 Catherine Louise Penman
Catherine Louise Penman Claire Faulkner
Claire Faulkner Stephen P. Lowis
Stephen P. Lowis Kathreena M. Kurian
Kathreena M. Kurian- 1Brain Tumour Research Group, Institute of Clinical Neurosciences, University of Bristol, Bristol, UK
- 2Bristol Genetics Laboratory, Pathology Sciences Southmead Hospital, Westbury on Trym, Bristol, UK
- 3Department of Paediatric Oncology, Bristol Royal Hospital for Children, Upper Maudlin Street, Bristol, UK
The mitogen-activated protein kinase (MAPK) pathway is known to play a key role in the initiation and maintenance of many tumors as well as normal development. This often occurs through mutation of the genes encoding RAS and RAF proteins which are involved in signal transduction in this pathway. BRAF is one of three RAF kinases which act as downstream effectors of growth factor signaling leading to cell cycle progression, proliferation, and survival. Initially reported as a point mutation (V600E) in the majority of metastatic melanomas, other alterations in the BRAF gene have now been reported in a variety of human cancers including papillary thyroid cancer, colon carcinomas, hairy cell leukemia, and more recently in gliomas. The identification of oncogenic mutations in the BRAF gene have led to a revolution in the treatment of metastatic melanoma using targeted molecular therapies that affect the MAPK pathway either directly through BRAF inhibition or downstream through inhibition of MEK. This review describes the molecular biology of BRAF in the context of pediatric low-grade gliomas, the role of BRAF as a diagnostic marker, the prognostic implications of BRAF, and evidence for therapeutic targeting of BRAF.
Introduction
Pediatric low-grade gliomas (LGGs) represent the most common central nervous system (CNS) tumors of childhood, with pilocytic astrocytomas (PAs) being the most prevalent, accounting for 17% of brain and spinal neoplasms in children age 0–14 years (1, 2) (see Table 1). For the purposes of this review, the term LGGs is used to describe a heterogeneous group of tumors including both WHO grade I and II neoplasms (see Table 1) (3). The incidence varies from 0.26 to 1.79/100,000 depending on the histology and geographical region, with an overall higher incidence in males (1). Survival is variable with a 5-year overall survival in PAs being reported as high as 100%, compared to 45% in diffuse fibrillary astrocytoma (DA) (1, 4). PAs are relatively benign slow growing tumors occurring most commonly within the cerebellum, but may also arise along the optic tract, in the hypothalamus and brain stem where they are difficult to fully resect (5). Complete resection has been reported in 94% of cerebellar PAs compared with only 3.2% of hypothalamic and chiasmatic tumors, with an overall tumor recurrence rate of 19% for PAs (6, 7). PAs of the optic tract are common in the hereditary syndrome neurofibromatosis type 1 and are commonly associated with defect in the NF1 gene (8). NF1 loss may activate the mitogen-activated protein kinase (MAPK) pathway through RAS, which in turn may activate BRAF and the PI3K pathway (Figure 1) (8–10). DAs have been reported to represent approximately 3% of pediatric gliomas, although they more commonly occur in adults (11). Within the pediatric population, the higher grade gliomas, anaplastic astrocytoma (AA), and glioblastoma multiforme (GBM) account for approximately 5 and 6.5% of pediatric gliomas, respectively (11).
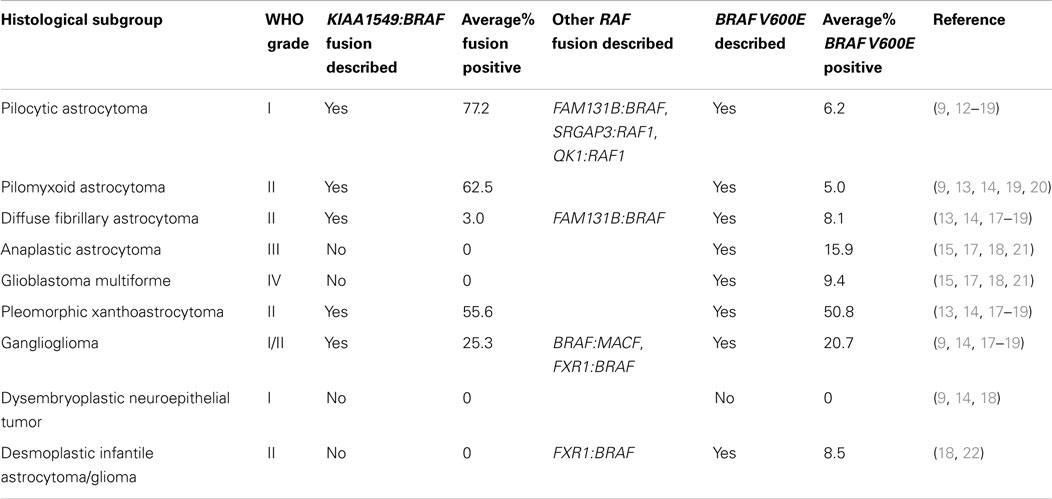
Table 1. Histological subgroups of low-grade and high-grade gliomas demonstrating in which tumors the BRAF gene fusion has been identified.
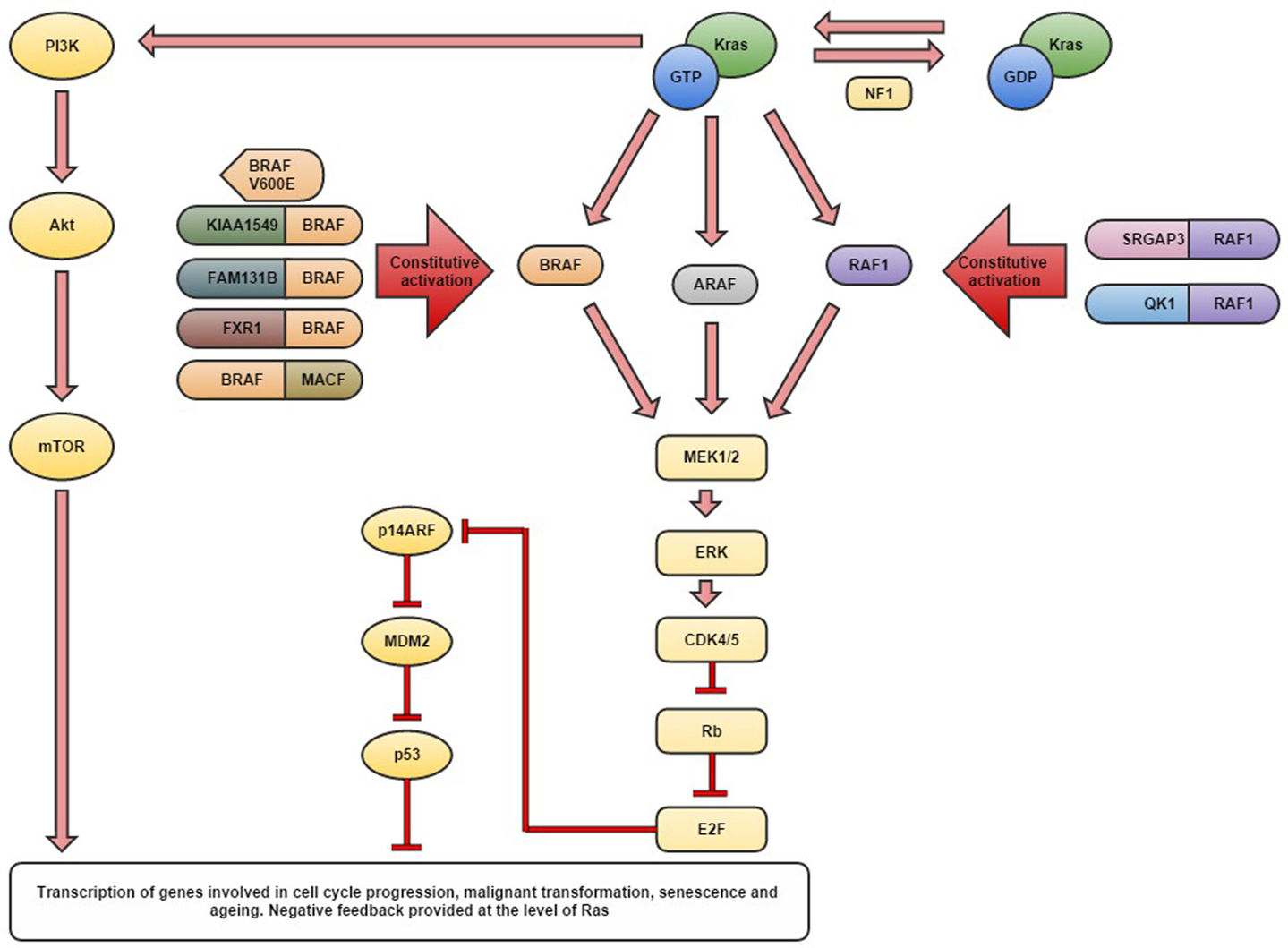
Figure 1. Schematic diagram detailing the currently identified aberrations in the MAPK pathway genes thought to be responsible for the development of pilocytic astrocytomas.
