 Christina Arieta Kuksin
Christina Arieta Kuksin Lisa M. Minter
Lisa M. Minter- 1Department of Veterinary and Animal Sciences, University of Massachusetts Amherst, Amherst, MA, USA
- 2Program in Molecular and Cellular Biology, University of Massachusetts Amherst, Amherst, MA, USA
An association between certain autoimmune conditions and increased risk of developing lymphoma is well documented. Recent evidence points to NOTCH signaling as a strong driver of autoimmunity. Furthermore, a role for NOTCH in various lymphomas, including classical Hodgkin lymphoma, non-Hodgkin lymphoma, and T cell lymphoma has also been described. In this mini-review, we will outline what is known about involvement of NOTCH signaling in those autoimmune conditions, such as rheumatoid arthritis and primary Sjörgren’s syndrome, which show an increased risk for subsequent diagnosis of lymphoma. Furthermore, we will detail what is known about the lymphomas associated with these autoimmune conditions and how aberrant or sustained NOTCH signaling in the immune cells that mediate these diseases may contribute to lymphoma.
It has long been recognized that patients with particular autoimmune disorders and inflammatory conditions have an increased risk for developing specific lymphomas. Generally, the link between autoimmune diseases and developing lymphoma is correlative; however, there exists a subset of autoimmune diseases for which the risk of subsequent lymphoma development is strong (1, 2). This review will focus on four autoimmune disorders that have a well-established association with lymphoma occurrence: rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), primary Sjörgren’s syndrome (pSS), and celiac disease. We will focus on the involvement of the NOTCH pathway in these disorders, as well as the developing lymphomas, and postulate how NOTCH signaling may be one of the pre-disposing factors in their development.
The NOTCH family comprises four transmembrane receptors (NOTCH1-4), which orchestrate multiple cell fate decisions. NOTCH receptors engage one of five ligands, Jagged (Jag) 1, 2 or Delta-like (DLL) 1, 3, 4, and are ultimately cleaved by γ-secretase, liberating the signaling-competent, intracellular domain from the membrane (NOTCHIC). NOTCHIC may then interact with proteins in the cytoplasm, such as NF-κB, or translocate to the nucleus to mediate downstream gene transcription, including the classical targets, Hairy enhancer of split (Hes), and Hes-related with YRPW motif [Hey; (3–5)]. NOTCH has been shown to be important in hematopoietic lineage decisions. During lymphocyte ontogeny, NOTCH1 promotes adoption of a T cell fate at the expense of B cell development (6). Dysregulated NOTCH signaling is implicated in many diseases, including autoimmune disorders and various malignancies (7, 8).
Rheumatoid arthritis is a chronic inflammatory condition characterized by painful swelling of affected joints. It has been most strongly associated with subsequent development of diffuse large B cell lymphoma (DLBCL), a specific type of non-Hodgkin’s lymphoma [NHL; (1)]. Extensive evidence indicates NOTCH signaling plays an important role in the pathogenesis of RA. Compared to healthy joints, all four NOTCH homologs show aberrant expression in the inflamed synovium of RA patients (9, 10). In particular, NOTCH1 and NOTCH3 are highly expressed, as are the ligands DLL1 and Jag1, on the synovial lining and sublining of synovial hyperplastic lesions. NOTCH1 and NOTCH3 expression overlapped with Jag1 expression (11). Moreover, in the lymphoid follicles of these patients, NOTCH1 was detected in T and B cells. Multiple studies have reported activated, cleaved NOTCH1IC in the synovium and in autoreactive T cells (10–12). Additionally, NOTCH1IC and NOTCH4IC have been found in RA synoviocytes (13).
Inhibiting NOTCH signaling ameliorates pro-inflammatory responses in RA. Blocking NOTCH cleavage using γ-secretase inhibitors (GSI) reduces Th1- and Th17-mediated inflammatory responses in vitro, as well as in a mouse model of RA (14). GSI or NOTCH3-neutralizing antibodies can reduce T cell proliferation and pro-inflammatory cytokine production (15). GSI also reduces TNF-induced IL-6 production and cell proliferation in RA synoviocytes (12, 16). These data provide evidence NOTCH signaling is important for RA progression by regulating cytokine production and cell proliferation.
The mechanism(s) by which NOTCH signaling is sustained or how it acts to facilitate RA progression remains an area of active investigation. Samples from RA patients incubated with TNF upregulated NOTCH and ligand expression (12, 13, 16). Interestingly, individual NOTCH ligands have differential effects on proliferation and cytokine production by RA cells. Culturing cells with DLL1 promotes T cell proliferation and pro-inflammatory cytokine production in RA (15, 16). Conversely, administering soluble Jag1 provides a negative signal to CD8+ T cells and reduces disease symptoms in a mouse model of RA (17). NOTCH may also be acting to regulate NF-κB activation in synoviocytes. Nuclear NOTCH1 has been shown to form a complex with RBP-Jκ and reverse NF-κB2 promoter suppression (18). Collectively, these date provide clear evidence NOTCH signaling is important for the pathogenesis of RA.
