 Ansgar Brüning
Ansgar Brüning Julia Jückstock
Julia Jückstock- Molecular Biology Laboratory, Ludwig-Maximilians-University, Munich, Germany
The application of cytostatic drugs targeting the high proliferation rates of cancer cells is currently the most commonly used treatment option in cancer chemotherapy. However, severe side effects and resistance mechanisms may occur as a result of such treatment, possibly limiting the therapeutic efficacy of these agents. In recent years, several therapeutic strategies have been developed that aim at targeting not the genomic integrity and replication machinery of cancer cells but instead their protein homeostasis. During malignant transformation, the cancer cell proteome develops vast aberrations in the expression of mutated proteins, oncoproteins, drug- and apoptosis-resistance proteins, etc. A complex network of protein quality-control mechanisms, including chaperoning by heat shock proteins (HSPs), not only is essential for maintaining the extravagant proteomic lifestyle of cancer cells but also represents an ideal cancer-specific target to be tackled. Furthermore, the high rate of protein synthesis and turnover in certain types of cancer cells can be specifically directed by interfering with the proteasomal and autophagosomal protein recycling and degradation machinery, as evidenced by the clinical application of proteasome inhibitors. Since proteins with loss of their native conformation are prone to unspecific aggregations and have proved to be detrimental to normal cellular function, specific induction of misfolded proteins by HSP inhibitors, proteasome inhibitors, hyperthermia, or inducers of endoplasmic reticulum stress represents a new method of cancer cell killing exploitable for therapeutic purposes. This review describes drugs – approved, repurposed, or under investigation – that can be used to accumulate misfolded proteins in cancer cells, and particularly focuses on the molecular aspects that lead to the cytotoxicity of misfolded proteins in cancer cells.
Introduction: How Do Proteins Fold and What Makes Misfolded Proteins Dangerous?
For an understanding of misfolded proteins, it is necessary to understand how cellular proteins attain and then further maintain their native conformation and how mature proteins and unfolded proteins are generated and converted into each other.
The principles and mechanisms of protein folding were one of the major research topics and achievements of biochemical research in the last century. For decades, Anfinsen’s model, which explained protein structure by thermodynamic principles applying to the polypeptide’s inherent amino acid sequence (1), was to be found in the introductory sections of all textbooks in protein biochemistry. According to Anfinsen’s thermodynamic hypothesis, the structure with the lowest conformational Gibbs free energy was finally taken by each single polypeptide due to a thermodynamic and stereochemical selection for side chain relations that form most stable and effective enzymes or structural proteins (1). Beyond this individual selection for the energetically most optimized conformation, evolution also selected for amino acid sequences that energetically allowed the smoothest and most “frustration-free” folding processes via a thermodynamic “folding funnel” (1–3).
Whereas Anfinsen’s model preferred the side chain elements as preferential organizing structures, recent hypotheses have inversely proposed the backbone hydrogen bonds as the driving force behind protein folding (4). According to the former theory, the finally folded protein was assumed to attain a single defined structure and shape (1, 4), and the unfolded conditions were described as being represented by a structureless statistical coil with nearly indefinite conformations – a so-called “featureless energy landscape” (4). The latter model assumes that a protein selects during its folding process from a limited repertoire of stable scaffolds of backbone hydrogen bond-satisfied α-helices and β-strands (4). This also implies that unfolded proteins are not structureless, shoelace-like linear amino acid alignments as often depicted in cartoons for graphical reasons, but actually, at least in part, retain discrete and stable scaffolds.
Once the protein has attained its final conformation, the problem of stabilizing this structure arises. Hydrophobic interactions that press non-polar side chains into the center of the protein are assumed to be a major force in protein stabilization (5, 6). At the protein surface, polar interactions, mainly by hydrogen bonds of polar side chains and backbone structure, are assumed to be of similar importance (6). Salt bridges and covalent disulfide bonds were identified as further forces supporting the stability of proteins (6). Accordingly, all conditions that interfere with these stabilizing forces, including extreme temperature, salt concentrations, and redox conditions, may lead to protein misfolding.
Another aspect that must be taken into account when studying protein folding relates to the very different conditions found in viable cells when compared to test tube conditions. Considering the life-cycle of a protein, each protein begins as a growing polypeptide chain protruding from the ribosomal exit tunnel and with several of its future interacting amino acid binding partners not even yet attached to the growing chain of the nascent polymer. In these ribosomal exit tunnels, first molecular interactions and helical structures are formed, and evidence exists to support the notion that the speed of translation is regulated by slow translating codon sequences just to optimize these first folding processes (7). After leaving the ribosomal tunnel, nascent polypeptides are also directly welcomed by chaperoning protein complexes, which facilitate and further guide the folding process of newly synthesized proteins (8). It is believed that a high percentage of nascent proteins are subject to immediate degradation due to early folding errors (9). Since many nascent proteins are synthesized in parallel at polysomes, the temporal and spatial proximity of unfolded peptides brings the additional risk of protein aggregation (10). Moreover, as mentioned above, even incomplete folding intermediates and partially folded states may form energetically but not physiologically active metastable structures (11, 12). An immediate, perinatal guidance and chaperoning of newborn proteins is therefore essential to creating functional, integrative proteins and to avoiding misfolded, function-less polypeptides with potentially cytotoxic features.
Since protein structure and function are coupled, misfolded proteins are, at first, loss-of-function proteins that might reduce cell viability, in particular when generated in larger quantities. A more dangerous feature of misfolded proteins, however, lies in their strong tendency toward abnormal protein–protein interactions or aggregations, which is reflected by the involvement of misfolded proteins and their aggregates in several amyloidotic diseases, including neurodegenerative syndromes such as Alzheimer’s disease and Parkinson’s disease (13, 14). The fact that several of these intracellular and extracellular protein aggregates contain β-sheet-like structures and form filamentous structures also supports the notion that misfolded proteins are not necessarily structureless protein coils or unspecific aggregates, at least when they are formed by homogenous proteins as in the case of several neurodegenerative diseases (13). Paradoxically, these larger aggregates appear to reflect a cell protective mechanism so as to sequester or segregate smaller, but highly reactive, nucleation cores of condensing protein aggregates (13).
Unspecific hydrophobic interactions, in particular, have been held responsible for protein aggregations that form when terminally folded proteins lose their native conformation and expose buried hydrophobic side chains on their surface (15, 16). These hydrophobic interactions are also believed to be the most problematic issues with newly synthesized polypeptides on single ribosomes or polysomes (12). Once exposed to the surface, the hydrophobic structures will quickly find possible interaction partners. The intracellular milieu can be regarded as a “crowded environment” (17), fully packed with proteins in close contact and near to their solubility limit (8, 12). Thus, misfolded proteins not only aggregate among each other but may also attach to normal native proteins and inhibit their function and activity. Since such misfolding effects and interactions can also include nuclear DNA replication and repair enzymes (18), misfolded proteins may not only exert proteotoxic but also genotoxic effects, thereby endangering the entire cellular “interactome” (19) by interfering both with the integrity of the proteome (proteostasis) and the genome. Therefore, a misfolded protein is not simply a loss-of-function protein but also a promiscuous little villain that might act like a free radical, exerting uncontrolled danger to the cell.
The way in which cells deal with misfolded proteins strongly depends on the nature, strength, length, and location of the damage induced by the various insults. Management of misfolded proteins can be achieved by heat shock protein (HSP)-mediated protein renaturation (repair); proteasomal, lysosomal, or autophagosomal degradation (recycling); intracellular disposal (aggregation); or – in its last consequence if overwhelmed – by programed cell death (despair). In the following paragraphs, the cellular management of misfolded proteins is described and therapeutic options to induce misfolded proteins in cancer cells are presented.
Hsp90 and Hsp90 Inhibitors
The best-known and evolutionarily most-conserved mechanism to protect against protein misfolding is the binding and refolding process mediated by so-called heat shock proteins (HSPs). HSPs recognize unfolded or misfolded proteins and facilitate their restructuring in either an ATP-dependent (large HSPs) or energy-independent manner (low weight HSPs). HSP of 90 kDa (hsp90) is a constitutively expressed HSP and is regarded as the most common and abundantly expressed HSP in eukaryotic cells (20, 21). Although commonly referred to as hsp90, it consists of a variety of isoforms that are encoding for cytosolic (hsp90α1, α2, β), mitochondrial (TRAP1), or endoplasmic reticulum (ER)-resident (GRP94) forms. Its primary function is less that of a stress response protein and more to bind to a certain group of client proteins unable to maintain a stable configuration without being assisted by hsp90 (20, 22, 23). Steroid hormone receptors (estrogen receptor, glucocorticoid receptor), cell cycle regulatory proteins (CDK4, cyclin D, polo-like kinase), and growth factor receptors and their downstream targets (epidermal growth factor receptor 1, HER2, AKT) are among the best-studied client proteins of hsp90 (20–22). Also, several cancer-specific mutations generating otherwise instable oncoproteins, such as mutant p53 or bcr-abl, rely on hsp90 chaperoning to keep them in a soluble form, thereby facilitating the extravagant but vulnerable “malignant lifestyle” of hsp90-addicted cancer cells (21, 24). Accordingly, hsp90 has been assumed to be a prominent target, in particular for hormone-responsive and growth factor receptor amplification-dependent cancer types.