Molecular Biology of BRAF
BRAF fusion
Until recently, the molecular biology of pediatric LGGs was relatively unknown (see Figure 1 and Table 2). The genetic aberrations commonly seen in adult gliomas including p53, PTEN, CDK4, and p16 have not been identified in low-grade pediatric gliomas suggesting that they may be genetically distinct (23).
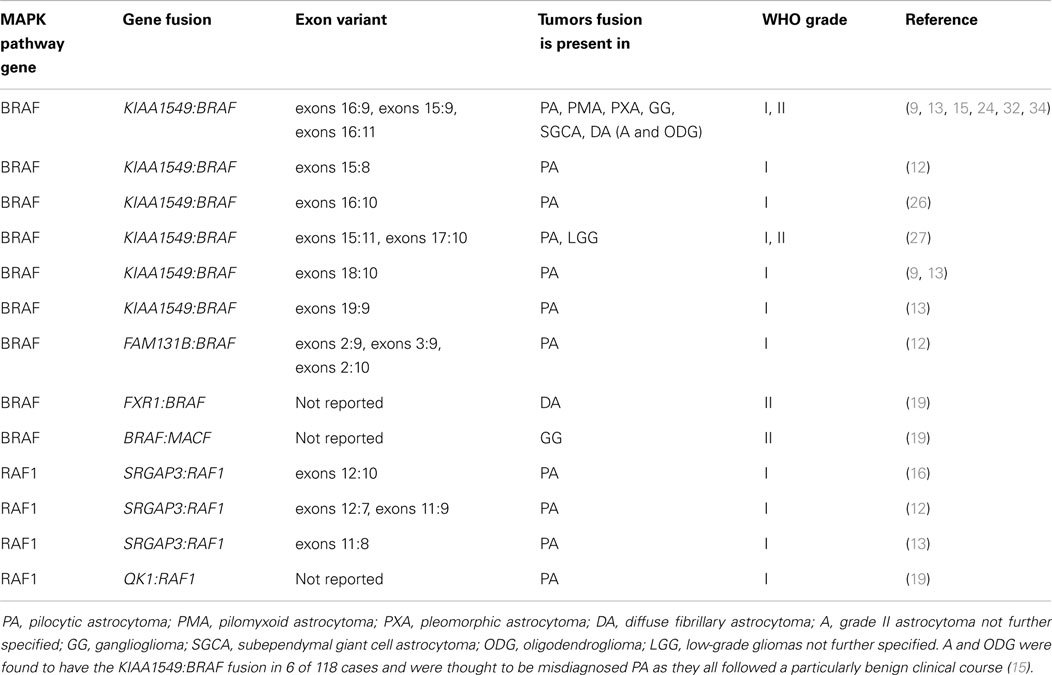
Table 2. Gene fusions involving members of the MAPK pathway including BRAF and in which low-grade gliomas the fusions have been identified.
In 2008, several papers identified a genetic defect in the BRAF gene thought to be responsible for constitutive activation of the MAPK pathway and thus development of PAs in children (15, 24, 25). Jones at al. described a tandem duplication at 7q34 resulting in fusion of the previously uncharacterized gene KIAA1549 and the BRAF gene to create a novel fusion oncogene in approximately 66% of PAs (15). Other less common fusion variants included exon 15:exon 9, exon 16:exon 11, exon 16:exon 10, exon 15:exon11, exon 17:exon 10, and exon 18:exon 10 (see Table 2) (9, 15, 26, 27). All fusions were found to have constitutive BRAF kinase activity and transforming ability in NIH3T3 cell lines (15). The constitutive kinase activity of KIAA1549:BRAF fusion oncoprotein is due to the loss of the BRAF N-terminal auto-inhibitory domain which usually regulates BRAF activity (15, 28). Lin et al. demonstrated that the KIAA1549:BRAF fusion is transcribed from the KIAA1549 gene promoter and thus is expressed at higher levels than wild-type BRAF, so the fusion oncoprotein is not only constitutively active but also over expressed giving two mechanisms of aberrant activity (27). Investigation into the effects of the oncogenic fusion protein formed by KIAA1549 and BRAF has revealed that the constitutive activation regulates neuroglial cell growth in a cell-type-specific manner causing proliferation of neural stem cells but not mature astrocytes (29). In certain studies, the KIAA1549:BRAF fusion gene was also found to function through MEK-dependent activation of both MAPK and mTOR pathways and the injection of neural stem cells containing the fusion were sufficient to induce glioma-like lesions in mice (29). Whereas other studies report that expression of the KIAA1549:BRAF fusion protein alone is not sufficient for gliomagenesis and instead results in senescence (29–31). More recently, several novel gene fusions have been discovered which may account for MAPK activation in PAs which do not harbor the KIAA1549:BRAF fusion variants, summarized in Table 2 (12, 13, 16, 26). A further novel mutation at codon 209 in the GNAQ gene has been described in a single case of PA (32). GNAQ encodes a Gα subunit of the guanine nucleotide-binding protein receptor involved in signaling upstream of RAS (33). Mutations in the GNAQ gene are thought to lead to constitutive activation of the MAPK pathway, independent of BRAF (33).
