 Hironori Kato
Hironori Kato Hideki Nishitoh*
Hideki Nishitoh*- Laboratory of Biochemistry and Molecular Biology, Department of Medical Sciences, University of Miyazaki, Miyazaki, Japan
The endoplasmic reticulum (ER) is a dynamic organelle that is essential for multiple cellular functions. During cellular stress conditions, including nutrient deprivation and dysregulation of protein synthesis, unfolded/misfolded proteins accumulate in the ER lumen, resulting in activation of the unfolded protein response (UPR). The UPR also contributes to the regulation of various intracellular signaling pathways such as calcium signaling and lipid signaling. More recently, the mitochondria-associated ER membrane (MAM), which is a site of close contact between the ER and mitochondria, has been shown to function as a platform for various intracellular stress responses including apoptotic signaling, inflammatory signaling, the autophagic response, and the UPR. Interestingly, in cancer, these signaling pathways from the ER are often dysregulated, contributing to cancer cell metabolism. Thus, the signaling pathway from the ER may be a novel therapeutic target for various cancers. In this review, we discuss recent research on the roles of stress responses from the ER, including the MAM.
Introduction
In eukaryotic cells, the endoplasmic reticulum (ER) is an organelle that extends throughout the cytoplasm as a vast membranous network. The shape of the ER reflects its many cellular functions, which include folding newly synthesized proteins, calcium homeostasis, and phospholipid synthesis, leading to the regulation of various intracellular signaling pathways (1–6). In particular, as a central platform of protein quality control, the ER contributes to adaptation to adverse synthetic, metabolic, and other conditions. When the integrity of the ER is perturbed by adverse conditions, unfolded/misfolded proteins accumulate in the ER lumen, a condition called ER stress, which in turn activates the unfolded protein response (UPR) (7, 8). Furthermore, some reports have suggested that perturbation of calcium homeostasis and dysregulation of lipid metabolism are also involved in activation of the UPR (9–14). UPR signaling consists of three major stress sensors that reside on the ER membrane: RNA-dependent protein kinase-like kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1α (IRE1α). The roles of these signaling pathways are to adapt to ER stress through translational attenuation, upregulation of ER chaperones, and protein degradation (8, 15) (Figure 1). Interestingly, activation of the UPR in cancer cells is sustained by exposure to various stresses, including hypoxia, oxidative stress, and nutrient starvation. Therefore, it is currently believed that the UPR may play critical roles in tumor progression, metastasis, tumorigenesis, and survival (16–18).
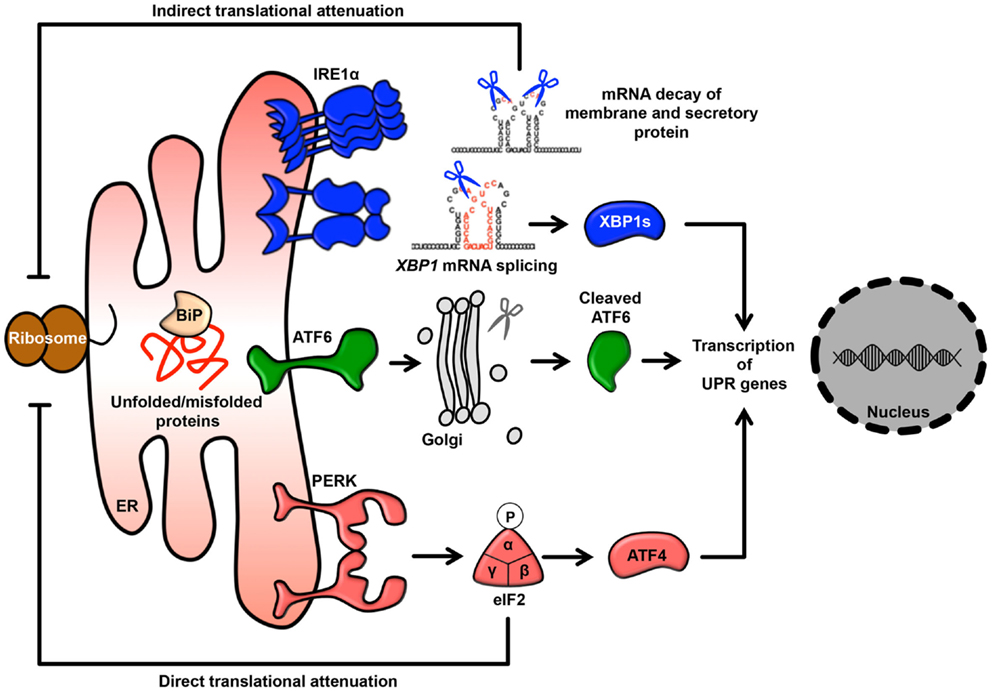
Figure 1. The UPR signaling pathway. Membrane and secretory proteins are synthesized by the rER and translocated into the ER lumen. Activated PERK phosphorylates eIF2α and causes general translational attenuation. The PERK-eIF2α pathway selectively induces expression of the transcription factor ATF4. Activated IRE1α undergoes dimerization and oligomerization, splices an intron from the XBP1 mRNA and produces the transcription factor XBP1s. IRE1α can also induce mRNA degradation. Activated ATF6 translocates to the Golgi and is sequentially cleaved by the Golgi-resident site-1 and site-2 proteases, thereby releasing cleaved ATF6.
