 Stefanie N. Linch
Stefanie N. Linch Michael J. McNamara†
Michael J. McNamara† William L. Redmond
William L. Redmond- Robert W. Franz Cancer Research Center, Earle A. Chiles Research Institute, Providence Portland Medical Center, Portland, OR, USA
Recent studies have highlighted the therapeutic efficacy of immunotherapy, a class of cancer treatments that utilize the patient’s own immune system to destroy cancerous cells. Within a tumor the presence of a family of negative regulatory molecules, collectively known as “checkpoint inhibitors,” can inhibit T cell function to suppress anti-tumor immunity. Checkpoint inhibitors, such as CTLA-4 and PD-1, attenuate T cell proliferation and cytokine production. Targeted blockade of CTLA-4 or PD-1 with antagonist monoclonal antibodies (mAbs) releases the “brakes” on T cells to boost anti-tumor immunity. Generating optimal “killer” CD8 T cell responses also requires T cell receptor activation plus co-stimulation, which can be provided through ligation of tumor necrosis factor receptor family members, including OX40 (CD134) and 4-1BB (CD137). OX40 is of particular interest as treatment with an activating (agonist) anti-OX40 mAb augments T cell differentiation and cytolytic function leading to enhanced anti-tumor immunity against a variety of tumors. When used as single agents, these drugs can induce potent clinical and immunologic responses in patients with metastatic disease. However, each of these agents only benefits a subset of patients, highlighting the critical need for more effective combinatorial therapeutic strategies. In this review, we will discuss our current understanding of the cellular and molecular mechanisms by which OX40 agonists synergize with checkpoint inhibitor blockade to augment T cell-mediated anti-tumor immunity and the potential opportunities for clinical translation of combinatorial immunotherapeutic strategies.
Introduction
Immunotherapy has become a major focus of anti-cancer therapy regimens and for good reason: when it works, patients can have long-lasting anti-tumor immune responses that not only eradicate primary tumors but metastatic lesions as well. Recently, investigators have focused on harnessing the power of checkpoint inhibitors, such as CTLA-4 and PD-1, to release the “brakes” on an anti-tumor T cell response through antibody-mediated antagonism of these receptors. However, checkpoint inhibition alone is not sufficient to promote tumor regression in a majority of patients. Generating a robust therapeutic immune response requires not only releasing the “brakes” but also stepping on the “gas.” T cell co-stimulation through receptors, like OX40 or 4-1BB, provides a potent “go” signal that actively promotes the expansion and proliferation of killer CD8 and helper CD4 T cells. Here, we discuss recent advances in the field of OX40 immunotherapy and the promise of triple combination therapy in the (not so distant) future.
OX40: Stepping on the Gas
OX40 (CD134; TNFRSF4) is a member of the TNFR super-family and was originally characterized as a receptor that was primarily expressed by rat CD4 T cells from the thymus and lymph nodes following stimulation with concanavalin A (1). Subsequent research demonstrated that in both mice and humans, OX40 is expressed by CD4 and CD8 T cells during antigen-specific priming (2–5). OX40 expression is induced following TCR/CD3 cross-linking, and by the presence of inflammatory cytokines, including IL-1, IL-2, and TNF-α. The expression of OX40 following antigen encounter is largely transient for both CD4 and CD8 T cells (24–72 h), with the duration of OX40 expression by CD8 T cells reported to be shorter than for CD4 T cells (6). In the absence of activating signals, relatively few mature T cell subsets have been shown to express OX40 at biologically relevant levels (7). However, the constitutive expression of OX40 by follicular helper CD4 T cells (Tfh) has been described in both mice and humans (8–11). Within germinal centers, the CD4+/CXCR5+/CCR7−subpopulation of Tfh cells have been shown to have the highest level of OX40 expression and are thought to be important regulators of antibody production (12–14). In mice, OX40 is also constitutively expressed on FoxP3+ regulatory T cells (Treg cells), in contrast to human Treg cells where its expression is inducible (7). In contrast, antigen-specific activation can induce OX40 expression by numerous subsets of differentiated CD4 and CD8 T cells. In a murine model system (OT-II), Th1 and Th17 cells were both capable of a similarly robust induction of OX40 in response to peptide-activation (15). In humans, a substantial proportion of tumor-infiltrating CD4 T cells express OX40, presumably due to recognition of tumor antigens, and the frequency of OX40+ CD4 T cells may be prognostic for patient outcomes (16, 17). Similarly, activated peripheral CD8 T cells have also been shown to express OX40 in mice and humans (18, 19).
Ligation of OX40 on CD8 and conventional (non-regulatory) CD4 T cells, using either its natural ligand (OX40L) or agonist antibodies, promotes their survival and expansion. Evidence of this comes from studies using OX40- and OX40L-deficient mice, which are discussed in detail in several recent reviews (7, 20). These studies demonstrated that OX40- or OX40L-knockout mice had reduced expansion of both CD4 and CD8 T cells, combined with defective memory responses following antigen challenge, indicating the importance of endogenous OX40 expression in regulating T cell expansion (20–25). Furthermore, treatment with agonist anti-OX40 monoclonal antibodies (mAbs) along with TCR stimulation in wild-type animals induced expansion, differentiation, and increased survival of CD4 and CD8 T cells. Likewise, depletion of CD8 or CD4 T cells eliminated the ability of anti-OX40 mAbs to induce tumor regression in several tumor models (23, 24, 26–28). One study demonstrated that anti-OX40 administration was sufficient to overcome CD8 T cell tolerance to a self-antigen and restored their cytotoxic activity, highlighting the therapeutic potential for OX40 agonists (29). This is of particular importance for patients with cancer, as T cell tolerance to the tumor is a major obstacle for therapeutic modalities. Another group has demonstrated that enhanced CD8 T cell function following anti-OX40 treatment was mediated by the induction of CD40L expression on effector T cells thereby promoting DC maturation, because CD40−/− mice have significantly fewer CD11c+ dendritic cells that migrate into the draining lymph nodes following anti-OX40 mAb (30). In fact, CD40−/− mice treated with anti-OX40 mAbs all succumb to their tumors in contrast to wild-type mice, which have a 60% survival rate, suggesting the importance of CD40 expression following OX40 stimulation. Collectively, these data suggest that exogenous manipulation of OX40 signaling can boost stagnant T cell responses.
Several investigators have conducted studies to determine the mechanism by which OX40 promotes T cell survival. It has been demonstrated that following activation, OX40-deficient CD4 T cells failed to sustain expression of the anti-apoptotic proteins Bcl-xL and Bcl-2. Moreover, the survival of activated CD4 T cells was rescued by retroviral transduction of Bcl-xL or Bcl-2 (23). Sustained expression of Bcl-xL was also necessary for the survival of tumor-reactive CD8 T cells following OX40 co-stimulation (31). Subsequent studies demonstrated that OX40 signaling in T cells induced expression of Survivin, and this was required to regulate and sustain T cell division over time. Survivin expression was maintained via the sustained activation of PI3K and PKB by OX40 signaling (32). However, Survivin expression does not supersede the requirement for Bcl-xL and Bcl-2 following OX40 signaling in order to inhibit T cell apoptosis. Enhanced expression of Survivin and Bcl-2 family members is mediated via activation of IκB kinase and NF-κB1 following OX40 signaling (33). Other investigators have shown that TRAF2 is required following OX40 signaling in antigen-specific CD4 T cells, as the expression of a dominant negative TRAF2 in CD4 T cells inhibited their expansion, survival, and cytokine production (34). One of the functions of TRAF2 appears to be to prevent CTLA-4 expression following T cell co-stimulation through OX40, as CTLA-4 blockade at the time of T cell priming with antigen and anti-OX40 mAbs partially restored defective expansion in mice expressing a dominant negative TRAF2 protein. It remains unknown whether the same TRAF adaptors and NF-κB pathways are activated in T cells following ligand binding by other TNFR family members, such as CD27 and GITR (35, 36). Similarities and differences in the signaling pathways activated by T cell co-stimulatory receptors, including both TNFR family members, like OX40 and CD27, and immunoglobulin super-family members, like CD28 and B7 families, has been reviewed extensively elsewhere (37). The activation of multiple pathways by both co-stimulatory receptor super-families results in enhanced cell growth and effector function, and improves survival (37). Numerous investigators are currently testing the modulation of these receptors for various clinical applications and immunotherapies.
Preclinical studies demonstrated that treatment of tumor-bearing hosts with OX40 agonists, including both anti-OX40 mAb and OX40L-Fc fusion proteins, resulted in tumor regression in several preclinical models (20, 26, 27, 38–40). Recent studies have investigated the mechanisms by which these agonists function. In addition to promoting effector T cell expansion, since OX40 is constitutively expressed on Treg cells, OX40 agonists have the ability to directly regulate Treg cells. There are conflicting reports on whether these agonists promote or diminish Treg cell responses. Some have observed that anti-OX40 mAbs blocked the suppressive function of Treg cells in vivo, while others have observed Treg cell expansion (27, 41–43). These studies suggest that anti-OX40 can push Treg cells in both directions, depending upon the context of stimulation and the cytokine milieu. Indeed, the importance of the OX40 co-stimulatory pathway in regulating immunity is exemplified by the presence of autoimmune-like disease in mice with constitutive expression of OX40L (44, 45).