Although not as tightly linked as for patients with RA, patients with SLE also display an increased risk of developing DLBCL and classical Hodgkin’s lymphoma [cHL; (1)]. Autoimmune SLE is characterized by the production of autoantibodies and immune-complex deposition that affect multiple organs. Increased numbers of macrophages, termed “M2b” that are defined by a signature profile of high IL-10/TNF/IL-1β/IL-6/MCP-1 production and low IL-12 secretion have been described in SLE (19). M2b macrophages also express high levels of NOTCH1IC as well as elevated Hes1 and Hey1. Furthermore, GSI treatment in a mouse model of SLE impaired macrophage differentiation into the M2b phenotype and ameliorated lupus-associated symptoms, which included reduced anti-dsDNA titers, decreased kidney scores, and attenuated IgG deposition (19). It is not exactly known how NOTCH signaling may be driving this pro-inflammatory response, but macrophages stimulated by activated, lymphocyte-derived self-apoptotic DNA (ALD-DNA) enhanced NOTCH1 signaling and was accompanied by increased nuclear translocation of NF-κB p50. In contrast, GSI treatment strongly downregulated NF-κB activity, supporting the notion that NOTCH1 is driving NF-κB associated pro-inflammatory responses in M2b macrophages (19).
In addition to its implication in M2b macrophage differentiation, NOTCH signaling may also play an important role in autoreactive lymphocytes associated with SLE. GSI treatment decreased CD4+ T cell proliferation in the lymph nodes and spleen, and lowered cytokine secretion and monocyte chemoattractant protein-1 (MCP-1) production (20). The authors of this report speculated that inhibiting NOTCH signaling altered disease-associated double negative T cells which, in turn, ameliorated disease symptoms. GSI treatment also decreased MCP-1 levels, which have been shown to contribute significantly to the Th1 response that drives the autoimmune manifestations in murine SLE (21–23). In human SLE, NOTCH1 expression is downregulated in T cells from patients with active SLE (24, 25). Decreased NOTCH1 mRNA and protein expression was determined to be due to epigenetic modification of the NOTCH1 promoter, including histone and CpG DNA methylation and transcriptional repression mediated by CREMα (24). Reduced NOTCH1 expression was associated with decreased proliferation and lower CD25 and Foxp3 expression following in vitro stimulation (25). NOTCH1 is important for generating de novo regulatory T cells (26), and it is intriguing to speculate that impaired NOTCH1 signaling may adversely affect regulatory T cells in SLE, either in number or function. Abrogated NOTCH1 signaling was also accompanied by an observed increase in IL-17A expression in SLE patients (24). It is therefore possible that NOTCH signaling drives SLE pathogenesis by inhibiting regulatory T cell development, thus allowing the generation of autoreactive, IL-17A-producing T cells.
Patients with pSS experience chronic autoimmune destruction of the exocrine glands, specifically the salivary and lacrimal glands. Cases of pSS are associated with an increased risk of DLBCL and marginal zone lymphoma, specifically, MALT lymphoma of the parotid gland (1). Not much is known about the role of NOTCH in pSS, although NOTCH2 mRNA has been detected in marginal zone B cells in the salivary gland and in tonsil germinal centers in pSS patients (27). Clusters of transitional type II B cells in salivary glands expressed NOTCH2 and BLIMP1 mRNA and these cells behaved like marginal zone B cells (28). Although evidence defining a contribution for NOTCH signaling in the pathogenesis Sjörgren’s syndrome is not abundant, NOTCH signaling is important in the development of salivary glands, a process, which is defective in pSS (29). Furthermore, abnormalities in NF-κB signaling have been observed, with polymorphisms in genes associated with the NF-κB pathways also documented in pSS patients (30–32). Given that NF-κB and NOTCH pathways are known to interact, it is possible that dysregulated NF-κB signaling may lead to dysregulated NOTCH signaling. Additional investigation of autoimmune mechanisms that potentiate pSS will be useful to further define any involvement of aberrant NOTCH signaling in this disease.