The microbial antibiotics geldanamycin and radicicol are the prototypes of hsp90 inhibitors. Based on intolerable toxicity, these molecules had to be chemically modified for application in humans, and most of the ongoing clinical studies with hsp90 inhibitors are aimed at identifying semi-synthetic derivatives of these lead compounds with an acceptable risk profile. Unfortunately, most recent studies using geldanamycin derivatives have provided disappointing results because of toxicities and insufficient efficacy (22, 25–27). Studies with radicicol (resorcinol) derivatives, in particular with ganetespib, appear to be more promising because of fewer adverse effects (22, 25–27). Liver and ocular (retinal) toxicities have been described as main adverse effects of hsp90 inhibition, and appeared to be experienced less with ganetespib than with most of the first generation hsp90 inhibitors (28).
Since both geldanamycin and radicicol target the highly conserved and unique ATP-binding domain of hsp90, new synthetic inhibitors have also been generated by rational drug design (22, 25–27). However, none of the various natural or synthetic hsp90 inhibitors under investigation have yet provided convincing clinical data, and future studies will show whether hsp90 can eventually be added to the list of effective cancer targets.
Hsp70, Hsp40, Hsp27, and HSF1
Hsp90 is assisted by several other HSPs and non-chaperoning co-factors, finally forming a large protein complex that recruits and releases client proteins in an energy-dependent manner (21, 22, 29). Client proteins for hsp90 are first bound to hsp70, which transfers the prospective client to hsp90 through the mediating help of an hsp70–hsp90 organizing protein (HOP). Binding of potential hsp90 client proteins to hsp70 is facilitated by its co-chaperone hsp40 (23, 30). Exposed hydrophobic amino acids, the typical feature of misfolded proteins, have been described as the main recognition signal for hsp70 proteins (15, 16, 31). Hsp70 proteins are not only supporter proteins for hsp90 but also represent a large chaperone family capable of acting independently of hsp90 and that can be found in all cellular compartments, including cytosol and nucleus (hsp70, hsp72, hsc70), mitochondria (GRP75 = mortalin), and the ER (GRP78 = BiP). Hsp70 chaperones may act on misfolded or nascent proteins either as “holders” or “folders” (31), which means that they prevent protein aggregation either by sheltering these aggregation-prone protein intermediates or by allowing these proteins to fold/refold into their native form in an assisted mechanism within a protected environment (31). Hsc70 (HSPA8) is a constitutively expressed major hsp70 isoform that is an essential factor for normal protein homeostasis even in unstressed cells (16). Misfolded proteins can also be destined by hsp70 proteins for their ultimate degradation. Proteins that expose KFERQ amino acid motifs on their surface during their unfolding process are preferentially bound by hsc70 and can be directed to lysosomes in a process called chaperone-mediated autophagy (CMA) (32, 33). In another mechanism of targeted protein degradation, interaction of hsc70 with the E3 ubiquitin ligase CHIP (carboxyl terminus of Hsc70-interacting protein) leads to ubiquitination of misfolded proteins and thus their destination of the ubiquitin-proteasome protein degradation pathway (34, 35). Since hsc70 is essential for normal protein homeostasis and its knock-out is lethal in mice (16, 36), hsc70 inhibition might not be an optimal target for cancer-specific induction of misfolded proteins. This contrasts with the inducible forms of hsp70 such as hsp72 (HSPA1), which are upregulated in a cell stress-specific manner and are often found to be constitutively overexpressed in cancer tissues (16, 36). Transcriptional activation of these inducible HSPs is mediated by the heat shock factor 1 (HSF1), which also regulates expression of hsp40 and the small HSP hsp27 by sharing a common promoter consensus sequence (heat shock response element) for HSF1 binding (37). HSF1 was also found to be constitutively activated in cancer tissues, modulating several cell cycle- and apoptosis-related pathways via its target genes (38–40). HSF1 itself is kept inactive in the cytosol by binding to hsp90, and the recruitment of hsp90 to misfolded proteins is considered a main activation mechanism to release monomeric HSF1 for its subsequent trimerization, post-translational activation, and nuclear translocation (24, 41). Also, since hsp90 inhibition causes hsp70 induction by HSF1 activation as a compensatory feed-back mechanism (24), combined inhibition of hsp90 and hsp70, or of hsp90 and HSF1 might be a more effective therapeutic approach for cancer treatment than single HSP targeting alone.
Indeed, several small-molecule inhibitors and aptamers for hsp70, hsp40, and hsp27 have been designed (16, 42–44), but most of them remain in pre-clinical development, or are either not applicable in humans or associated with intolerable side effects (16, 42–44). Notably, the natural bioflavonoid quercetin was shown to inhibit phosphorylation and transcriptional activity of the heat shock transcription factor HSF1, thus reducing HSP expression at its most basal level (45–48). This HSP and HSF1 inhibition may also contribute to the observed cancer-preventing effects of a flavonoid-rich diet, which includes fruits and vegetables. However, due to their low bioavailability, the concentrations of flavonoids needed to induce direct cytotoxic effects in cancer cells for (chemo-)therapeutic reasons are obviously not achievable in humans, even when applied as nutritional supplements (49). More effective and clinically more easily applicable inhibitors of HSF1 are therefore urgently sought. Promising HSF1 targeting strategies are currently under development, although are apparently not yet suited for clinical applications (24, 50, 51).
Protein Ubiquitination and Proteasomal Degradation
Ubiquitin is a 76 amino acid polypeptide that can covalently be attached via its carboxy-terminus to free (lysyl) amino groups of proteins. Ubiquitination of proteins generates a cellular recognition motif that is involved in various functions ranging from transcription factor and protein kinase activation to DNA repair and protein degradation – depending on the extent and exact location of this post-translational modification (52, 53). Monoubiquitination of peptides of more than 20 amino acids was found to be a minimal requirement for protein degradation, but the canonical fourfold (poly-)ubiquitination with three further lysine (K48) side chain-linked ubiquitins appears to be most apt for an effective and rapid substrate recognition by the proteasome (54). This canonical polyubiquitin structure, as well as several other mixed polyubiquitin structures, can be recognized by the external 19S subunits of the 26S proteasome complex (54, 55). Prior to degradation of ubiquitinated proteins by the proteasomal 20S core subunit, the attached ubiquitin chains are released by the external 19S subunits for recycling, although they can also be co-degraded by the proteasome (56). After first passing the 19S subunit, the proteasomal target proteins are then unfolded in an energy-dependent manner and introduced into the narrow enzymatic cavity of proteasome for degradation. The barrel-shaped 20S proteasomal core complex contains three different proteolytic activities in duplicate (β1: caspase-like-, β2: tryptic-, and β5: chymotryptic activity), which initiate an efficient cleavage of the proteasomal target proteins into smaller peptides (57).
It is important to note that specific ubiquitination and ensuing proteasomal degradation is not an exclusive degradation mechanism of misfolded proteins but is also used to regulate the expression level of several native cell cycle regulatory proteins [cyclins, proliferating cell nuclear antigen (PCNA), p53], signaling pathway molecules (β-catenin, IκB), and survival factors (mcl-1) during the course of normal protein homeostasis and cell cycle progression (53, 55, 57, 58). Moreover, proteasomes are involved in protein maturation, including the processing and maturation of the NF-κB transcription factor subunit p50 and the drug-resistant protein MDR1 (57). Therefore, targeting proteasomal activity has not only been of interest for the generation of misfolded, cytotoxic proteins but also for interfering with the expression of proteins involved in several hallmarks of cancer, including cell cycle progression, signal transduction, and apoptosis.
Proteasome Inhibitors
Bortezomib (PS-341, Velcade ™) has long been known as a paragon of a clinically applicable proteasome inhibitor. Bortezomib has been approved for the treatment of multiple myeloma and mantle cell lymphoma (55, 59, 60). The great expectations of transferring the success of bortezomib to non-hematological solid cancer types have unfortunately not yet been fulfilled. It has been suggested that the high antibody-producing capacity of myeloma cells and thus the need for an efficient proteasomal degradation system to cope with the recycling process of misfolded ER-generated antibodies [ER-associated degradation process (ERAD); see below] might contribute to the high sensitivity of myeloma cells to bortezomib (9, 60, 61). Originally, bortezomib was developed to inhibit the proteasomal degradation of the NF-κB inhibitor IκB, thus targeting the pro-inflammatory, but also cancer-promoting, effect of the NF-κB transcription factor (55, 60, 62). Recent insights indicate that the anti-tumoral effect of bortezomib is not only mediated by its NF-κB inhibitory activity but also by its ability to induce accumulation of misfolded proteins in the cytosol and the ER (60, 62–65). However, the use of bortezomib, even for highly sensitive multiple myeloma, is limited by its strong tendency to induce a proteasome inhibition-independent peripheral neuropathy by acting on neuronal mitochondria (61). Since neurodegenerative diseases are associated with protein misfolding and aggregation, the neuropathological effects of bortezomib might also be assumed to be mediated by the possible proteotoxic effects of bortezomib in neuronal cells. However, although proteasome inhibitor-induced neurodegeneration and inclusion body formation have been described in animal models, similarities between proteasome inhibitor-induced neurodegeneration and Parkinson’s disease-like histopathological features could not be established (66).