It has been observed that approximately 5–20% of the mitochondrial surface directly contacts the ER (19, 20). This site of close contact between the ER and mitochondria is called the mitochondria-associated ER membrane (MAM) and is formed by several molecular bridges. The mitochondrial outer membrane proteins mitofusin 1 and 2 (MFN1 and MFN2) are required for mitochondrial fusion. MFN1 contributes to mitochondrial docking and fusion, whereas MFN2 stabilizes associations between mitochondria. This maintenance of mitochondrial morphology is regulated by hetero-oligomeric MFN complexes on the mitochondrial outer membrane. Interestingly, MFN2 is also located on the ER membrane and forms homotypic or heterotypic complexes with mitochondrial MFNs, resulting in an interaction between the ER and mitochondria (21) (Figure 2). Acting as a similar molecular bridge, the inositol triphosphate receptor (IP3R) on the ER indirectly interacts with the mitochondrial outer membrane-resident voltage-dependent anion channel 1 (VDAC1) via the cytosolic chaperone glucose-regulated protein 75 (GRP75) (22). In addition, recent reports have demonstrated that the B-cell receptor-associated protein 31 (Bap31)-mitochondrial Fission-1 homolog (Fis1) complex (23) and vesicle-associated membrane protein-associated protein B (VAPB)-protein-tyrosine phosphatase interacting protein 51 (PTPIP51) complex act as tethering complexes for the ER-mitochondrion bridge (24) (Figure 2).
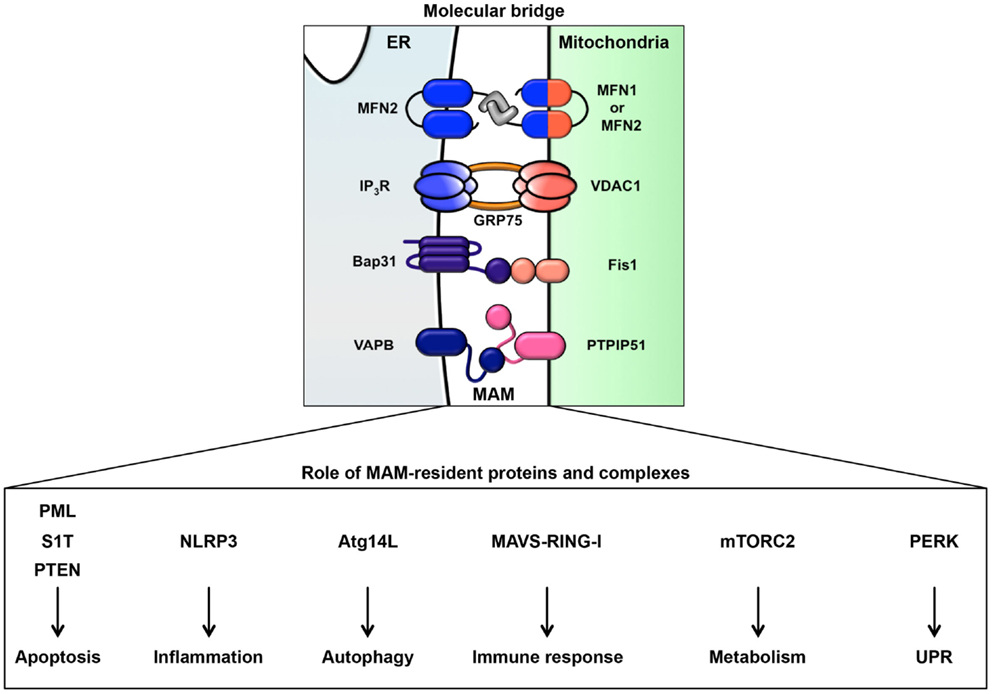
Figure 2. ER-mitochondrion tethering. In mammalian cells, four types of molecular bridges for ER-mitochondrion tethering have been identified. ER-resident MFN2 interacts with mitochondrial MFN1 and MFN2. The ER calcium channel IP3R associates with the mitochondrial calcium channel VDAC1 through GRP75. The ER protein Bap31 connects with the mitochondrial protein Fis1. The ER protein VAPB interacts with the mitochondrial protein PTPIP51. Indicated MAM-resident proteins and complexes regulate various signaling pathway from MAM as described in figure.