As with pSS, little is known about NOTCH signaling in celiac disease, which results from chronic immune stimulation in response to dietary proteins in patients with a genetic predisposition. Celiac disease is a distinctly T cell-mediated condition and the TCRs responsible for recognizing and responding to the inciting antigen, gliadin, a component of the gluten protein, have been recently identified (33). Celiac disease is also associated with development of non-Hodgkin’s T cell lymphoma. Intestinal biopsies from patients with active celiac disease, including those on a gluten free diet, revealed decreased NOTCH1 and HES1 mRNA expression in goblet cells, compared to healthy patients (34). Furthermore, dysregulated NOTCH activity is related to the immune-mediated pathogenesis of irritable bowel diseases, such as ulcerative colitis and Crohn’s disease (35). Polymorphisms in the NF-κB-related genes, REL and OLIG3/TNFAIP3 have been identified and were shown to contribute to celiac disease (36). Should NOTCH signaling in gut T cells prove to be pathogenic in celiac disease, a significant treatment challenge will likely result: that is how to balance decreasing aberrant NOTCH signaling in autoreactive T cells with maintaining a healthy level of NOTCH signaling in intestinal goblet cells (37).
A diagnosis of any of the four autoimmune diseases described above leaves patients at increased risk of developing lymphoma (Figure 1). It is interesting to note, however, that aggressive lymphocyte neoplasms are associated with these autoimmune diseases, rather than more the indolent chronic lymphocytic leukemias (1). DLBCL is the most common type of NHL. This fast-growing lymphoma is characterized by large B cells that grow diffusely throughout the lymph nodes. While NOTCH signaling has a clear role in T cell proliferation and T cell malignancies, dysregulated NOTCH signaling in B cell lymphoma is more controversial (38). The TMD8 cell line was developed from a patient with DLBCL. These cells express NOTCH1, NOTCH2, Jagged1, DLL4, and constitutively express HES1 mRNA. Most DLBCLs express Epstein–Barr proteins (EBNA2), which mimic NOTCH signaling to promote HES expression. However, TMD8 cells do not express EBNA2; therefore, the constitutive HES1 expression is thought to be driven by NOTCH signaling. Treating this cell line with GSI impaired cell growth and reduced expression of HES1. However, one caveat of this study is that GSI did not fully inhibit NOTCH1 cleavage, which might indicate a slow rate of NOTCH1 protein degradation. Alternatively, the biological effects seen may result from GSI acting on another target of γ-secretase, such as NOTCH2; however, NOTCH2 cleavage after GSI treatment was not assessed (39). Although NOTCH1IC could be detected in the TMD8 cell line, a tissue microarray containing 68 well-characterized DLBCLs revealed none that stained positively for NOTCH1IC (40). This may suggest that family members other than NOTCH1 are dysregulated in these tumors, but more studies are needed to ascertain the degree to which activated NOTCH drives DLBCL.
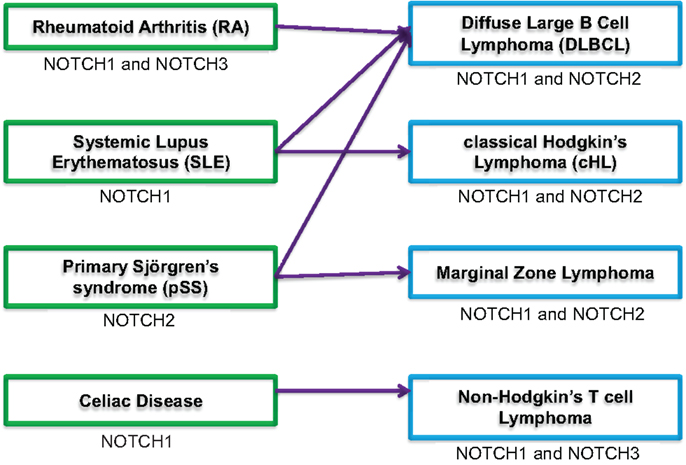
Figure 1. Potential association of autoimmune conditions and lymphoma. Schematic showing four autoimmune diseases (green boxes, left side) associated with increased risk for developing lymphomas [blue boxes, right side; (1, 2)]. The arrows indicate correlation between specific autoimmune diseases and most frequently associated lymphoma. NOTCH signaling component(s) dysregulated in autoimmune conditions or lymphoma is indicated under each box.
In studies of patients with DLBCL, mutations in the heterodimerization and PEST domains of NOTCH2 were found in a small (8%) percentage. Of this cohort, 40% had increased copy numbers of the NOTCH2 allele carrying the PEST domain mutation, and in one patient the total copy number of NOTCH2 was increased. Furthermore, mutant NOTCH2 receptors showed increased activity when stimulated with NOTCH ligands in vitro (41). A SNP variation in 13q12, which contains the NOTCH signaling mediator, LNX2, has also been identified. This gene encodes a PDZ domain-containing zinc finger 1 protein, which may function as an E3 ubiquitin ligase. Elevated levels of LNX family members were shown to promote ubiquitin–proteasomal degradation of the NOTCH negative regulator, NUMB, resulting in enhanced NOTCH signaling (42). NF-κB expression and activation is also high in DLBCL tumors (43–46). Thus, both NOTCH and NF-κB may play a role in DLBCL progression.