BRAF V600E mutation
Characterization of mutant proteins has revealed a mutation hotspot resulting in a valine to glutamate substitution at position 600, often referred to as BRAF V600E in a range of tumor types (20, 35–39). The BRAF protein and its close relation RAF1 (CRAF) are usually subject to auto-regulation through different N-terminal auto-inhibitory domains (28). The oncogenic V600E mutation lies within the activation segment disrupting the auto-inhibitory mechanism and converting BRAF into its active form thus allowing constitutive activation of the MAPK pathway (40). Gronych et al. demonstrated that overexpression of the mutant BRAF V600E kinase domain alone induced tumor formation with clinical and histological features of PAs whereas the full length V600E mutant protein which still contained the autoregulatory domain did not give rise to tumors (31). In inkk4a/ARF-deficient mice, the full length BRAF V600E could induce tumorigenesis but these more closely resembled high-grade astrocytomas (31). Lyustikman et al. demonstrated a causal relationship between constitutive activation of RAF1, and thus the downstream MAPK signaling pathway, and glioma formation in mice (30). Activation of RAF1 alone induced hyperplastic lesions in Ntv-a mice, yet such lesions did not progress without concomitant loss of ARF (see Figure 1). With loss of ARF and the RAF1 mutation, mice developed lesions similar to glioblastoma, yet without ARF loss, small hyperplastic lesions developed which may represent tumors more similar to PAs (30). Huillard et al. reported that the presence of BRAF V600E alone was insufficient for gliomagenesis and a concomitant homozygous deletion of CDKN2A (which encodes P14ARF and P16INK4A) was required for the development of astrocytomas from neural progenitor cells (Figure 1) (41). It was demonstrated that expression of BRAFV600E may cause transformation when combined with loss of CDKN2A in human neural progenitor cells and the resultant tumors displayed the histology of malignant astrocytomas (41).
Outwith the glioma biology field, it has been proposed that the BRAF V600E mutation may be responsible for the induction of growth arrest and senescence in melanocytic naevi, in a process termed “oncogene-induced senescence” whereby melanocytic naevi can remain in growth arrest for a lifetime (42). Oncogene-induced senescence has been described when BRAF V600E mutation was introduced with a lentiviral vector into human neurospheres derived from the cerebral cortex of first trimester human fetal autopsy specimens (43). Neurosphere cells initially underwent transformation, subsequently followed by senescence with expression of senescence-associated markers (44). Interestingly, Jacob et al. have described that the majority of PAs are senescent and that this effect is triggered through the p16ink4a pathway following aberrant MAPK activity (45). Moreover, Hawkins et al. overexpressed BRAF in hTERT immortalized human astrocytes and found that this caused growth arrest and senescence with associated DNA damage (32).
BRAF as a Diagnostic Marker
BRAF fusion
The KIAA1549:BRAF fusion has been reported in a range of PAs (59–90%) and so it is increasingly used as a diagnostic marker for PAs, where neuropathological distinction from malignant glioma can be difficult (46, 47). The literature is divided on the incidence of BRAF fusions in other pediatric LGGs which has implications for testing of these entities (9, 14, 22, 43, 46, 47). A few studies have shown a lack of BRAF fusions in ganglioglioma (GG), desmoplastic infantile GG/astrocytoma, dysembryoplastic neuroepithelial tumor, pilomyxoid astrocytoma (PMA), and pleomorphic xanthoastrocytoma (PXA) (9, 22, 43, 46, 47). By contrast, Horbinski et al. describe BRAF rearrangements in up to 15% of non-pilocytic LGGs including GG, PMA, and PXA, but not DAs (14). In a larger study by Hawkins et al. of pediatric low-grade astrocytomas including 105 PAs, 6 PMAs, 71 diffuse astrocytomas, and 4 unspecified low-grade astrocytomas, BRAF fusions were described in 62% of PAs, 67% of PMAs, 37% of diffuse astrocytomas, and 50% of unspecified low-grade astrocytomas.
Cin et al. screened 125 primary PAs for the known KIAA1549:BRAF fusion, the SRGAP3:RAF1 fusion, and also described a novel fusion between the otherwise uncharacterized gene product FAM131B and BRAF (12). In this study, fusions were identified in 82% of cerebellar PAs and 57% of non-cerebellar PAs, and a further 4.8% of tumors contained the BRAF V600E mutation (12). Cykowski et al. report the use of the KIAA1549:BRAF fusion and TP53 to distinguish PAs with atypical features from the highly malignant glioblastoma (48). Korshunov et al. describe the use of the KIAA1549:BRAF fusion to distinguish PAs from R132H IDH1 mutation positive diffuse astrocytoma counterparts with high specificity, with a consistent lack of R132H IDH1 mutation positivity in PAs (49).
BRAF V600E mutation
In a large cohort study of CNS tumors ranging from grade I to grade IV cancers, BRAF V600E mutation occurred most frequently in 66.7% of PXAs, but was also found at lower levels in PAs, GGs, and malignant gliomas (17). In another cohort study of over 1,300 CNS tumors, 66.7% of PXAs, 18% of GGs, and 9% of extra-cerebellar PAs harbored the BRAF V600E mutation (4). Further mutations have been found in the BRAF gene in gliomas including a 3bp insertion at codon 598 which mimics the V600E mutation (16). Further findings indicate that aberrations of MYB and MYBL1 may help distinguish LGGs from PAs as these aberrations were found in 68% of diffuse astrocytomas but 0% pilocytic tumors (42). The BRAF V600E mutant and the KIAA1549:BRAF fusion are generally mutually exclusive with only a few cases reported with both the fusion and the V600E mutation (12, 14, 32).
In summary, although currently assessment of the BRAF fusion is of most diagnostic use in posterior fossa PAs, and the BRAF V600E mutation is more prevalent in PXAs, both of these alterations have been described to varying degrees in other pediatric LGGs including DA.
Prognostic Implications of BRAF Status
Although there is much debate over the association of the fusion status with outcome, it remains generally accepted that the patient age, location of the tumor, and extent of resection are the most important prognostic indicators (6, 7, 14, 50). However, the presence of the KIAA1549:BRAF fusion positive compared to fusion negative pediatric LGGs has been associated with improved outcome in two studies (14, 32) and reported to have no effect on outcome in four (6, 15, 27, 34). For example, in one of the positive studies, the KIAA1549:BRAF fusion was associated with better clinical outcome in the large cohort by Hawkins et al. of pediatric low-grade astrocytomas including PAs, PMAs, and diffuse astrocytomas (32). In this study, all LGGs included were extra-cerebellar, incompletely resected (less than 75% resection), and followed up for more than 1 year (32). The overall findings were that the 5-year progression-free survival (PFS) was 61% in BRAF fusion positive tumors compared to 18% in fusion negative tumors (with PFS defined as greater than 25% increase in tumor volume on consecutive MRI scans) (32). Moreover, multivariate analysis revealed that BRAF fusion was an independent prognostic factor in incompletely resected LGGs (32). Significantly, all fusion negative PA patients under the age of 18 months experienced progression of their tumors within 8 years of diagnosis, and three of five patients in this group died within the study period of 16 years (32). In the second positive study, Horbinski et al. also report the presence of BRAF rearrangements as a positive prognostic marker in a cohort of LGGs including PAs, GGs, PMAs, PXAs, diffuse astrocytomas, oligodendrogliomas (ODGs), subependymal giant cell astrocytomas (SGCAs), dysembryoplastic neuroepithelial tumors, and LGGs not otherwise specified (14). BRAF rearrangement was found to be associated with longer PFS and decreased risk of death, only one fusion positive patient died within the 20-year follow-up period (14).
