 Christina Lutz-Nicoladoni
Christina Lutz-Nicoladoni Dominik Wolf3
Dominik Wolf3 Sieghart Sopper
Sieghart Sopper- 1Department of Hematology and Oncology, Medical University Innsbruck, Innsbruck, Austria
- 2Tumor Immunology Laboratory, Tyrolean Cancer Research Institute, Innsbruck, Austria
- 3Medical Clinic III for Oncology, Haematology and Rheumatology, University Clinic Bonn (UKB), Bonn, Germany
Maintenance of immunological tolerance is a critical hallmark of the immune system. Several signaling checkpoints necessary to balance activating and inhibitory input to immune cells have been described so far, among which the E3 ligase Cbl-b appears to be a central player. Cbl-b is expressed in all leukocyte subsets and regulates several signaling pathways in T cells, NK cells, B cells, and different types of myeloid cells. In most cases, Cbl-b negatively regulates activation signals through antigen or pattern recognition receptors and co-stimulatory molecules. In line with this function, cblb-deficient immune cells display lower activation thresholds and cblb knockout mice spontaneously develop autoimmunity and are highly susceptible to experimental autoimmunity. Interestingly, genetic association studies link CBLB-polymorphisms with autoimmunity also in humans. Vice versa, the increased activation potential of cblb-deficient cells renders them more potent to fight against malignancies or infections. Accordingly, several reports have shown that cblb knockout mice reject tumors, which mainly depends on cytotoxic T and NK cells. Thus, targeting Cbl-b may be an interesting strategy to enhance anti-cancer immunity. In this review, we summarize the findings on the molecular function of Cbl-b in different cell types and illustrate the potential of Cbl-b as target for immunomodulatory therapies.
Modulation of Immune Cell Functions by the E3 Ligase Cbl-b
Ubiquitination represents an important post-transcriptional regulatory mechanism of various cellular pathways. The specificity of this process is conferred by E3 ubiquitin ligases. Over the last decade, many E3 ubiquitin ligases involved in the fine-tuning of immunological responses have been described (1–3). Among these, Casitas B-lineage lymphoma proto-oncogene-b (Cbl-b) stands out as one of the most important gate keepers of immune activation due to its function as a non-redundant negative regulator of immune activation (4–7). Here, we report on the recent advances in our understanding of the molecular function of Cbl-b and discuss potential clinical implications of Cbl-b targeting for cancer immunotherapy.
Protein Ubiquitination
Post-transcriptional protein modification such as ubiquitination adds an additional important layer to the regulation of cellular processes. Ubiquitin (Ub) is a 76 amino acid peptide, which is tagged to proteins through a process involving three enzymes: E1, E2, and E3. Ubiquitination is initiated by an ubiquitin activating enzyme (E1) forming a thiol ester bond with the C-terminal glycine residue of ubiquitin. The activated Ub can then be transferred to one of the Ub conjugating enzymes (E2) via a similar thioester linkage. In a final step, E3 ligases catalyze the formation of an isopeptide bond between Ub and a specific lysine residue of the target protein (8, 9). In addition to 2 E1 and 35 E2 ligases, the mammalian genome encodes over 1000 E3 proteins, which are responsible for the broad range of target proteins regulated by ubiquitin modification (10).
Poly ubiquitination via lysine residue K48 serves as a proteasome-targeting signal leading to degradation of the substrate protein via the 26S proteasome while mono-ubiquitination or poly ubiquitination via other lysine residues, e.g., K63, mainly modifies protein function by altered protein trafficking and subcellular localization (11, 12). Cytoplasmic signaling proteins and nuclear transcription factors tend to be ubiquitinated for proteasomal degradation while surface receptors like receptor tyrosine kinases, G-protein-coupled receptors, and the TCR are more often regulated by endocytosis and subsequent lysosomal degradation (13–15).
Molecular Mechanisms of Cbl-Family E3 Ligases
The Cbl proteins are encoded by a highly conserved gene family, which can be found from nematodes to mammals (16). The name is derived from the retroviral oncoprotein v-Cbl promoting development of B cell leukemia in mice. V-Cbl represents a dominant mutant antagonizing the function of its cellular homolog c-Cbl (17, 18). The mammalian genome encodes three Cbl proteins (Figure 1): c-Cbl (also termed Cbl2, Cbl-SL, or RNF55) (18), Cbl-b (also termed RNF56) (19), and Cbl-c (also called Cbl-3) (20, 21). While c-Cbl and Cbl-b are expressed in a wide range of tissues, expression of Cbl-3 and another structurally similar E3 ligase, Cbl-like protein-1 (CBLL1, also called HAKAI) is restricted to epithelia (17, 19–23). Recently, ZNF645, a new E3 ligase with structural homologies to Cbl proteins, has been described as testis specific gene (24). Cbl proteins interact with target proteins via their protein–protein interaction domains (Figure 1), allowing regulation of multiple pathways (25). This interaction involves recognition of specific phosphotyrosine-containing sequence motifs that are generated on protein tyrosine kinases (PTK). Thus, the current paradigm holds that Cbl-family E3 ligases are selective regulators of activated PTKs (26).
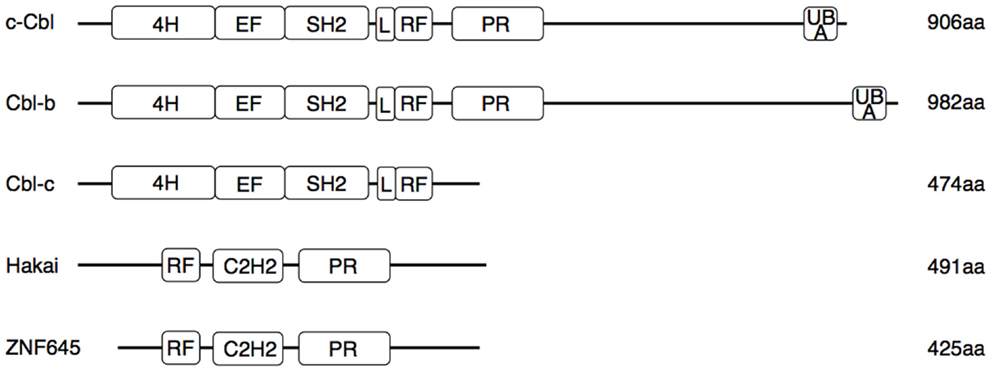
Figure 1. Functional domains of human Cbl and Cbl-like proteins. Three N-terminal regions [4-helical bundle (4H), EF-hand domain, and SH2 domain] constitute the tyrosine kinase binding (TKB) domain. Separated by a conserved linker (L) region follows a RING finger (RF) domain, which binds to E2 enzymes. The proline-rich sequence (PR) interacts with SH3 domains of various proteins involved in signaling and endocytosis. In the Cbl-like proteins, the order of the RF and TKB (here comprising a single C3H2 domain) is reversed. Only c-Cbl and Cbl-b contain an ubiquitin-associated domain (UBA) involved in binding and dimerization of ubiquitin. Several tyrosine residues in the L region and in the less conserved C-terminal half can be phosphorylated, resulting in conformational changes necessary for the ligase activity or interaction with SH2 domains of signaling proteins. Mutations associated with myeloproliferative disorders are located in the RF and L regions.
All three mammalian Cbl proteins are RING-type E3 ligases containing an N-terminal tyrosine kinase binding (TKB) domain consisting of a four-helical bundle, a calcium binding EF-hand and a Src homology (SH2) domain, followed by a linker helical region and the RING domain (Figure 1), responsible for their catalytic function (16, 27–32). The evolutionary highly conserved N-terminal RING-type zinc finger domains forming the basic E3 module of Cbl proteins are essential and sufficient for activated PTK directed E3 ligase activity (33, 34) as evidenced by a loss of function mutation in the E3 ligase domain of cblb, which phenocopies the cblb knockout phenotype in vivo (35). Additionally, c-Cbl and Cbl-b contain proline-rich regions mediating the association with tyrosine- and serine phosphorylation sites, and an ubiquitin-associated (UBA)/leucine zipper domain for dimerization (Figure 1) (16, 36–38). Via their protein interaction domains Cbl proteins interact with a large number of target proteins either as E3 ligases or adaptor molecules, e.g., with Src family kinases, SH2-domain containing proteins of the PTK-dependent signaling network including Vav guanine exchange factors, the p85 subunit of phosphatidylinositol 3-kinase (PI3K), and adaptor proteins of the Crk-family allowing the regulation of multiple pathways (26). Proteins ubiquitinated by Cbl proteins are either degraded in the proteasome or sequestered to specific cellular locations. Of the three Cbl proteins in mammals, Cbl-b is preferentially expressed in peripheral lymphoid organs suggesting a prominent function for adaptive immune responses. Specifically, Cbl-b seems to be central for maintenance of peripheral tolerance as cblb knock out mice develop spontaneous autoimmunity characterized by auto-antibody production and infiltration of activated T and B cells into multiple organs (4, 5). Cbl-linked networks (Figure 2) have been implicated in the control of the immune system, cell proliferation, differentiation, and cell morphology (25, 39). Spatial or temporal dysregulation of Cbl proteins results in autoimmunity or increased tumor progression.
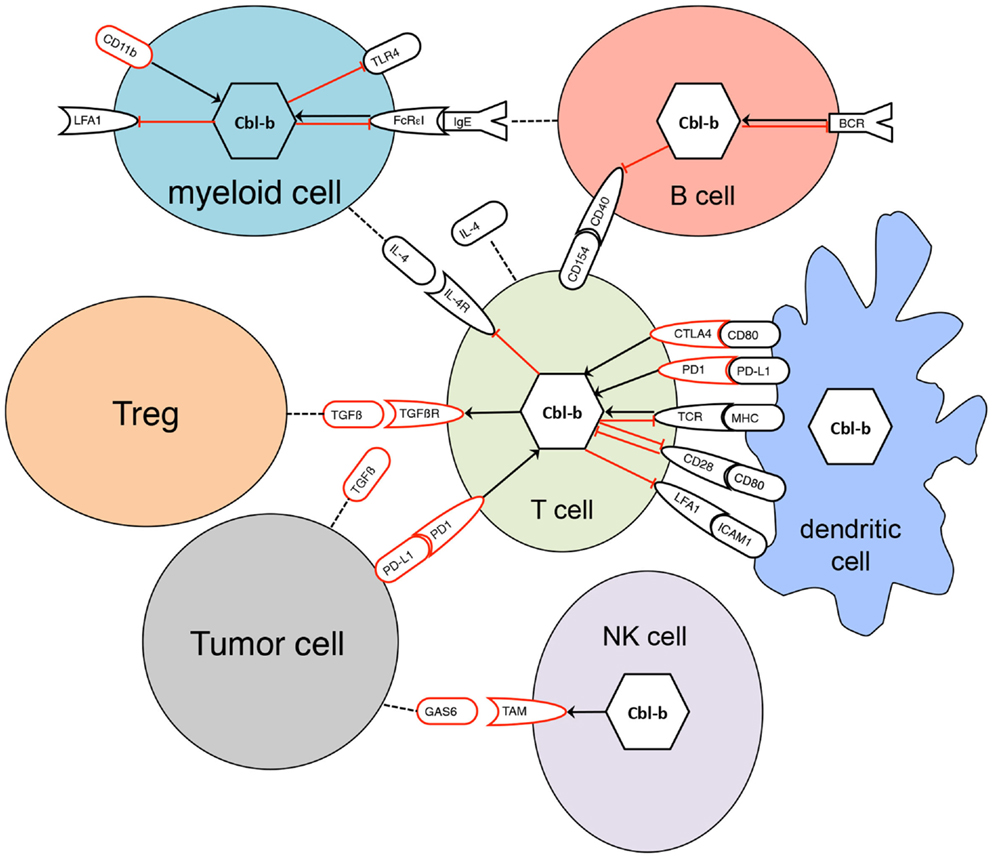
Figure 2. Interaction of Cbl-b with signaling pathways in diverse cells. Black receptors represent activating signaling pathways, red receptors inhibitory pathways. Black arrows indicate positive regulation, red bar-headed lines are representative for negative regulation. Dotted lines indicate secretion of proteins. Regulation of Cbl-b occurs not only at the transcriptional level but also by post-transcriptional mechanisms such as phosphorylation, degradation, or sequestration to specific protein complexes.