Classical Hodgkin’s lymphoma is characterized by the presence of very large cells called Hodgkin and Reed-Sternberg (HRS) cells, although other abnormal cell types may also be present. NOTCH signaling has been shown to play a prominent role in cHL pathogenesis. NOTCH1 and NOTCH2 are aberrantly expressed in HRS cells and are stimulated through Jagged1 to promote proliferation and protect from apoptosis (47). HRS cells are known to express Jagged1, Jagged2, DLL1, DLL3, and DLL4 (47, 48). Finally, an absence of the NOTCH1 inhibitor DELTEX1, together with increased expression of the NOTCH co-activator, MAML, likely drive NOTCH signaling in HRS cells (48, 49).
NOTCH and NF-κB signaling pathways have been shown to cooperate, resulting in cHL pathogenesis. HRS cells are characterized by high levels of nuclear NF-κB p50/p65 dimers, which are required for proliferation, protection from apoptosis, and growth in lymphoma mouse models (50–52). In addition, mutations in NF-κB regulators, such as IκBα, IκBε, or IκB kinase (IKK)α, contribute to constitutive activation of canonical NF-κB signaling in HRS cells (51, 53, 54). Inhibiting NOTCH activity in HRS cells using GSI induces apoptosis through simultaneous targeting of the NOTCH and alternative NF-κB signaling pathways (55). Thus, NOTCH signaling conspires to drive cHL pathology and inhibiting NOTCH signaling may have positive implications for treatment.
MALT lymphoma of the parotid gland represents a small subset of head and neck NHL. A diagnosis of Sjörgren’s syndrome is associated with a 1000-fold increase in the risk of parotid gland marginal zone lymphoma (56). Although no studies have explored how NOTCH signaling may contribute to development of MALT lymphoma of the parotid gland, there have been studies examining MALT lymphoma in the spleen (SMZL). In 30% of SMZL cases, alterations in genes of the NOTCH pathway emerged as highly recurrent. NOTCH2, a key regulator of marginal zone B cell development, was identified as the most frequently mutated gene, occurring in approximately 20% of cases, often resulting in disruption of the PEST domain (57–59). In addition to NOTCH2, genetic lesions in other modulators or members of the NOTCH pathway were also observed in SMZL, including SPEN, DELTEX1, and NOTCH1 (58). Mutations in members of the NF-κB pathway have also been implicated in MALT lymphoma (60–62). In fact, somatic mutations of TNFAIP3 (A20) protein, which plays a key role in controlling NF-κB activation, have been observed in the MALT lymphoma subtype frequently associated with SS (60). More studies are needed to ascertain whether and how NOTCH signaling drives MALT lymphomas associated with pSS.
The majority of systemic autoimmune diseases predispose to developing lymphomas of B cell origin. However, patients with autoimmune diseases of the gastrointestinal tract, such as celiac disease, exhibit a higher risk of NHL T cell lymphoma (63), a subset of NHL arising from a single T cell clone. Mutations in NOTCH1 have been found with high frequency in all major oncogenic subclasses of human T cell lymphomas, suggesting NOTCH1 plays a significant role in lymphomagenesis (64). In a small study, half of the T cell lymphomas examined carried activating NOTCH1 mutations and/or mutations in the NOTCH-inhibiting FBXW7, which encodes an ubiquitin ligase that degrades NOTCH (65, 66). In addition, NOTCH3 mRNA has also been noted in patients with T cell lymphoma, and thymocyte-specific NOTCH3IC transgenic mice develop T cell lymphoma of the spleen and lymph nodes (67, 68). Interestingly, NOTCH2 mRNA has been shown to be decreased in γ-irradiation-induced T cell lymphomas (69). T cell lymphomas treated in vitro with GSI showed growth inhibition and caspase-mediated apoptosis (65, 70). A role for NOTCH–NF-κB interaction has been described in mouse models of T cell lymphoma (67), and elevated NF-κB activity in the primary cells of patients with T cell lymphoma has been described in cases of acute but not chronic disease (71). As with other types of lymphomas discussed here, NOTCH signaling appears to play an important role in T cell survival in lymphomas (47).