Due to the neurotoxic side effects of bortezomib and an increasing occurrence of refractory myelomas, much effort has been made to generate and identify new proteasome inhibitors with fewer adverse effects. In 2012, the irreversible proteasome inhibitor and epoxomicin derivate carfilzomib (Kyprolis™) was approved for treatment of bortezomib-resistant and relapsed multiple myeloma patients (62, 67). Although sharing the same target with bortezomib, the chymotryptic activity of the proteasome, carfilzomib was shown to overcome bortezomib resistance and to be associated with fewer neuropathological adverse effects than bortezomib (62, 67, 68) (Table 1). Since carfilzomib has the advantage of being an irreversible proteasome inhibitor, a more patient-friendly administration schedule with less frequent application and a better compliance may add to the advantages of carfilzomib.
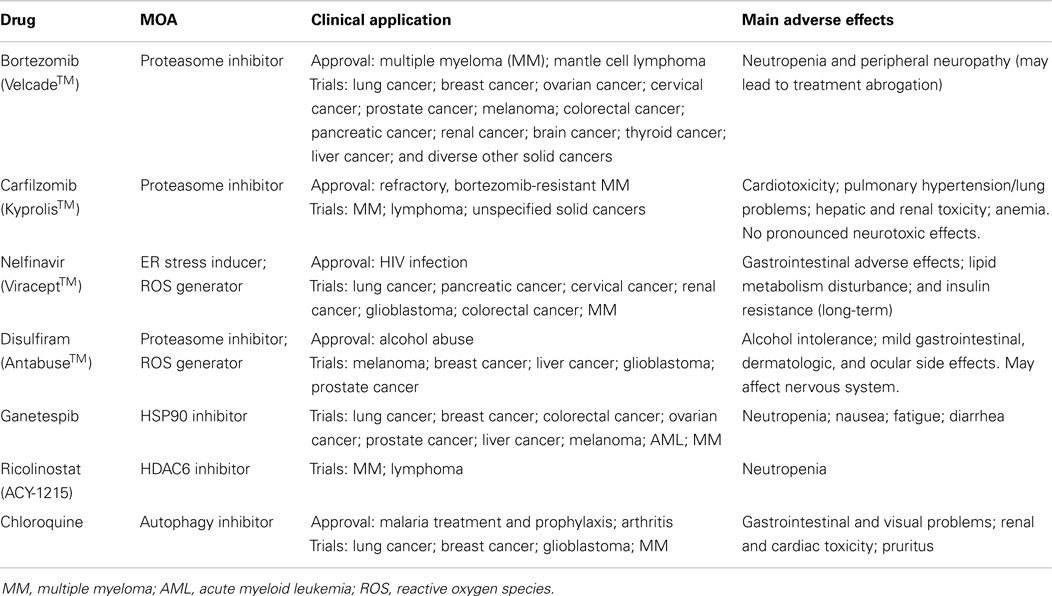
Table 1. Drugs described in this review and their mechanism of action (MOA), status of approval, and main adverse effects.
Additional newly generated proteasome inhibitors have been developed, which are currently being tested in clinical trials (53, 61, 65, 69) and will hopefully soon expand our limited arsenal of clinically applicable proteasome inhibitors. Unfortunately, both bortezomib and carfilzomib are extremely expensive medications and result in high therapy costs (68), so more cost-effective proteasome inhibitors are urgently needed. Interestingly, several natural polyphenols have been described as exerting proteasome-inhibitory activity (70, 71). As mentioned, however, the clinical use of flavonoids in cancer treatment is limited due to their poor bioavailability (49, 72), although the compounds may be used as lead molecules for the development of proteasome inhibitors. Notably, natural flavonoids have also been identified as reducing the therapeutic efficacy of bortezomib by a direct chemical interaction of vicinal hydroxyl groups of flavonoids with the boronic acid residue of bortezomib, thus leading to disregard of a combination therapy of bortezomib with flavonoids (73).
Instead, other combination therapies of bortezomib, in particular with hsp90 inhibitors appear to be more appropriate for therapeutic applications. A clinical study with the geldanamycin-derivate 17-AAG and bortezomib, however, revealed no clinical response but severe adverse effects resulted from this combination in acute myeloid leukemia patients (74). By contrast, a study with bortezomib and the hsp90 inhibitor tanespimycin in patients with relapsed and refractory multiple myeloma (75) revealed an objective response rate of 27% and was described to be well tolerated (75). In patients with diverse solid cancer receiving similar doses of this drug combination, however, no objective response was observed (76).
Disulfiram
Another prospective tool for cost-effective proteasome inhibition is the repurposed use of approved and (if ever possible) patent-free drugs that have also proved to cause proteasome inhibition as an off-target effect. The alcohol-deterring drug disulfiram (Antabuse) has been shown to be a highly active proteasome inhibitor (77, 78), in particular when combined with endogenous or exogenous copper ions (79). Treatment of cancer cells with disulfiram caused all characteristics of efficient proteasome inhibition, including NF-κB inhibition and accumulation of polyubiquitinated proteins and protein aggregates (77, 78, 80). Notably, non-malignant cells responded less to disulfiram treatment, in accordance with the low toxicity profile of this well-tolerated drug, as also experienced in course of its use for anti-alcoholism for over 60 years (79). In addition to its proteasome-inhibitory effect, disulfiram/copper has been shown to generate reactive oxygen species in cancer cells, further contributing to the large amount of misfolded and aggregated proteins seen after disulfiram treatment (78, 80). Disulfiram is degraded in vivo into various reactive metabolites, one of which, diethyldithiocarbonate, is able to covalently bind to reactive thiol groups of proteins and to inactivate cancer-promoting kinases and drug-resistance-conferring enzymes such as protein kinase C (PK-C), P-glycoprotein (MDR1), DNA methyltransferases (DNMT), and aldehyde dehydrogenase (ALDH) (81). Inhibition of ALDH has long been held responsible for the alcohol-deterring effect of disulfiram, but ALDH is also known to belong to a group of cancer-related genes referred to as cancer stem cell genes (81). Since cancer stem cell genes have been held responsible for drug resistance and cancer recurrence, targeting of ALDH1 by disulfiram may also tackle the highly drug-resistant cancer stem cell subpopulation. These promising pleiotropic, but mostly cancer cell specific, proteotoxic effects of disulfiram recently instigated several clinical trials with disulfiram in cancer patients (79, 82).
Aggresome Formation and Re-Solubilization: Role of HDAC6
As depicted above, proteasome and HSP inhibition will eventually lead to the accumulation of misfolded and polyubiquitinated proteins. Based on their inherent cohesive properties mediated by their exposed hydrophobic surfaces, both ubiquitinated and non-ubiquitinated misfolded proteins tend to adhere as small aggregates (Figure 1). Individual ubiquitinated proteins and small ubiquitinated aggregates can be recognized by specific ubiquitin-binding proteins such as HDAC6 via its zinc finger ubiquitin-binding domain. HDAC6 is an unusual histone deacetylase located in the cytosol that regulates microtubule acetylation and is also able to bind ubiquitinated proteins. Based on HDAC6’s additional ability to bind to microtubule motor protein dynein, these aggregates are actively transported along the microtubular system into perinuclear aggregates around the microtubule organizing center (MTOC) (10, 83, 84). Recognition of small, scattered ubiquitinated aggregates by HDAC6 has been described as being mediated by unanchored ubiquitin chains, which are generated by aggregate-attached ubiquitin ligase ataxin-3 (85). Whereas proteasomal target proteins are primarily tagged by K-48 (lysine-48) linked ubiquitins; K-63 linked ubiquitin chains appear to be a preferential modification for aggresomal targeting by HDAC6 and were assumed to mediate a redirection from proteasomal degradation to aggresome formation in the case of proteasomal inhibition or overload (86). Accordingly, aggresome formation is not an unspecific protein aggregation but a specific, ubiquitin-controlled sorting process. Furthermore, these aggresomes consist not only of misfolded and deposited proteins but have also been shown to contain a large amount of associated HSPs and ubiquitin-binding proteins, including HDAC6 [Figure 1; (10, 83, 84)]. Aggresomes contain, and are also surrounded by, large numbers of proteasomes (10, 83, 84), which help to resolubilize these aggregates not only through their intrinsic proteasomal digestion but also by generating unanchored K63-branched polyubiquitin chains, which then stimulate HDAC6-mediated autophagy, another cellular disposal mechanism in involving HDAC6 (87). Notably, HDAC6 has also been shown to control further maturation of autophagic vesicles by stimulating autophagosome–lysosome fusion (Figure 1) in a manner different from the normal autophagosome–lysosome fusion process (88).