Cbl-b Function in T Cells
Cbl-b is highly expressed in murine and human CD4+ and CD8+ T cells (GFN SymAtlas, http://biogps.org) and its expression levels are tightly regulated by CD28 and CTLA-4 stimulation (40, 41) and other co-stimulatory and inhibitory signals (Figure 2) (42). Over the years, work by several groups has documented an essential role of Cbl-b in the negative regulation of T cell activation (6, 7, 39).
T cell activation and tolerance induction are tightly controlled processes regulating immune responses to pathogens and tumors while preventing autoimmunity. Autoimmunity is mainly averted through central tolerance by negative selection of thymocytes carrying TCR for self-antigens (43, 44). However, mechanisms of peripheral tolerance are needed for T cells that escaped thymic selection, and include tolerance of low level expressed or low-affinity antigens, immunosuppression mediated by regulatory T cells (Treg), and other suppressive cell populations, induction of anergy, e.g., in the absence of co-stimulation and activation-induced cell death (43, 45, 46). While maintaining tolerance prevents autoimmunity on one hand, tumor induced anergy induction of T cells is critical and hazardous on the other hand. Immunosuppression by cancer cells involves induction and expansion of various immunosuppressive cell types such as Tregs and myeloid derived suppressor cells as well as production of inhibiting cytokines, e.g., transforming growth factor-beta (TGF-β), and finally supports tumor cells to escape the immune system (47).
T cells require two signals for proper activation: the first provided by interaction of the TCR complex with the cognate peptide antigen presented by major histocompatibility complex molecules and a second through co-stimulatory molecules on antigen presenting cells. Following initial triggering of the antigen receptor, the Src family kinases Lck and Fyn are recruited to phosphorylate ZAP-70, which subsequently phosphorylates SLP-76 and LAT. Subsequently, a multi-subunit protein complex containing inducible T cell kinase, PI3K, phospholipase C-γ (PLCγ), and Vav1 is formed resulting in PLCγ-regulated calcium influx, cytoskeletal rearrangements via Vav1, Rac, WASP, and activation of protein kinase C-theta (PKCθ) (48). PKCθ is a key molecule of the immunological synapse responsible for cytokine responses, proliferation of T cells (49), and TH17-mediated autoimmunity (50). The sustained calcium influx following TCR engagement activates the phosphatase calcineurin, which subsequently dephosphorylates the NFAT family of transcription factors. Dephosphorylated NFAT proteins then translocate to the nucleus to induce expression of various genes (51).
Final activation of T cells is achieved by a second signal delivered by several co-stimulatory molecules, of which CD28 is the most prominent. CD28 is activated by binding to its ligands CD80 or CD86 to enhance TCR proximal signaling and to induce additional pathways, leading to activation of NF-κB, NFAT, and AP-1 transcription factors (52). Cooperation of NFAT with AP-1 is required for IL-2 gene transcription and mRNA stability (51). Furthermore, CD28 co-stimulation leads to cytoskeletal reshuffle in order to form and stabilize the immunological synapse. Strong TCR stimulation without co-stimulation or TCR stimulation by a low-affinity antigen results in T cell anergy instead of activation (53) and anergic T cells remain in a viable, but unresponsive state.
After receiving tolerizing signals, e.g., TCR stimulation in the absence of co-stimulation, Cbl-b is up-regulated (40, 41) and inhibits signaling events downstream of PI3K. In Jurkat cells, this was mediated by ubiquitination of the p85 subunit of PI3K leading to reduced interaction of p85 with CD28 (54). However, ubiquitination of p85 was never shown in primary cells, and these findings have recently been challenged by a report that Cbl-b does not directly inhibit PI3K, but suppresses PTEN inactivation independently of its ubiquitin ligase activity (55). Cbl-b associates with Nedd4 inhibiting its autophosphorylation and subsequent ubiquitination of PTEN, thereby inhibiting down-regulation of PTEN. In addition, introducing nedd4 deficiency into cblb-deficient mice abrogates the hyperresponsive phenotype of cblb−/− T cells. As these experiments have been exclusively performed in murine models, it is still possible that direct effects of Cbl-b on PI3K contribute to the modulation of CD28 signaling in human T cells.
Furthermore, Cbl-b reduces phosphorylation and activation of PLCγ-1 in anergic cells. Accordingly, loss of Cbl-b rescues reduced calcium mobilization of anergic T cells, resulting in impaired induction of T cell tolerance (56). Cbl-b regulates calcium influx and IL-2 production through Vav-1 activation (57). Cbl-b directly interacts with Vav1 through its N-terminal proline-rich region, thereby controlling phosphorylation of Vav1, but not by targeting it for degradation. Whereas in wild-type cells TCR plus CD28 coreceptor stimulation is required for Vav1 activation, in cblb-deficient cells sole TCR stimulation is sufficient for Vav1 activation (57), which is important for formation and stability of the immunologic synapse. Cbl-b and c-Cbl together promote antigen-induced down-regulation of the TCR (58). Moreover, Cbl-b was shown to repress NF-κB transactivation (59). Following stimulation of CD28, Cbl-b is post-transcriptionally eliminated by ubiquitination and proteasomal degradation, which is a prerequisite for the maintenance of T cell proliferation and IL-2 production (41). Gruber et al. described an antagonistic cross-talk between PKCθ and Cbl-b, which colocalize upon TCR and CD28 co-stimulation, resulting in phosphorylation of Cbl-b by PKCθ probably inducing a conformational change of Cbl-b (60). Subsequently, Cbl-b is ubiquitinated by the E3 ligase Nedd4 mediating proteasomal degradation (61). In summary, genetic loss of Cbl-b in naive T cells uncouples TCR stimulation from the requirement of CD28 co-stimulation for effective proliferation and IL-2 secretion. Cblb-deficient T cells are hyperresponsive, and even upon TCR stimulation in the absence of CD28 co-stimulation they proliferate comparable to wild-type T cells after double stimulation with anti-CD3 and anti-CD28 antibodies (4, 5).
Cbl-b is also involved in activation-induced cell death. Cblb-deficient CD4+ TH1 cells are resistant to apoptosis induction by CD3 stimulation in the absence of CD28+ co-stimulation (62) and also clonal deletion of CD8+ T cells was shown to depend on Cbl-b (63, 64). This is important in the context of chronic viral infection where virus-specific CD8+ T cells become exhausted. It was shown that functional impairment (exhaustion) and to a lesser extent also elimination of virus-specific T cell clones was delayed in Cbl-b deficient animals persistently infected with LCMV leading to immunopathology and decreased survival (63).
The central role of Cbl-b in the maintenance of the balance between T cell tolerance and T cell activation is mirrored in the autoimmune phenotype of cblb knock out mice. Despite normal T cell development cblb knock out mice are highly susceptible to experimental autoimmune encephalomyelitis (5). Furthermore, depending on the genetic background cblb-deficient mice develop spontaneous autoimmunity characterized by auto-antibody production, infiltration of activated T and B lymphocytes into multiple organs, increased size of submandibular gland, and parenchymal damage starting at 6 months of age (4). Along this line, increased collagen-induced autoimmune arthritis induced by immunization and subsequent boosting with collagen is found in cblb knock out DBA/1 mice compared to wildtype (wt) DBA/1 mice (56). In another model of anergy induction in T cells p14/TCRVaVb8.2 transgenic mice recognizing the LCMV p33 peptide presented by MHC class I were injected with the cognate p33 peptide. On a cblb-deficient background increased in vivo expansion of antigen-specific T cells was detected, and instead of anergy the rechallenge with p33 induced a cytokine storm followed by massive edemas in multiple organs and high mortality of the animals (56).
Cbl-b controls the threshold for T cell activation (40, 41) and regulates the affinity of antigens to their TCR (65). Gronski et al. demonstrated increased development of autoimmune diabetes by crossing p14/Rip-gp transgenic mice onto a cblb−/− background. About 100% of cblb-deficient p14/Rip-gp mice versus <50% of cblb+/+ p14/Rip-gp mice developed diabetes when infected with LCMV expressing the low-affinity ligand LF6, accompanied by enhanced T cell proliferation and enhanced cytotoxic T-lymphocyte function in cblb-deficient mice (65).
Transforming growth factor-β plays a central role in T cell differentiation (66) by inhibiting TH1 and TH2 polarization (67) and triggering differentiation of inducible Tregs or, in cooperation with IL-6, induction of TH17 cells (68). Additionally, TGF-β exerts immune suppressive functions and inhibits T cell activity averting development of autoimmunity (69). TGF-β secretion by tumor cells and tumor-infiltrating Tregs therefore contributes to immune evasion of tumor cells (70). Cbl-b deficiency renders peripheral CD4+ and CD8+ T cells partially resistant to inhibitory effects exerted by Tregs via disruption of the TGF-β pathway (56, 71–76). The interaction of CD4+CD25+ regulatory T cells and CD4+CD25− effector cells in cblb−/− mice was extensively studied by Wohlfert et al. (75), demonstrating normal function of cblb-deficient Tregs, but resistance of cbl-b deficient CD4+CD25− T cells to inhibition by either Tregs or soluble TGF-β. In the presence of TGF-β, naive CD4+CD25+ T cells can be converted into CD4+CD25+FoxP3+ inducible Tregs (77). This differentiation is negatively regulated by the PI3K/Akt/mTOR and the FOXO1/3a pathway (78, 79). Cbl-b promotes TGF-β-mediated iTreg conversion by tuning the threshold of T cell activation via an Akt2-dependent mechanism (80). Recently, SMAD7, a negative regulator of TGF-β receptor signaling, was identified as a critical target of Cbl-b (81), thus regulating sensitivity toward TGF-β effects. On a molecular level, Cbl-b directly interacts with and ubiquitinates SMAD7, targeting it for degradation and thereby increasing TGF-β/SMAD signaling in T cells. Accordingly, resistance of cblb-deficient T cells to TGF-β is abrogated by conditional deletion of SMAD7 (81).
Moreover, Cbl-b seems to be involved in the negative regulation of CD8+ T cells via the interaction of programed cell death ligand 1 (PD-L1) on dendritic cells (DCs) programed cell death 1 (PD1). It was recently shown that PD-L1 silencing in DC blocks up-regulation of Cbl-b in CD8+ T cells after antigen presentation resulting in hyperactive pro-inflammatory TCRhigh CD8+ T cells and accelerated anti-tumor immune responses in the EG.7 mouse tumor model (42). Regulation of Cbl-b by PD1 possibly contributes to the clinical benefit seen in patients with advanced cancer after treatment with antibodies blocking the interaction between PD1 and PD-L1 (82, 83). Similarly, the improved survival after treatment of melanoma patients with CTLA-4 blocking antibodies (84) may also involve Cbl-b down-regulation as activation of CTLA-4 increases Cbl-b levels by transcriptional regulation (Figure 2) (40).