OX40 signaling has also been shown to inhibit the production of IL-10 by and suppressive function of Treg cells (46). Supporting these data, administration of anti-OX40 mAbs prior to tumor engraftment rendered Treg cells functionally inactive through inhibition of IL-10 production and elimination of Treg cell-mediated suppression of CD8 T cell responses (27, 30, 41). One recent report observed that cells expressing activating FcγR were required for the selective depletion of Treg cells from tumors, while there was no change in Treg cells in the draining lymph nodes at day 5 following anti-OX40 therapy (47). Other studies confirm that even at later time points following anti-OX40 treatment, there is no change in the frequency of Treg cells in the draining lymph nodes, so this effect may be localized to the tumor (27). In fact, this effect may be transient, as another report showed that at day 7 there was no difference in Treg cell frequency in the tumor between control-treated and anti-OX40-treated mice using the same CT26 colon cancer model (28). This study in particular also suggests that the immunological effects of anti-OX40 therapy can vary based on the tumor model examined; thus, one must be cautious of making generalizations regarding the precise mechanism of OX40 agonists. Other studies report that anti-OX40 mAbs reduce the suppressive activity of Treg cells in vitro and in vivo (27, 41). Whether anti-OX40 functions via Treg cell suppression, deletion, or both, treatment with these agonists should diminish the inhibitory effects mediated by Treg cells and thereby promote anti-tumor CD8 T cell responses necessary to maintain long-term anti-tumor immune responses. It is likely that multiple mechanisms are important for the anti-tumor activity of OX40 agonists.
The ability of OX40 agonists to regulate immune responses, as well as the expression of OX40 on CD4 and CD8 lymphocytes from the tumors and tumor-draining lymph nodes in mice and humans (38, 40, 48), led investigators to examine OX40 manipulation as a treatment for cancer patients. Recently, the use of anti-OX40 monotherapy was tested in a Phase 1 trial in patients with solid tumors, with promising results (49). Twelve out of 30 patients receiving an OX40 agonist had regression of at least 1 metastatic lesion with only 1 cycle of treatment. Patient toxicities were much milder for anti-OX40 mAbs compared to more severe toxicities, i.e., autoimmune-like disease, colitis, etc., caused by treatment with CTLA-4 blockade (ipilimumab), and most frequently included a temporary lymphopenia. Patients receiving the OX40 agonist had an expansion of CD4 (non-Treg cells) and CD8 T cells following drug infusion with concomitant expression of activation markers CD38 and HLA-DR. Unlike treatment with ipilimumab, treatment with an OX40 agonist did not induce expansion of Treg cells either in the blood or the tumor (49, 50). What investigators did observe was that two out of three patients had IFN-γ-producing CD8 T cells following stimulation with autologous tumor cell lines in vitro, suggesting a tumor-specific T cell response, though the antigens they recognize remain unknown. Unfortunately, the development of human anti-mouse antibodies to the drug precluded continued treatment (49). MedImmune has several Phase 1 clinical trials investigating OX40 agonists including NCT02318394, NCT02205333, and NCT02221960. Indeed, the use of OX40 agonists in the clinic represents an exciting new chapter in cancer immunotherapy. Further studies and patient immune monitoring will provide further insight into the mechanisms by which OX40 agonists enhance an anti-tumor immune response. Nevertheless, despite all the positive data supporting the use of OX40 agonists in cancer, it is unlikely that anti-OX40 alone will be sufficient to cure all patients or all tumor types. However, there is great promise that combination immunotherapy incorporating both OX40 and checkpoint inhibition may be able to do what single agents alone cannot (Figure 1; Table 1).
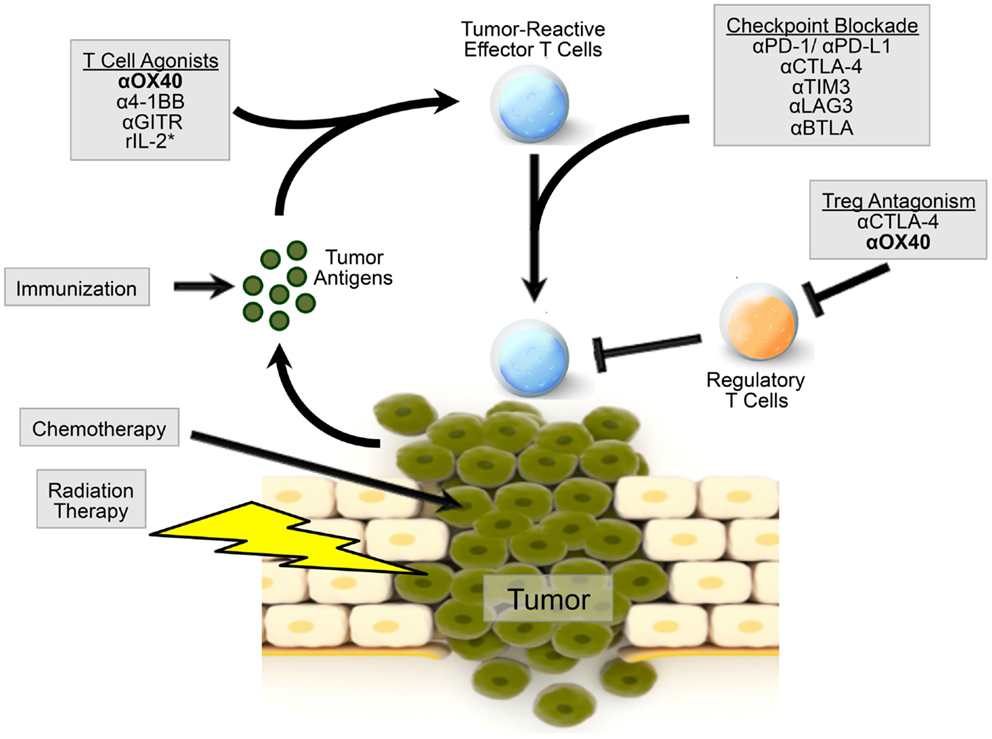
Figure 1. Model of OX40 agonism in combination immunotherapy radiation and chemotherapy can induce the release of tumor-associated antigens by the tumor. Patients can be immunized against these tumor-associated antigens to induce a robust immune response. Agonism of OX40, as well as other co-stimulatory molecules, can boost the generation of tumor-reactive effector T cells. OX40 agonism combined with checkpoint inhibition, via CTLA-4 or PD-1 blockade, or additional immunotherapy can further augment an effector T cell response. OX40 agonism can also inhibit Treg cell function, alleviating immunosuppression in the tumor microenvironment. OX40 agonism with combination therapy may provide a microenvironment more amenable to direct killing by effector T cells. *r denotes recombinant IL-2.
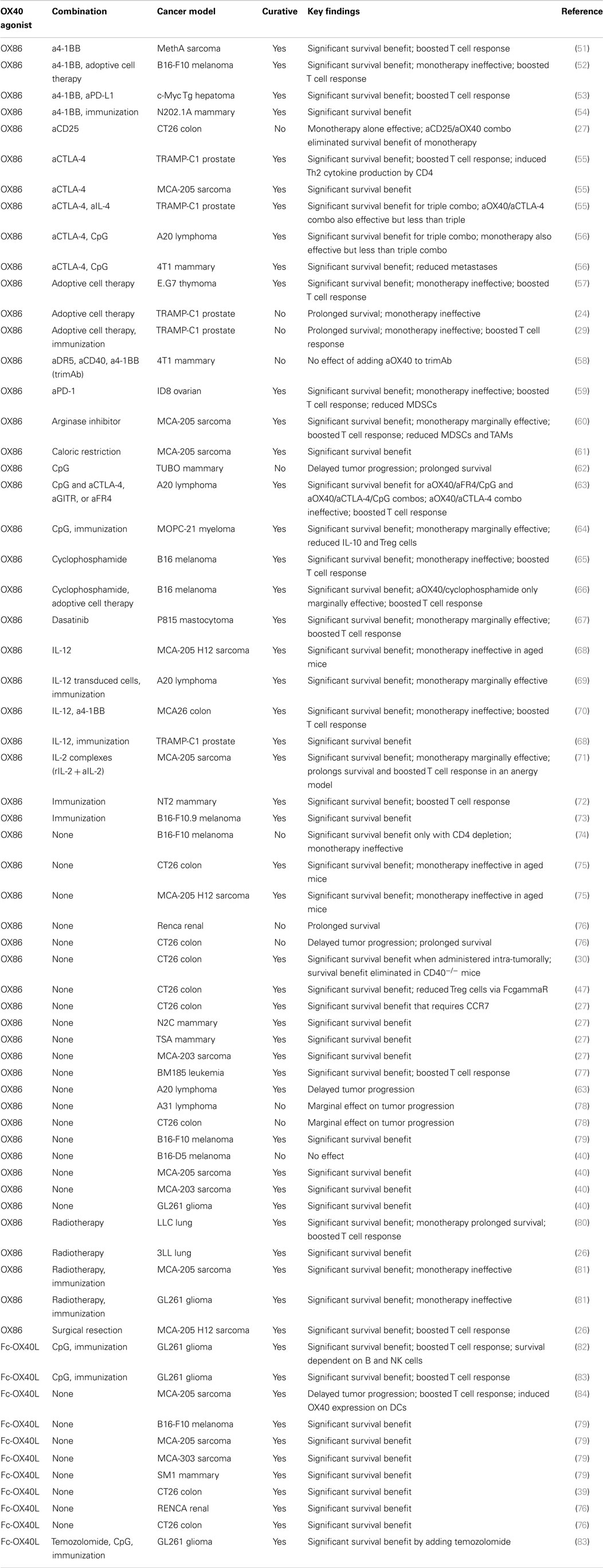
Table 1. Summary of animal studies using OX40 agonists alone or in combination.