Patients with certain autoimmune diseases face an increased risk of developing aggressive lymphomas. Based on the evidence presented here, we suggest that dysregulated NOTCH signaling is implicated both in the autoimmune responses and the associated lymphomas. Chronic antigenic stimulation and inflammation define the immunological environment of autoimmunity and are factors that can precipitate the onset of lymphoma. Furthermore, convergence of the NOTCH and NF-κB signaling pathways likely promote both the pro-inflammatory environment and transformation of stimulated clones. Inhibiting the NOTCH pathway in autoimmune disease may act to attenuate both conditions since NOTCH signaling may be a mediator both of the autoimmune process and the lymphomas that arise subsequently.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Anderson LA, Gadalla S, Morton LM, Landgren O, Pfeiffer R, Warren JL, et al. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int J Cancer (2009) 125:398–405. doi: 10.1002/ijc.24287
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Baecklund E, Smedby KE, Sutton LA, Askling J, Rosenquist R. Lymphoma development in patients with autoimmune and inflammatory disorders – what are the driving forces? Semin Cancer Biol (2014) 24:61–70. doi:10.1016/j.semcancer.2013.12.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Iso T, Kedes L, Hamamori Y. HES and HERP families: multiple effectors of the Notch signaling pathway. J Cell Physiol (2003) 194:237–55. doi:10.1002/jcp.10208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137:216–33. doi:10.1016/j.cell.2009.03.045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Shin HM, Tilahun ME, Cho OH, Chandiran K, Kuksin CA, Keerthivasan S, et al. NOTCH1 can initiate NF-kappaB activation via cytosolic interactions with components of the T cell signalosome. Front Immunol (2014) 5:249. doi:10.3389/fimmu.2014.00249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity (2010) 32:14–27. doi:10.1016/j.immuni.2010.01.004
7. Jundt F, Schwarzer R, Dorken B. Notch signaling in leukemias and lymphomas. Curr Mol Med (2008) 8:51–9. doi:10.2174/156652408783565540
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Palaga T, Minter L. Notch signaling and its emerging role in autoimmunity. Front Biol (2013) 8:279–94. doi:10.1007/s11515-012-1209-z
9. Ishii H, Nakazawa M, Yoshino S, Nakamura H, Nishioka K, Nakajima T. Expression of Notch homologues in the synovium of rheumatoid arthritis and osteoarthritis patients. Rheumatol Int (2001) 21:10–4. doi:10.1007/s002960100119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Jiao Z, Wang W, Guo M, Zhang T, Chen L, Wang Y, et al. Expression analysis of Notch-related molecules in peripheral blood T helper cells of patients with rheumatoid arthritis. Scand J Rheumatol (2010) 39:26–32. doi:10.3109/03009740903124424
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Yabe Y, Matsumoto T, Tsurumoto T, Shindo H. Immunohistological localization of Notch receptors and their ligands Delta and Jagged in synovial tissues of rheumatoid arthritis. J Orthop Sci (2005) 10:589–94. doi:10.1007/s00776-005-0943-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Nakazawa M, Ishii H, Aono H, Takai M, Honda T, Aratani S, et al. Role of Notch-1 intracellular domain in activation of rheumatoid synoviocytes. Arthritis Rheum (2001) 44:1545–54. doi:10.1002/1529-0131(200107)44:7<1545::AID-ART278>3.0.CO;2-Q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Ando K, Kanazawa S, Tetsuka T, Ohta S, Jiang X, Tada T, et al. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene (2003) 22:7796–803. doi:10.1038/sj.onc.1206965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Jiao Z, Wang W, Hua S, Liu M, Wang H, Wang X, et al. Blockade of Notch signaling ameliorates murine collagen-induced arthritis via suppressing Th1 and Th17 cell responses. Am J Pathol (2014) 184:1085–93. doi:10.1016/j.ajpath.2013.12.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Jiao Z, Wang W, Xu H, Wang S, Guo M, Chen Y, et al. Engagement of activated Notch signalling in collagen II-specific T helper type 1 (Th1)- and Th17-type expansion involving Notch3 and Delta-like1. Clin Exp Immunol (2011) 164:66–71. doi:10.1111/j.1365-2249.2010.04310.