Recent evidence suggests that Cbl-b is also involved in the polarization of T helper cells into functionally distinct subsets. Levels of Cbl-b are significantly lower in TH2 and TH9 cells compared to TH1 and TH17 cells, and in vitro differentiation of naïve T cells from cblb knock out mice yielded more TH2 and TH9 cells in comparison to those derived from wt mice (85). In addition, absence of Cbl-b resulted in stronger TH2 and TH9 responses and was associated with severe airway inflammation in a mouse model of asthma (85). This, however, is somewhat in contrast to a previous report where, using a different mouse strain and different immunization protocols, increased susceptibility to asthma induction in cblb-deficient mice was mediated by a TH1 response in the lung (86). IL-4 signaling through the JAKs/Stat6 pathway is essential for both TH2 and TH9 differentiation (87). Cbl-b was shown to bind to Stat6 via its TKB domain leading to ubiquitination and degradation though the proteasome, explaining increased signaling through the Stat6 pathway in cblb−/− cells, and finally increased TH2 differentiation (85). The molecular mechanism of increased TH9 differentiation, however, remains completely unclear, especially because TH9 cells require an active TGF-β signal to be differentiated, which, however, is blocked due to the accumulation of the inhibitory SMAD7, mentioned above.
Targeting Cbl-b in T Cells for Tumor Immunotherapy
As already shown for various other checkpoint regulators, genetic loss of the Cbl-b protein leads to autoimmunity but is linked to improved tumor immune surveillance. A recent report using a shRNA screen for major immune regulators governing anti-cancer immune responses supports the importance of Cbl-b as checkpoint molecule (88). Along this line cblb-deficient animals reject tumors in various autochthonous and transplanted tumor models (4, 5). Moreover, mice carrying a cblb E3 ligase-defective mutation are resistant to tumor formation in vivo and reject experimental and spontaneous malignancies (35, 72, 73). As demonstrated by several groups, including our own, this effect is mainly mediated by CD8+ cytotoxic T cells and NK cells (73, 89).
In more detail, TC-1 cells, c-H-ras-transformed syngenic fibroblasts expressing the human papilloma virus 16-derived oncoproteins E6 and E7 were subcutaneously injected into wild-type, heterozygous, and cblb knockout C57BL/6 mice (73). Tumor growth and histology of tumors were unaltered between the groups during the first 2 weeks after tumor cell inoculation, however, after 2 weeks cblb-deficient recipients started to reject tumors spontaneously and tumor masses were continuously reduced until tumors even became undetectable in some animals. This strong anti-tumor response was paralleled by an increased tumor infiltration rate of CD8+ T cells while numbers of tumor-infiltrating CD4+ T cells were unaltered. Moreover, increased numbers of INF-γ secreting CD8+ T cells could be detected in the draining lymph nodes. Treg cells infiltrated TC-1 tumors in both cblb+/+ and cblb−/− mice; however, loss of Cbl-b dramatically changed the ratio of CD8+ T cells to Tregs, which is also a strong predictor of improved outcome in human cancer (90). These findings are supported by the work of Chiang et al. (72), who demonstrated that cblb-deficient mice efficiently rejected subcutaneously inoculated EL4 and EG7 lymphoma cells lacking B7 co-stimulatory molecules. EL4 T linage lymphoma cells represent a tumor model with weak immunogenicity as well as reported TGF-β secretion, while EG7 cells are highly immunogenic EL4 transfectants expressing chicken ovalbumin (OVA) as model tumor antigen. Tumor growth of both cell lines was dramatically reduced in cblb-deficient animals demonstrating that genetic ablation of Cbl-b provokes the ability to reject or attenuate growths of weak and highly immunogenic tumors independent of CD28-co-stimulation of effector cells. Furthermore, when crossing ataxia telangiectasia mutated (ATM) deficient mice onto a cblb-deficient background, 50% of ATM−/− cblb+/+ mice died within 6 months of age of spontaneous T cell lymphomas while no death was observed among ATM−/− cblb−/− double knockout mice. Further studies using Cbl-b E3 ligase-defective (C373Aki/Ki) knock in mice (91) revealed that selective genetic inactivation of Cbl-b ligase activity phenocopies the total loss of Cbl-b, resulting in hyperactivation of T cells, impaired induction of T cell anergy, spontaneous autoimmunity, and tumor rejection demonstrating that the catalytic function of Cbl-b is essential for negative regulation of T cells in vivo (35).
The observed increased anti-tumor response seems to be in part the result of TGF-β insensitivity due to SMAD7 degradation after Cbl-b ubiquitination. To evaluate the in vivo relevance of Cbl-b in SMAD7 regulation, cblb knockout mice were crossed with CD4Cre-smad7fl/fl knockout mice to generate T cell-specific cblb/smad7 double knockout animals, which were investigated concerning their in vivo anti-tumor responses (81). While subcutaneously injected TC-1 tumors are efficiently rejected in cblb single knockout mice, they substantially grow in T cell specific double knockout mice that display comparable survival rates to wildtype animals indicating that concomitant loss of smad7 abrogates the survival benefit of cblb-deficient mice. This insensitivity of cblb-deficient T cells to negative cues from the tumor microenvironment contributes to their increased anti-tumor efficacy.
The above studies also investigated a possible therapeutic efficacy of adoptively transferred cblb-deficient T cells in tumor-bearing hosts. Immunodeficient recombination activating gene-2 (rag2)−/− mice, lacking both T and B cells, were inoculated with TC1 tumor cells and treated with adoptive cell transfer (ACT) of CD8+ T cells from either wt or cblb-deficient donor mice. In contrast to adoptively transferred wt cells, cblb-deficient naive CD8+ T cells were able to markedly delay tumor growth (73). Nevertheless, ACT of polyclonal cblb-deficient CD8+ T cells into immunocompetent wild-type mice yielded contradictory results. While Chiang et al. showed that ACT of cblb-deficient CD8+ T cells into EG.7 tumor-bearing C57Bl/6 wt mice led to delayed tumor outgrowth or even eradication of established tumors (72), we found that sole ACT of polyclonal cblb-deficient CD8+ T cells in immunocompetent wildtype mice challenged with either EG.7 lymphoma or B16.ova melanoma, was not sufficient to significantly delay tumor growth (74). In contrast to other reports (92–95), we did not transfer TCR transgenic tumor-antigen specific but polyclonal T cells. Moreover, we used immunocompetent wildtype recipient mice and did not combine the ACT with induction of lymphopenia as often approached (92). As monotherapy with cblb-deficient polyclonal CD8+ T cells did not provoke any benefit in neither B16.ova melanoma nor EG.7 lymphoma injected immunocompetent mice in our hands, we established a combination therapy of ACT of cblb-deficient CD8+ T cells and DC vaccination, resulting in delayed tumor outgrowth and significantly prolonged survival rates when DC vaccination was combined with ACT of cblb-deficient instead of wt CD8+ T cells (74). The enhanced anti-tumor activity was accompanied by a significantly higher CD8+ T cell infiltration into the tumor and significantly increased numbers of tumor-antigen-positive OVA-specific CD8+ T cells as well as IFN-γ secreting cells in the tumor draining lymph node. Furthermore, specific killing of peptide-loaded target cells was significantly increased in B16.ova bearing mice treated with antigen-pulsed wt DCs and ACT of cblb−/− CD8+ T cells compared to animals challenged with the DC vaccine in combination with wt CD8+ T cells. Vaccination with tumor antigen-pulsed DCs thereby provide a second stimulus leading to an efficient accumulation and in vivo selection of tumor antigen-specific CD8+ T cells. Our data indicate that cblb targeting in T cells alone is not sufficient to counteract cancer-associated immune suppression, but that reactivation of the engineered T cells by a DC vaccine induces a profound anti-tumor immune response in non-TCR-transgenic mice (74).
Cbl-b Function in NK Cells
Cbl proteins not only regulate adaptive immune cell functions but are also critically involved in the regulation of innate lymphocyte populations, such as NK cells (89). NK cells are among the first cells to arrive at the inflamed tissue where they exert potent cytotoxic effector functions and modulate the local immune response (96). In addition, NK cells are critically involved in immunosurveillance of cancer (97). The cytotoxic effects of NK cells and their ability to secrete cytokines is regulated by a multitude of activating and inhibiting receptors, which are expressed in a variety of combinations in the different NK cell subsets (98). Initial work on the role of Cbl proteins in NK cells was restricted to c-Cbl, with early reports demonstrating phosphorylation of c-Cbl after activation of NK cells by various stimuli (99–101). It was shown that this leads to interaction with adaptor molecules of several signal transduction pathways (99, 102) but the functional consequences remained unclear until the group of Long showed that c-Cbl imposes a block, which can only be released by two strong synergistic activating signals. Signals through activating NK cell receptors lead to integration of c-Cbl into a macromolecular complex consisting of the adaptor Crk, the scaffold protein p130CAS and the guanine-nucleotide exchange factor C3G, which is critical for signaling in a wide variety of pathways. Inhibitory signals through KIR receptors inhibit formation and result in disassembly of this complex (103). Interestingly, there was no evidence that E3 ligase function of c-Cbl plays a role in this process. In follow up publications, it was shown that two synergistic activating signals, for example, through NKG2D and CD244 are needed to overcome blockade by c-Cbl (104) and that this synergistic action is mediated by a combined phosphorylation of separate tyrosine residues of the adaptor protein SLP-76 (105). On the other hand, c-Cbl is involved in a kind of negative feedback loop in the down regulation of NKG2D after prolonged exposure to one of its ligands, MICA (106).
Very recently, an elegant study involving genetic ablation of cblb demonstrated a prominent role also of Cbl-b for activation and effector functions of NK cells. Knockout of cblb resulted in increased cytotoxicity as well as higher production of IFN-γ and perforin (89). Site directed mutagenesis resulting in functional inactivation of the catalytic domain further confirmed that the E3 ligase activity is necessary for this effect. Moreover, cblb-deficient NK cells were able to prolong survival and reduce metastasis in several tumor models. Members of the TAM family of cell surface tyrosine kinase receptors (107) were identified as prime targets for Cbl-b-mediated ubiquitination in NK cells (Figure 2). Apparently, Cbl-b ubiquitinates all three TAM family members, Tyro-3, Axl, and Mer, and controls TAM receptor internalization, which is necessary for their functional activity. Small molecule inhibitors of TAM receptor kinases increased NK cell cytotoxicity to the same extent as seen in Cbl-b-deficient animals. Together, these data show that Cbl-b is a key regulator of NK cell effector mechanisms and that down modulating Cbl-b function or inhibiting its substrates is a promising approach in cancer immunotherapy.
