui N, Vicente C, Rifon J, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood (2010) 115(3):615–25. doi:10.1182/blood-2009-06-227363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Cristobal I, Garcia-Orti L, Cirauqui C, Cortes-Lavaud X, Garcia-Sanchez MA, Calasanz MJ, et al. Overexpression of SET is a recurrent event associated with poor outcome and contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica (2012) 97(4):543–50. doi:10.3324/haematol.2011.050542
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Piazza R, Valletta S, Winkelmann N, Redaelli S, Spinelli R, Pirola A, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet (2013) 45(1):18–24. doi:10.1038/ng.2495
39. Jiang L, Stanevich V, Satyshur KA, Kong M, Watkins GR, Wadzinski BE, et al. Structural basis of protein phosphatase 2A stable latency. Nat Commun (2013) 4:1699. doi:10.1038/ncomms2663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Prickett TD, Brautigan DL. The alpha4 regulatory subunit exerts opposing allosteric effects on protein phosphatases PP6 and PP2A. J Biol Chem (2006) 281(41):30503–11. doi:10.1074/jbc.M601054200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Cristobal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia (2011) 25(4):606–14. doi:10.1038/leu.2010.294
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Cristobal I, Rincon R, Manso R, Madoz-Gurpide J, Carames C, del Puerto-Nevado L. Hyperphosphorylation of PP2A in colorectal cancer and the potential therapeutic value showed by its forskolin-induced dephosphorylation and activation. Biochim Biophys Acta (2014) 1842(9):1823–9. doi:10.1016/j.bbadis.2014.06.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med (2013) 5(1):105–21. doi:10.1002/emmm.201201283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Roberts KG, Smith AM, McDougall F, Carpenter H, Horan M, Neviani P, et al. Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers. Cancer Res (2010) 70(13):5438–47. doi:10.1158/0008-5472.CAN-09-2544
45. Yang Y, Huang Q, Lu Y, Li X, Huang S. Reactivating PP2A by FTY720 as a novel therapy for AML with C-KIT tyrosine kinase domain mutation. J Cell Biochem (2012) 113(4):1314–22. doi:10.1002/jcb.24003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest (2013) 123(10):4144–57. doi:10.1172/JCI68951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Switzer CH, Cheng RY, Vitek TM, Christensen DJ, Wink DA, Vitek MP. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene (2011) 30(22):2504–13. doi:10.1038/onc.2010.622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res (2014) 20(8):2092–103. doi:10.1158/1078-0432.CCR-13-2575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Gutierrez A, Pan L, Groen RW, Baleydier F, Kentsis A, Marineau J, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest (2014) 124(2):644–55. doi:10.1172/JCI65093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Sangodkar J. Targeting the FOXO1/KLF6 axis regulates EGFR signaling and treatment response. J Clin Invest (2012) 122(7):2637–51. doi:10.1172/JCI62058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: protein phosphatase 2A, gene expression, enzyme activation, leukemia, kinase signaling
Citation: Ramaswamy K, Spitzer B and Kentsis A (2015) Therapeutic re-activation of protein phosphatase 2A in acute myeloid leukemia. Front. Oncol. 5:16. doi: 10.3389/fonc.2015.00016
Received: 30 December 2014; Paper pending published: 07 January 2015;
Accepted: 13 January 2015; Published online: 02 February 2015.
Edited by:
Peter Ruvolo, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Peter Ruvolo, The University of Texas MD Anderson Cancer Center, USAAlejandro Gutierrez, Boston Children’s Hospital, USA
Copyright: © 2015 Ramaswamy, Spitzer and Kentsis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alex Kentsis, Molecular Pharmacology and Chemistry Program, Department of Pediatrics, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, Weill Medical College of Cornell University, 1275 York Avenue, New York, NY 10021, USA e-mail:a2VudHNpc3Jlc2VhcmNoZ3JvdXBAZ21haWwuY29t