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Jiao Z, Wang W, Ma J, Wang S, Su Z, Xu H. Notch signaling mediates TNF-alpha-induced IL-6 production in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Clin Dev Immunol (2012) 2012:350209. doi:10.1155/2012/350209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Kijima M, Iwata A, Maekawa Y, Uehara H, Izumi K, Kitamura A, et al. Jagged1 suppresses collagen-induced arthritis by indirectly providing a negative signal in CD8+ T cells. J Immunol (2009) 182:3566–72. doi:10.4049/jimmunol.0803765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Nakazawa M, Ishii H, Nakamura H, Yoshino SI, Fukamizu A, Nishioka K, et al. NFkappaB2 (p52) promoter activation via Notch signaling pathway in rheumatoid synoviocytes. Int J Mol Med (2001) 7:31–6. doi:10.3892/ijmm.7.1.31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Zhang W, Xu W, Xiong S. Blockade of Notch1 signaling alleviates murine lupus via blunting macrophage activation and M2b polarization. J Immunol (2010) 184:6465–78. doi:10.4049/jimmunol.0904016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Teachey DT, Seif AE, Brown VI, Bruno M, Bunte RM, Chang YJ, et al. Targeting Notch signaling in autoimmune and lymphoproliferative disease. Blood (2008) 111:705–14. doi:10.1182/blood-2007-05-087353
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Hasegawa H, Kohno M, Sasaki M, Inoue A, Ito MR, Terada M, et al. Antagonist of monocyte chemoattractant protein 1 ameliorates the initiation and progression of lupus nephritis and renal vasculitis in MRL/lpr mice. Arthritis Rheum (2003) 48:2555–66. doi:10.1002/art.11231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Shimizu S, Nakashima H, Karube K, Ohshima K, Egashira K. Monocyte chemoattractant protein-1 activates a regional Th1 immunoresponse in nephritis of MRL/lpr mice. Clin Exp Rheumatol (2005) 23:239–42. doi:10.1093/ndt/gfn270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR. Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med (1999) 190:1813–24. doi:10.1084/jem.190.12.1813
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Rauen T, Grammatikos AP, Hedrich CM, Floege J, Tenbrock K, Ohl K, et al. cAMP-responsive element modulator alpha (CREMalpha) contributes to decreased Notch-1 expression in T cells from patients with active systemic lupus erythematosus (SLE). J Biol Chem (2012) 287:42525–32. doi:10.1074/jbc.M112.425371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Sodsai P, Hirankarn N, Avihingsanon Y, Palaga T. Defects in Notch1 upregulation upon activation of T Cells from patients with systemic lupus erythematosus are related to lupus disease activity. Lupus (2008) 17:645–53. doi:10.1177/0961203308089406
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Samon JB, Champhekar A, Minter LM, Telfer JC, Miele L, Fauq A, et al. Notch1 and TGFbeta1 cooperatively regulate Foxp3 expression and the maintenance of peripheral regulatory T cells. Blood (2008) 112:1813–21. doi:10.1182/blood-2008-03-144980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Le Pottier L, Devauchelle V, Fautrel A, Daridon C, Saraux A, Youinou P, et al. Ectopic germinal centers are rare in Sjogren’s syndrome salivary glands and do not exclude autoreactive B cells. J Immunol (2009) 182:3540–7. doi:10.4049/jimmunol.0803588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Guerrier T, Le Pottier L, Devauchelle V, Pers JO, Jamin C, Youinou P. Role of Toll-like receptors in primary Sjogren’s syndrome with a special emphasis on B-cell maturation within exocrine tissues. J Autoimmun (2012) 39:69–76. doi:10.1016/j.jaut.2012.01.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Liu F, Wang S. Molecular cues for development and regeneration of salivary glands. Histol Histopathol (2014) 29:305–12.
30. Musone SL, Taylor KE, Nititham J, Chu C, Poon A, Liao W, et al. Sequencing of TNFAIP3 and association of variants with multiple autoimmune diseases. Genes Immun (2011) 12:176–82. doi:10.1038/gene.2010.64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Nordmark G, Wang C, Vasaitis L, Eriksson P, Theander E, Kvarnstrom M, et al. Association of genes in the NF-kappaB pathway with antibody-positive primary Sjogren’s syndrome. Scand J Immunol (2013) 78:447–54. doi:10.1111/sji.12101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Sun F, Li P, Chen H, Wu Z, Xu J, Shen M, et al. Association studies of TNFSF4, TNFAIP3 and FAM167A-BLK polymorphisms with primary Sjogren’s syndrome in Han Chinese. J Hum Genet (2013) 58:475–9. doi:10.1038/jhg.2013.26
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Petersen J, Montserrat V, Mujico JR, Loh KL, Beringer DX, van Lummel M, et al. T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat Struct Mol Biol (2014) 21:480–8. doi:10.1038/nsmb.2817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Capuano M, Iaffaldano L, Tinto N, Montanaro D, Capobianco V, Izzo V, et al. MicroRNA-449a overexpression, reduced NOTCH1 signals and scarce goblet cells characterize the small intestine of celiac patients. PLoS One (2011) 6:e29094. doi:10.1371/journal.pone.0029094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Jeon MK, Klaus C, Kaemmerer E, Gassler N. Intestinal barrier: molecular pathways and modifiers. World J Gastrointest Pathophysiol (2013) 4:94–9. doi:10.4291/wjgp.v4.i4.94