Cbl-b Function in B Cells
Production of autoantibodies is a prominent feature of cblb−/− mice (4, 108) leading to the hypothesis that Cbl-b participates in negative regulation of B cell activation pathways (Figure 2). Moreover, B cell-specific ablation of both c-Cbl and Cbl-b in mice displays only slight developmental changes of B cells such as an increase in B1 cells and marginal zone B cells but causes aberrant B cell receptor (BCR) signaling and impaired anergy in mature B cells culminating in the development of a systemic lupus erythematosus like disease (109). Many of the interaction partners of Cbl-b documented for T cells such as PLCγ2, p85/PI3K, or the nucleotide exchange factor Vav are also involved in the normal signal cascade in B cells after activation of the BCR. Therefore, it is likely that they are also targeted by Cbl-b in B cells. Indeed, early work with a chicken B cell line documented direct association of Cbl-b with PLCγ2 (110). However, in contrast to c-cbl (111), knock down of cblb resulted in reduced PLCγ2 activation in chicken B cells whereas overexpression of Cbl-b in a mouse B cell line lead to increased Ca++ mobilization. Together with PLCγ2, Cbl-b formed a complex with BTK and the BLNK and was indispensable for sustained intracytoplasmic Ca++ concentrations, suggesting a positive regulation of BCR-mediated signaling by Cbl-b (110). These findings from B cell lines is in strong contrast to work with primary mature mouse B cells, where cblb-deficient cells display enhanced proliferative responses upon stimulation via BCR or CD40 (Figure 2) (4) and sustained phosphorylation of components of the BCR signaling complex as well as prolonged Ca++ fluxes (112). The regulation of B cell activation is seemingly mediated by an interaction of Cbl-b with the spleen tyrosine kinase Syk leading to its ubiquitination and possibly degradation (112). Syk is also targeted by c-Cbl (113). Another important signaling pathway regulated by Cbl-b is the interaction between CD40L on T cells and CD40 on B cells, which plays a central role in homeostatic regulation of B cell function. Cblb-deficient B cells display hyperproliferation and increased survival in response to CD40 stimulation (108). In addition, T cell independent antibody production is increased in cblb−/− mice. Elegant experiments, where rag−/− mice were reconstituted with cblb-deficient B cells and wt T cells, demonstrated that also T cell dependent antibody titers against the keyhole limpet hemocyanin (KLH) model antigen are increased (108). Nevertheless, reconstitution with a combination of cblb-deficient T and B cells further increased KLH-specific humoral immune response. In the absence of Cbl-b, IκBα and JNK were selectively hyperphosphorylated suggesting that of the different signaling pathways downstream of CD40, NF-κB seems to be specifically targeted by Cbl-b (108). TNF receptor-associated factors (TRAF) are important adaptor molecules and their recruitment to the cytoplasmic tail of CD40 is essential for CD40-mediated signal transduction. It was demonstrated that Cbl-b binds to TRAF-2, another E3 ligase leading to its ubiquitination thereby facilitating access of the negative regulator TRAF-3 (108), which results in NF-κB activation.
In human B cells, both c-Cbl and Cbl-b are constitutively associated with CIN85 forming a complex with BLNK (114). This interaction with CIN85 also promotes degradation of Syk. However, it was so far not investigated whether c-Cbl or Cbl-b is responsible for this molecular effect.
B cells not only produce antibodies but are also involved in antigen processing and presentation to T cells. Recently, it was shown that Cbl-b but not c-Cbl is required for endocytic trafficking of the BCR complex together with the captured antigen to the MHC II containing late endosomes (115). Although BCR is ubiquitinated following stimulation, this seems not to be mediated by Cbl-b as Cbl-b constructs lacking E3 ligase activity were able to reconstitute BCR sorting to late endosomes in cblb−/− cells. Rather the UBA domain is required for endocytic trafficking, possibly acting as a scaffold.
Taken together, there is evidence that both c-Cbl and Cbl-b regulate the B cell activation process and thus fine-tune humoral immune response. The relative contribution of the two Cbl proteins in the various physiological and pathological settings still awaits rigorous investigation.
Cbl-b Function in Dendritic Cells
Dendritic cells are leukocytes whose main function is to capture and process antigens and to present them to cells of the adaptive immune system in order to activate or tolerize them. Immature DCs are characterized by high endocytotic activity and sense and uptake pathogens through pattern recognition receptors (116). Antigens are then presented to T cells in the tumor draining lymphnode. In the same time, DCs up-regulate co-receptors such as CD80, CD86, and CD40, thereby enhancing their T cell stimulation capacity. This results in the differentiation and activation of antigen-specific T-lymphocytes, enabling them to exert their specific effector functions (117).
The E3 ligase c-Cbl has been identified as a modulator of DC activation (118). Toll-like receptor (TLR)-induced expression of pro-inflammatory cytokines and chemokines, such as IL-1α, IL-1β, IL-6, IL-12p70, and CXCL1/KC was enhanced in ccbl-deficient DCs upon LPS exposure. Up-regulation of cytokines was accompanied by enhanced NF-κB transactivation. Loss of c-Cbl did not only up-regulate expression of TLR-triggered pro-inflammatory cytokines but also increased basal expression under steady-state conditions. By overexpressing several mutants of c-Cbl in ccbl knockout DCs before LPS stimulation, the authors proved that the RING domain of c-Cbl is required for IL-12 inhibition. Furthermore, in vivo proliferation of CD4+ and CD8+ T cells was enhanced in mice vaccinated with OVA-pulsed DCs from ccbl negative donors compared to the group treated with wt DCs. Splenocytes from mice primed with ccbl-deficient DCs demonstrated higher peptide specific cytotoxicity, higher numbers of IFN-γ secreting CD4+ and CD8+ T cells, whereas IL-4 secretion was unaltered. Thus, DC deficient for ccbl is more potent inducers of TH1 polarization. To assess whether ccbl-deficiency in DCs would increase their ability to reject tumors, immune competent C57BL/6 wt mice were subcutaneously injected with OVA-expressing EG.7 lymphoma cells and vaccinated with either wt or ccbl-deficient bone marrow derived DCs (BMDCs). Tumor growth in mice treated with ccbl-deficient DCs was significantly delayed compared to the control groups suggesting that inhibition of c-Cbl could probably improve DC based vaccines (118).
Besides c-Cbl, DCs also highly express Cbl-b (119), which might be involved in a negative feed back loop regulating TLR signaling (120). Along this line, it was demonstrated that low-avidity outside-in signaling through CD11b negatively regulates TLR signaling in macrophages through increased Cbl-b-mediated degradation of MyD88 and TRIF. In addition, activation of Akt kinase after binding of either RANK or CD40 to their cognate ligands is dependant on Cbl-b, whereas in B cells c-Cbl is involved (121).
Since cblb-deficient mice are largely protected from tumor formation (72, 73) and adoptive transfer of cblb negative or transiently silenced CD8+ T cells in combination with DC vaccination delays tumor growth in several mouse models (74, 122) it was interesting to know whether loss of Cbl-b in DCs also contributes to tumor resistance of cblb knockout animals (119). We confirmed high expression of Cbl-b in murine BMDCs and showed that cblb-deficiency did not alter BMDC differentiation in vitro. While expression of several functionally important surface proteins on immature and LPS-stimulated DCs was unchanged between wt and cblb negative DCs, we found increased DEC-205 expression on cblb knockout cells (119). Since DEC-205 expression is associated with Treg induction (123), this could at least in part explain the increased frequency of tumor-infiltrating Treg in cbl-b negative mice (72). When comparing secretion of pro-inflammatory cytokines and chemokines in wt versus cblb-deficient BMDCs using different stimulation regimens, we found that LPS-induced TLR4 stimulation resulted in significantly increased TNF-α, IL-6, and MIP-1α secretion in cblb knockout DC. Similarly, TNF-α, together with IL-1α and MCP-1, was increased upon stimulation by the TLR-9 agonist CpG in cblb negative DCs compared to wt DCs. Both, TLR4 and TLR9 stimulation, induce a MyD88-dependent signaling pathway leading to production of pro-inflammatory cytokines and chemokines. This is in accordance with the role of Cbl-b in MyD88 degradation and subsequent suppression of MyD88 inflammatory responses (120). Stimulation with TLR3 agonists did not result in different cytokine profiles of wt and cblb-deficient DCs consistent with a predominantly MyD88-independent signaling by this TLR (124). While cblb-deficient DCs induce stronger allogeneic T cell responses, antigen-specific T cell proliferation was unaltered in vitro and in vivo (119). Increased cytokine production of cblb negative DCs seems to be insufficient to influence peptide-induced proliferation of OT-I tg CD8+ and OT-II tg CD4+ T cells, maybe due to the fact that OT-I and OT-II T cells proliferate even upon stimulation with very low antigen concentrations. Also, cross-presentation capacity of BMDCs was not affected by loss of Cbl-b. According to our in vitro findings, the anti-tumor responses in mice vaccinated with OVA-peptide-loaded DC were not different between the various genotypes (119). Therefore, and in contrast to c-Cbl targeting, silencing of Cbl-b in DC seems to be less promising for improving the T cell priming capacity of DC vaccines.
Cbl-b Function in Myeloid/Monocytic Cells
In comparison to other cell types, less is known on the role of Cbl-b in cells of myeloid origin. In the initial description of Cbl-b, it was already shown that it is up-regulated in human myeloid cell lines upon differentiation (19), however, only recently evidence is emerging how Cbl-b controls several aspects of myeloid cell function. It is involved in negative feed back loops of TLR signaling (120) and regulates macrophage activation by fatty acids (125). Cblb-deficient mice display enhanced infiltration and activation of macrophages, resulting in peripheral insulin resistance (126) and the aggravated lung inflammation seen after LPS induced sepsis in cblb-knockout mice is at least in part mediated by increased activation of macrophages (127). In this context, it is also interesting to note that specific mutations of Cbl proteins, mainly c-Cbl and to a lesser extent also Cbl-b, have been associated with certain myeloproliferative disorders (128, 129), already pointing out to an important role of these proteins for myeloid cell biology. In addition, c-Cbl is frequently mutated in advanced mastocytosis or mast cell leukemia (130). The mutations found not only in ccbl but also in cblb are located mainly in the RING finger (RF) and linker domains, clustering in and around the binding pocket for E2 interactions, suggesting that loss of E3 ligase activity is important for the oncogenic process (131). These clinical findings are endorsed by studies using mice with hematopoietic cells deficient of both ccbl and cblb, which display rapidly fatal myeloproliferative disease (132).
However, Cbl proteins are also relevant in the non-cancer setting in myeloid cells. As an example, challenging mice with LPS lead to severe acute lung inflammation, which is more pronounced in cblb knockout animals (127). In this TLR4-dependent model of sepsis stimulation with LPS increased activation of neutrophils as evidenced by augmented CD11b expression and CD62L shedding. In addition, production of TNF-α and MIP-1α after LPS stimulation was higher in cblb-deficient animals, whereas other cytokines such as RANTES and IL-6 remained unchanged but showed Cbl-b dependent regulation when polymicrobial sepsis was induced after cecal ligation and puncture (127), or after cardiotoxin-induced muscle damage (133). Genetic ablation of cblb, however, had no obvious effect on cytokine production after stimulation with TLR3 agonists (127), which is in line with our observation using BMDC (119). It was also demonstrated that Cbl-b controls TLR4 signaling (Figure 2) via rapid down regulation of TLR4 and by regulating the association between TLR4 and the adaptor molecule MyD88. Although Cbl-b seems to be required for TLR4 ubiquitination under resting conditions, down regulation of TLR4 after activation was not due to Cbl-b-mediated ubiquitination and degradation (127). Later, it was shown that Cbl-b regulates LPS-induced TLR4 signaling by ubiquitination of the adaptor molecules MyD88 and TRIF (120). Stimulation of TLR4 with saturated fatty acids, however, induced ubiquitination and degradation of TLR4 in a Cbl-b dependant manner without ubiquitination of MyD88 or TRIF (125). Thus, Cbl-b is involved in the control of TLR4-mediated activation of myeloid cells through different mechanisms depending on the actual stimulus.
Toll-like receptor-triggered responses are regulated through several negative feedback loops. Signaling through the integrin CD11b activates Syk leading to degradation of the TLR adaptor molecules MyD88 and TRIF by Cbl-b. For this process, the E3 ligase activity is required (120). In contrast, another integrin-mediated pathway to dampen TLR induced activation is independent of this Cbl-b/MyD88 regulatory axis (134). Adhesion to endothelial cells is an important step in the recruitment of mononuclear phagocytes to inflamed tissue. Of the various adhesion molecule interactions involved, association of LFA-1 with ICAM-1 but not VLA-4-mediated adhesion is regulated by Cbl-b. Cblb-deficient cells showed increased association of the LFA-1 β chain with 14-3-3β, one of the cytoplasmic factors transmitting inside-out activating stimuli (135).