Combination OX40 Agonism and Checkpoint Blockade: Releasing the Brakes Only Goes so Far
CTLA-4 Blockade with OX40 Agonism
In contrast to OX40, CTLA-4 is a negative regulatory surface molecule on T cells that competitively inhibits the CD28 co-stimulatory pathway by binding to B7-1 and B7-2. CTLA-4 is constitutively expressed on Treg cells and absent on naïve T cells, though expression is induced upon T cell activation as a means to attenuate and restrict T cell responses. This negative regulator is vital to prevent expansion of autoreactive T cells, as evidence by overt lymphoproliferative disease in CTLA-4 knockout mice (85, 86). Along these lines, inhibition of CTLA-4 using mAbs boosted effector CD4 and CD8 T cell function while inhibiting the suppressive function of Treg cells (87–91). These data led investigators to hypothesize that removing the “brakes” on a T cell response via CTLA-4 blockade would effectively allow the immune system to eliminate cancer cells and induce long-lasting anti-tumor immunity. Indeed, investigators have demonstrated the potency of checkpoint inhibition in cancer (90, 92–96). Clinical use of ipilimumab to block CTLA-4 has demonstrated improved survival in patients with metastatic melanoma (97–99). However, only a subset of patients treated with ipilimumab exhibit an objective clinical response (97). Thus, it is clear that additional strategies are necessary to improve patient outcomes and reduce lymphocyte dysfunction in cancer.
Investigators have tested whether combination immunotherapy, targeting both co-inhibitory and co-stimulatory molecules, is capable of overcoming tumor immune tolerance to induce a potent CD8 T cell response and ultimately tumor regression. The hypothesis behind this research is that because these molecules target distinct and also complementary pathways that tumor regression and the induction of a cytolytic T cell response may be amplified. Recent data from our laboratory indicated that combined anti-OX40/anti-CTLA-4 mAb therapy dramatically improved survival in the poorly immunogenic TRAMP-C1 prostate and the more immunogenic MCA-205 sarcoma models. Specifically, this combination therapy induced robust effector CD4 and CD8 T cell responses necessary to induce tumor regression (55). Likewise, Marabelle et al. recently demonstrated that combined anti-OX40/anti-CTLA-4 (with adjuvant CpG) was capable of inducing regression of local and distant tumors using several aggressive tumor models when administered intra-tumorally. The mechanism for intratumoral administration of combination therapy appears to be through depletion of Treg cells at the tumor site, allowing for a greater influx of CD8 T cells into the tumor (56). In contrast, when this combination therapy is given systemically, we observed no change in the frequency of Treg cells in the tumor, while the frequency actually increased in the draining lymph node (55). The route of administration may explain these differences as the same study showed that intratumoral administration of combined anti-OX40/anti-CTLA-4 did not affect Treg cells at a distant tumor site (56). However, Houot and Levy observed a reduction in Treg cells following systemic administration of combination therapy (63). These differences should be investigated further, but may be linked to the tumor model (lymphoma versus prostate-derived tumor), antibody clone (9D9 versus 9H10 for CTLA-4 blockade), dosing, or timing of therapy. In fact, the treatment regimen for combination immunotherapy, whether it is concurrent, staggered, or sequential, may prove very important in determining survival and immunological outcomes in cancer.
Another interesting observation from preclinical studies using combined anti-OX40/anti-CTLA-4 therapy was the induction of a population of Th2-cytokine secreting (IL-4+, IL-5+, IL-13+, IL-2−, TNF-α−) CD4 T cells (55). IL-4 was the primary driver of Th2 CD4 T cell differentiation as IL-4 blockade augmented the efficacy of combined anti-OX40/anti-CTLA-4 therapy in the TRAMP-C1 model. However, it remains unclear what immune subsets are affected by IL-4 or anti-IL-4 therapy. Furthermore, it is not known whether the expansion of Th2 CD4 T cells limits the efficacy of combination therapy. Previous work has demonstrated that low-affinity TCR signaling preferentially elicits Th2 CD4 T cell responses (100, 101). Whether this holds true following combined anti-OX40/anti-CTLA-4 therapy is currently an active area of investigation in our laboratory. If this hypothesis is correct, then one might speculate that a Th2 response could be redirected toward a more favorable Th1 response when combined with an antigen-specific vaccine, thereby boosting the efficacy of combination therapy without the need to block IL-4 in patients. Further studies to elucidate the connection between anti-OX40/anti-CTLA-4 combination therapy and IL-4 will be of interest as anti-OX40 progresses in the clinic.
PD-1 Blockade with OX40 Agonism
A structural relative of CD28 and CD33, PD-1 is a transmembrane protein that plays a fundamental role in the inhibition of activated lymphocytes (102–106). High levels of PD-1 expression are frequently associated with populations of exhausted T cells, but robust expression of PD-1 has been observed in multiple subsets of activated lymphocytes, including T, B, and NK cells. PD-1 has two closely related ligands, PD-L1 and PD-L2, which are expressed by multiple cell types, and PD-L1 has a soluble isoform that can be secreted by some cancer cell lines and is detectable in the sera of some tumor-bearing hosts (107–109). PD-L1 is also abundantly expressed by numerous tumors and can be induced by exposure to both Type I and Type II interferons (110–113). Engagement of PD-1 on activated T cells decreases their capacity for a cytotoxic response following antigen recognition, suppresses proliferation, and potentiates apoptosis (107, 114–118). Both PD-1 and PD-L1 blockade have shown significant efficacy in a range of murine and human cancer models (103, 112, 119). Multiple studies have demonstrated that disruption of PD-1 signaling induced the expansion and cytolytic capacity of effector T cell populations and increased their infiltration into tumors (119–131). PD-1 blockade has also been shown to reduce the frequency and function of Treg cells within some tumors (124, 127, 128, 130, 132).
In clinical trials, checkpoint inhibition via PD-1 or PD-L1 blockade has shown substantial promise with respect to both efficacy and safety (133–136). In September 2014, The FDA approved the first anti-PD-1 mAb (pembrolizumab) for use in the treatment of advanced metastatic melanomas (137). Two additional anti-PD-1 mAbs, nivolumab and pidilizumab, and two anti-PD-L1 mAbs have also demonstrated encouraging efficacy in ongoing clinical trials (138–143). The unique mechanism of action responsible for the efficacy of PD-1/PD-L1 blockade makes this pathway an excellent candidate for combinatorial immunotherapies (58, 144). Indeed, combined blockade of PD-1 and CTLA-4 has generated impressive results in both preclinical and clinical trials (145). Disruption of PD-1 signaling can also synergize with LAG3 blockade, radiotherapy, BRAF inhibitors, and many other treatment strategies (146–150). However, one of the limitations of PD-1/PD-L1 blockade has been a difficulty in initiating a protective immune response against poorly immunogenic or large tumors (110, 112). Because of this, PD-1 blockade may be uniquely well suited for combinatorial immunotherapy strategies incorporating agents that support the development of tumor-reactive effector lymphocytes – treatments such as T and NK cell mAb agonists or tumor-specific immunization (119, 151, 152).
Administration of an OX40 agonist has multiple immunological effects that may complement the activity of PD-1/PD-L1 blockade. First, ligation of OX40 supports the expansion, survival, and effector function of activated CD4 and CD8 T cells, populations that express the PD-1 receptor. Second, OX40 co-stimulation has been reported to enhance the ability of T cells to respond productively to lower affinity antigens and OX40 ligation can enhance IFN-γ production by T cells in response to TCR stimulation. Because many cancerous cells up-regulate PD-L1 in response to IFN-γ exposure, PD-1/PD-L1 blockade may uniquely complement the therapeutic efficacy of OX40-driven effector lymphocytes within the tumor microenvironment. Furthermore, because cancerous cells may also up-regulate antigen presentation in response to IFN-γ exposure, the combination of PD-1 blockade and OX40 agonism may support a pro-inflammatory feedback loop within the tumor microenvironment that further augments anti-tumor immunity.
In a recent report, Guo and colleagues observed that PD-1 blockade synergized with an agonistic anti-OX40 mAb to promote regression of an implantable murine ovarian cancer, ID8, which was non-responsive to either monotherapy (59). The authors reported that the combination significantly increased the ratio of CD8 T cells at the tumor site (peritoneal cavity), relative to both Treg cells and myeloid-derived suppressor cells (MDSCs). They also reported that the combination of anti-OX40 and anti-PD-1 mAbs dramatically expanded peritoneal CD4 and CD8 effector memory cells, while reducing the frequency of the naïve T cells. They observed that tumor-resident T cells from the anti-PD-1/anti-OX40 group produced significantly higher levels of IFN-γ in response to PMA stimulation. In addition, splenocytes from these mice showed increased reactivity toward an ID8-specific antigen, mesothelin. Unsurprisingly, T cell depletion experiments indicated that the therapeutic effect was entirely dependent on the presence of CD8 T cells and partially dependent on CD4 T cells. Notably, the authors reported that cultured ID8 cancer cells expressed minimal PD-L1 or PD-L2, which may support the hypothesis that OX40-stimulated T cells induce PD-L1 expression at the tumor site via enhanced IFN-γ production. A separate study reported that triple combination therapy, using co-stimulatory anti-OX40 and anti-4-1BB mAbs and an inhibitory anti-PD-1 mAb, was uniquely effective in a murine hepatocellular carcinoma model, with enhanced tumor infiltration of cytotoxic effector T cells (153). Moving forward, a deeper understanding of the immunological interplay between PD-1/PD-L1 and OX40-targeted therapies will help identify and refine complementary therapeutic interventions. Although currently somewhat sparse, the existing body of evidence suggests PD-1 blockade is likely to synergize with OX40 agonists and may be particularly well suited for tumors that are naturally immunogenic and/or express high levels of PD-L1.