However, it is possible that the BRAF fusion is more a diagnostic marker of the PA which has an inherent better prognosis, as other studies of PA demonstrate no survival advantage in BRAF fusion positive cases compared with negative cases (6, 15, 27, 51). In a small cohort of PAs, the presence of the BRAF V600E mutation has been reported to be significantly associated with both diffusely infiltrating architecture and increased risk of progression in LGGs (14, 52).
The association with location is less controversial with mounting evidence that the BRAF V600E mutation is more common in supratentorial PAs and the KIAA1549:BRAF fusion being more common in posterior fossa PAs (14, 33, 34, 53, 54). However, further evidence from Hawkins et al. looking specifically at supratentorial LGGs found that midline tumors which are usually unresectable were more likely to harbor the KIAA1549:BRAF fusion (65% cases) compared to only 11% of lobar tumors which were found to be BRAF fusion positive (32). In this study, it was also found that BRAF fusion positive tumors had better 5-year PFS irrespective of tumor histotype or location with 5-year PFS of 65% in fusion positive PAs compared to 17% for fusion negative tumors (32).
Therapeutic Targeting of BRAF and MAPK Pathway
BRAF and MAPK inhibitors in cell lines and animal models
Non-central nervous system cell lines and xenografts. Therapeutic manipulation of the BRAF and MAPK pathway has been extensively investigated in many tumor types (see Figure 1 and Table 3). For example, sorafenib is a potent RAF1 inhibitor with action against BRAF, PDGFRβ, and VEGFR-3 (55). Sorafenib has been shown to act against both wild-type and BRAF V600E mutant cell lines and colon, breast, and non-small cell lung cancer xenograft models (55, 56). Inhibition of tumor growth in these models was not associated with any apparent toxicities and appeared to be elicited through abrogation of MAPK signaling; however, the drug did not discriminate between those cells with aberrant MAPK activity and wild-type cells (55). Tsai et al. report the discovery of a selective inhibitor PLX4720 of BRAF V600E whose cytotoxic effects are specific to mutant cells only (57). This PLX4720 drug induced cell cycle arrest and apoptosis in V600E mutant cell lines derived from colon carcinoma, and melanoma cells and yet had minimal effects in other cancer cell lines which did not possess the V600E mutant, including RAS-mutant colon carcinoma cells, large cell lung cancer cells, and metastatic melanoma (57). Moreover, a dose of 20 mg/kg PLX4720 was given orally for 14 days to mice with colorectal carcinoma xenografts which harbored the V600E mutation, achieving tumor regression below palpable levels in four of nine mice (57). Solit et al. demonstrated that BRAF mutant cells, unlike their wild-type relatives, are dependent on MEK signaling for growth and survival and therefore BRAF V600E mutant cells had enhanced sensitivity to MEK inhibitors compared to wild-type BRAF cells and NRAS mutant cells (58). Furthermore, daily treatment with the MEK inhibitor PD0325901 in BRAF V600E mice xenografts showed complete suppression of growth, whereas wild-type xenografts were insensitive to MEK inhibition (58).
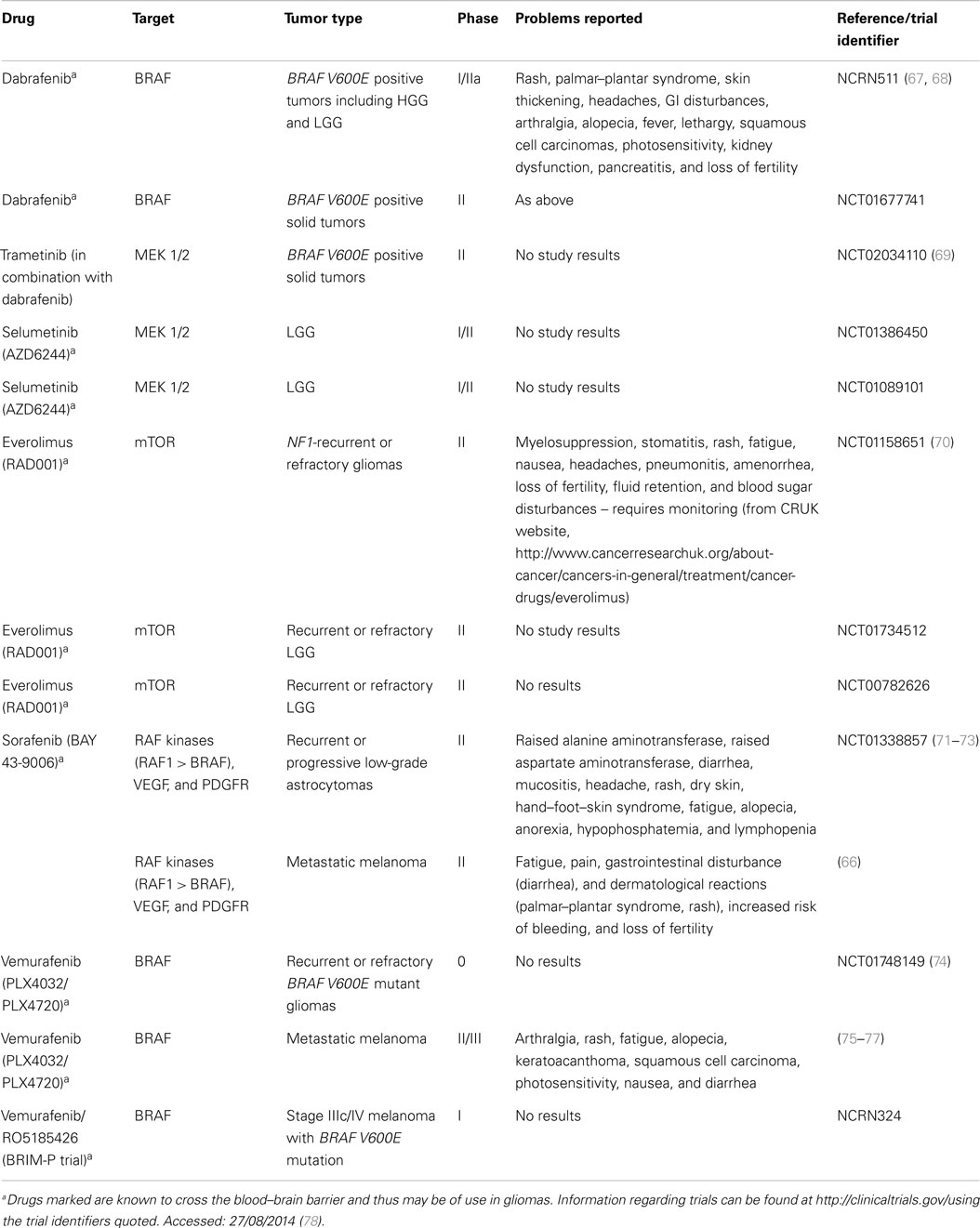
Table 3. Small molecule inhibitors currently in clinical trials to evaluate safety and efficacy in pediatric low-grade gliomas and other tumors.