Using bone marrow-derived mast cells from mice and the rat basophilic leukemia cell line RBL-2H3 quite some information has been gathered on the role of Cbl-b in mast cells. While both c-Cbl and Cbl-b are expressed in rodent mast cells and become tyrosine phosphorylated upon FcεRI engagement (136) for human cells only data on c-cbl exist. In the rodent models, Cbl-b but not c-Cbl has been associated with reduced phosphorylation of various proteins involved in the signaling cascade such as Syk (136, 137), resulting in a negative regulation of FcRεI-induced degranulation (136) and cytokine production (138). Mice genetically engineered to express Cbl-b with a dysfunctional RF domain revealed increased phosphorylation of tyrosine kinases Syk and Lyn and downstream substrates such as LAT (linker for activation of T cells) as well as PLCγ1 (91), similar to that found in mast cells of Cbl-b-deficient mice indicating that the E3 ligase activity is necessary. However, downstream effects seemed to be differentially affected. Phosphorylation of IKKα/β and cytokine production were much stronger in cells where the complete Cbl-b protein was missing compared to cells with a Cbl-b protein mutated in the RF. This shows that the regulatory function of Cbl-b for IgE-induced inflammatory cytokine production is largely independent of the RF domain (91).
Cbl-b is also involved in the regulation of osteoclast activity as cblb-deficient mice display osteopenia (139). It was later shown that bone resorption is predominantly regulated by Cbl-b, whereas osteoclast survival is dependent on interaction between c-Cbl and PI3K (140). Thus, Cbl-b may also serve as potential target to modify bone metabolism in diseases such as osteoporosis.
Cbl-b in Autoimmunity
As genetic ablation of cblb is associated with spontaneous development of autoimmunity and increased susceptibility to experimental induction of autoimmune diseases (4, 5) it is not surprising that several groups later found links between genetic cblb variants and susceptibility to autoimmunity in an animal model for diabetes (141) as well as in various human autoimmune diseases such as type 1 diabetes (142), lupus erythematodes (143), asthma (144), and multiple sclerosis (145–147). Some of the single nucleotide polymorphisms have been found in the promoter region, possibly influencing transcriptional regulation. Indeed, reduced expression of Cbl-b in PBMC, or more specifically in CD4+ T cells, was a common finding in several autoimmune diseases (143, 148–150) and CBLB mRNA levels in T cells were inversely correlated with relapse rates in multiple sclerosis patients (151). Consistent with reduced Cbl-b levels, several studies also demonstrated increased proliferation of T cells from autoimmune patients especially under anergic conditions (149, 150) and reduced susceptibility to inhibition by Treg (148). A recent study demonstrated that the transcription factor C/EBPβ displayed stronger binding to a disease associated variant and that the negative effect of IFN-β stimulation on T cell proliferation was compromised in CBLB risk allele carriers (150). The functional consequences of other variants, especially in the coding region of CBLB still await elucidation.
Improving Cancer Immunotherapy by Targeting Cbl-b in Clinical Settings
Translation of Cbl-b targeting to clinically applicable concepts requires transient and reversible inhibition of Cbl-b activity. Therefore, we established a synthetic siRNA transfection protocol and demonstrated that transient siRNA-mediated cblb silencing results in hyperresponsiveness, decreased TGF-β sensitivity, and increased IFN-γ production (122). Repeated adoptive cell therapy of cblb silenced T cells in combination with the DC vaccine resulted in delayed tumor outgrowth and prolonged survival rates without overt signs of autoimmunity. In agreement with the phenotype of cblb-deficient murine CD8+ T cells, siRNA-mediated knockdown of CBLB in naive human CD8+ T cells similarly shows markedly enhanced IFN-γ production, even in the absence of CD28 co-stimulation. CBLB mRNA was almost undetectable for 1 week suggesting that nucleofected T cells should stay hyperreactive for several days within the patient upon adoptive transfer (122, 152). We here envision using CBLB silenced autologous cells as combination partner for other immune activating therapies, such as checkpoint monoclonal antibodies, DNA-based vaccines, and DC vaccination (153, 154).
Adoptive cell therapies have achieved promising results in clinical trials. Ex vivo expanded tumor-infiltrating lymphocytes (TILs) have induced regression in patients with metastatic melanoma (155, 156) and virus-specific T cells are used in virus-associated hematologic malignancies (157). Efficacy of adoptive immunotherapy can be increased by ex vivo gene transfer in order to bypass the need of the ex vivo expansion and accumulation of tumor-specific T cells prior the ACT (158–160). Nevertheless, although tumor-specific immune responses are often induced, the therapeutic efficacy of these approaches is limited because of insufficient in vivo activation, expansion, and survival of transferred effector immune cells (159). In addition, the restricted knowledge of tumor antigens that are capable to induce potent anti-tumor immune responses limits the applicability of ACT therapies so far. Besides the unsatisfactory clinical benefit, another disadvantage of common ACT approaches is that they generally require time- and cost-consuming protocols to generate enough tumor-reactive CD8+ T cells. Moreover, the use of lentiviral vectors to generate TCR-transgenic T cells harbors the risk of insertional mutagenesis, potentially causing leukemogenesis (161). Therefore, using hyperreactive polyclonal non-TCR-transgenic T cells instead of tumor-specific cells for ACT in combination with a second stimulus as, for example, DC vaccination would simplify the standard operating procedure. Strategies rendering adoptively transferred effector cells resistant to inhibitory cues from the microenvironment are highly attractive for improvement of the efficacy of cancer immunotherapy. Our results obtained in preclinical mouse models suggest that transient siRNA-mediated CBLB targeting in polyclonal CD8+ T cells may improve adoptive T cell therapy in the clinic (74, 122, 152). Moreover, decreasing Cbl-b expression of NKT cells stimulated by α-galactosylceramide for ACT (162) may render them resistant to anergy (163) thus improving ACT (164). Alternatively, other stimulating ligands, which do not up-regulate Cbl-b in NKT cells may be useful in the clinical setting (163). As recently shown, also NK cell-based immunotherapies are amenable to improvement by modulation of Cbl-b. Adoptive transfer of cbl-b deficient NK cells significantly reduced tumor burden in a melanoma mouse model (89). Similar effects can also be achieved by inhibiting the most prominent targets of Cbl-b ubiquitination in NK cells, the TAM receptor kinases with small molecule compounds.
Final Comments
Although relatively little information is available regarding the mechanisms of Cbl-b in human cells ample evidence indicates that Cbl-b exerts important regulatory functions in diverse cells of hematopoietic origin, resulting in a balanced immune response. Depending on the clinical situation, either inhibiting or strengthening Cbl-b functions might be desirable. In the autoimmune setting, inhibition of degradation of Cbl-b interaction partners by proteasome inhibitors could constitute a rationale approach (1). Vice versa, anti-cancer immunity could be enhanced by direct or indirect inhibition of Cbl-b function. Small molecule inhibitors specific for Cbl-b have not yet been developed. However, inhibition of molecular substrates of Cbl-b ubiquitination such as TAM receptors that successfully increased NK cell-mediated cytotoxicity and reduced metastatic spread are promising candidates to be tested in clinical trials (89). Also, the recent promising results of clinical studies with immune checkpoint antibodies may be based on Cbl-b. Another way to harness this regulatory pathway for reinforcing tumor-specific cells is to down modulate Cbl-b by molecular means (e.g., by siRNA) for adoptive cellular immunotherapy. The feasibility of such an approach will soon be investigated in a phase I clinical study (NCT02166255). In summary, targeting the immunological gate keeper Cbl-b opens new avenues to treat human diseases, such as cancer and autoimmunity.
Conflict of Interest Statement
The authors declare that the manuscript was prepared in the absence of any conflict of interest. Dominik Wolf is co-applicant of the patents WO/2009/073905 and WO/2010/119061 A1 related to Cbl-b.
Acknowledgments
CLN is supported by a Hertha Firnberg fellowship (FWF T550-B19) and the Österreichische Krebshilfe Tirol. DW received grants from FWF and OeNB. SS is supported by the Gesellschaft für AIDS Forschung.
Abbreviations
ACT, adoptive cell transfer; AP-1, activator protein-1; APC, antigen presenting cell; BCR, B cell receptor; BLNK, B cell linker protein (=SLP-65, BASH); BMDC, bone marrow derived dendritic cell; BTK, Bruton’s tyrosine kinase; Cbl-b, Casitas B-lineage lymphoma proto-oncogene B; C/EBP-b, CCAAT-enhancer binding protein beta; CIN85, Cbl-interacting protein of 85 kDa; CTLA-4, cytotoxic T-lymphocyte antigen 4; DC, dendritic cell; DEC-205, CD205; EAE, experimental autoimmune encephalomyelitis; FOX, forkhead box; Fyn, proto-oncogene; ICAM-1, intercellular adhesion molecule 1; IFN-γ, interferon gamma; IKK α/β, inhibitor of NF-κB kinase alpha/beta; IL-2, interleukin-2; KIR, killer-Ig-like receptor; LAT, linker for activation of T cells; Lck, lymphocyte specific tyrosine kinase; LCMV, lymphocytic choriomeningitis virus; LFA-1, leukocyte function-associated antigen 1; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MICA, MHC I polypeptide related sequence A; MIP-1, macrophage inflammatory protein-1; MCP-1, monocyte chemoattractant protein-1; mTOR, mammalian target of rapamycin; MyD88, myeloid differentiation primary response gene; Nedd4, neural precursor cell expressed developmentally down-regulated protein 4; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor of κB; NKG2D, killer cell lectin-like receptor; OVA, ovalbumin; PAMP, pathogen-associated molecular patterns; PD1, programed cell death 1; PD-L1, programed cell death ligand 1; PKCθ, protein kinase C-theta; PI3K, phosphatidylinositol 3-kinase; PLCγ, phospholipase C-gamma; PTEN, phosphatase and tensin homolog; PTK, phospho tyrosine kinase; Rac, member of Rho family GTPases; rag2, recombination activating gene-2; RANK, receptor activator of NF-κB; RANTES, regulated on activation, normal T cell expressed and secreted; Ras, member of family of small GTPases; RING, really interesting new gene; SH2, Src homology domain; SIINFEKL, OVA257–264 peptide; SLP76, SH2 domain containing leukocyte protein of 76kD; SMAD7, mothers against decapentaplegic homolog 7; Src, proto-oncogene Src; TAM, tyro3, axel, mer family of TAM receptor tyrosine kinases; TCR, T cell receptor; TNF-α, tumor necrosis factor-alpha; TKB, tyrosine kinase binding domain; TRAF, TNF receptor-associated factor; TRIF, TIR-domain containing adapter; TGF-β, transforming growth factor-beta; TLR, toll-like receptor; Treg, regulatory T cell; TRK, receptor tyrosine kinase; Ub, ubiquitin; UBA, ubiquitin-associated domain; Vav1, protooncogene Vav1; VLA-4, very late antigen 4; WASP, Wiskott–Aldrich syndrome protein; Wt, wildtype; ZAP-70, zeta-associated protein of 70 kDa.