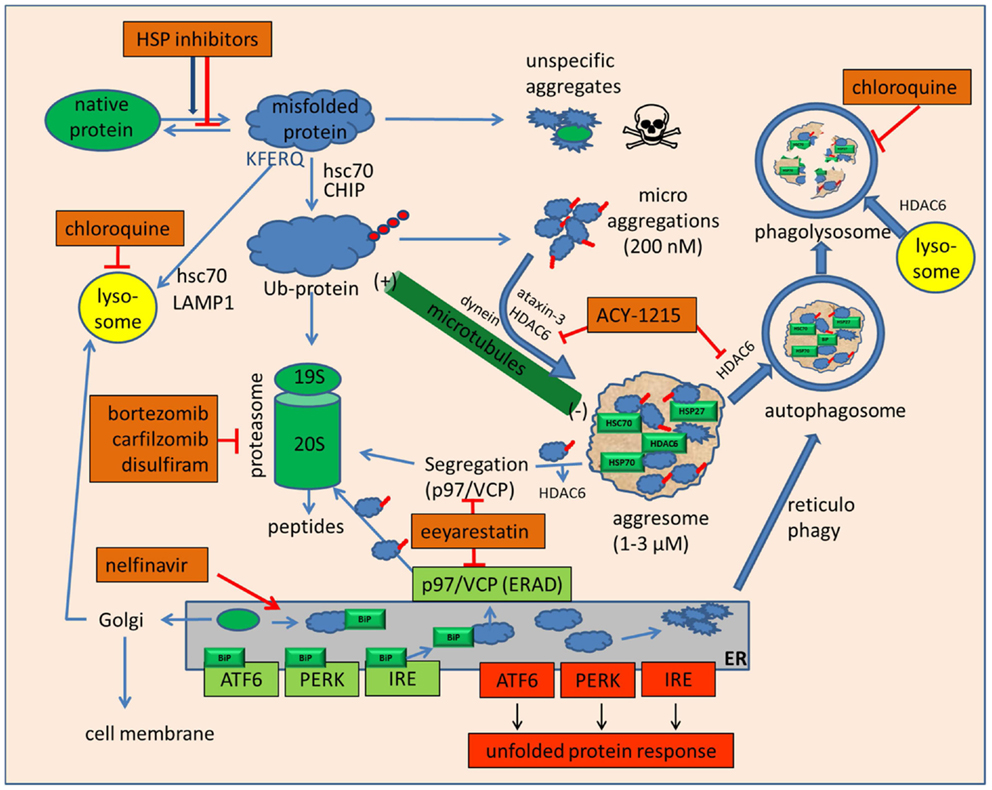
Figure 1. Drugs that inhibit folding or disposal of misfolded proteins. Native mature proteins, nascent proteins, or misfolded proteins can be prevented from folding or refolding by small and large heat shock protein inhibitors, of which the hsp90 inhibitors based on geldanamycin and radicicol are currently the most advanced in clinical studies. To avoid accumulation of misfolded proteins, their degradation can be mediated by hsc70, which may divert these proteins either to lysosomes to be degraded by chaperone-mediated autophagy or, by specific ubiquitination, to proteasomes. In case of unmanageable amounts of misfolded proteins or proteasome inhibition, unspecific aggregation of these proteins may occur. These highly cytotoxic small protein aggregates can be sequestered in an HDAC6-dependent manner with the help of microtubules into large, perinuclear aggresomes near the microtubule organizing center. Inhibition of HDAC6 by tubacin, tubastatin, or ACY-1215 inhibits disposal of microaggregates and may enhance the toxicity of bortezomib and HSP inhibitors. Aggresomes are not final deposits but may be re-dissolved either en bloc by macroautophagy or by molecular segregation via p97/VCP and final degradation by proteasomes. The p97/VCP inhibitor eeyarestatin may inhibit this degradation pathway but also aggresome formation by interfering with ataxin-3. Eeyarestatin also inhibits both anterograde and retrograde transport of ER proteins. Induction of ER stress induces the unfolded protein response by sequestering BiP from membrane receptors ATF6, PERK, and IRE. The unfolded protein response leads to cytoprotective chaperone synthesis but also to the expression of pro-apoptotic CHOP, NOXA, and c-JUN-kinase activation in case of prolonged or unmanageable ER stress. ER stress may also be alleviated by autophagy, whose efficacy can be inhibited by the lysosomotropic anti-malaria drug chloroquine.
The HDAC6 multitalent also exerts its deacetylase activity on hsp90 and modifies hsp90 client binding by facilitating its chaperoning of steroid hormone receptors and HSF1 (89–91). Recruitment of HDAC6 to ubiquitinated proteins leads to the dissociation of the repressive HDAC6/hsp90/HSF1 complex (91) and allows the release of transcriptionally active HSF1 to the nucleus. The engagement of HDAC6 at the aggresome–autophagy pathway hence also indirectly facilitates HSF1 activity. p97/VCP (valosin-containing protein), another binding partner of HDAC6 and itself a multi-interactive, ATP-dependent chaperone (92–94), is assumed to be involved not only in the specific separation of hsp90 and HSF1 by its “segregase” activity but also in the binding and remodeling of polyubiquitinated proteins before their delivery to the proteasome (93–95). Additionally, p97/VCP dissociates polyubiquitinated proteins bound to HDAC6 (91). Accumulation of polyubiquitinated proteins thus leads to HDAC6-dependent HSF1 activation and HSP induction, p97/VCP-dependent recruitment and “preparation” of polyubiquitinated proteins to proteasomes, and, in the case of pharmacological proteasome inhibition or physiological overload, to an HDAC6-dependent detoxification of polyubiquitinated proteins by the aggresome/autophagy pathway.
Pharmacological Inhibition of Aggresome Formation: HDAC6 Inhibitors
The central involvement of HDAC6 in aggresome formation and clearance makes HDAC6 one of the most interesting druggable targets for the induction of proteotoxicity in cancer cells. Also, HDAC6 has been found to be overexpressed in various cancer tissues, associated with advanced cancer stages and increased neoplastic transformation (96). Several pan-histone deacetylase inhibitors have been developed and tested in clinical studies for a variety of diseases, including different types of cancer (97, 98). Although hematological malignancies responded best to most of the already clinically tested pan-histone deacetylase inhibitors, the efficacy on solid cancer types was disappointingly poor and also associated with intolerable side effects (98). The unforeseeable pleiotropic epigenetic mechanism caused by non-specific (nuclear) histone deacetylase inhibitors may also limit their application for use in cancer treatment or HDAC6 inhibition, and has led to the search for selective HDAC6 inhibitors with no inhibitory effects on transcription modifying histone deacetylases. Through screening of small molecules under the rationale of selecting for tubulin deacetylase inhibitors with no cross-reactive histone deacetylase activity, the HDAC6 inhibitor tubacin was identified, and suggested for use in the treatment of neurodegenerative diseases or to reduce cancer cell migration and angiogenesis (99). Hideshima et al. then proved the hypothesis that the combined use of bortezomib with tubacin leads to an accumulation of non-disposed cytotoxic proteins and aggregates in cancer cells (100). Indeed, a synergistic effect of these two drugs against multiple myeloma cells could be observed with no detectable toxic effect on peripheral blood mononuclear cells (100). This and follow-up studies also revealed the efficacy of tubacin as a single agent against leukemia cells (100, 101) and a chemo-sensitizing effect on cytotoxic drugs in breast- and prostate-cancer cells (102).
Another specific HDAC6 inhibitor, tubastatin A (103) revealed promising effects in neurodegenerative diseases (104) but displayed apparently limited efficacy on solid cancer cells (105). A very interesting new and orally applicable HDAC6 inhibitor, ACY-1215 (ricolinostat), has recently been developed, which causes ER stress and caspase-dependent apoptosis in synergy with bortezomib in multiple myeloma cells (106). The promising pre-clinical data and the oral applicability have instigated several clinical studies to test the efficacy of ACY-1215 either as a single agent or in combination with bortezomib in patients with multiple myeloma and other lymphoid malignancies1.
Endoplasmic Reticulum Stress
Besides the cytosol, the ER is a major site for protein synthesis, in particular for those proteins destined for extracellular secretion, the cell membrane, or their retention within the endomembrane system. At the rough ER, nascent proteins are co-translationally transported across the ER membrane into the ER lumen (107), where they immediately encounter ER-resident chaperones, most prominently represented by hsp70 family member BiP/GRP78 and hsp90 family member GRP94 to help proper protein folding (15, 108). Most of these proteins also undergo post-translational modifications, including N- or O-linked glycosylation or protein disulfide bridge-building (109, 110), thereby adding further mechanisms of protein stabilization but also challenges for proper protein folding.