Central nervous system cell lines and xenografts. In gliomas, Huillard et al. report significantly increased survival in mice transplanted with human V600E mutated astrocytoma cells when treated with the first generation BRAF inhibitor vemurafenib (PLX4720) (41). In these intracranial astrocytoma models, the overall tumor size was decreased with BRAF inhibitor treatment, whereas no response was observed when administered with the wild-type BRAF control treatment (41). Moreover, Nicolaides et al. demonstrated efficacy of the BRAFV600E inhibitor PLX4720 in reducing tumor growth and increasing survival in the BRAFV600E mouse model, whilst being ineffective in the wild-type xenograft models (21). A MEK inhibitor AZD6244 was tested against the pediatric pre-clinical testing in vitro panel with the highest in vitro concentrations inhibiting growth by 50% in 5 of 23 cell lines derived from a variety of pediatric cancers including glioblastoma, rhabdomyosarcoma, rhabdoid tumors, Ewing’s sarcoma, neuroblastoma, acute lymphoblastic leukemia, acute myeloid leukemia, anaplastic large cell lymphoma, and non-Hodgkin’s lymphoma (59). When the MEK inhibitor AZD6244 was tested against PA xenograft models, complete regression was observed in the BRAF V600E mutant xenograft whereas tumor progression was observed in wild-type BRAF xenografts (59).
See et al. further report the use of MEK inhibitors (AZD6244 or PD0325901) in NF1-deficient GBM cell lines with resultant growth inhibition in a subset of cells (both agents) and in vivo in nude mouse xenograft models (PD03125901) (8). This may indicate that other tumors driven by NF1 loss, including PA associated with neurofibromatosis, may be amenable to small molecule inhibition of downstream targets in the MAPK pathway (8).
Paradoxical activation, whereby the BRAF inhibitor activates the MAPK pathway, has been described in cells with wild-type BRAF treated with a first generation BRAF inhibitor and appears to be caused by transactivation of RAF dimers to initiate ERK signaling (60). Moreover, Hatzivassilliou et al. have called for careful patient selection in RAF inhibitor trials due to the finding of paradoxical activation of the MAPK pathway in V600E negative cells through RAF1 priming (61). Second generation BRAF inhibitors, such as PLX PB3, on the other hand, have been found to have equal action against both the mutant and fusion protein (62). These BRAF inhibitors do not cause paradoxical activation in cells containing wild-type BRAF resulting in the desired effects of MAPK pathway inhibition and decreased proliferation (62).
BRAF and MAPK inhibitors in clinical cohorts
Currently, there are few trials involving MAPK inhibiting agents and pediatric LGGs (ongoing trials are summarized in Table 3).
BRAF and MAPK inhibitors in melanoma. Much of the clinical research into agents such as BRAF or MEK inhibitors has been performed on V600E positive metastatic melanomas in adults (63–66). Although these tumors are distinct from pediatric gliomas, information gained on safety, toxicity, dosage, and efficacy of these novel agents can help us understand the potential of these new drugs in treating these LGGs arising with similar molecular alterations (39, 43).
Bollag et al. report 26/36 metastatic melanoma patients achieving a partial or complete response to maximum tolerated dose of vemurafenib in a phase I clinical trial (78). The main problem reported in this trial was the development of multiple skin squamous cell carcinomas in 31% patients, a finding which has been replicated in several other BRAF inhibitor trials (64, 75, 78). A further phase II study in patients with metastatic melanoma reported an overall response rate of 53% with vemurafenib, with a median overall survival 15.9 months compared to 6–10 months with standard therapy (77). A phase III clinical trial of vemurafenib compared to the standard therapy dacarbazine in previously untreated metastatic melanoma with known BRAF V600E mutation demonstrated an increase in overall survival of 20%, and a reduction of 63% in the risk of death in the vemurafenib group (75). Common adverse effects reported with the BRAF inhibitor were again cutaneous squamous cell carcinoma, as well as those detailed in Table 3. The overwhelming success of treatment of metastatic melanoma with this novel agent led to the decision that patients originally randomized to dacarbazine should be allowed to cross over into the other treatment group (75).
BRAF and MAPK inhibitors in pediatric glioma. In pediatric low-grade astrocytomas, a recent phase II trial of the multikinase inhibitor sorafenib (Table 3) was discontinued early due to the unexpected acceleration of tumor growth (73). Eleven patients with progressive LGG following at least one chemotherapy treatment were recruited, three of which were positive for NF1 and five who possessed the BRAF gene alteration KIAA1549:BRAF (73). The pathology included pilocytic and PMAs, DA, GG, and LGG not otherwise specified (73). The median time to tumor progression was 2.8 months in nine patients, and after three cycles of treatment, all but two participants had experiences disease progression (73). Sorafenib was well tolerated but the striking finding was the rapid progression of these usually slow growing tumors leading to the discontinuation of this trial (73).
Resistance to inhibitors
Resistance to inhibitors in melanoma. Duration of response to BRAF inhibitors in metastatic melanoma has been recorded as ranging from 2 to 18 months (64). Several diverse mechanisms for this resistance have been proposed, including receptor tyrosine kinase and NRAS upregulation (79), dimerization of wild-type or fused BRAF (60, 62, 80), and through the other RAF kinases most notably RAF1 (60, 61, 81–83). RAF1 mutations lead to increased homodimerization and heterodimerization with BRAF following exposure to the RAF inhibitor PLX4032 (82). Increased dimerization leads to the development of resistance to PLX4032, known clinically as vemurafenib, in A725 cells (82). Emery et al. report novel MEK 1 mutations arising as a consequence of treatment of metastatic melanoma with the MEK inhibitor AZD6244 (84). MEK mutations were found either in the allosteric drug binding site or in one of the function domains and were shown to cause pharmacological resistance to both AZD6244 and cross resistance to the BRAF inhibitor PLX4720 (84). Other MEK1 mutations have been described as a mechanism conferring resistance in metastatic melanoma from a patient with an initial near-complete response to PLX4032 with subsequent relapse in week 16 of treatment leading to rapid disease progression and death (85). Further mechanisms of developing resistance have been described including increased COT expression (a gene transcribed from MAP3K8 which acts as a MAPK agonist) which increases phosphorylation of ERK and MEK in a RAF-independent fashion leading to PLX4720 resistance in metastatic melanoma cell lines (86). Furthermore, this resistance extended to MEK inhibition with AZD6244 indicating that COT may act through both MEK-dependent and -independent mechanisms (86).