References
1. Lin AE, Mak TW. The role of E3 ligases in autoimmunity and the regulation of autoreactive T cells. Curr Opin Immunol (2007) 19(6):665–73. doi: 10.1016/j.coi.2007.10.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Moynagh PN. The roles of Pellino E3 ubiquitin ligases in immunity. Nat Rev Immunol (2014) 14(2):122–31. doi:10.1038/nri3599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Rajsbaum R, Garcia-Sastre A, Versteeg GA. TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J Mol Biol (2014) 426(6):1265–84. doi:10.1016/j.jmb.2013.12.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature (2000) 403(6766):211–6. doi:10.1038/35003228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, et al. Cbl-b regulates the CD28 dependence of T-cell activation. Nature (2000) 403(6766):216–20. doi:10.1038/35003235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Paolino M, Penninger JM. Cbl-b in T-cell activation. Semin Immunopathol (2010) 32(2):137–48. doi:10.1007/s00281-010-0197-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Wallner S, Gruber T, Baier G, Wolf D. Releasing the brake: targeting Cbl-b to enhance lymphocyte effector functions. Clin Dev Immunol (2012) 2012:692639. doi:10.1155/2012/692639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochim Biophys Acta (2014) 1843(1):47–60. doi:10.1016/j.bbamcr.2013.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem (2001) 70:503–33. doi:10.1146/annurev.biochem.70.1.503
10. van Wijk SJ, Timmers HT. The family of ubiquitin-conjugating enzymes (E2s): deciding between life and death of proteins. FASEB J (2010) 24(4):981–93. doi:10.1096/fj.09-136259
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Haglund K, Di Fiore PP, Dikic I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci (2003) 28(11):598–603. doi:10.1016/j.tibs.2003.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Weissman AM. Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol (2001) 2(3):169–78. doi:10.1038/35056563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Heissmeyer V, Rao A. E3 ligases in T cell anergy – turning immune responses into tolerance. Sci STKE (2004) 2004(241):e29. doi:10.1126/stke.2412004pe29
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Sorkin A, Von Zastrow M. Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol (2002) 3(8):600–14. doi:10.1038/nrm883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Tanno H, Komada M. The ubiquitin code and its decoding machinery in the endocytic pathway. J Biochem (2013) 153(6):497–504. doi:10.1093/jb/mvt028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Nau MM, Lipkowitz S. Comparative genomic organization of the cbl genes. Gene (2003) 308:103–13. doi:10.1016/S0378-1119(03)00471-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Blake TJ, Shapiro M, Morse HC III, Langdon WY. The sequences of the human and mouse c-cbl proto-oncogenes show v-cbl was generated by a large truncation encompassing a proline-rich domain and a leucine zipper-like motif. Oncogene (1991) 6(4):653–7.
18. Langdon WY, Hartley JW, Klinken SP, Ruscetti SK, Morse HC III. v-cbl, an oncogene from a dual-recombinant murine retrovirus that induces early B-lineage lymphomas. Proc Natl Acad Sci U S A (1989) 86(4):1168–72. doi:10.1073/pnas.86.4.1168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Keane MM, Rivero-Lezcano OM, Mitchell JA, Robbins KC, Lipkowitz S. Cloning and characterization of cbl-b: a SH3 binding protein with homology to the c-cbl proto-oncogene. Oncogene (1995) 10(12):2367–77.
20. Keane MM, Ettenberg SA, Nau MM, Banerjee P, Cuello M, Penninger J, et al. cbl-3: a new mammalian cbl family protein. Oncogene (1999) 18(22):3365–75. doi:10.1038/sj.onc.1202753
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Kim M, Tezuka T, Suziki Y, Sugano S, Hirai M, Yamamoto T. Molecular cloning and characterization of a novel cbl-family gene, cbl-c. Gene (1999) 239(1):145–54. doi:10.1016/S0378-1119(99)00356-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol (2002) 4(3):222–31. doi:10.1038/ncb758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Griffiths EK, Sanchez O, Mill P, Krawczyk C, Hojilla CV, Rubin E, et al. Cbl-3-deficient mice exhibit normal epithelial development. Mol Cell Biol (2003) 23(21):7708–18. doi:10.1128/MCB.23.21.7708-7718.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Liu YQ, Bai G, Zhang H, Su D, Tao DC, Yang Y, et al. Human RING finger protein ZNF645 is a novel testis-specific E3 ubiquitin ligase. Asian J Androl (2010) 12(5):658–66. doi:10.1038/aja.2010.54
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Schmidt MH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol (2005) 6(12):907–18. doi:10.1038/nrm1762
26. Mohapatra B, Ahmad G, Nadeau S, Zutshi N, An W, Scheffe S, et al. Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim Biophys Acta (2013) 1833(1):122–39. doi:10.1016/j.bbamcr.2012.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature (1999) 398(6722):84–90. doi:10.1038/18050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Rao N, Dodge I, Band H. The Cbl family of ubiquitin ligases: critical negative regulators of tyrosine kinase signaling in the immune system. J Leukoc Biol (2002) 71(5):753–63.
29. Sanjay A, Horne WC, Baron R. The Cbl family: ubiquitin ligases regulating signaling by tyrosine kinases. Sci STKE (2001) 2001(110):e40. doi:10.1126/stke.2001.110.pe40
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Thien CB, Walker F, Langdon WY. RING finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol Cell (2001) 7(2):355–65. doi:10.1016/S1097-2765(01)00183-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Tsygankov AY, Teckchandani AM, Feshchenko EA, Swaminathan G. Beyond the RING: CBL proteins as multivalent adapters. Oncogene (2001) 20(44):6382–402. doi:10.1038/sj.onc.1204781
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science (1999) 286(5438):309–12. doi:10.1126/science.286.5438.309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Lill NL, Douillard P, Awwad RA, Ota S, Lupher ML Jr, Miyake S, et al. The evolutionarily conserved N-terminal region of Cbl is sufficient to enhance down-regulation of the epidermal growth factor receptor. J Biol Chem (2000) 275(1):367–77. doi:10.1074/jbc.275.1.367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Ota S, Hazeki K, Rao N, Lupher ML Jr, Andoniou CE, Druker B, et al. The RING finger domain of Cbl is essential for negative regulation of the Syk tyrosine kinase. J Biol Chem (2000) 275(1):414–22. doi:10.1074/jbc.275.1.414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Paolino M, Thien CB, Gruber T, Hinterleitner R, Baier G, Langdon WY, et al. Essential role of E3 ubiquitin ligase activity in Cbl-b-regulated T cell functions. J Immunol (2011) 186(4):2138–47. doi:10.4049/jimmunol.1003390
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Bartkiewicz M, Houghton A, Baron R. Leucine zipper-mediated homodimerization of the adaptor protein c-Cbl. A role in c-Cbl’s tyrosine phosphorylation and its association with epidermal growth factor receptor. J Biol Chem (1999) 274(43):30887–95. doi:10.1074/jbc.274.43.30887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Davies GC, Ettenberg SA, Coats AO, Mussante M, Ravichandran S, Collins J, et al. Cbl-b interacts with ubiquitinated proteins; differential functions of the UBA domains of c-Cbl and Cbl-b. Oncogene (2004) 23(42):7104–15. doi:10.1038/sj.onc.1207952
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Liu J, DeYoung SM, Hwang JB, O’Leary EE, Saltiel AR. The roles of Cbl-b and c-Cbl in insulin-stimulated glucose transport. J Biol Chem (2003) 278(38):36754–62. doi:10.1074/jbc.M300664200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Loeser S, Penninger JM. Regulation of peripheral T cell tolerance by the E3 ubiquitin ligase Cbl-b. Semin Immunol (2007) 19(3):206–14. doi:10.1016/j.smim.2007.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Li D, Gal I, Vermes C, Alegre ML, Chong AS, Chen L, et al. Cutting edge: Cbl-b: one of the key molecules tuning CD28- and CTLA-4-mediated T cell costimulation. J Immunol (2004) 173(12):7135–9. doi:10.4049/jimmunol.173.12.7135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Zhang J, Bardos T, Li D, Gal I, Vermes C, Xu J, et al. Cutting edge: regulation of T cell activation threshold by CD28 costimulation through targeting Cbl-b for ubiquitination. J Immunol (2002) 169(5):2236–40. doi:10.4049/jimmunol.169.5.2236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med (2011) 3(10):581–92. doi:10.1002/emmm.201100165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature (2005) 435(7042):590–7. doi:10.1038/nature03724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Mathis D, Benoist C. Back to central tolerance. Immunity (2004) 20(5):509–16. doi:10.1016/S1074-7613(04)00111-6
45. Borde M, Barrington RA, Heissmeyer V, Carroll MC, Rao A. Transcriptional basis of lymphocyte tolerance. Immunol Rev (2006) 210:105–19. doi:10.1111/j.0105-2896.2006.00370.x
46. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev (2006) 212:8–27. doi:10.1111/j.0105-2896.2006.00427.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol (2006) 6(4):295–307. doi:10.1038/nri1806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Samelson LE. Signal transduction mediated by the T cell antigen receptor: the role of adapter proteins. Annu Rev Immunol (2002) 20:371–94. doi:10.1146/annurev.immunol.20.092601.111357
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Altman A, Isakov N, Baier G. Protein kinase Ctheta: a new essential superstar on the T-cell stage. Immunol Today (2000) 21(11):567–73. doi:10.1016/S0167-5699(00)01749-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Marsland BJ, Kopf M. T-cell fate and function: PKC-theta and beyond. Trends Immunol (2008) 29(4):179–85. doi:10.1016/j.it.2008.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol (2005) 5(6):472–84. doi:10.1038/nri1632
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Bour-Jordan H, Esensten JH, Martinez-Llordella M, Penaranda C, Stumpf M, Bluestone JA. Intrinsic and extrinsic control of peripheral T-cell tolerance by costimulatory molecules of the CD28/B7 family. Immunol Rev (2011) 241(1):180–205. doi:10.1111/j.1600-065X.2011.01011.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Sadegh-Nasseri S, Dalai SK, Korb Ferris LC, Mirshahidi S. Suboptimal engagement of the T-cell receptor by a variety of peptide-MHC ligands triggers T-cell anergy. Immunology (2010) 129(1):1–7. doi:10.1111/j.1365-2567.2009.03206.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Fang D, Liu YC. Proteolysis-independent regulation of PI3K by Cbl-b-mediated ubiquitination in T cells. Nat Immunol (2001) 2(9):870–5. doi:10.1038/ni0901-870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Guo H, Qiao G, Ying H, Li Z, Zhao Y, Liang Y, et al. E3 ubiquitin ligase Cbl-b regulates Pten via Nedd4 in T cells independently of its ubiquitin ligase activity. Cell Rep (2012) 1(5):472–82. doi:10.1016/j.celrep.2012.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity (2004) 21(2):167–77. doi:10.1016/j.immuni.2004.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Krawczyk CM, Jones RG, Atfield A, Bachmaier K, Arya S, Odermatt B, et al. Differential control of CD28-regulated in vivo immunity by the E3 ligase Cbl-b. J Immunol (2005) 174(3):1472–8. doi:10.4049/jimmunol.174.3.1472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Naramura M, Jang IK, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol (2002) 3(12):1192–9. doi:10.1038/ni855
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Qiao G, Li Z, Molinero L, Alegre ML, Ying H, Sun Z, et al. T-cell receptor-induced NF-kappaB activation is negatively regulated by E3 ubiquitin ligase Cbl-b. Mol Cell Biol (2008) 28(7):2470–80. doi:10.1128/MCB.01505-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Gruber T, Hermann-Kleiter N, Hinterleitner R, Fresser F, Schneider R, Gastl G, et al. PKC-theta modulates the strength of T cell responses by targeting Cbl-b for ubiquitination and degradation. Sci Signal (2009) 2(76):ra30. doi:10.1126/scisignal.2000046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Yang B, Gay DL, MacLeod MK, Cao X, Hala T, Sweezer EM, et al. Nedd4 augments the adaptive immune response by promoting ubiquitin-mediated degradation of Cbl-b in activated T cells. Nat Immunol (2008) 9(12):1356–63. doi:10.1038/ni.1670