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Trynka G, Zhernakova A, Romanos J, Franke L, Hunt KA, Turner G, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut (2009) 58:1078–83. doi:10.1136/gut.2008.169052
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem (2004) 279:12876–82. doi:10.1074/jbc.M311652200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Chiaramonte R. Still puzzling Notch signaling in B-cell malignancies. Leuk Res (2006) 30:1331–2. doi:10.1016/j.leukres.2006.07.009
39. Tohda S, Sato T, Kogoshi H, Fu L, Sakano S, Nara N. Establishment of a novel B-cell lymphoma cell line with suppressed growth by gamma-secretase inhibitors. Leuk Res (2006) 30:1385–90. doi:10.1016/j.leukres.2006.05.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Kluk MJ, Ashworth T, Wang H, Knoechel B, Mason EF, Morgan EA, et al. Gauging NOTCH1 activation in cancer using immunohistochemistry. PLoS One (2013) 8:e67306. doi:10.1371/journal.pone.0067306
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Lee SY, Kumano K, Nakazaki K, Sanada M, Matsumoto A, Yamamoto G, et al. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci (2009) 100:920–6. doi:10.1111/j.1349-7006.2009.01130.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kumar V, Matsuo K, Takahashi A, Hosono N, Tsunoda T, Kamatani N, et al. Common variants on 14q32 and 13q12 are associated with DLBCL susceptibility. J Hum Genet (2011) 56:436–9. doi:10.1038/jhg.2011.35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med (2001) 194:1861–74. doi:10.1084/jem.194.12.1861
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Feuerhake F, Kutok JL, Monti S, Chen W, LaCasce AS, Cattoretti G, et al. NFkappaB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood (2005) 106:1392–9. doi:10.1182/blood-2004-12-4901
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Odqvist L, Montes-Moreno S, Sanchez-Pacheco RE, Young KH, Martin-Sanchez E, Cereceda L, et al. NFkappaB expression is a feature of both activated B-cell-like and germinal center B-cell-like subtypes of diffuse large B-cell lymphoma. Mod Pathol (2014) 27(10):1331–7. doi:10.1038/modpathol.2014.34
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Thomas RK, Wickenhauser C, Tawadros S, Diehl V, Kuppers R, Wolf J, et al. Mutational analysis of the IkappaBalpha gene in activated B cell-like diffuse large B-cell lymphoma. Br J Haematol (2004) 126:50–4. doi:10.1111/j.1365-2141.2004.05000.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Jundt F, Anagnostopoulos I, Forster R, Mathas S, Stein H, Dorken B. Activated Notch1 signaling promotes tumor cell proliferation and survival in Hodgkin and anaplastic large cell lymphoma. Blood (2002) 99:3398–403. doi:10.1182/blood.V99.9.3398
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Kochert K, Ullrich K, Kreher S, Aster JC, Kitagawa M, Johrens K, et al. High-level expression of Mastermind-like 2 contributes to aberrant activation of the NOTCH signaling pathway in human lymphomas. Oncogene (2011) 30:1831–40. doi:10.1038/onc.2010.544
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Jundt F, Acikgoz O, Kwon SH, Schwarzer R, Anagnostopoulos I, Wiesner B, et al. Aberrant expression of Notch1 interferes with the B-lymphoid phenotype of neoplastic B cells in classical Hodgkin lymphoma. Leukemia (2008) 22:1587–94. doi:10.1038/leu.2008.101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, et al. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin’s disease tumor cells. J Clin Invest (1997) 100:2961–9. doi:10.1172/JCI119849
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood (1999) 94:3129–34.
52. Mathas S, Lietz A, Janz M, Hinz M, Jundt F, Scheidereit C, et al. Inhibition of NF-kappaB essentially contributes to arsenic-induced apoptosis. Blood (2003) 102:1028–34. doi:10.1182/blood-2002-04-1154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Emmerich F, Theurich S, Hummel M, Haeffker A, Vry MS, Dohner K, et al. Inactivating I kappa B epsilon mutations in Hodgkin/Reed-Sternberg cells. J Pathol (2003) 201:413–20. doi:10.1002/path.1454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Krappmann D, Emmerich F, Kordes U, Scharschmidt E, Dorken B, Scheidereit C. Molecular mechanisms of constitutive NF-kappaB/Rel activation in Hodgkin/Reed-Sternberg cells. Oncogene (1999) 18:943–53. doi:10.1038/sj.onc.1202351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Schwarzer R, Dorken B, Jundt F. Notch is an essential upstream regulator of NF-kappaB and is relevant for survival of Hodgkin and Reed-Sternberg cells. Leukemia (2012) 26:806–13. doi:10.1038/leu.2011.265
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Ekstrom Smedby K, Vajdic CM, Falster M, Engels EA, Martinez-Maza O, Turner J, et al. Autoimmune disorders and risk of non-Hodgkin lymphoma subtypes: a pooled analysis within the InterLymph Consortium. Blood (2008) 111:4029–38. doi:10.1182/blood-2007-10-119974