Similar to the situation in cytosolic protein biosynthesis, a large proportion of nascent proteins in the ER are assumed to misfold and to go “off-pathway” even under normal physiological conditions. Furthermore, the ER lumen, narrowly sandwiched between two phospholipid membranes, has been described as an even more densely crowded environment than the cytosol, additionally facilitating unspecific protein attachments and aggregations (15). Since, with the exception of bulk reticulophagy, the lumen of the ER contains no endogenous protein degradation system, and the anterograde transport of ER proteins to the Golgi, lysosomes, endosomes, or the extracellular environment requires properly folded proteins, a retrograde transport of ER proteins into the cytosol remains the only possible mechanism of preventing misfolded protein accumulation within the ER. In this ERAD, misfolded proteins are re-exported across the ER membrane by a specific multi protein complex, ubiquitinated by ER membrane-integrated ubiquitin ligases, and finally become degraded by cytosolic proteasomes (111, 112). Notably, association of the cytosolic p97/VCP protein, an important interacting partner with HDAC6, has also been described as being an essential factor for driving the luminal proteins through the ER membrane pore complex into the cytosol (92, 112).
Accordingly, all agents and conditions that interfere with these folding, maturation, and retranslocation processes can lead to protein misfolding and aggregation within this sensitive organelle. Chemicals that act as glycosylation inhibitors (tunicamycin), calcium ionophore inhibitors (A23187, thapsigargin), heavy metal ions (cadmium, lead), reducing agents (dithiothreitol), as well as conditions like hypoxia or oxidative stress, all lead to a phenomenon called ER stress (113–116). In the ER-stress response, a triad of ER membrane-resident signaling receptors and transducers, IRE1, ATF6, and PERK1, become activated and lead to the transcriptional activation of cytosolic and ER-resident chaperones to cope with the increasing number of misfolded proteins. Induction of autophagy (reticulophagy; ER-phagy) may also occur and supports the removal of damaged regions of the ER (117). Under very intensive or even unmanageable ER-stress conditions, a variety of pro-apoptotic pathways ensue, including CHOP induction, c-JUN-kinase activation, and caspase cleavage (118–120), which eventually prevails over the cytoprotective arm of the ER-stress response and may lead to apoptosis. Targeting of protein folding within the ER is therefore a very promising strategy to induce apoptosis in cancer cells, in particular in those cancer cells characterized by an unphysiologically high protein secretion rate, such as, for example, multiple myeloma cells. Whereas the above-mentioned drugs such as tunicamycin or thapsigargin are valuable tools for cell biology studies, they display unacceptable toxicities in humans and are not suited for therapeutic applications. Interestingly, several already established drugs used for non-cancerous diseases have been described as inducing ER stress at pharmacologically relevant concentrations in humans as an off-target effect (113, 116). The non-steroidal anti-inflammatory COX-2 inhibitor celecoxib is an approved drug to treat various forms of arthritis and pain, but has also been described as exerting ER stress by functioning as a SERCA (sarco/ER Ca2+ ATPase) inhibitor (113, 116). However, although well tolerated in humans, the ER-stress-inducing ability of celecoxib seems to be weaker than that of direct SERCA inhibitors such as thapsigargin, and the usefulness of celecoxib against advanced cancer has been questioned (116). Various HIV protease inhibitors have been described as inducing ER stress in human tissue cells as a side effect (121–123). In particular the HIV drugs lopinavir, saquinavir, and nelfinavir appear to be potent inducers of the ER-stress reaction, leading to a focused interest in these drugs for the induction of ER stress and apoptosis in cancer cells (116, 124–128). In fact, with currently over 27 clinical studies in cancer patients2, nelfinavir, either used as a single agent or in combination therapy, is on the list of the most promising prospective candidates to induce selective proteotoxicity in cancer cells at pharmacologically relevant concentrations. Although the exact mechanism by which nelfinavir induces ER stress is not yet clear, it was shown that nelfinavir causes the upregulation of cytosolic and ER-resident HSPs, and induces apoptosis in cancer cells associated with caspase activation and induction of the pro-apoptotic transcription factor CHOP (125, 126). Nelfinavir was also shown to be combinable with bortezomib to enhance its activity on cancer cells (129). Since the retrograde transport of misfolded ER proteins is inhibited by the p97/VCP inhibitor eeyarestatin (130, 131), we recently tested the combination of eeyarestatin with nelfinavir but found no synergistic effect between these two agents in cervical cancer cells (132). In contrast, eeyarestatin markedly sensitized cervical cancer cells to bortezomib treatment (132), which was also observed in preceding studies in which eeyarestatin was used to augment the ER-stress-inducing ability of bortezomib in leukemia cells (131). The combination of bortezomib with eeyarestatin massively increased the accumulation of polyubiquitinated proteins in cancer cells, induced macroautophagy, and activated the pro-apoptotic ATF4/CHOP/NOXA-mediated pathway of the ER-stress response (131, 132). These cytotoxic effects were also observed when eeyarestatin was applied as a single agent to cancer cells (131, 132), but not in normal blood cells (131). Eeyarestatin as a single agent might therefore be of interest as an alternative to bortezomib treatment, and has also successfully been applied in a xenograft model (133), although little is known of its pharmacological effects in humans. In combination with bortezomib, eeyarestatin might help to reduce the amount of bortezomib needed to induce cytotoxic effects in solid cancer cells (132).
Therapeutic Hyperthermia
Heat application is the most direct method to generate misfolded proteins in cancer cells. Indeed, local, regional, and whole body hyperthermia is a practiced cancer treatment modality, although in most cases is used to support and improve the efficacy of radiotherapy and chemotherapy (134, 135). Thanks to the basic or inducible expression of HSPs, normal body cells, and cancer cells can withstand temperatures well above the core body temperature of 37°C. In the oncologically relevant therapeutic temperature range of 40–47°C, a time and temperature relation exists that defines whether cells will survive or undergo cell death after heat exposure (136, 137). Temperatures between 40.5 and 42°C were described to cause only weak heat stress and no extensive apoptosis in cancer cells, with the extent of apoptosis rapidly increasing when temperatures were elevated from 42 to 45°C (137, 138). Further temperature elevation to 46–47°C then mainly causes unspecific, heat-induced necrosis alone (138). Interestingly, at these elevated temperatures, the quantity of HSPs is not further increased but instead decreased (135). Instead, some HSPs, in particular hsp70, are translocated to the cell membrane or are released by necrotic cells into the extracellular milieu and are able to elicit an immunologic response against surviving cancer cells (135, 139). The cellular response to heat is also highly variable among cell types. Temperatures of more than 48–50°C are believed to be generally intolerable to cancer as well as healthy body cells and can be used for local thermal ablation (137) in cases where surgery is either not possible or not wanted by the patient.
Although the whole cell and all its organelles are subjected to the detrimental heat impact, the nucleus seems to be one of the organelles most affected by hyperthermia. Non-histone nuclear proteins appear to be particularly sensitive to heat inactivation, resulting in denaturation of these nuclear proteins and their aggregation among each other, but also to correctly folded, aggregation-sensitive native proteins within the nucleus (18). Overall, this may lead to nuclear enzyme inactivation, DNA masking by aggregated proteins, impairment of DNA synthesis and repair, and a mitotic catastrophe in case cells continue to divide (18, 136, 137). In addition to an additive pro-apoptotic effect of heat to the cytotoxic insults of radiation or chemotherapeutic drugs on cancer cells (136), the supportive effect of hyperthermia is probably also due to the inhibitory effect of heat on DNA repair and drug detoxifying enzymes. However, despite several precious and specific anti-cancer effects, hyperthermia, as mentioned above, has not been established as a single cancer treatment but is primarily used as a supportive modality in conjunction with chemo- and radiotherapy (134, 135).
Conclusion and Perspectives
Induction of proteotoxicity through the accumulation of misfolded proteins has evolved as a new treatment modality in the fight against cancer. Clinically approved drugs such as bortezomib and carfilzomib provide evidence of the functionality of this approach. Newly developed agents like the HDAC6 inhibitor ACY-1215 or repurposed drugs like nelfinavir or disulfiram are currently being tested in clinical trials with cancer patients and will hopefully further broaden our arsenal of anti-cancer drugs. Notably, most proteotoxic agents that have been approved or are in clinical trials target the ubiquitin-proteasome-system (UPS) and are mainly effective in multiple myeloma cells, which rely on a functional ER/ERAD/UPS for excessive and proper antibody production. Similarly, it can be assumed that other cancer cell types with a marked secretory phenotype may also be affected by ER/ERAD/UPS inhibitors. In accordance with this notion, a recent dose-escalating Phase Ia study with nelfinavir as a single agent, that covered a large variety of solid cancer entities, revealed response rates primarily in patients with neuroendocrine tumors (140). In most other solid cancer types, however, the chemo-sensitizing or combination effects of proteotoxic drugs may prevail, and have become the focus of an increasing number of very promising clinical and pre-clinical studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully thank the Wilhelm-Vaillant Stiftung, Munich, Germany, for supporting the authors.