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Hanlon A, Jang S, Salgame P. Cbl-b differentially regulates activation-induced apoptosis in T helper 1 and T helper 2 cells. Immunology (2005) 116(4):507–12. doi:10.1111/j.1365-2567.2005.02252.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Ou R, Zhang M, Huang L, Moskophidis D. Control of virus-specific CD8+ T-cell exhaustion and immune-mediated pathology by E3 ubiquitin ligase Cbl-b during chronic viral infection. J Virol (2008) 82(7):3353–68. doi:10.1128/JVI.01350-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Parish IA, Rao S, Smyth GK, Juelich T, Denyer GS, Davey GM, et al. The molecular signature of CD8+ T cells undergoing deletional tolerance. Blood (2009) 113(19):4575–85. doi:10.1182/blood-2008-10-185223
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Gronski MA, Boulter JM, Moskophidis D, Nguyen LT, Holmberg K, Elford AR, et al. TCR affinity and negative regulation limit autoimmunity. Nat Med (2004) 10(11):1234–9. doi:10.1038/nm1114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Tran DQ. TGF-beta: the sword, the wand, and the shield of FOXP3(+) regulatory T cells. J Mol Cell Biol (2012) 4(1):29–37. doi:10.1093/jmcb/mjr033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Gorelik L, Constant S, Flavell RA. Mechanism of transforming growth factor beta-induced inhibition of T helper type 1 differentiation. J Exp Med (2002) 195(11):1499–505. doi:10.1084/jem.20012076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441(7090):235–8. doi:10.1038/nature04753
69. Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity (2000) 12(2):171–81. doi:10.1016/S1074-7613(00)80170-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8(5):369–80. doi:10.1016/j.ccr.2005.10.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Adams CO, Housley WJ, Bhowmick S, Cone RE, Rajan TV, Forouhar F, et al. Cbl-b(-/-) T cells demonstrate in vivo resistance to regulatory T cells but a context-dependent resistance to TGF-beta. J Immunol (2010) 185(4):2051–8. doi:10.4049/jimmunol.1001171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest (2007) 117(4):1029–36. doi:10.1172/JCI29472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med (2007) 204(4):879–91. doi:10.1084/jem.20061699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Lutz-Nicoladoni C, Wallner S, Stoitzner P, Pircher M, Gruber T, Wolf AM, et al. Reinforcement of cancer immunotherapy by adoptive transfer of cblb-deficient CD8+ T cells combined with a DC vaccine. Immunol Cell Biol (2012) 90(1):130–4. doi:10.1038/icb.2011.11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Wohlfert EA, Callahan MK, Clark RB. Resistance to CD4+CD25+ regulatory T cells and TGF-beta in Cbl-b-/- mice. J Immunol (2004) 173(2):1059–65. doi:10.4049/jimmunol.173.2.1059
76. Wohlfert EA, Gorelik L, Mittler R, Flavell RA, Clark RB. Cutting edge: deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-beta sensitivity in vitro and in vivo. J Immunol (2006) 176(3):1316–20. doi:10.4049/jimmunol.176.3.1316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi:10.1084/jem.20030152
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Harada Y, Harada Y, Elly C, Ying G, Paik JH, DePinho RA, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med (2010) 207(7):1381–91. doi:10.1084/jem.20100004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Haxhinasto S, Mathis D, Benoist C. The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med (2008) 205(3):565–74. doi:10.1084/jem.20071477
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Qiao G, Zhao Y, Li Z, Tang PQ, Langdon WY, Yang T, et al. T cell activation threshold regulated by E3 ubiquitin ligase Cbl-b determines fate of inducible regulatory T cells. J Immunol (2013) 191(2):632–9. doi:10.4049/jimmunol.1202068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Gruber T, Hinterleitner R, Hermann-Kleiter N, Meisel M, Kleiter I, Wang CM, et al. Cbl-b mediates TGFbeta sensitivity by downregulating inhibitory SMAD7 in primary T cells. J Mol Cell Biol (2013) 5(6):358–68. doi:10.1093/jmcb/mjt017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi:10.1056/NEJMoa1200694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi:10.1056/NEJMoa1200690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Qiao G, Ying H, Zhao Y, Liang Y, Guo H, Shen H, et al. E3 ubiquitin ligase Cbl-b suppresses proallergic T cell development and allergic airway inflammation. Cell Rep (2014) 6(4):709–23. doi:10.1016/j.celrep.2014.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Oh SY, Park JU, Zheng T, Kim YK, Wu F, Cho SH, et al. Cbl-b regulates airway mucosal tolerance to aeroallergen. Clin Exp Allergy (2011) 41(3):434–42. doi:10.1111/j.1365-2222.2010.03593.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Kaplan MH. Th9 cells: differentiation and disease. Immunol Rev (2013) 252(1):104–15. doi:10.1111/imr.12028
88. Zhou P, Shaffer DR, Alvarez Arias DA, Nakazaki Y, Pos W, Torres AJ, et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature (2014) 506(7486):52–7. doi:10.1038/nature12988
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature (2014) 507(7493):508–12. doi:10.1038/nature12998
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Wolf D, Wolf AM, Rumpold H, Fiegl H, Zeimet AG, Muller-Holzner E, et al. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin Cancer Res (2005) 11(23):8326–31. doi:10.1158/1078-0432.CCR-05-1244
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Oksvold MP, Dagger SA, Thien CB, Langdon WY. The Cbl-b RING finger domain has a limited role in regulating inflammatory cytokine production by IgE-activated mast cells. Mol Immunol (2008) 45(4):925–36. doi:10.1016/j.molimm.2007.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Brode S, Cooke A. Immune-potentiating effects of the chemotherapeutic drug cyclophosphamide. Crit Rev Immunol (2008) 28(2):109–26. doi:10.1615/CritRevImmunol.v28.i2.20
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Shafer-Weaver KA, Watkins SK, Anderson MJ, Draper LJ, Malyguine A, Alvord WG, et al. Immunity to murine prostatic tumors: continuous provision of T-cell help prevents CD8 T-cell tolerance and activates tumor-infiltrating dendritic cells. Cancer Res (2009) 69(15):6256–64. doi:10.1158/0008-5472.CAN-08-4516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res (2009) 69(22):8693–9. doi:10.1158/0008-5472.CAN-09-1456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Wada S, Yoshimura K, Hipkiss EL, Harris TJ, Yen HR, Goldberg MV, et al. Cyclophosphamide augments antitumor immunity: studies in an autochthonous prostate cancer model. Cancer Res (2009) 69(10):4309–18. doi:10.1158/0008-5472.CAN-08-4102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Shi FD, Ljunggren HG, La Cava A, Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol (2011) 11(10):658–71. doi:10.1038/nri3065
97. Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity (2008) 28(4):571–80. doi:10.1016/j.immuni.2008.02.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Montaldo E, Del Zotto G, Della Chiesa M, Mingari MC, Moretta A, De Maria A, et al. Human NK cell receptors/markers: a tool to analyze NK cell development, subsets and function. Cytometry A (2013) 83(8):702–13. doi:10.1002/cyto.a.22302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Huang JY, Umehara H, Inoue H, Tabassam FH, Okazaki T, Kono T, et al. Differential interaction of Cbl with Grb2 and CrkL in CD2-mediated NK cell activation. Mol Immunol (2000) 37(17):1057–65. doi:10.1016/S0161-5890(01)00020-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Mallone R, Funaro A, Zubiaur M, Baj G, Ausiello CM, Tacchetti C, et al. Signaling through CD38 induces NK cell activation. Int Immunol (2001) 13(4):397–409. doi:10.1093/intimm/13.4.397
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. McVicar DW, Burshtyn DN. Intracellular signaling by the killer immunoglobulin-like receptors and Ly49. Sci STKE (2001) 2001(75):re1. doi:10.1126/stke.2001.75.re1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Umehara H, Inoue H, Huang J, Kono T, Minami Y, Tanaka Y, et al. Role for adapter proteins in costimulatory signals of CD2 and IL-2 on NK cell activation. Mol Immunol (2002) 38(8):587–96. doi:10.1016/S0161-5890(01)00099-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Peterson ME, Long EO. Inhibitory receptor signaling via tyrosine phosphorylation of the adaptor Crk. Immunity (2008) 29(4):578–88. doi:10.1016/j.immuni.2008.07.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Kim HS, Das A, Gross CC, Bryceson YT, Long EO. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c-Cbl ubiquitin ligase. Immunity (2010) 32(2):175–86. doi:10.1016/j.immuni.2010.02.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Kim HS, Long EO. Complementary phosphorylation sites in the adaptor protein SLP-76 promote synergistic activation of natural killer cells. Sci Signal (2012) 5(232):ra49. doi:10.1126/scisignal.2002754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Molfetta R, Quatrini L, Capuano C, Gasparrini F, Zitti B, Zingoni A, et al. c-Cbl regulates MICA- but not ULBP2-induced NKG2D down-modulation in human NK cells. Eur J Immunol (2014) 44(9):2761–70. doi:10.1002/eji.201444512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res (2008) 100:35–83. doi:10.1016/S0065-230X(08)00002-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Qiao G, Lei M, Li Z, Sun Y, Minto A, Fu YX, et al. Negative regulation of CD40-mediated B cell responses by E3 ubiquitin ligase casitas-B-lineage lymphoma protein-B. J Immunol (2007) 179(7):4473–9. doi:10.4049/jimmunol.179.7.4473
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Kitaura Y, Jang IK, Wang Y, Han YC, Inazu T, Cadera EJ, et al. Control of the B cell-intrinsic tolerance programs by ubiquitin ligases Cbl and Cbl-b. Immunity (2007) 26(5):567–78. doi:10.1016/j.immuni.2007.03.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Yasuda T, Tezuka T, Maeda A, Inazu T, Yamanashi Y, Gu H, et al. Cbl-b positively regulates Btk-mediated activation of phospholipase C-gamma2 in B cells. J Exp Med (2002) 196(1):51–63. doi:10.1084/jem.20020068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Yasuda T, Maeda A, Kurosaki M, Tezuka T, Hironaka K, Yamamoto T, et al. Cbl suppresses B cell receptor-mediated phospholipase C (PLC)-gamma2 activation by regulating B cell linker protein-PLC-gamma2 binding. J Exp Med (2000) 191(4):641–50. doi:10.1084/jem.191.4.641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Sohn HW, Gu H, Pierce SK. Cbl-b negatively regulates B cell antigen receptor signaling in mature B cells through ubiquitination of the tyrosine kinase Syk. J Exp Med (2003) 197(11):1511–24. doi:10.1084/jem.20021686
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Katkere B, Rosa S, Drake JR. The Syk-binding ubiquitin ligase c-Cbl mediates signaling-dependent B cell receptor ubiquitination and B cell receptor-mediated antigen processing and presentation. J Biol Chem (2012) 287(20):16636–44. doi:10.1074/jbc.M112.357640
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Niiro H, Jabbarzadeh-Tabrizi S, Kikushige Y, Shima T, Noda K, Ota S, et al. CIN85 is required for Cbl-mediated regulation of antigen receptor signaling in human B cells. Blood (2012) 119(10):2263–73. doi:10.1182/blood-2011-04-351965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Veselits M, Tanaka A, Lipkowitz S, O’Neill S, Sciammas R, Finnegan A, et al. Recruitment of Cbl-b to B cell antigen receptor couples antigen recognition to toll-like receptor 9 activation in late endosomes. PLoS One (2014) 9(3):e89792. doi:10.1371/journal.pone.0089792
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol (2012) 30:1–22. doi:10.1146/annurev-immunol-100311-102839