One issue that needs to be considered when moving these combinations to the clinic is the potential for increased toxicity and immune-related adverse events (irAEs). As described above, the results from a Phase 1 clinical trial (NCT01644968) conducted in patients with late-stage cancer indicated that OX40 immunotherapy was generally well tolerated. The majority of irAEs were relatively minor (Grade 1 and 2), while all of moderate to severe (Grade 3 and 4) irAEs were due to treatment-induced lymphopenia that was shown to be temporary (49). In comparison, a larger Phase 1 study of melanoma and renal cancer patients receiving anti-PD-1 observed moderate or severe irAEs in 14% (41/296) of patients (143). For anti-CTLA-4 monotherapy, the incidence of irAEs is highly dose-dependent but the frequency of moderate or severe irAEs for patients being treated with anti-CTLA-4 tends to be between 20 and 40% (154). Importantly, patients receiving a combination of anti-CTLA-4 and anti-PD-1 were more likely to experience Grade 3 or 4 irAEs (53%), although this combination yielded a substantially higher objective response rate than either monotherapy (155). The efficacy and toxicity of combining anti-OX40 therapy with either CTLA-4 or PD-1 has not been evaluated in humans, although Phase 1 clinical trials for both combinations are currently accruing patients (NCT02205333). Because anti-OX40 therapy augments the development, activity, and survival of effector lymphocyte populations, it is possible that the combination of anti-OX40 with checkpoint blockade will produce a higher frequency of irAEs than the respective individual treatments. However, synergy between anti-OX40 therapy and checkpoint blockade may yield objective responses at lower dosages than are required when each drug is used as a monotherapy. Overall, the ongoing clinical experience with anti-CTLA-4 suggests that modulating dosages and clinically managing irAEs can be an effective strategy to alleviate symptoms and maintain patients on treatment (156).
On the Horizon: Other Checkpoint Inhibitors with Potential Synergy
Like OX40, CTLA-4, and PD-1, T cells express numerous cell surface receptors capable of modulating an anti-tumor immune response. Here, we touch on several surface receptors that when targeted may synergize with OX40 agonists (Figure 1). A member of the immunoglobulin super-family, Lymphocyte Activation Gene 3 (LAG3) is expressed by subsets of both activated and exhausted lymphocytes and plasmacytoid dendritic cells, and plays a key role in regulating the effector activity of tumor-associated lymphocytes (145, 157, 158). In some tumors, LAG3 is highly expressed by infiltrating effector T cells. LAG3 is known to interact with MHC-II receptors, and blockade of LAG3 has been shown to support effector T cell activity both in vitro and in vivo (159–164). Experiments suggest a synergistic interaction between combination anti-PD-1 and anti-LAG3 therapies that appears to enhance anti-tumor immunity, in part by preventing exhaustion and anergy in effector T cell populations (146, 149, 165). Although no studies have yet been published that directly evaluate the efficacy of combined anti-LAG3/anti-OX40 treatment, the current understanding of the mechanisms that underlie each therapy suggest the potential for cooperative activity. Specifically, OX40 agonists induce expansion and infiltration of effector T cells into the tumor, and the cytotoxic activity of these cells in the tumor microenvironment may be supported by LAG3 blockade. Additionally, many tumor cells and APCs up-regulate MHC-II expression in response to IFN-γ exposure and because OX40 therapy increases IFN-γ production by infiltrating T cells, there may be a rational basis for evaluating this combination.
Another possible target for combination therapy with anti-OX40 mAb is through targeted blockade of killer immunoglobulin-like receptors (KIRs). Primarily expressed by NK cells, KIRs are a class of transmembrane proteins that are important regulators of antigen-specificity and cytotoxic activity (166, 167). KIR family receptors are known to interact with MHC molecules on adjacent cells, with each KIR having specificity for different MHC subsets (168). KIRs can transduce either activating or inhibitory signals, and the balance between these signals is critical for mediating both self-tolerance and cytolytic activity. As a cancer immunotherapy, blockade of inhibitory KIRs using mAbs has demonstrated promise in murine tumor models and is being evaluated in early-stage clinical trials (169–172). Although the direct combination of OX40 agonism and inhibitory KIR blockade has not been reported, one might surmise that this combination might induce a potent anti-tumor response. Inhibitory KIR blockade, which can induce NK-cell-mediated tumor lysis, may promote the release of tumor-associated antigens. These antigens may provide TCR stimulation to OX40-stimulated T cells, thereby enhancing the effects of anti-OX40 therapy. It will be interesting to see what clinical trials unfold, and whether these combinations will be tested in the near future.
T cell immunoglobulin mucin 3 (TIM3) and B- and T-lymphocyte attenuator (BTLA, CD272) are also cell surface receptors that are expressed by effector T cell populations and transduce inhibitory signals (145). TIM3 is expressed on tumor-reactive CD8 T cells and antibody-mediated blockade of TIM3 enhanced their ability to produce IFN-γ (173). Galectin-9, which is highly expressed by some tumors, has been reported to be the natural ligand for TIM3, although this relationship is somewhat controversial (174, 175). Blockade of TIM3 promoted tumor regression, both as a monotherapy and in combination with anti-PD-1 (173, 176, 177). BTLA is expressed at a high level by tumor-reactive CD8 T cells and interaction with its ligand, HVEM, can inhibit the functional activity of this population (178, 179). Because HVEM is expressed by some tumors, and BTLA-deficient mice mount more robust T cell responses, blockade of this target has been proposed as a cancer immunotherapy (180–182). Similar to LAG3 blockade, targeting TIM3 and/or BTLA may augment the efficacy of OX40 therapy by supporting the expansion, survival, and cytotoxic effector function of lymphocytes, particularly within the microenvironment of the tumor (183–186).
Beyond checkpoint inhibition, several other therapeutic interventions have been reported to complement the anti-tumor activity of OX40 stimulation, including other immune-stimulatory mAbs, recombinant IL-2, immunization, radiotherapy, intratumoral TLR ligands, chemotherapeutics, and more (26, 53, 54, 63, 65, 70, 71, 80, 153, 187–192). One study investigated the combination of anti-OX40 mAbs and the chemotherapeutic cyclophosphamide, which is known to activate tumor-reactive T cells and selectively deplete Treg cells. This combination initiated tumor regression in the poorly immunogenic B16 melanoma model and induced a potent anti-tumor T cell response (65). Mechanistic studies revealed that anti-OX40 and cyclophosphamide induced a memory CD4 T cell population capable of producing both Th1 and Th2 cytokines (66). Other investigators have looked at the use of anti-OX40 surgical resection or radiation of the tumor. These experiments demonstrated that an OX40 agonist administered at the time of resection prevented local disease recurrence, and when combined with radiation prolonged survival and the frequency of disease-free animals (26). These studies contributed to the design of two clinical trials; one examining radiation, cyclophosphamide, and anti-OX40 in patients with metastatic prostate cancer (NCT01303705); and the other using combination stereotactic body radiation and anti-OX40 in patients with metastatic breast cancer (NCT01862900).
Triple Combinations and Beyond…
Because anti-tumor immunity is directed by a dynamic constellation of signals, maximizing the therapeutic benefit of lymphocyte agonists, such as anti-OX40 mAbs, will likely depend on incorporating multiple complementary interventions. The most viable candidates for combinatorial therapies are those that have already achieved FDA-approval, especially since anti-OX40 itself remains experimental. One particularly intriguing possibility is a triple combination of OX40 agonism with concomitant PD-1 and CTLA-4 blockade. The distinct mechanisms underlying PD-1 and CTLA-4 blockade have already been shown to synergize in the treatment of many murine and human cancers (135, 140, 150, 155, 193, 194). A “triple threat” immunotherapy approach that includes OX40 stimulation may help augment the efficacy of dual PD-1/CTLA-4 blockade by enhancing the expansion, survival, and cytolytic activity of tumor-reactive effector T cells. Other FDA-approved therapies, such as recombinant IL-2 and radiotherapy, may also be well suited for multiplex immunotherapy approaches that incorporate both OX40 stimulation and checkpoint inhibitor blockade. Administration of IL-2 can augment the activity of OX40 stimulation, in part because it stimulates the proliferation of T cells and their up-regulation of the OX40 receptor through a JAK3/STAT5-dependent mechanism (71, 195–199). Radiotherapy can also complement OX40 treatment and may be a valuable tool for disrupting the immune suppressive microenvironment of established tumors and releasing tumor-associated antigens for recognition by the immune system (26, 80, 147, 153, 200, 201). Because each of the aforementioned approaches utilizes a distinct mechanism, varied combinations of these strategies may yield unique therapeutic efficacy when combined with OX40 agonism (Table 1). Indeed, overcoming the challenge of mounting a curative immune response in a diverse population of patients will almost certainly require multiple complementary therapeutic modalities to overcome the immunosuppressive tumor microenvironment of established tumors and provide a protective anti-tumor immune response.
Author Contributions
SL and MM reviewed the relevant literature and drafted the manuscript. SL, MM, and WR revised the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
William L. Redmond receives research support from Bristol-Myers Squibb and Galectin Therapeutics. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the NIH (5R00CA136678), the Safeway Foundation, and the Providence Portland Medical Foundation.
References
1. Mallett S, Fossum S, Barclay AN. Characterization of the MRC OX40 antigen of activated CD4 positive T lymphocytes – a molecule related to nerve growth factor receptor. EMBO J (1990) 9(4):1063–8.
2. Lane P. Role of OX40 signals in coordinating CD4 T cell selection, migration, and cytokine differentiation in T helper (Th)1 and Th2 cells. J Exp Med (2000) 191(2):201–6. doi: 10.1084/jem.191.2.201
3. Baum PR, Gayle RB III, Ramsdell F, Srinivasan S, Sorensen RA, Watson ML, et al. Molecular characterization of murine and human OX40/OX40 ligand systems: identification of a human OX40 ligand as the HTLV-1-regulated protein gp34. EMBO J (1994) 13(17):3992–4001.