Footnotes
References
1. Anfinsen CB. Principles that govern the folding of protein chains. Science (1973) 181(4096):223–30. doi: 10.1126/science.181.4096.223
2. Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struct Biol (2004) 14(1):70–5. doi:10.1016/j.sbi.2004.01.009
3. Contessoto VG, Lima DT, Oliveira RJ, Bruni AT, Chahine J, Leite VB. Analyzing the effect of homogeneous frustration in protein folding. Proteins (2013) 81(10):1727–37. doi:10.1002/prot.24309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Rose GD, Fleming PJ, Banavar JR, Maritan A. A backbone-based theory of protein folding. Proc Natl Acad Sci U S A (2006) 103(45):16623–33. doi:10.1073/pnas.0606843103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Tsai CJ, Maizel JV Jr, Nussinov R. The hydrophobic effect: a new insight from cold denaturation and a two-state water structure. Crit Rev Biochem Mol Biol (2002) 37(2):55–69. doi:10.1080/10409230290771456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Pace NC, Scholtz JM, Grimsley GR. Forces stabilizing proteins. FEBS Lett (2014) 588(14):2177–84. doi:10.1016/j.febslet.2014.05.006
7. Zhang G, Ignatova Z. Folding at the birth of the nascent chain: coordinating translation with co-translational folding. Curr Opin Struct Biol (2011) 21(1):25–31. doi:10.1016/j.sbi.2010.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Wolff S, Weissman JS, Dillin A. Differential scales of protein quality control. Cell (2014) 157(1):52–64. doi:10.1016/j.cell.2014.03.007
9. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature (2003) 426(6968):895–9. doi:10.1038/nature02263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Garcia-Mata R, Gao YS, Sztul E. Hassles with taking out the garbage: aggravating aggresomes. Traffic (2002) 3(6):388–96. doi:10.1034/j.1600-0854.2002.30602.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Bartlett AI, Radford SE. An expanding arsenal of experimental methods yields an explosion of insights into protein folding mechanisms. Nat Struct Mol Biol (2009) 16(6):582–8. doi:10.1038/nsmb.1592
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem (2013) 82:323–55. doi:10.1146/annurev-biochem-060208-092442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Selkoe DJ. Folding proteins in fatal ways. Nature (2003) 426(6968):900–4. doi:10.1038/nature02264
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Aigelsreiter A, Janig E, Stumptner C, Fuchsbichler A, Zatloukal K, Denk H. How a cell deals with abnormal proteins. Pathogenetic mechanisms in protein aggregation diseases. Pathobiology (2007) 74(3):145–58. doi:10.1159/000103374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Gidalevitz T, Stevens F, Argon Y. Orchestration of secretory protein folding by ER chaperones. Biochem Biophys Acta (2013) 1833(11):2410–24. doi:10.1016/j.bbamcr.2013.03.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Murphy ME. The HSP70 family and cancer. Carcinogenesis (2013) 34(6):1181–8. doi:10.1093/carcin/bgt111
17. Ellis RJ, Minton AP. Protein aggregation in crowded environments. Biol Chem (2006) 387:485–97. doi:10.1515/BC.2006.064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Lepock JR. Role of nuclear protein denaturation and aggregation in thermal radiosensitization. Int J Hyperthermia (2004) 20(2):115–30. doi:10.1080/02656730310001637334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Tompa P, Rose GD. The Levinthal paradox of the interactome. Protein Sci (2011) 20(12):2074–9. doi:10.1002/pro.747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Taipale M, Jarosz DF, Lindquist S. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol (2010) 11(7):515–28. doi:10.1038/nrm2918
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Curr Pharm Des (2013) 19(3):347–65. doi:10.2174/1381612811306030347
22. Garcia-Carbonero R, Carnero A, Paz-Ares L. Inhibition of HSP90 molecular chaperones: moving into the clinic. Lancet Oncol (2013) 14(9):e358–69. doi:10.1016/S1470-2045(13)70169-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Röhl A, Rohrberg J, Buchner J. The chaperone Hsp90: changing partners for demanding clients. Trends Biochem Sci (2013) 38(5):253–62. doi:10.1016/j.tibs.2013.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Whitesell L, Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin Ther Targets (2009) 13(4):469–78. doi:10.1517/14728220902832697
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Jhaveri K, Taldone T, Modi S, Chiosis G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta (2012) 1823(3):742–55. doi:10.1016/j.bbamcr.2011.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat Rev (2013) 39(4):375–87. doi:10.1016/j.ctrv.2012.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos MA, Psaltopoulou T. Hsp90 inhibitors in breast cancer: a systematic review. Breast (2013) 22(5):569–78. doi:10.1016/j.breast.2013.06.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Zhou D, Liu Y, Ye J, Ying W, Ogawa LS, Inoue T, et al. A rat retinal damage model predicts for potential clinical visual disturbances induced by Hsp90 inhibitors. Toxicol Appl Pharmacol (2013) 273:401–9. doi:10.1016/j.taap.2013.09.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Li J, Buchner J. Structure, function and regulation of the hsp90 machinery. Biomed J (2013) 36(3):106–17. doi:10.4103/2319-4170.113230
30. Hernández MP, Sullivan WP, Toft DO. The assembly and intermolecular properties of the hsp70-Hop-hsp90 molecular chaperone complex. J Biol Chem (2002) 277(41):38294–304. doi:10.1074/jbc.M206566200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci (2005) 62(6):670–84. doi:10.1007/s00018-004-4464-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol (2012) 22(8):407–17. doi:10.1016/j.tcb.2012.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Benbrook DM, Long A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp Oncol (2012) 34(3):286–97.
34. McDonough H, Patterson C. CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones (2003) 8(4):303–8. doi:10.1379/1466-1268(2003)008<0303:CALBTC>2.0.CO;2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Stankiewicz M, Nikolay R, Rybin V, Mayer MP. CHIP participates in protein triage decisions by preferentially ubiquitinating Hsp70-bound substrates. FEBS J (2010) 277(16):3353–67. doi:10.1111/j.1742-4658.2010.07737.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Daugaard M, Rohde M, Jäättelä M. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett (2007) 581(19):3702–10. doi:10.1016/j.febslet.2007.05.039
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. de Thonel A, Le Mouël A, Mezger V. Transcriptional regulation of small HSP-HSF1 and beyond. Int J Biochem Cell Biol (2012) 44(10):1593–612. doi:10.1016/j.biocel.2012.06.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Beere HM. ‘The stress of dying’: the role of heat shock proteins in the regulation of apoptosis. J Cell Sci (2004) 117:2641–51. doi:10.1242/jcs.01284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Mendillo ML, Santagata S, Koeva M, Bell GW, Hu R, Tamimi RM, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell (2012) 150(3):549–62. doi:10.1016/j.cell.2012.06.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kumar S, Tomar MS, Acharya A. HSF1-mediated regulation of tumor cell apoptosis: a novel target for cancer therapeutics. Future Oncol (2013) 9(10):1573–86. doi:10.2217/fon.13.106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol (2010) 11:545–55. doi:10.1038/nrm2938
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, Workman P. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle (2010) 9(8):1542–50. doi:10.4161/cc.9.8.11204
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. McConnell JR, McAlpine SR. Heat shock proteins 27, 40, and 70 as combinational and dual therapeutic cancer targets. Bioorg Med Chem Lett (2013) 23(7):1923–8. doi:10.1016/j.bmcl.2013.02.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Zhang L, Fok JH, Davies FE. Heat shock proteins in multiple myeloma. Oncotarget (2014) 5(5):1132–48.
45. Hosokawa N, Hirayoshi K, Kudo H, Takechi H, Aoike A, Kawai K, et al. Inhibition of the activation of heat shock factor in vivo and in vitro by flavonoids. Mol Cell Biol (1992) 12(8):3490–8.
46. Elia G, Santoro MG. Regulation of heat shock protein synthesis by quercetin in human erythroleukaemia cells. Biochem J (1994) 300:201–9.
47. Nagai N, Nakai A, Nagata K. Quercetin suppresses heat shock response by down regulation of HSF1. Biochem Biophys Res Commun (1995) 208(3):1099–105. doi:10.1006/bbrc.1995.1447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Aalinkeel R, Bindukumar B, Reynolds JL, Sykes DE, Mahajan SD, Chadha KC, et al. The dietary bioflavonoid, quercetin, selectively induces apoptosis of prostate cancer cells by down-regulating the expression of heat shock protein 90. Prostate (2008) 68:1773–89. doi:10.1002/pros.20845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Bruning A. Inhibition of mTOR signaling by quercetin in cancer treatment and prevention. Anticancer Agents Med Chem (2013) 13(7):1025–31. doi:10.2174/18715206113139990114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Heimberger T, Andrulis M, Riedel S, Stühmer T, Schraud H, Beilhack A, et al. The heat shock transcription factor 1 as a potential new therapeutic target in multiple myeloma. Br J Haematol (2013) 160(4):465–76. doi:10.1111/bjh.12164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Chen Y, Chen J, Loo A, Jaeger S, Bagdasarian L, Yu J, et al. Targeting HSF1 sensitizes cancer cells to HSP90 inhibition. Oncotarget (2013) 4(6):816–29.