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Martinez N, Almaraz C, Vaque JP, Varela I, Derdak S, Beltran S, et al. Whole-exome sequencing in splenic marginal zone lymphoma reveals mutations in genes involved in marginal zone differentiation. Leukemia (2014) 28:1334–40. doi:10.1038/leu.2013.365
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med (2012) 209:1537–51. doi:10.1084/jem.20120904
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Troen G, Wlodarska I, Warsame A, Hernandez Llodra S, De Wolf-Peeters C, Delabie J. NOTCH2 mutations in marginal zone lymphoma. Haematologica (2008) 93:1107–9. doi:10.3324/haematol.11635
60. Nocturne G, Boudaoud S, Miceli-Richard C, Viengchareun S, Lazure T, Nititham J, et al. Germline and somatic genetic variations of TNFAIP3 in lymphoma complicating primary Sjogren’s syndrome. Blood (2013) 122:4068–76. doi:10.1182/blood-2013-05-503383
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Thieblemont C, Bertoni F, Copie-Bergman C, Ferreri AJ, Ponzoni M. Chronic inflammation and extra-nodal marginal-zone lymphomas of MALT-type. Semin Cancer Biol (2014) 24:33–42. doi:10.1016/j.semcancer.2013.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Zhu J, Wei RL, Pi YL, Guo Q. Significance of Bcl10 gene mutations in the clinical diagnosis of MALT-type ocular adnexal lymphoma in the Chinese population. Genet Mol Res (2013) 12:1194–204. doi:10.4238/2013.April.12.6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Tarella C, Gueli A, Ruella M, Cignetti A. Lymphocyte transformation and autoimmune disorders. Autoimmun Rev (2013) 12:802–13. doi:10.1016/j.autrev.2012.11.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Weng AP, Ferrando AA, Lee W, Morris JP, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science (2004) 306:269–71. doi:10.1126/science.1102160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Kamstrup MR, Biskup E, Gjerdrum LM, Ralfkiaer E, Niazi O, Gniadecki R. The importance of Notch signaling in peripheral T-cell lymphomas. Leuk Lymphoma (2014) 55:639–44. doi:10.3109/10428194.2013.807510
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Shimizu D, Taki T, Utsunomiya A, Nakagawa H, Nomura K, Matsumoto Y, et al. Detection of NOTCH1 mutations in adult T-cell leukemia/lymphoma and peripheral T-cell lymphoma. Int J Hematol (2007) 85:212–8. doi:10.1532/IJH97.06165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, et al. Constitutive activation of NF-kappaB and T-cell leukemia/lymphoma in Notch3 transgenic mice. EMBO J (2000) 19:3337–48. doi:10.1093/emboj/19.13.3337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Bellavia D, Campese AF, Checquolo S, Balestri A, Biondi A, Cazzaniga G, et al. Combined expression of pTalpha and Notch3 in T cell leukemia identifies the requirement of preTCR for leukemogenesis. Proc Natl Acad Sci U S A (2002) 99:3788–93. doi:10.1073/pnas.062050599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Lopez-Nieva P, Santos J, Fernandez-Piqueras J. Defective expression of Notch1 and Notch2 in connection to alterations of c-Myc and Ikaros in gamma-radiation-induced mouse thymic lymphomas. Carcinogenesis (2004) 25:1299–304. doi:10.1093/carcin/bgh124
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Ramakrishnan V, Ansell S, Haug J, Grote D, Kimlinger T, Stenson M, et al. MRK003, a gamma-secretase inhibitor exhibits promising in vitro pre-clinical activity in multiple myeloma and non-Hodgkin’s lymphoma. Leukemia (2012) 26:340–8. doi:10.1038/leu.2011.192
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Arima N, Matsushita K, Obata H, Ohtsubo H, Fujiwara H, Arimura K, et al. NF-kappaB involvement in the activation of primary adult T-cell leukemia cells and its clinical implications. Exp Hematol (1999) 27:1168–75. doi:10.1016/S0301-472X(99)00053-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: NOTCH, NF-κB, autoimmunity, lymphoma
Citation: Kuksin CA and Minter LM (2015) The link between autoimmunity and lymphoma: does NOTCH signaling play a contributing role? Front. Oncol. 5:51. doi: 10.3389/fonc.2015.00051
Received: 08 August 2014; Accepted: 11 February 2015;
Published online: 24 February 2015.
Edited by:
Aleksandra Filipovic, Imperial College London, UKReviewed by:
Massimiliano Agostini, Medical Research Council, UKSo-Ichiro Fukada, Osaka University, Japan
Copyright: © 2015 Kuksin and Minter. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M. Minter, Department of Veterinary and Animal Sciences, University of Massachusetts Amherst, 427K Integrated Sciences Building, 661 North Pleasant Street, Amherst, MA 01003, USA e-mail:bG1pbnRlckB2YXNjaS51bWFzcy5lZHU=