4. Al-Shamkhani A, Birkeland ML, Puklavec M, Brown MH, James W, Barclay AN. OX40 is differentially expressed on activated rat and mouse T cells and is the sole receptor for the OX40 ligand. Eur J Immunol (1996) 26(8):1695–9. doi:10.1002/eji.1830260805
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Bansal-Pakala P, Halteman BS, Cheng MH, Croft M. Costimulation of CD8 T cell responses by OX40. J Immunol (2004) 172(8):4821–5. doi:10.4049/jimmunol.172.8.4821
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Cannons JL, Lau P, Ghumman B, DeBenedette MA, Yagita H, Okumura K, et al. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol (2001) 167(3):1313–24. doi:10.4049/jimmunol.167.3.1313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu Rev Immunol (2010) 28:57–78. doi:10.1146/annurev-immunol-030409-101243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi:10.1146/annurev-immunol-031210-101400
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Vinuesa CG, Tangye SG, Moser B, Mackay CR. Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol (2005) 5(11):853–65. doi:10.1038/nri1714
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Abdoli R, Najafian N. T Helper cells fate mapping by co-stimulatory molecules and its functions in allograft rejection and tolerance. Int J Organ Transplant Med (2014) 5(3):97–110.
11. Fazilleau N, Mark L, McHeyzer-Williams LJ, McHeyzer-Williams MG. Follicular helper T cells: lineage and location. Immunity (2009) 30(3):324–35. doi:10.1016/j.immuni.2009.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol (2004) 173(1):68–78. doi:10.4049/jimmunol.173.1.68
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Ma CS, Suryani S, Avery DT, Chan A, Nanan R, Santner-Nanan B, et al. Early commitment of naive human CD4(+) T cells to the T follicular helper (T(FH)) cell lineage is induced by IL-12. Immunol Cell Biol (2009) 87(8):590–600. doi:10.1038/icb.2009.64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, et al. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol (2005) 175(4):2340–8. doi:10.4049/jimmunol.175.4.2340
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic differences between Th1 and Th17 cells and negative regulation of Th1 cell differentiation by IL-17. J Leukoc Biol (2007) 81(5):1258–68. doi:10.1189/jlb.1006610
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Ladanyi A, Somlai B, Gilde K, Fejos Z, Gaudi I, Timar J. T-cell activation marker expression on tumor-infiltrating lymphocytes as prognostic factor in cutaneous malignant melanoma. Clin Cancer Res (2004) 10(2):521–30. doi:10.1158/1078-0432.CCR-1161-03
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Petty JK, He K, Corless CL, Vetto JT, Weinberg AD. Survival in human colorectal cancer correlates with expression of the T-cell costimulatory molecule OX-40 (CD134). Am J Surg (2002) 183(5):512–8. doi:10.1016/S0002-9610(02)00831-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Wang HC, Klein JR. Multiple levels of activation of murine CD8(+) intraepithelial lymphocytes defined by OX40 (CD134) expression: effects on cell-mediated cytotoxicity, IFN-gamma, and IL-10 regulation. J Immunol (2001) 167(12):6717–23. doi:10.4049/jimmunol.167.12.6717
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Serghides L, Bukczynski J, Wen T, Wang C, Routy JP, Boulassel MR, et al. Evaluation of OX40 ligand as a costimulator of human antiviral memory CD8 T cell responses: comparison with B7.1 and 4-1BBL. J Immunol (2005) 175(10):6368–77. doi:10.4049/jimmunol.175.10.6368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol (2009) 29(3):187–201. doi:10.1615/CritRevImmunol.v29.i3.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol (2000) 165(6):3043–50. doi:10.4049/jimmunol.165.6.3043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Gramaglia I, Weinberg AD, Lemon M, Croft M. Ox-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol (1998) 161(12):6510–7.
23. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity (2001) 15(3):445–55. doi:10.1016/S1074-7613(01)00191-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Redmond WL, Gough MJ, Charbonneau B, Ratliff TL, Weinberg AD. Defects in the acquisition of CD8 T cell effector function after priming with tumor or soluble antigen can be overcome by the addition of an OX40 agonist. J Immunol (2007) 179(11):7244–53. doi:10.4049/jimmunol.179.11.7244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Hendriks J, Xiao Y, Rossen JW, van der Sluijs KF, Sugamura K, Ishii N, et al. During viral infection of the respiratory tract, CD27, 4-1BB, and OX40 collectively determine formation of CD8+ memory T cells and their capacity for secondary expansion. J Immunol (2005) 175(3):1665–76. doi:10.4049/jimmunol.175.3.1665
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Gough MJ, Crittenden MR, Sarff M, Pang P, Seung SK, Vetto JT, et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother (2010) 33(8):798–809. doi:10.1097/CJI.0b013e3181ee7095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med (2008) 205(4):825–39. doi:10.1084/jem.20071341
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Gough MJ, Ruby CE, Redmond WL, Dhungel B, Brown A, Weinberg AD. OX40 agonist therapy enhances CD8 infiltration and decreases immune suppression in the tumor. Cancer Res (2008) 68(13):5206–15. doi:10.1158/0008-5472.CAN-07-6484
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Redmond WL, Gough MJ, Weinberg AD. Ligation of the OX40 co-stimulatory receptor reverses self-Ag and tumor-induced CD8 T-cell anergy in vivo. Eur J Immunol (2009) 39(8):2184–94. doi:10.1002/eji.200939348
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Burocchi A, Pittoni P, Gorzanelli A, Colombo MP, Piconese S. Intratumor OX40 stimulation inhibits IRF1 expression and IL-10 production by Treg cells while enhancing CD40L expression by effector memory T cells. Eur J Immunol (2011) 41(12):3615–26. doi:10.1002/eji.201141700
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Song A, Tang X, Harms KM, Croft M. OX40 and Bcl-xL promote the persistence of CD8 T cells to recall tumor-associated antigen. J Immunol (2005) 175(6):3534–41. doi:10.4049/jimmunol.175.6.3534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Song J, So T, Cheng M, Tang X, Croft M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity (2005) 22(5):621–31. doi:10.1016/j.immuni.2005.03.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Song J, So T, Croft M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J Immunol (2008) 180(11):7240–8. doi:10.4049/jimmunol.180.11.7240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Prell RA, Evans DE, Thalhofer C, Shi T, Funatake C, Weinberg AD. OX40-mediated memory T cell generation is TNF receptor-associated factor 2 dependent. J Immunol (2003) 171(11):5997–6005. doi:10.4049/jimmunol.171.11.5997
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Croft M. The TNF family in T cell differentiation and function – unanswered questions and future directions. Semin Immunol (2014) 26(3):183–90. doi:10.1016/j.smim.2014.02.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Mbanwi AN, Watts TH. Costimulatory TNFR family members in control of viral infection: outstanding questions. Semin Immunol (2014) 26(3):210–9. doi:10.1016/j.smim.2014.05.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol (2013) 13(4):227–42. doi:10.1038/nri3405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Morris A, Vetto JT, Ramstad T, Funatake CJ, Choolun E, Entwisle C, et al. Induction of anti-mammary cancer immunity by engaging the OX-40 receptor in vivo. Breast Cancer Res Treat (2001) 67(1):71–80. doi:10.1023/A:1010649303056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Ali SA, Ahmad M, Lynam J, McLean CS, Entwisle C, Loudon P, et al. Anti-tumour therapeutic efficacy of OX40L in murine tumour model. Vaccine (2004) 22(27–28):3585–94. doi:10.1016/j.vaccine.2004.03.041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kjaergaard J, Tanaka J, Kim JA, Rothchild K, Weinberg A, Shu S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res (2000) 60(19):5514–21.
41. Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP. Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood (2005) 105(7):2845–51. doi:10.1182/blood-2004-07-2959
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, et al. OX40 costimulation turns off Foxp3+ Tregs. Blood (2007) 110(7):2501–10. doi:10.1182/blood-2007-01-070748
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Ruby CE, Yates MA, Hirschhorn-Cymerman D, Chlebeck P, Wolchok JD, Houghton AN, et al. Cutting Edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J Immunol (2009) 183(8):4853–7. doi:10.4049/jimmunol.0901112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Murata K, Nose M, Ndhlovu LC, Sato T, Sugamura K, Ishii N. Constitutive OX40/OX40 ligand interaction induces autoimmune-like diseases. J Immunol (2002) 169(8):4628–36. doi:10.4049/jimmunol.169.8.4628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Sugamura K, Ishii N, Weinberg AD. Therapeutic targeting of the effector T-cell co-stimulatory molecule OX40. Nat Rev Immunol (2004) 4(6):420–31. doi:10.1038/nri1371
46. Ito T, Wang YH, Duramad O, Hanabuchi S, Perng OA, Gilliet M, et al. OX40 ligand shuts down IL-10-producing regulatory T cells. Proc Natl Acad Sci U S A (2006) 103(35):13138–43. doi:10.1073/pnas.0603107103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol Cell Biol (2014) 92(6):475–80. doi:10.1038/icb.2014.26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Vetto JT, Lum S, Morris A, Sicotte M, Davis J, Lemon M, et al. Presence of the T-cell activation marker OX-40 on tumor infiltrating lymphocytes and draining lymph node cells from patients with melanoma and head and neck cancers. Am J Surg (1997) 174(3):258–65. doi:10.1016/S0002-9610(97)00139-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Curti BD, Kovacsovics-Bankowski M, Morris N, Walker E, Chisholm L, Floyd K, et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res (2013) 73(24):7189–98. doi:10.1158/0008-5472.CAN-12-4174
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Kavanagh B, O’Brien S, Lee D, Hou Y, Weinberg V, Rini B, et al. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood (2008) 112(4):1175–83. doi:10.1182/blood-2007-11-125435
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lee SJ, Myers L, Muralimohan G, Dai J, Qiao Y, Li Z, et al. 4-1BB and OX40 dual costimulation synergistically stimulate primary specific CD8 T cells for robust effector function. J Immunol (2004) 173(5):3002–12. doi:10.4049/jimmunol.173.5.3002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Gray JC, French RR, James S, Al-Shamkhani A, Johnson PW, Glennie MJ. Optimising anti-tumour CD8 T-cell responses using combinations of immunomodulatory antibodies. Eur J Immunol (2008) 38(9):2499–511. doi:10.1002/eji.200838208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Morales-Kastresana A, Sanmamed MF, Rodriguez I, Palazon A, Martinez-Forero I, Labiano S, et al. Combined immunostimulatory monoclonal antibodies extend survival in an aggressive transgenic hepatocellular carcinoma mouse model. Clin Cancer Res (2013) 19(22):6151–62. doi:10.1158/1078-0432.CCR-13-1189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Cuadros C, Dominguez AL, Lollini PL, Croft M, Mittler RS, Borgstrom P, et al. Vaccination with dendritic cells pulsed with apoptotic tumors in combination with anti-OX40 and anti-4-1BB monoclonal antibodies induces T cell-mediated protective immunity in Her-2/neu transgenic mice. Int J Cancer (2005) 116(6):934–43. doi:10.1002/ijc.21098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of co-stimulatory (OX40) and co-inhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust anti-tumor immunity. Cancer Immunol Res (2014) 2(2):142–53. doi:10.1158/2326-6066.CIR-13-0031-T
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G, et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest (2013) 123(6):2447–63. doi:10.1172/JCI64859
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Song A, Song J, Tang X, Croft M. Cooperation between CD4 and CD8 T cells for anti-tumor activity is enhanced by OX40 signals. Eur J Immunol (2007) 37(5):1224–32. doi:10.1002/eji.200636957
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Takeda K, Kojima Y, Uno T, Hayakawa Y, Teng MW, Yoshizawa H, et al. Combination therapy of established tumors by antibodies targeting immune activating and suppressing molecules. J Immunol (2010) 184(10):5493–501. doi:10.4049/jimmunol.0903033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Guo Z, Wang X, Cheng D, Xia Z, Luan M, Zhang S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS One (2014) 9(2):e89350. doi:10.1371/journal.pone.0089350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Gough MJ, Killeen N, Weinberg AD. Targeting macrophages in the tumour environment to enhance the efficacy of alphaOX40 therapy. Immunology (2012) 136(4):437–47. doi:10.1111/j.1365-2567.2012.03600.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Farazi M, Nguyen J, Goldufsky J, Linnane S, Lukaesko L, Weinberg AD, et al. Caloric restriction maintains OX40 agonist-mediated tumor immunity and CD4 T cell priming during aging. Cancer Immunol Immunother (2014) 63(6):615–26. doi:10.1007/s00262-014-1542-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Smith SE, Hoelzinger DB, Dominguez AL, Van Snick J, Lustgarten J. Signals through 4-1BB inhibit T regulatory cells by blocking IL-9 production enhancing antitumor responses. Cancer Immunol Immunother (2011) 60(12):1775–87. doi:10.1007/s00262-011-1075-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Houot R, Levy R. T-cell modulation combined with intratumoral CpG cures lymphoma in a mouse model without the need for chemotherapy. Blood (2009) 113(15):3546–52. doi:10.1182/blood-2008-07-170274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Qian J, Zheng Y, Zheng C, Wang L, Qin H, Hong S, et al. Active vaccination with Dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood (2012) 119(1):161–9. doi:10.1182/blood-2011-07-368472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Hirschhorn-Cymerman D, Rizzuto GA, Merghoub T, Cohen AD, Avogadri F, Lesokhin AM, et al. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. J Exp Med (2009) 206(5):1103–16. doi:10.1084/jem.20082205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, et al. Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med (2012) 209(11):2113–26. doi:10.1084/jem.20120532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Yang Y, Liu C, Peng W, Lizee G, Overwijk WW, Liu Y, et al. Antitumor T-cell responses contribute to the effects of dasatinib on c-KIT mutant murine mastocytoma and are potentiated by anti-OX40. Blood (2012) 120(23):4533–43. doi:10.1182/blood-2012-02-407163
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Ruby CE, Montler R, Zheng R, Shu S, Weinberg AD. IL-12 is required for anti-OX40-mediated CD4 T cell survival. J Immunol (2008) 180(4):2140–8. doi:10.4049/jimmunol.180.4.2140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Curti A, Parenza M, Colombo MP. Autologous and MHC class I-negative allogeneic tumor cells secreting IL-12 together cure disseminated A20 lymphoma. Blood (2003) 101(2):568–75. doi:10.1182/blood-2002-03-0991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Pan PY, Zang Y, Weber K, Meseck ML, Chen SH. OX40 ligation enhances primary and memory cytotoxic T lymphocyte responses in an immunotherapy for hepatic colon metastases. Mol Ther (2002) 6(4):528–36. doi:10.1006/mthe.2002.0699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Redmond WL, Triplett T, Floyd K, Weinberg AD. Dual anti-OX40/IL-2 therapy augments tumor immunotherapy via IL-2R-mediated regulation of OX40 expression. PLoS One (2012) 7(4):e34467. doi:10.1371/journal.pone.0034467
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Murata S, Ladle BH, Kim PS, Lutz ER, Wolpoe ME, Ivie SE, et al. OX40 costimulation synergizes with GM-CSF whole-cell vaccination to overcome established CD8+ T cell tolerance to an endogenous tumor antigen. J Immunol (2006) 176(2):974–83. doi:10.4049/jimmunol.176.2.974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Dollins CM, Nair S, Boczkowski D, Lee J, Layzer JM, Gilboa E, et al. Assembling OX40 aptamers on a molecular scaffold to create a receptor-activating aptamer. Chem Biol (2008) 15(7):675–82. doi:10.1016/j.chembiol.2008.05.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Fujiwara S, Nagai H, Shimoura N, Oniki S, Yoshimoto T, Nishigori C. Intratumoral CD4+ T lymphodepletion sensitizes poorly immunogenic melanomas to immunotherapy with an OX40 agonist. J Invest Dermatol (2014) 134(7):1884–92. doi:10.1038/jid.2014.42
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Ruby CE, Weinberg AD. OX40-enhanced tumor rejection and effector T cell differentiation decreases with age. J Immunol (2009) 182(3):1481–9. doi:10.4049/jimmunol.182.3.1481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Sadun RE, Hsu WE, Zhang N, Nien YC, Bergfeld SA, Sabzevari H, et al. Fc-mOX40L fusion protein produces complete remission and enhanced survival in 2 murine tumor models. J Immunother (2008) 31(3):235–45. doi:10.1097/CJI.0b013e31816a88e0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Lustgarten J, Dominguez AL, Thoman M. Aged mice develop protective antitumor immune responses with appropriate costimulation. J Immunol (2004) 173(7):4510–5. doi:10.4049/jimmunol.173.7.4510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Taraban VY, Rowley TF, O’Brien L, Chan HT, Haswell LE, Green MH, et al. Expression and costimulatory effects of the TNF receptor superfamily members CD134 (OX40) and CD137 (4-1BB), and their role in the generation of anti-tumor immune responses. Eur J Immunol (2002) 32(12):3617–27. doi:10.1002/1521-4141(200212)32:12<3617::AID-IMMU3617>3.0.CO;2-M
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Weinberg AD, Rivera MM, Prell R, Morris A, Ramstad T, Vetto JT, et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. J Immunol (2000) 164(4):2160–9. doi:10.4049/jimmunol.164.4.2160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Yokouchi H, Yamazaki K, Chamoto K, Kikuchi E, Shinagawa N, Oizumi S, et al. Anti-OX40 monoclonal antibody therapy in combination with radiotherapy results in therapeutic antitumor immunity to murine lung cancer. Cancer Sci (2008) 99(2):361–7. doi:10.1111/j.1349-7006.2007.00664.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Kjaergaard J, Wang LX, Kuriyama H, Shu S, Plautz GE. Active immunotherapy for advanced intracranial murine tumors by using dendritic cell-tumor cell fusion vaccines. J Neurosurg (2005) 103(1):156–64. doi:10.3171/jns.2005.103.1.0156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Murphy KA, Erickson JR, Johnson CS, Seiler CE, Bedi J, Hu P, et al. CD8+ T cell-independent tumor regression induced by Fc-OX40L and therapeutic vaccination in a mouse model of glioma. J Immunol (2014) 192(1):224–33. doi:10.4049/jimmunol.1301633
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Murphy KA, Lechner MG, Popescu FE, Bedi J, Decker SA, Hu P, et al. An in vivo immunotherapy screen of costimulatory molecules identifies Fc-OX40L as a potent reagent for the treatment of established murine gliomas. Clin Cancer Res (2012) 18(17):4657–68. doi:10.1158/1078-0432.CCR-12-0990
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Pardee AD, McCurry D, Alber S, Hu P, Epstein AL, Storkus WJ. A therapeutic OX40 agonist dynamically alters dendritic, endothelial, and T cell subsets within the established tumor microenvironment. Cancer Res (2010) 70(22):9041–52. doi:10.1158/0008-5472.CAN-10-1369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity (1995) 3(5):541–7. doi:10.1016/1074-7613(95)90125-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science (1995) 270(5238):985–8. doi:10.1126/science.270.5238.985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Pedicord VA, Montalvo W, Leiner IM, Allison JP. Single dose of anti-CTLA-4 enhances CD8+ T-cell memory formation, function, and maintenance. Proc Natl Acad Sci U S A (2011) 108(1):266–71. doi:10.1073/pnas.1016791108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med (2009) 206(8):1717–25. doi:10.1084/jem.20082492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest (2006) 116(7):1935–45. doi:10.1172/JCI27745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Shrikant P, Khoruts A, Mescher MF. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity (1999) 11(4):483–93. doi:10.1016/S1074-7613(00)80123-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Sutmuller RP, van Duivenvoorde LM, van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med (2001) 194(6):823–32. doi:10.1084/jem.194.6.823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A (1997) 94(15):8099–103. doi:10.1073/pnas.94.15.8099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (1996) 271(5256):1734–6. doi:10.1126/science.271.5256.1734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Pentcheva-Hoang T, Corse E, Allison JP. Negative regulators of T-cell activation: potential targets for therapeutic intervention in cancer, autoimmune disease, and persistent infections. Immunol Rev (2009) 229(1):67–87. doi:10.1111/j.1600-065X.2009.00763.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Sotomayor EM, Borrello I, Tubb E, Allison JP, Levitsky HI. In vivo blockade of CTLA-4 enhances the priming of responsive T cells but fails to prevent the induction of tumor antigen-specific tolerance. Proc Natl Acad Sci U S A (1999) 96(20):11476–81. doi:10.1073/pnas.96.20.11476
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Yang YF, Zou JP, Mu J, Wijesuriya R, Ono S, Walunas T, et al. Enhanced induction of antitumor T-cell responses by cytotoxic T lymphocyte-associated molecule-4 blockade: the effect is manifested only at the restricted tumor-bearing stages. Cancer Res (1997) 57(18):4036–41.
97. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
98. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, Phase 2, dose-ranging study. Lancet Oncol (2010) 11(2):155–64. doi:10.1016/S1470-2045(09)70334-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. O’Day SJ, Maio M, Chiarion-Sileni V, Gajewski TF, Pehamberger H, Bondarenko IN, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm Phase II study. Ann Oncol (2010) 21(8):1712–7. doi:10.1093/annonc/mdq013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Milner JD, Fazilleau N, McHeyzer-Williams M, Paul W. Cutting edge: lack of high affinity competition for peptide in polyclonal CD4+ responses unmasks IL-4 production. J Immunol (2010) 184(12):6569–73. doi:10.4049/jimmunol.1000674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Tao X, Grant C, Constant S, Bottomly K. Induction of IL-4-producing CD4+ T cells by antigenic peptides altered for TCR binding. J Immunol (1997) 158(9):4237–44.
102. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J (1992) 11(11):3887–95.
103. Flies DB, Sandler BJ, Sznol M, Chen L. Blockade of the B7-H1/PD-1 pathway for cancer immunotherapy. Yale J Biol Med (2011) 84(4):409–21.
104. Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature (2011) 480(7378):480–9. doi:10.1038/nature10673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Riella LV, Paterson AM, Sharpe AH, Chandraker A. Role of the PD-1 pathway in the immune response. Am J Transplant (2012) 12(10):2575–87. doi:10.1111/j.1600-6143.2012.04224.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Zhou G, Levitsky H. Towards curative cancer immunotherapy: overcoming posttherapy tumor escape. Clin Dev Immunol (2012) 2012:124187. doi:10.1155/2012/124187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi:10.1146/annurev.immunol.26.021607.090331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2(3):261–8. doi:10.1038/85330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Frigola X, Inman BA, Lohse CM, Krco CJ, Cheville JC, Thompson RH, et al. Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin Cancer Res (2011) 17(7):1915–23. doi:10.1158/1078-0432.CCR-10-0250
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A (2002) 99(19):12293–7. doi:10.1073/pnas.192461099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A (2003) 100(9):5336–41. doi:10.1073/pnas.0931259100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol (2012) 24(2):207–12. doi:10.1016/j.coi.2011.12.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, et al. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation (2002) 9(2):133–45. doi:10.1080/713774061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J (2014) 20(4):265–71. doi:10.1097/PPO.0000000000000059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Wei F, Zhong S, Ma Z, Kong H, Medvec A, Ahmed R, et al. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc Natl Acad Sci U S A (2013) 110(27):E2480–9. doi:10.1073/pnas.1305394110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Gianchecchi E, Delfino DV, Fierabracci A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun Rev (2013) 12(11):1091–100. doi:10.1016/j.autrev.2013.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qiao H, Fahl SP, et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood (2012) 120(24):4772–82. doi:10.1182/blood-2012-04-427013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. West EE, Jin HT, Rasheed AU, Penaloza-Macmaster P, Ha SJ, Tan WG, et al. PD-L1 blockade synergizes with IL-2 therapy in reinvigorating exhausted T cells. J Clin Invest (2013) 123(6):2604–15. doi:10.1172/JCI67008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res (2005) 65(3):1089–96.
120. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med (2003) 9(5):562–7. doi:10.1038/nm863
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi:10.1038/nm0902-1039c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol (2005) 17(2):133–44. doi:10.1093/intimm/dxh194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Li B, VanRoey M, Wang C, Chen TH, Korman A, Jooss K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor – secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin Cancer Res (2009) 15(5):1623–34. doi:10.1158/1078-0432.CCR-08-1825
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Mkrtichyan M, Najjar YG, Raulfs EC, Abdalla MY, Samara R, Rotem-Yehudar R, et al. Anti-PD-1 synergizes with cyclophosphamide to induce potent anti-tumor vaccine effects through novel mechanisms. Eur J Immunol (2011) 41(10):2977–86. doi:10.1002/eji.201141639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Zhang Y, Huang S, Gong D, Qin Y, Shen Q. Programmed death-1 upregulation is correlated with dysfunction of tumor-infiltrating CD8+ T lymphocytes in human non-small cell lung cancer. Cell Mol Immunol (2010) 7(5):389–95. doi:10.1038/cmi.2010.28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Blank C, Kuball J, Voelkl S, Wiendl H, Becker B, Walter B, et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer (2006) 119(2):317–27. doi:10.1002/ijc.21775
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Okudaira K, Hokari R, Tsuzuki Y, Okada Y, Komoto S, Watanabe C, et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int J Oncol (2009) 35(4):741–9. doi:10.3892/ijo_00000387
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Rosenblatt J, Glotzbecker B, Mills H, Vasir B, Tzachanis D, Levine JD, et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J Immunother (2011) 34(5):409–18. doi:10.1097/CJI.0b013e31821ca6ce
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res (2003) 63(19):6501–5.
130. Wang W, Lau R, Yu D, Zhu W, Korman A, Weber J. PD1 blockade reverses the suppression of melanoma antigen-specific CTL by CD4+ CD25(Hi) regulatory T cells. Int Immunol (2009) 21(9):1065–77. doi:10.1093/intimm/dxp072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Zhou Q, Xiao H, Liu Y, Peng Y, Hong Y, Yagita H, et al. Blockade of programmed death-1 pathway rescues the effector function of tumor-infiltrating T cells and enhances the antitumor efficacy of lentivector immunization. J Immunol (2010) 185(9):5082–92. doi:10.4049/jimmunol.1001821
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Mangsbo SM, Sandin LC, Anger K, Korman AJ, Loskog A, Totterman TH. Enhanced tumor eradication by combining CTLA-4 or PD-1 blockade with CpG therapy. J Immunother (2010) 33(3):225–35. doi:10.1097/CJI.0b013e3181c01fcb
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Dolan DE, Gupta S. PD-1 pathway inhibitors: changing the landscape of cancer immunotherapy. Cancer Control (2014) 21(3):231–7.