52. Wang G, Gao Y, Li L, Jin G, Cai Z, Chao JI, et al. K63-linked ubiquitination in kinase activation and cancer. Front Oncol (2012) 2:5. doi:10.3389/fonc.2012.00005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Johnson DE. The ubiquitin-proteasome system: opportunities for therapeutic intervention in solid tumors. Endocr Relat Cancer (2015) 22(1):T1–17. doi:10.1530/ERC-14-0005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Ciechanover A, Stanhill A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim Biophys Acta (2014) 1843(1):86–96. doi:10.1016/j.bbamcr.2013.07.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer (2004) 4(5):349–60. doi:10.1038/nrc1361
56. Shabek N, Ciechanover A. Degradation of ubiquitin: the fate of the cellular reaper. Cell Cycle (2010) 9(3):523–30. doi:10.4161/cc.9.3.11152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Navon A, Ciechanover A. The 26 S proteasome: from basic mechanisms to drug targeting. J Biol Chem (2009) 284(49):33713–8. doi:10.1074/jbc.R109.018481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Yamaguchi H, Hsu JL, Hung MC. Regulation of ubiquitination-mediated protein degradation by survival kinases in cancer. Front Oncol (2012) 2:15. doi:10.3389/fonc.2012.00015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Kouroukis TC, Baldassarre FG, Haynes AE, Imrie K, Reece DE, Cheung MC. Bortezomib in multiple myeloma: systematic review and clinical considerations. Curr Oncol (2014) 21(4):e573–603. doi:10.3747/co.21.1798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Skrott Z, Cvek B. Linking the activity of bortezomib in multiple myeloma and autoimmune diseases. Crit Rev Oncol Hematol (2014) 92(2):61–70. doi:10.1016/j.critrevonc.2014.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Kisselev AF, van der Linden WA, Overkleeft HS. Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol (2012) 19(1):99–115. doi:10.1016/j.chembiol.2012.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Crawford LJ, Irvine AE. Targeting the ubiquitin proteasome system in haematological malignancies. Blood Rev (2013) 27(6):297–304. doi:10.1016/j.blre.2013.10.002
63. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood (2006) 107:4907–16. doi:10.1182/blood-2005-08-3531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Brüning A, Burger P, Vogel M, Rahmeh M, Friese K, Lenhard M, et al. Bortezomib treatment of ovarian cancer cells mediates endoplasmic reticulum stress, cell cycle arrest, and apoptosis. Invest New Drugs (2009) 27(6):543–51. doi:10.1007/s10637-008-9206-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Kubiczkova L, Pour L, Sedlarikova L, Hajek R, Sevcikova S. Proteasome inhibitors – molecular basis and current perspectives in multiple myeloma. J Cell Mol Med (2014) 18(6):947–61. doi:10.1111/jcmm.12279
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Kordower JH, Kanaan NM, Chu Y, Suresh Babu R, Stansell J III, Terpstra BT, et al. Failure of proteasome inhibitor administration to provide a model of Parkinson’s disease in rats and monkeys. Ann Neurol (2006) 60(2):264–8. doi:10.1002/ana.20935
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Redic K. Carfilzomib: a novel agent for multiple myeloma. J Pharm Pharmacol (2013) 65(8):1095–106. doi:10.1111/jphp.12072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Thompson JL. Carfilzomib: a second-generation proteasome inhibitor for the treatment of relapsed and refractory multiple myeloma. Ann Pharmacother (2013) 47(1):56–62. doi:10.1345/aph.1R561
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Allegra A, Alonci A, Gerace D, Russo S, Innao V, Calabrò L, et al. New orally active proteasome inhibitors in multiple myeloma. Leuk Res (2014) 38(1):1–9. doi:10.1016/j.leukres.2013.10.018
70. Chang TL. Inhibitory effect of flavonoids on 26S proteasome activity. J Agric Food Chem (2009) 57:9706–15. doi:10.1021/jf9017492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Klappan AK, Hones S, Mylonas I, Brüning A. Proteasome inhibition by quercetin triggers macroautophagy and blocks mTOR activity. Histochem Cell Biol (2012) 137(1):25–36. doi:10.1007/s00418-011-0869-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Russo M, Spagnuolo C, Tedesco I, Bilotto S, Russo GL. The flavonoid quercetin in disease prevention and therapy: facts and fancies. Biochem Pharmacol (2012) 83:6–15. doi:10.1016/j.bcp.2011.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Jia L, Liu FT. Why bortezomib cannot go with ‘green’? Cancer Biol Med (2013) 10(4):206–13. doi:10.7497/j.issn.2095-3941.2013.04.004
74. Walker AR, Klisovic R, Johnston JS, Jiang Y, Geyer S, Kefauver C, et al. Pharmacokinetics and dose escalation of the heat shock protein inhibitor 17-allyamino-17-demethoxygeldanamycin in combination with bortezomib in relapsed or refractory acute myeloid leukemia. Leuk Lymphoma (2013) 54(9):1996–2002. doi:10.3109/10428194.2012.760733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Richardson PG, Chanan-Khan AA, Lonial S, Krishnan AY, Carroll MP, Alsina M, et al. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study. Br J Haematol (2011) 153:729–40. doi:10.1111/j.1365-2141.2011.08664.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Schenk E, Hendrickson AE, Northfelt D, Toft DO, Ames MM, Menefee M, et al. Phase I study of tanespimycin in combination with bortezomib in patients with advanced solid malignancies. Invest New Drugs (2013) 31(5):1251–6. doi:10.1007/s10637-013-9946-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Lövborg H, Oberg F, Rickardson L, Gullbo J, Nygren P, Larsson R. Inhibition of proteasome activity, nuclear factor-KappaB translocation and cell survival by the antialcoholism drug disulfiram. Int J Cancer (2006) 118(6):1577–80. doi:10.1002/ijc.21534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Chen D, Cui QC, Yang H, Dou QP. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res (2006) 66(21):10425–33. doi:10.1158/0008-5472.CAN-06-2126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Kast RE, Boockvar JA, Brüning A, Cappello F, Chang WW, Cvek B, et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the international initiative for accelerated improvement of glioblastoma care. Oncotarget (2013) 4(4):502–30.
80. Papaioannou M, Mylonas I, Kast RE, Brüning A. Disulfiram/copper causes redox-related proteotoxicity and concomitant heat shock response in ovarian cancer cells that is augmented by auranofin-mediated thioredoxin inhibition. Oncoscience (2014) 1:21–9.