Overcoming limitations through combination therapy
Combined BRAF and MEK inhibition has been attempted as a method of overcoming resistance (83). Combined therapy using both a BRAF inhibitor (PLX4720) and a MEK inhibitor (AZD6244) has demonstrated the ability to overcome resistance to MEK inhibition in metastatic melanoma cell lines (78, 80). Fragomeni et al. demonstrated the ability to induce complete regression of V600E positive xenograft metastatic melanomas through combining CRM1 and BRAF inhibition (87). CRM1 is known to play a role in melanoma proliferation, but has not been found to be part of tumor development or progression in LGGs to date (87). Flexible switching between RAF isoforms as a mechanism of resistance to RAF inhibitors may be overcome through co-targeting of MEK and IGF1R or PI3K in BRAF inhibitor-resistant melanoma cells (BRAF inhibitor-SB-590885) (25). A single RAF isoform was found to be sufficient to activate downstream signaling and knock down of two isoforms was not sufficient to induce cell cycle arrest seen when all three isoforms were blocked (25).
Huillard et al. demonstrated the increased efficacy of dual therapy combining the BRAF V600E inhibitor PLX4720 and the CDK4/6 inhibitor PD0332991 in human GBM xenograft models compared to either agent as a monotherapy (41). Combination of these two agents directly targeted two distinct enzymatic activities and was shown to suppress the paradoxical stimulation of Akt (Figure 1) which occurs with treatment of xenografts with PLX4720 alone (41).
In clinical cohorts, the timing and order of combination therapy has been found to impact on outcome, with prolonged survival being seen in patients receiving MEK inhibitors before BRAF inhibitors compared to the reverse sequence of drug administration (88). Moreover, there was increased tolerability of combination therapies in this sequence, with a lower incidence of the development of neoplastic cutaneous skin lesions (88). The testing of agents in combination to overcome resistance is interesting in the pre-clinical setting. However, at present, they cannot be considered for clinical trials in pediatric patients due to regulatory issues.
Conclusion
Currently, assessment of the KIAA1549 BRAF fusion is most useful as a diagnostic biomarker in posterior fossa PAs. Although the BRAF V600E mutation is more prevalent in PXAs, it has been described to varying degrees in other pediatric LGGs and is of less diagnostic use. In terms of prognosis, the KIAA1549 BRAF fusion has been described as an independent positive prognostic biomarker in low-grade pediatric gliomas, irrespective of tumor type. As more targeted molecular therapies become available, there will be increased pressure for testing for both the BRAF V600E and the KIAA1549 BRAF fusion in order to predict treatment options in pediatric LGGs. However, it will be important to learn from lessons from therapeutic targeting of BRAF in metastatic melanoma, whereby targeting one pathway may induce resistance through upregulation of other pathways. Initial cell line work suggests that a combinatorial approach may be more successful. To date although clinical trials are underway, the benefit of targeting BRAF in pediatric LGG is as yet unproven.
Author Contributions
CP and KK: manuscript research and writing. CF and SL: manuscript review and revision.
Conflict of Interest Statement
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
Abbreviations
A, grade II astrocytoma; AA, anaplastic astrocytoma; CNS, central nervous system; DA, diffuse fibrillary astrocytoma; GBM, glioblastoma multiforme; GG, ganglioglioma; HGG, high-grade glioma; LGG, low-grade glioma; MAPK, mitogen-activated protein kinase; ODG, oligodendroglioma; PA, pilocytic astrocytoma; PFS, progression-free survival; PMA, pilomyxoid astrocytoma; PXA, pleomorphic xanthoastrocytoma; SGCA, subependymal giant cell astrocytoma.
References
1. Johnson KJ, Cullen J, Barnholtz-Sloan JS, Ostrom QT, Langer CE, Turner MC, et al. Childhood brain tumor epidemiology: a brain tumor epidemiology consortium review. Cancer Epidemiol Biomarkers Prev (2014) 23(12):2716–36. doi:10.1158/1055-9965.EPI-14-0207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. McKean-Cowdin R, Razavi P, Barrington-Trimis J, Baldwin RT, Asgharzadeh S, Cockburn M, et al. Trends in childhood brain tumor incidence, 1973-2009. J Neurooncol (2013) 115(2):153–60. doi:10.1007/s11060-013-1212-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol (2007) 114(2):97–109. doi:10.1007/s00401-007-0278-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol (2005) 109(1):93–108. doi:10.1007/s00401-005-0991-y
5. Dirven CMF, Mooij JJA, Molenaar WM. Cerebellar pilocytic astrocytoma: a treatment protocol based upon analysis of 73 cases and a review of the literature. Childs Nerv Syst (1997) 13(1):17–23. doi:10.1007/s003810050033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Colin C, Padovani L, Chappe C, Mercurio S, Scavarda D, Loundou A, et al. Outcome analysis of childhood pilocytic astrocytomas: a retrospective study of 148 cases at a single institution. Neuropathol Appl Neurobiol (2013) 39(6):693–705. doi:10.1111/nan.12013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Stokland T, Liu J-F, Ironside JW, Ellison DW, Taylor R, Robinson KJ, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol (2010) 12(12):1257–68. doi:10.1093/neuonc/noq092
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. See WL, Tan IL, Mukherjee J, Nicolaides T, Pieper RO. Sensitivity of glioblastomas to clinically available MEK inhibitors is defined by neurofibromin 1 deficiency. Cancer Res (2012) 72(13):3350–9. doi:10.1158/0008-5472.CAN-12-0334
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Dimitriadis E, Alexiou GA, Tsotsou P, Simeonidi E, Stefanaki K, Patereli A, et al. BRAF alterations in pediatric low grade gliomas and mixed neuronal-glial tumors. J Neurooncol (2013) 113(3):353–8. doi:10.1007/s11060-013-1131-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Yu J, Deshmukh H, Gutmann RJ, Emnett RJ, Rodriguez FJ, Watson MA, et al. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology (2009) 73(19):1526–31. doi:10.1212/WNL.0b013e3181c0664a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Qaddoumi I, Sultan I, Gajjar A. Outcome and prognostic features in pediatric gliomas a review of 6212 cases from the surveillance, epidemiology, and end results database. Cancer (2009) 115(24):5761–70. doi:10.1002/cncr.24663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Cin H, Meyer C, Herr R, Janzarik WG, Lambert S, Jones DTW, et al. Oncogenic FAM131B-BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol (2011) 121(6):763–74. doi:10.1007/s00401-011-0817-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Forshew T, Tatevosslan RG, Lawson ARJ, Ma J, Neale G, Ogunkolade BW, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol (2009) 218(2):172–81. doi:10.1002/path.2558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Horbinski C, Nikiforova MN, Hagenkord JM, Hamilton RL, Pollack IF. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol (2012) 14(6):777–89. doi:10.1093/neuonc/nos077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Jones DTW, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res (2008) 68(21):8673–7. doi:10.1158/0008-5472.CAN-08-2097
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Jones DTW, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene (2009) 28(20):2119–23. doi:10.1038/onc.2009.73
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Myung JK, Cho H, Park C-K, Kim S-K, Lee S-H, Park S-H. Analysis of the BRAF(V600E) mutation in central nervous system tumors. Transl Oncol (2012) 5(6):430–6. doi:10.1593/tlo.12328
18. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol (2011) 121(3):397–405. doi:10.1007/s00401-011-0802-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nature Gene (2013) 45(6):602–12. doi:10.1038/ng.2611
20. Skrypek M, Foreman N, Guillaume D, Moertel C. Pilomyxoid astrocytoma treated successfully with vemurafenib. Pediatr Blood Cancer (2014) 61(11):2099–100. doi:10.1002/pbc.25084
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Nicolaides TP, Li H, Solomon DA, Hariono S, Hashizume R, Barkovich K, et al. Targeted therapy for BRAF(V600E) malignant astrocytoma. Clin Cancer Res (2011) 17(24):7595–604. doi:10.1158/1078-0432.CCR-11-1456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Koelsche C, Sahm F, Paulus W, Mittelbronn M, Giangaspero F, Antonelli M, et al. BRAF V600E expression and distribution in desmoplastic infantile astrocytoma/ganglioglioma. Neuropathol Appl Neurobiol (2014) 40(3):337–44. doi:10.1111/nan.12072
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Cheng Y, Pang JCS, Ng HK, Ding M, Zhang SF, Zheng J, et al. Pilocytic astrocytomas do not show most of the genetic changes commonly seen in diffuse astrocytomas. Histopathology (2000) 37(5):437–44. doi:10.1046/j.1365-2559.2000.01005.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest (2008) 118(5):1739–49. doi:10.1172/JCI33656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Sievert AJ, Jackson EM, Gai X, Hakonarson H, Judkins AR, Resnick AC, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol (2009) 19(3):449–58. doi:10.1111/j.1750-3639.2008.00225.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Dahiya S, Yu J, Kaul A, Leonard JR, Gutmann DH. Novel BRAF alteration in a sporadic pilocytic astrocytoma. Case Report Med (2012) 2012:418672. doi:10.1155/2012/418672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Lin A, Rodriguez FJ, Karajannis MA, Williams SC, Legault G, Zagzag D, et al. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol (2012) 71(1):66–72. doi:10.1097/NEN.0b013e31823f2cb0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Tran NH, Wu XC, Frost JA. B-Raf and Raf-1 are regulated by distinct autoregulatory mechanisms. J Biol Chem (2005) 280(16):16244–53. doi:10.1074/jbc.M501185200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kaul A, Chen Y-H, Emnett RJ, Dahiya S, Gutmann DH. Pediatric glioma-associated KIAA1549:BRAF expression regulates neuroglial cell growth in a cell type-specific and mTOR-dependent manner. Genes Dev (2012) 26(23):2561–6. doi:10.1101/gad.200907.112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Lyustikman Y, Momota H, Pao W, Holland EC. Constitutive activation of Raf-1 induces glioma formation in mice. Neoplasia (2008) 10(5):501–10. doi:10.1593/neo.08206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Gronych J, Korshunov A, Bageritz J, Milde T, Jugold M, Hambardzumyan D, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest (2011) 121(4):1344–8. doi:10.1172/JCI44656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Hawkins C, Walker E, Mohamed N, Zhang C, Jacob K, Shirinian M, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res (2011) 17(14):4790–8. doi:10.1158/1078-0432.CCR-11-0034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Laviv Y, Toledano H, Michowiz S, Dratviman-Storobinsky O, Turm Y, Fichman-Horn S, et al. BRAF, GNAQ, and GNA11 mutations and copy number in pediatric low-grade glioma. FEBS Open Bio (2012) 2:129–34. doi:10.1016/j.fob.2012.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Horbinski C, Hamilton RL, Nikiforov Y, Pollack IF. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol (2010) 119(5):641–9. doi:10.1007/s00401-009-0634-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature (2002) 417(6892):949–54. doi:10.1038/nature00766
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Sinicrope FA, Smyrk TC, Tougeron D, Thibodeau SN, Singh S, Muranyi A, et al. Mutation-specific antibody detects mutant BRAF(V600E) protein expression in human colon carcinomas. Cancer (2013) 119(15):2765–70. doi:10.1002/cncr.28133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Kebebew E, Weng J, Bauer J, Ranvier G, Clark OH, Duh Q-Y, et al. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann Surg (2007) 246(3):466–71. doi:10.1097/SLA.0b013e318148563d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Kurt B, Yalcin S, Alagoz E, Karslioglu Y, Yigit N, Gunal A, et al. The relationship of the BRAF(V600E) mutation and the established prognostic factors in papillary thyroid carcinomas. Endocr Pathol (2012) 23(3):135–40. doi:10.1007/s12022-012-9218-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Busam KJ, Hedvat C, Pulitzer M, von Deimling A, Jungbluth AA. Immunohistochemical analysis of BRAF(V600E) expression of primary and metastatic melanoma and comparison with mutation status and melanocyte differentiation antigens of metastatic lesions. Am J Surg Pathol (2013) 37(3):413–20. doi:10.1097/PAS.0b013e318271249e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell (2004) 116(6):855–67. doi:10.1016/S0092-8674(04)00215-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Huillard E, Hashizume R, Phillips JJ, Griveau A, Ihrie RA, Aoki Y, et al. Cooperative interactions of BRAF(V600E) kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci U S A (2012) 109(22):8710–5. doi:10.1073/pnas.1117255109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst C, et al. BRAF(E600)-associated senescence-like cell cycle arrest of human naevi. Nature (2005) 436(7051):720–4. doi:10.1038/nature03890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, et al. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol (2010) 12(7):621–30. doi:10.1093/neuonc/noq007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Raabe EH, Lim KS, Kim JM, Meeker A, Mao X-G, Nikkhah G, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res (2011) 17(11):3590–9. doi:10.1158/1078-0432.CCR-10-3349
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Jacob K, Quang-Khuong D-A, Jones DTW, Witt H, Lambert S, Albrecht S, et al. Genetic aberrations leading to MAPK pathway activation mediate oncogene-induced senescence in sporadic pilocytic astrocytomas. Clin Cancer Res (2011) 17(14):4650–60. doi:10.1158/1078-0432.CCR-11-0127
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Jacob K, Albrecht S, Sollier C, Faury D, Sader E, Montpetit A, et al. Duplication of 7q34 is specific to juvenile pilocytic astrocytomas and a hallmark of cerebellar and optic pathway tumours. Br J Cancer (2009) 101(4):722–33. doi:10.1038/sj.bjc.6605179
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Lawson ARJ, Tatevossian RG, Phipps KP, Picker SR, Michalski A, Sheer D, et al. RAF gene fusions are specific to pilocytic astrocytoma in a broad paediatric brain tumour cohort. Acta Neuropathol (2010) 120(2):271–3. doi:10.1007/s00401-010-0693-y
48. Cykowski MD, Allen RA, Kanaly AC, Fung K-M, Marshall R, Perry A, et al. The differential diagnosis of pilocytic astrocytoma with atypical features and malignant glioma: an analysis of 16 cases with emphasis on distinguishing molecular features. J Neurooncol (2013) 115(3):477–86. doi:10.1007/s11060-013-1249-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol (2009) 118(3):401–5. doi:10.1007/s00401-009-0550-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Tihan T, Ersen A, Qaddoumi I, Sughayer MA, Tolunay S, Al-Hussaini M, et al. Pathologic characteristics of pediatric intracranial pilocytic astrocytomas and their impact on outcome in 3 countries: a multi-institutional study. Am J Surg Pathol (2012) 36(1):43–55. doi:10.1097/PAS.0b013e3182329480