134. Naidoo J, Page DB, Wolchok JD. Immune modulation for cancer therapy. Br J Cancer (2014) 111(12):2214–9. doi:10.1038/bjc.2014.348
135. Weber J. Immune checkpoint proteins: a new therapeutic paradigm for cancer – preclinical background: CTLA-4 and PD-1 blockade. Semin Oncol (2010) 37(5):430–9. doi:10.1053/j.seminoncol.2010.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res (2012) 19(2):462–8. doi:10.1158/1078-0432.CCR-12-2625
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a Phase 1 trial. Lancet (2014) 384(9948):1109–17. doi:10.1016/S0140-6736(14)60958-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res (2008) 14(10):3044–51. doi:10.1158/1078-0432.CCR-07-4079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Callahan MK, Wolchok JD. At the bedside: CTLA-4- and PD-1-blocking antibodies in cancer immunotherapy. J Leukoc Biol (2013) 94(1):41–53. doi:10.1189/jlb.1212631
141. Harvey RD. Immunologic and clinical effects of targeting PD-1 in lung cancer. Clin Pharmacol Ther (2014) 96(2):214–23. doi:10.1038/clpt.2014.74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res (2013) 19(5):1021–34. doi:10.1158/1078-0432.CCR-12-2063
143. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res (2013) 73(23):6900–12. doi:10.1158/0008-5472.CAN-13-1550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi:10.1038/nrc3239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res (2011) 72(4):917–27. doi:10.1158/0008-5472.CAN-11-1620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Verbrugge I, Galli M, Smyth MJ, Johnstone RW, Haynes NM. Enhancing the antitumor effects of radiotherapy with combinations of immunostimulatory antibodies. Oncoimmunology (2012) 1(9):1629–31. doi:10.4161/onci.21652
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, et al. Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J Clin Invest (2013) 123(3):1371–81. doi:10.1172/JCI66236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Turnis ME, Korman AJ, Drake CG, Vignali DA. Combinatorial immunotherapy: PD-1 may not be LAG-ing behind any more. Oncoimmunology (2012) 1(7):1172–4. doi:10.4161/onci.20593
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Perez-Gracia JL, Labiano S, Rodriguez-Ruiz ME, Sanmamed MF, Melero I. Orchestrating immune check-point blockade for cancer immunotherapy in combinations. Curr Opin Immunol (2014) 27:89–97. doi:10.1016/j.coi.2014.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol (2010) 10(8):580–93. doi:10.1038/nri2817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Abken H, Hombach A, Heuser C, Kronfeld K, Seliger B. Tuning tumor-specific T-cell activation: a matter of costimulation? Trends Immunol (2002) 23(5):240–5. doi:10.1016/S1471-4906(02)02180-4
153. Melero I, Grimaldi AM, Perez-Gracia JL, Ascierto PA. Clinical development of immunostimulatory monoclonal antibodies and opportunities for combination. Clin Cancer Res (2013) 19(5):997–1008. doi:10.1158/1078-0432.CCR-12-2214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Di Giacomo AM, Biagioli M, Maio M. The emerging toxicity profiles of anti-CTLA-4 antibodies across clinical indications. Semin Oncol (2010) 37(5):499–507. doi:10.1053/j.seminoncol.2010.09.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol (2012) 30(21):2691–7. doi:10.1200/JCO.2012.41.6750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med (1990) 171(5):1393–405. doi:10.1084/jem.171.5.1393
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol (2010) 344:269–78. doi:10.1007/82_2010_114
159. Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunol Lett (2012) 150(1–2):116–22. doi:10.1016/j.imlet.2012.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol (2009) 182(11):6659–69. doi:10.4049/jimmunol.0804211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest (2007) 117(11):3383–92. doi:10.1172/JCI31184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J (2007) 26(2):494–504. doi:10.1038/sj.emboj.7601520
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Huard B, Prigent P, Pages F, Bruniquel D, Triebel F. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur J Immunol (1996) 26(5):1180–6. doi:10.1002/eji.1830260533
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A (2010) 107(17):7875–80. doi:10.1073/pnas.1003345107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21(4):503–13. doi:10.1016/j.immuni.2004.08.010
166. Thielens A, Vivier E, Romagne F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. Curr Opin Immunol (2012) 24(2):239–45. doi:10.1016/j.coi.2012.01.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Campbell KS, Purdy AK. Structure/function of human killer cell immunoglobulin-like receptors: lessons from polymorphisms, evolution, crystal structures and mutations. Immunology (2011) 132(3):315–25. doi:10.1111/j.1365-2567.2010.03398.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. van Bergen J, Trowsdale J. Ligand specificity of killer cell immunoglobulin-like receptors: a brief history of KIR. Front Immunol (2012) 3:394. doi:10.3389/fimmu.2012.00394
169. Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, Chanuc F, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood (2014) 123(5):678–86. doi:10.1182/blood-2013-08-519199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Romagne F, Andre P, Spee P, Zahn S, Anfossi N, Gauthier L, et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood (2009) 114(13):2667–77. doi:10.1182/blood-2009-02-206532
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A, et al. A Phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood (2012) 120(22):4317–23. doi:10.1182/blood-2012-06-437558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Mentlik James A, Cohen AD, Campbell KS. Combination immune therapies to enhance anti-tumor responses by NK cells. Front Immunol (2014) 4:481. doi:10.3389/fimmu.2013.00481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
173. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res (2011) 71(10):3540–51. doi:10.1158/0008-5472.CAN-11-0096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Leitner J, Rieger A, Pickl WF, Zlabinger G, Grabmeier-Pfistershammer K, Steinberger P. TIM-3 does not act as a receptor for galectin-9. PLoS Pathog (2013) 9(3):e1003253. doi:10.1371/journal.ppat.1003253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol (2005) 6(12):1245–52. doi:10.1038/ni1271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med (2010) 207(10):2187–94. doi:10.1084/jem.20100643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Baghdadi M, Nagao H, Yoshiyama H, Akiba H, Yagita H, Dosaka-Akita H, et al. Combined blockade of TIM-3 and TIM-4 augments cancer vaccine efficacy against established melanomas. Cancer Immunol Immunother (2012) 62(4):629–37. doi:10.1007/s00262-012-1371-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol (2005) 6(1):90–8. doi:10.1038/ni1144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Derre L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, et al. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest (2009) 120(1):157–67. doi:10.1172/JCI40070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol (2003) 4(7):670–9. doi:10.1038/ni944
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Paulos CM, June CH. Putting the brakes on BTLA in T cell-mediated cancer immunotherapy. J Clin Invest (2010) 120(1):76–80. doi:10.1172/JCI41811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Cai G, Freeman GJ. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev (2009) 229(1):244–58. doi:10.1111/j.1600-065X.2009.00783.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res (2012) 72(4):887–96. doi:10.1158/0008-5472.CAN-11-2637
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Haymaker C, Wu R, Bernatchez C, Radvanyi L. PD-1 and BTLA and CD8(+) T-cell “exhaustion” in cancer: “exercising” an alternative viewpoint. Oncoimmunology (2012) 1(5):735–8. doi:10.4161/onci.20823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol (2013) 25(2):214–21. doi:10.1016/j.coi.2012.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Lasaro MO, Sazanovich M, Giles-Davis W, Mrass P, Bunte RM, Sewell DA, et al. Active immunotherapy combined with blockade of a coinhibitory pathway achieves regression of large tumor masses in cancer-prone mice. Mol Ther (2011) 19(9):1727–36. doi:10.1038/mt.2011.88
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Schaer DA, Hirschhorn-Cymerman D, Wolchok JD. Targeting tumor-necrosis factor receptor pathways for tumor immunotherapy. J Immunother Cancer (2014) 2:7. doi:10.1186/2051-1426-2-7
188. Kjaergaard J, Peng L, Cohen PA, Drazba JA, Weinberg AD, Shu S. Augmentation versus inhibition: effects of conjunctional OX-40 receptor monoclonal antibody and IL-2 treatment on adoptive immunotherapy of advanced tumor. J Immunol (2001) 167(11):6669–77. doi:10.4049/jimmunol.167.11.6669
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer (2012) 12(4):237–51. doi:10.1038/nrc3237
190. Jensen SM, Maston LD, Gough MJ, Ruby CE, Redmond WL, Crittenden M, et al. Signaling through OX40 enhances antitumor immunity. Semin Oncol (2010) 37(5):524–32. doi:10.1053/j.seminoncol.2010.09.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Redmond WL, Weinberg AD. Targeting OX40 and OX40L for the treatment of autoimmunity and cancer. Crit Rev Immunol (2007) 27(5):415–36. doi:10.1615/CritRevImmunol.v27.i5.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res (2009) 69(6):2514–22. doi:10.1158/0008-5472.CAN-08-4709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A (2010) 107(9):4275–80. doi:10.1073/pnas.0915174107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
194. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi:10.1128/MCB.25.21.9543-9553.2005
195. Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL-2 to enhance antiviral T-cell responses in vivo. Nat Med (2003) 9(5):540–7. doi:10.1038/nm866
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol (2004) 172(7):3983–8. doi:10.4049/jimmunol.172.7.3983
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
197. Waldmann TA, Dubois S, Tagaya Y. Contrasting roles of IL-2 and IL-15 in the life and death of lymphocytes: implications for immunotherapy. Immunity (2001) 14(2):105–10. doi:10.1016/S1074-7613(01)00093-0
198. Hofer T, Krichevsky O, Altan-Bonnet G. Competition for IL-2 between regulatory and effector T cells to chisel immune responses. Front Immunol (2012) 3:268. doi:10.3389/fimmu.2012.00268
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. McNamara MJ, Kasiewicz MJ, Linch SN, Dubay C, Redmond WL. Common gamma chain (γc) cytokines differentially potentiate TNFR family signaling in antigen-activated CD8 T cells. J Immunother Cancer (2014) 2(1):1–15. doi:10.1186/s40425-014-0028-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. de la Cruz-Merino L, Illescas-Vacas A, Grueso-Lopez A, Barco-Sanchez A, Miguez-Sanchez C. Radiation for awakening the dormant immune system, a promising challenge to be explored. Front Immunol (2014) 5:102. doi:10.3389/fimmu.2014.00102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Burnette B, Fu YX, Weichselbaum RR. The confluence of radiotherapy and immunotherapy. Front Oncol (2012) 2:143. doi:10.3389/fonc.2012.00143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: OX40, CTLA-4, PD-1, co-stimulation, immunotherapy, cancer
Citation: Linch SN, McNamara MJ and Redmond WL (2015) OX40 agonists and combination immunotherapy: putting the pedal to the metal. Front. Oncol. 5:34. doi: 10.3389/fonc.2015.00034
Received: 12 November 2014; Accepted: 30 January 2015;
Published online: 16 February 2015.
Edited by:
Michael Andrew Curran, University of Texas MD Anderson Cancer Center, USAReviewed by:
Sandra Demaria, New York University School of Medicine, USAChrystal M. Paulos, Hollings Cancer Center, USA
Copyright: © 2015 Linch, McNamara and Redmond. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William L. Redmond, Robert W. Franz Cancer Research Center, Earle A. Chiles Research Institute, Providence Portland Medical Center, 4805 NE Glisan Street, 2N35, Portland, OR 97213, USA e-mail:d2lsbGlhbS5yZWRtb25kQHByb3ZpZGVuY2Uub3Jn
†Stefanie N. Linch and Michael J. McNamara have contributed equally to this work.