81. Brüning A, Kast RE. Oxidizing to death: disulfiram for cancer cell killing. Cell Cycle (2014) 13(10):1513–4. doi:10.4161/cc.28959
82. Kona FR, Buac D, Burger MA. Disulfiram, and disulfiram derivatives as novel potential anticancer drugs targeting the ubiquitin-proteasome system in both preclinical and clinical studies. Curr Cancer Drug Targets (2011) 11(3):338–46. doi:10.2174/156800911794519798
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Kopito RR. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol (2000) 10(12):524–30. doi:10.1016/S0962-8924(00)01852-3
84. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell (2003) 115(6):727–38. doi:10.1016/S0092-8674(03)00939-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Ouyang H, Ali YO, Ravichandran M, Dong A, Qiu W, MacKenzie F, et al. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J Biol Chem (2012) 287(4):2317–27. doi:10.1074/jbc.M111.273730
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Chin LS, Olzmann JA, Li L. Parkin-mediated ubiquitin signalling in aggresome formation and autophagy. Biochem Soc Trans (2010) 38:144–9. doi:10.1042/BST0380144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Hao R, Nanduri P, Rao Y, Panichelli RS, Ito A, Yoshida M, et al. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol Cell (2013) 51(6):819–28. doi:10.1016/j.molcel.2013.08.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J (2010) 29(5):969–80. doi:10.1038/emboj.2009.405
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem (2005) 280(29):26729–34. doi:10.1074/jbc.C500186200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Kovacs JJ, Cohen TJ, Yao TP. Chaperoning steroid hormone signaling via reversible acetylation. Nucl Recept Signal (2005) 3:e004. doi:10.1621/nrs.03004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev (2007) 21(17):2172–81. doi:10.1101/gad.436407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature (2001) 414(6864):652–6. doi:10.1038/414652a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Yamanaka K, Sasagawa Y, Ogura T. Recent advances in p97/VCP/Cdc48 cellular functions. Biochim Biophys Acta (2012) 1823(1):130–7. doi:10.1016/j.bbamcr.2011.07.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol (2012) 14(2):117–23. doi:10.1038/ncb2407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, et al. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J (2006) 25(14):3357–66. doi:10.1038/sj.emboj.7601210
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol (2011) 2011:875824. doi:10.1155/2011/875824
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Martínez-Iglesias O, Ruiz-Llorente L, Sánchez-Martínez R, García L, Zambrano A, Aranda A. Histone deacetylase inhibitors: mechanism of action and therapeutic use in cancer. Clin Transl Oncol (2008) 10(7):395–8. doi:10.1007/s12094-008-0221-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Slingerland M, Guchelaar HJ, Gelderblom H. Histone deacetylase inhibitors: an overview of the clinical studies in solid tumors. Anticancer Drugs (2014) 25(2):140–9. doi:10.1097/CAD.0000000000000040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci U S A (2003) 100(8):4389–94. doi:10.1073/pnas.0430973100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A (2005) 102(24):8567–72. doi:10.1073/pnas.0503221102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Aldana-Masangkay GI, Rodriguez-Gonzalez A, Lin T, Ikeda AK, Hsieh YT, Kim YM, et al. Tubacin suppresses proliferation and induces apoptosis of acute lymphoblastic leukemia cells. Leuk Lymphoma (2011) 52(8):1544–55. doi:10.3109/10428194.2011.570821
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci U S A (2010) 107(46):20003–8. doi:10.1073/pnas.1013754107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc (2010) 132(31):10842–6. doi:10.1021/ja102758v
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Zhang L, Liu C, Wu J, Tao JJ, Sui XL, Yao ZG, et al. Tubastatin A/ACY-1215 improves cognition in Alzheimer’s disease transgenic mice. J Alzheimers Dis (2014) 41(4):1193–205. doi:10.3233/JAD-140066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Rosik L, Niegisch G, Fischer U, Jung M, Schulz WA, Hoffmann MJ. Limited efficacy of specific HDAC6 inhibition in urothelial cancer cells. Cancer Biol Ther (2014) 15(6):742–57. doi:10.4161/cbt.28469
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood (2012) 119(11):2579–89. doi:10.1182/blood-2011-10-387365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature (2007) 450:663–9. doi:10.1038/nature06384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem (2011) 80:71–99. doi:10.1146/annurev-biochem-062209-093836
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Swanton E, Bulleid NJ. Protein folding and translocation across the endoplasmic reticulum membrane. Mol Membr Biol (2003) 20(2):99–104. doi:10.1080/0968768031000069241
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell (2010) 40(2):238–52. doi:10.1016/j.molcel.2010.10.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science (2011) 334(6059):1086–90. doi:10.1126/science.1209235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol (2013) 5(9):a013185. doi:10.1101/cshperspect.a013185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Strasser A, Puthalakath H. Fold up or perish: unfolded protein response and chemotherapy. Cell Death Differ (2008) 15(2):223–5. doi:10.1038/sj.cdd.4402279
114. Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol (2009) 625(1–3):234–46. doi:10.1016/j.ejphar.2009.06.064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Gorman AM, Healy SJ, Jäger R, Samali A. Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther (2012) 134(3):306–16. doi:10.1016/j.pharmthera.2012.02.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Schönthal AH. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem Pharmacol (2013) 85(5):653–66. doi:10.1016/j.bcp.2012.09.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Cebollero E, Reggiori F, Kraft C. Reticulophagy and ribophagy: regulated degradation of protein production factories. Int J Cell Biol (2012) 2012:182834. doi:10.1155/2012/182834
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol (2011) 13(3):184–90. doi:10.1038/ncb0311-184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta (2013) 1833(12):3460–70. doi:10.1016/j.bbamcr.2013.06.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta (2013) 1833(12):3507–17. doi:10.1016/j.bbamcr.2013.07.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Djedaini M, Peraldi P, Drici MD, Darini C, Saint-Marc P, Dani C, et al. Lopinavir co-induces insulin resistance and ER stress in human adipocytes. Biochem Biophys Res Commun (2009) 386:96–100. doi:10.1016/j.bbrc.2009.05.148
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Wu X, Sun L, Zha W, Studer E, Gurley E, Chen L, et al. HIV protease inhibitors induce endoplasmic reticulum stress and disrupt barrier integrity in intestinal epithelial cells. Gastroenterology (2010) 138:197–209. doi:10.1053/j.gastro.2009.08.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Taura M, Kariya R, Kudo E, Goto H, Iwawaki T, Amano M, et al. Comparative analysis of ER stress response into HIV protease inhibitors: lopinavir but not darunavir induces potent ER stress response via ROS/JNK pathway. Free Radic Biol Med (2013) 65:778–88. doi:10.1016/j.freeradbiomed.2013.08.161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Gupta AK, Li B, Cerniglia GJ, Ahmed MS, Hahn SM, Maity A. The HIV protease inhibitor nelfinavir downregulates Akt phosphorylation by inhibiting proteasomal activity and inducing the unfolded protein response. Neoplasia (2007) 9(4):271–8. doi:10.1593/neo.07124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res (2007) 13(17):5183–94. doi:10.1158/1078-0432.CCR-07-0161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Pyrko P, Kardosh A, Wang W, Xiong W, Schönthal AH, Chen TC. HIV-1 protease inhibitors nelfinavir and atazanavir induce malignant glioma death by triggering endoplasmic reticulum stress. Cancer Res (2007) 67(22):10920–8. doi:10.1158/0008-5472.CAN-07-0796
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. McLean K, VanDeVen NA, Sorenson DR, Daudi S, Liu JR. The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. Gynecol Oncol (2009) 112(3):623–30. doi:10.1016/j.ygyno.2008.11.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Brüning A, Gingelmaier A, Friese K, Mylonas I. New prospects for nelfinavir in non-HIV-related diseases. Curr Mol Pharmacol (2010) 3(2):91–7. doi:10.2174/1874-470211003020091
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Brüning A, Vogel M, Mylonas I, Friese K, Burges A. Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr Cancer Drug Targets (2011) 11(7):799–809. doi:10.2174/156800911796798913
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Fiebiger E, Hirsch C, Vyas JM, Gordon E, Ploegh HL, Tortorella D. Dissection of the dislocation pathway for type I membrane proteins with a new small molecule inhibitor, eeyarestatin. Mol Biol Cell (2004) 15(4):1635–46. doi:10.1091/mbc.E03-07-0506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A (2009) 106(7):2200–5. doi:10.1073/pnas.0807611106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Brem GJ, Mylonas I, Brüning A. Eeyarestatin causes cervical cancer cell sensitization to bortezomib treatment by augmenting ER stress and CHOP expression. Gynecol Oncol (2013) 128(2):383–90. doi:10.1016/j.ygyno.2012.10.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Valle CW, Min T, Bodas M, Mazur S, Begum S, Tang D, et al. Critical role of VCP/p97 in the pathogenesis and progression of non-small cell lung carcinoma. PLoS One (2011) 6(12):e29073. doi:10.1371/journal.pone.0029073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Wust P, Hildebrandt B, Sreenivasa G, Rau B, Gellermann J, Riess H, et al. Hyperthermia in combined treatment of cancer. Lancet Oncol (2002) 3(8):487–97. doi:10.1016/S1470-2045(02)00818-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Issels RD. Hyperthermia adds to chemotherapy. Eur J Cancer (2008) 44(17):2546–54. doi:10.1016/j.ejca.2008.07.038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Hildebrandt B, Wust P, Ahlers O, Dieing A, Sreenivasa G, Kerner T, et al. The cellular and molecular basis of hyperthermia. Crit Rev Oncol Hematol (2002) 43(1):33–56. doi:10.1016/S1040-8428(01)00179-2
137. Roti Roti JL. Cellular responses to hyperthermia (40-46 degrees C): cell killing and molecular events. Int J Hyperthermia (2008) 24(1):3–15. doi:10.1080/02656730701769841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Harmon BV, Corder AM, Collins RJ, Gobé GC, Allen J, Allan DJ, et al. Cell death induced in a murine mastocytoma by 42-47 degrees C heating in vitro: evidence that the form of death changes from apoptosis to necrosis above a critical heat load. Int J Radiat Biol (1990) 58(5):845–58. doi:10.1080/09553009014552221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Multhoff G. Activation of natural killer cells by heat shock protein 70. Int J Hyperthermia (2009) 25(3):169–75. doi:10.1080/02656730902902001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: proteasome, autophagy, bortezomib, nelfinavir, endoplasmic reticulum stress, aggresome, HDAC6
Citation: Brüning A and Jückstock J (2015) Misfolded proteins: from little villains to little helpers in the fight against cancer. Front. Oncol. 5:47. doi: 10.3389/fonc.2015.00047
Received: 07 January 2015; Paper pending published: 26 January 2015;
Accepted: 11 February 2015; Published online: 24 February 2015.
Edited by:
Matiullah Khan, AIMST University, MalaysiaReviewed by:
Diego Ruano, University of Seville, SpainYun Dai, Virginia Commonwealth University and Massey Cancer Center, USA
Copyright: © 2015 Brüning and Jückstock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ansgar Brüning, Molecular Biology Laboratory, University Hospital Munich, Maistrasse 11, Munich 80337, Germany e-mail:YW5zZ2FyLmJydWVuaW5nQG1lZC51bmktbXVlbmNoZW4uZGU=