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Cruz GR, Oliveira ID, Moraes L, Paniago MDG, de Seixas Alves MT, Capellano AM, et al. Analysis of KIAA1549-BRAF fusion gene expression and IDH1/IDH2 mutations in low grade pediatric astrocytomas. J Neurooncol (2014) 117(2):235–42. doi:10.1007/s11060-014-1398-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Kandala G, Bannykh S, Fan S, Baden K, Pau A, Baden L, et al. Pilocytic astrocytomas with infiltrating patterns of growth carry a high rate of BRAF V600E mutation. Modern Pathology (2012) 25:428–38. doi:10.1038/modpathol.2012.7
53. Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG. Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol (2008) 67(9):878–87. doi:10.1097/NEN.0b013e3181845622
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Lin FM, Cordes K, Li LH, Hood L, Couser WG, Shankland SJ, et al. Hematopoietic stem cells contribute to the regeneration of renal tubules after renal ischemia-reperfusion injury in mice. J Am Soc Nephrol (2003) 14(5):1188–99. doi:10.1097/01.ASN.0000061595.28546.A0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Wilhelm SM, Carter C, Tang LY, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res (2004) 64(19):7099–109. doi:10.1158/0008-5472.CAN-04-1443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther (2008) 7(10):3129–40. doi:10.1158/1535-7163.MCT-08-0013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A (2008) 105(8):3041–6. doi:10.1073/pnas.0711741105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature (2006) 439(7074):358–62. doi:10.1038/nature04304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, et al. Initial testing (Stage 1) of AZD6244 (ARRY-142886) by the pediatric preclinical testing program. Pediatr Blood Cancer (2010) 55(4):668–77. doi:10.1002/pbc.22576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature (2010) 464(7287):427–U126. doi:10.1038/nature08902
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature (2010) 464(7287):431–U132. doi:10.1038/nature08833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Lang S-S, Sievert AJ, Boucher KL, Madsen PJ, Slaunwhite E, Brewington D, et al. Development of pediatric glioma models for BRAF-targeted therapy. Neurosurgery (2012) 71(2):E575-E. doi:10.1227/01.neu.0000417783.83750.a5
63. Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (2012) 380(9839):358–65. doi:10.1016/S0140-6736(12)60868-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med (2010) 363(9):809–19. doi:10.1056/NEJMoa1002011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Larkin J, Del Vecchio M, Ascierto PA, Krajsova I, Schachter J, Neyns B, et al. Vemurafenib in patients with BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol (2014) 15(4):436–44. doi:10.1016/S1470-2045(14)70051-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Eisen T, Ahmad T, Flaherty KT, Gore M, Kaye S, Marais R, et al. Sorafenib in advanced melanoma: a phase II randomised discontinuation trial analysis. Br J Cancer (2006) 95(5):581–6. doi:10.1038/sj.bjc.6603291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Cancer Research UK. Dabrafenib (Tafinlar). (2014). Available from: http://www.cancerresearchuk.org/about-cancer/cancers-in-general/treatment/cancer-drugs/dabrafenib
68. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet (2012) 379(9829):1893–901. doi:10.1016/S0140-6736(12)60398-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res (2011) 17(5):989–1000. doi:10.1158/1078-0432.CCR-10-2200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Fouladi M, Laningham F, Wu J, O’Shaughnessy MA, Molina K, Broniscer A, et al. Phase I study of everolimus in pediatric patients with refractory solid tumors. J Clin Oncol (2007) 25(30):4806–12. doi:10.1200/JCO.2007.11.4017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Cancer Research UK. Sorafenib (Nexavar). (2014). Available from: http://www.cancerresearchuk.org/about-cancer/cancers-in-general/treatment/cancer-drugs/sorafenib
72. Kane RC, Farrell AT, Saber H, Tang S, Williams G, Jee JM, et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res (2006) 12(24):7271–8. doi:10.1158/1078-0432.CCR-06-1249
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol (2014) 16(10):1408–16. doi:10.1093/neuonc/nou059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Rochet NM, Kottschade LA, Markovic SN. Vemurafenib for melanoma metastases to the brain. N Engl J Med (2011) 365(25):2439–41. doi:10.1056/NEJMc1111672
75. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med (2011) 364(26):2507–16. doi:10.1056/NEJMoa1103782
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol (2014) 15(3):323–32. doi:10.1016/S1470-2045(14)70012-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med (2012) 366(8):707–14. doi:10.1056/NEJMoa1112302
78. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature (2010) 467(7315):596–9. doi:10.1038/nature09454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature (2010) 468(7326):973–U377. doi:10.1038/nature09626
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature (2011) 480(7377):387–U144. doi:10.1038/nature10662
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell (2010) 18(6):683–95. doi:10.1016/j.ccr.2010.11.023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Antony R, Emery CM, Sawyer AM, Garraway LA. C-RAF mutations confer resistance to RAF inhibitors. Cancer Res (2013) 73(15):4840–51. doi:10.1158/0008-5472.CAN-12-4089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med (2012) 367(18):1694–703. doi:10.1056/NEJMoa1210093
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A (2009) 106(48):20411–6. doi:10.1073/pnas.0905833106
85. Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol (2011) 29(22):3085–96. doi:10.1200/JCO.2010.33.2312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature (2010) 468(7326):968–U370. doi:10.1038/nature09627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Fragomeni RAS, Chung HW, Landesman Y, Senapedis W, Saint-Martin J-R, Tsao H, et al. CRM1 and BRAF inhibition synergize and induce tumor regression in BRAF-mutant melanoma. Mol Cancer Ther (2013) 12(7):1171–9. doi:10.1158/1535-7163.MCT-12-1171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Goldinger SM, Zimmer L, Schulz C, Ugurel S, Hoeller C, Kaehler KC, et al. Upstream mitogen-activated protein kinase (MAPK) pathway inhibition: MEK inhibitor followed by a BRAF inhibitor in advanced melanoma patients. Eur J Cancer (2014) 50(2):406–10. doi:10.1016/j.ejca.2013.09.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: BRAF, glioma, brain tumor, diagnostic biomarker, prognostic biomarker, therapeutic targeting
Citation: Penman CL, Faulkner C, Lowis SP and Kurian KM (2015) Current understanding of BRAF alterations in diagnosis, prognosis, and therapeutic targeting in pediatric low-grade gliomas. Front. Oncol. 5:54. doi: 10.3389/fonc.2015.00054
Received: 17 December 2014; Paper pending published: 23 January 2015;
Accepted: 16 February 2015; Published online: 03 March 2015.
Edited by:
Nader Sanai, Barrow Neurological Institute, USAReviewed by:
Alexandre Arcaro, University of Bern, SwitzerlandJohn Howard Sampson, Duke University Medical Center, USA
Theodore Nicolaides, University of California San Francisco, USA
Copyright: © 2015 Penman, Faulkner, Lowis and Kurian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathreena M. Kurian, Brain Tumour Research Group, Institute of Clinical Neurosciences, Southmead Hospital, Level 1 Learning and Research Building, Westbury on Trym, Bristol BS10 5NB, UK e-mail:a2F0aHJlZW5hLmt1cmlhbkBuYnQubmhzLnVr