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol (2003) 4(6):579–85. doi:10.1038/ni928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Chiou SH, Shahi P, Wagner RT, Hu H, Lapteva N, Seethammagari M, et al. The E3 ligase c-Cbl regulates dendritic cell activation. EMBO Rep (2011) 12(9):971–9. doi:10.1038/embor.2011.143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Wallner S, Lutz-Nicoladoni C, Tripp CH, Gastl G, Baier G, Penninger JM, et al. The role of the e3 ligase cbl-B in murine dendritic cells. PLoS One (2013) 8(6):e65178. doi:10.1371/journal.pone.0065178
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol (2010) 11(8):734–42. doi:10.1038/ni.1908
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Arron JR, Vologodskaia M, Wong BR, Naramura M, Kim N, Gu H, et al. A positive regulatory role for Cbl family proteins in tumor necrosis factor-related activation-induced cytokine (trance) and CD40L-mediated Akt activation. J Biol Chem (2001) 276(32):30011–7. doi:10.1074/jbc.M100414200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Hinterleitner R, Gruber T, Pfeifhofer-Obermair C, Lutz-Nicoladoni C, Tzankov A, Schuster M, et al. Adoptive transfer of siRNA Cblb-silenced CD8+ T lymphocytes augments tumor vaccine efficacy in a B16 melanoma model. PLoS One (2012) 7(9):e44295. doi:10.1371/journal.pone.0044295
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Mahnke K, Qian Y, Knop J, Enk AH. Induction of CD4+/CD25+ regulatory T cells by targeting of antigens to immature dendritic cells. Blood (2003) 101(12):4862–9. doi:10.1182/blood-2002-10-3229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4(7):499–511. doi:10.1038/nri1391
125. Abe T, Hirasaka K, Kagawa S, Kohno S, Ochi A, Utsunomiya K, et al. Cbl-b is a critical regulator of macrophage activation associated with obesity-induced insulin resistance in mice. Diabetes (2013) 62(6):1957–69. doi:10.2337/db12-0677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Hirasaka K, Kohno S, Goto J, Furochi H, Mawatari K, Harada N, et al. Deficiency of Cbl-b gene enhances infiltration and activation of macrophages in adipose tissue and causes peripheral insulin resistance in mice. Diabetes (2007) 56(10):2511–22. doi:10.2337/db06-1768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Bachmaier K, Toya S, Gao X, Triantafillou T, Garrean S, Park GY, et al. E3 ubiquitin ligase Cblb regulates the acute inflammatory response underlying lung injury. Nat Med (2007) 13(8):920–6. doi:10.1038/nm1607
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ, et al. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood (2007) 110(3):1022–4. doi:10.1182/blood-2006-12-061176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, et al. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol (2009) 27(36):6109–16. doi:10.1200/JCO.2009.23.7503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Schwaab J, Schnittger S, Sotlar K, Walz C, Fabarius A, Pfirrmann M, et al. Comprehensive mutational profiling in advanced systemic mastocytosis. Blood (2013) 122(14):2460–6. doi:10.1182/blood-2013-04-496448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Nadeau S, An W, Palermo N, Feng D, Ahmad G, Dong L, et al. Oncogenic signaling by leukemia-associated mutant Cbl proteins. Biochem Anal Biochem (2012) 1(Suppl 6):7921. doi:10.4172/2161-1009.S6-001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Naramura M, Nandwani N, Gu H, Band V, Band H. Rapidly fatal myeloproliferative disorders in mice with deletion of casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc Natl Acad Sci U S A (2010) 107(37):16274–9. doi:10.1073/pnas.1007575107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Kohno S, Ueji T, Abe T, Nakao R, Hirasaka K, Oarada M, et al. Rantes secreted from macrophages disturbs skeletal muscle regeneration after cardiotoxin injection in Cbl-b-deficient mice. Muscle Nerve (2011) 43(2):223–9. doi:10.1002/mus.21829
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Yee NK, Hamerman JA. Beta(2) integrins inhibit TLR responses by regulating NF-kappaB pathway and p38 MAPK activation. Eur J Immunol (2013) 43(3):779–92. doi:10.1002/eji.201242550
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Choi EY, Orlova VV, Fagerholm SC, Nurmi SM, Zhang L, Ballantyne CM, et al. Regulation of LFA-1-dependent inflammatory cell recruitment by Cbl-b and 14-3-3 proteins. Blood (2008) 111(7):3607–14. doi:10.1182/blood-2007-07-103077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Zhang J, Chiang YJ, Hodes RJ, Siraganian RP. Inactivation of c-Cbl or Cbl-b differentially affects signaling from the high affinity IgE receptor. J Immunol (2004) 173(3):1811–8. doi:10.4049/jimmunol.173.3.1811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Qu X, Sada K, Kyo S, Maeno K, Miah SM, Yamamura H. Negative regulation of FcepsilonRI-mediated mast cell activation by a ubiquitin-protein ligase Cbl-b. Blood (2004) 103(5):1779–86. doi:10.1182/blood-2003-07-2260
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Gustin SE, Thien CB, Langdon WY. Cbl-b is a negative regulator of inflammatory cytokines produced by IgE-activated mast cells. J Immunol (2006) 177(9):5980–9. doi:10.4049/jimmunol.177.9.5980
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Nakajima A, Sanjay A, Chiusaroli R, Adapala NS, Neff L, Itzsteink C, et al. Loss of Cbl-b increases osteoclast bone-resorbing activity and induces osteopenia. J Bone Miner Res (2009) 24(7):1162–72. doi:10.1359/jbmr.090205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Adapala NS, Barbe MF, Tsygankov AY, Lorenzo JA, Sanjay A. Loss of Cbl-PI3K interaction enhances osteoclast survival due to p21-Ras mediated PI3K activation independent of Cbl-b. J Cell Biochem (2014) 115(7):1277–89. doi:10.1002/jcb.24779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Yokoi N, Komeda K, Wang HY, Yano H, Kitada K, Saitoh Y, et al. Cblb is a major susceptibility gene for rat type 1 diabetes mellitus. Nat Genet (2002) 31(4):391–4. doi:10.1038/ng927
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Bergholdt R, Taxvig C, Eising S, Nerup J, Pociot F. CBLB variants in type 1 diabetes and their genetic interaction with CTLA4. J Leukoc Biol (2005) 77(4):579–85. doi:10.1189/jlb.0904524
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Doniz-Padilla L, Martinez-Jimenez V, Nino-Moreno P, Abud-Mendoza C, Hernandez-Castro B, Gonzalez-Amaro R, et al. Expression and function of Cbl-b in T cells from patients with systemic lupus erythematosus, and detection of the 2126 A/G Cblb gene polymorphism in the Mexican mestizo population. Lupus (2011) 20(6):628–35. doi:10.1177/0961203310394896
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. DeWan AT, Egan KB, Hellenbrand K, Sorrentino K, Pizzoferrato N, Walsh KM, et al. Whole-exome sequencing of a pedigree segregating asthma. BMC Med Genet (2012) 13:95. doi:10.1186/1471-2350-13-95
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature (2011) 476(7359):214–9. doi:10.1038/nature10251
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Sanna S, Pitzalis M, Zoledziewska M, Zara I, Sidore C, Murru R, et al. Variants within the immunoregulatory CBLB gene are associated with multiple sclerosis. Nat Genet (2010) 42(6):495–7. doi:10.1038/ng.584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. International Multiple Sclerosis Genetics Consortium, Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, et al. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med (2007) 357(9):851–62. doi:10.1056/NEJMoa073493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Banica LM, Besliu AN, Pistol GC, Stavaru C, Vlad V, Predeteanu D, et al. Dysregulation of anergy-related factors involved in regulatory T cells defects in systemic lupus erythematosus patients: rapamycin and vitamin D efficacy in restoring regulatory T cells. Int J Rheum Dis (2014). doi:10.1111/1756-185X.12509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Gomez-Martin D, Ibarra-Sanchez M, Romo-Tena J, Cruz-Ruiz J, Esparza-Lopez J, Galindo-Campos M, et al. Casitas B lineage lymphoma b is a key regulator of peripheral tolerance in systemic lupus erythematosus. Arthritis Rheum (2013) 65(4):1032–42. doi:10.1002/art.37833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Sturner KH, Borgmeyer U, Schulze C, Pless O, Martin R. A multiple sclerosis-associated variant of CBLB links genetic risk with type I IFN function. J Immunol (2014) 193(9):4439–47. doi:10.4049/jimmunol.1303077
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Zhou WB, Wang R, Deng YN, Ji XB, Huang GX, Xu YZ. Study of Cbl-b dynamics in peripheral blood lymphocytes isolated from patients with multiple sclerosis. Neurosci Lett (2008) 440(3):336–9. doi:10.1016/j.neulet.2008.05.089
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Gruber T, Hinterleitner R, Pfeifhofer-Obermair C, Wolf D, Baier G. Engineering effective T-cell based antitumor immunity. Oncoimmunology (2013) 2(2):e22893. doi:10.4161/onci.22893
153. den Brok MH, Nierkens S, Figdor CG, Ruers TJ, Adema GJ. Dendritic cells: tools and targets for antitumor vaccination. Expert Rev Vaccines (2005) 4(5):699–710. doi:10.1586/14760584.4.5.699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med (2010) 363(5):411–22. doi:10.1056/NEJMoa1001294
155. Powell DJ Jr, Dudley ME, Hogan KA, Wunderlich JR, Rosenberg SA. Adoptive transfer of vaccine-induced peripheral blood mononuclear cells to patients with metastatic melanoma following lymphodepletion. J Immunol (2006) 177(9):6527–39. doi:10.4049/jimmunol.177.9.6527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer (2008) 8(4):299–308. doi:10.1038/nrc2355
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Leen AM, Rooney CM, Foster AE. Improving T cell therapy for cancer. Annu Rev Immunol (2007) 25:243–65. doi:10.1146/annurev.immunol.25.022106.141527
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Coccoris M, Straetemans T, Govers C, Lamers C, Sleijfer S, Debets R. T cell receptor (TCR) gene therapy to treat melanoma: lessons from clinical and preclinical studies. Expert Opin Biol Ther (2010) 10(4):547–62. doi:10.1517/14712591003614756
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314(5796):126–9. doi:10.1126/science.1129003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Ngo MC, Rooney CM, Howard JM, Heslop HE. Ex vivo gene transfer for improved adoptive immunotherapy of cancer. Hum Mol Genet (2011) 20(R1):R93–9. doi:10.1093/hmg/ddr102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science (2003) 302(5644):415–9. doi:10.1126/science.1088547
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Pilones KA, Aryankalayil J, Demaria S. Invariant NKT cells as novel targets for immunotherapy in solid tumors. Clin Dev Immunol (2012) 2012:720803. doi:10.1155/2012/720803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Huang JR, Tsai YC, Chang YJ, Wu JC, Hung JT, Lin KH, et al. alpha-galactosylceramide but not phenyl-glycolipids induced NKT cell anergy and IL-33-mediated myeloid-derived suppressor cell accumulation via upregulation of egr2/3. J Immunol (2014) 192(4):1972–81. doi:10.4049/jimmunol.1302623
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Singh AK, Gaur P, Das SN. Natural killer T cell anergy, co-stimulatory molecules and immunotherapeutic interventions. Hum Immunol (2014) 75(3):250–60. doi:10.1016/j.humimm.2013.12.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cancer, autoimmunity, immunotherapy, ubiquitination, adoptive cell transfer
Citation: Lutz-Nicoladoni C, Wolf D and Sopper S (2015) Modulation of immune cell functions by the E3 ligase Cbl-b. Front. Oncol. 5:58. doi: 10.3389/fonc.2015.00058
Received: 12 December 2014; Paper pending published: 09 January 2015;
Accepted: 24 February 2015; Published online: 11 March 2015.
Edited by:
Amir Sharabi, Tel-Aviv University, IsraelReviewed by:
Xinhui Wang, Massachusetts General Hospital and Harvard Medical School, USADoug Palmer, National Cancer Institute, USA
Copyright: © 2015 Lutz-Nicoladoni, Wolf and Sopper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sieghart Sopper, Tumor Immunology Laboratory, Department of Hematology and Oncology, Medical University Innsbruck, Anichstr. 35, Innsbruck 6020, Austria e-mail:c2llZ2hhcnQuc29wcGVyQGktbWVkLmFjLmF0