The MAM is crucial for the regulation of numerous cellular functions including lipid trafficking (25–27), calcium cycling (28, 29), and energy metabolism (30). This subdomain of the ER also directly contributes to biogenesis through the synthesis of phospholipids and sphingolipids, the levels of which are enriched at the MAM (25, 31–35). Interestingly, most cancer cells show characteristic alterations in de novo lipid biosynthesis, lipogenic phenotype, and lipid metabolism (36, 37). Therefore, current reports have considered that lipogenesis- and lipolysis-related pathways are involved in tumorigenesis, migration, invasion, and survival (36–38). The MAM has also been shown to act as a platform for various intracellular signaling pathways, including oncogenic signaling (39–41). Although several groups have addressed the pathophysiological relevance between the MAM and diseases, the role of the MAM, especially in cancer, has not been clearly elucidated. In this review, we will summarize some emerging roles of stress responses from the ER, particularly from the MAM, and discuss whether the MAM might be a potential therapeutic target in cancer.
Signaling Pathways from the ER
Endoplasmic reticulum stress activates the UPR, which is the most thoroughly characterized stress signaling pathway from the ER. The UPR also plays important roles in tumor development and tumor growth through adaptation to microenvironments. In cancer cells, the UPR is constitutively activated because abnormal cell proliferation requires elevated protein synthesis during tumor development and tumor growth (42, 43). In addition, cancer cells are exposed to various stresses such as hypoxia, low glucose, low pH, and nutrient starvation, which are well known to induce the UPR (42–45). Recent research in oncology indicates that these microenvironmental challenges are associated with the constitutive activation of the UPR. Additionally, the perturbation of calcium homeostasis in cancer cells is correlated with their abnormal phenotypes, including sustained cell proliferation and avoidance of cell death, through the remodeling of calcium signaling (46–48). Furthermore, cancer cells undergo changes in lipid metabolism via the dysregulation of lipogenic and lipolytic enzymes, leading to lipid stress signaling. This is closely correlated with tumorigenesis, malignancy, and growth (36–38). Abnormalities in calcium homeostasis and lipid metabolism directly trigger ER stress (9–14). This section focuses on the cancer-related roles of the ER stress signaling pathways, including UPR signaling, calcium signaling, and lipid signaling.
The UPR
Unfolded protein response signaling is mediated by three major transmembrane transducers: PERK, ATF6, and IRE1α (Figure 1). These sensors are maintained in an inactive state by binding to an ER chaperone, the 78 kDa glucose-regulated protein (GRP78), which is known as a master regulator of the UPR. During ER stress, GRP78 dissociates from sensor proteins in response to the accumulation of unfolded/misfolded proteins and reduced ER calcium content, resulting in the activation of distinct UPR signaling branches (8, 13, 49).
The PERK Branch
Protein kinase-like kinase is an ER type I transmembrane kinase that has a PEK-like catalytic domain in its cytosolic C-terminal region. The activated PERK pathway phosphorylates eukaryotic translation initiation factor 2α (eIF2α) and leads to the inhibition of protein translation into the ER (8, 15, 50). In addition, phosphorylation of eIF2α selectively induces the expression of activating transcription factor 4 (ATF4), thereby inducing growth arrest and upregulating UPR genes including CCAAT/enhancer-binding protein-homologous protein (CHOP), ER oxidoreductase 1α, and several pro-apoptotic factors (8, 15). Phosphorylation of eIF2α is also induced by multiple kinases, including protein kinase R, general control non-repressed 2, and haem-regulated eIF2α kinase. eIF2α phosphorylation-related signaling is described as the integrated stress response (ISR) (43, 51–53). The ISR is activated in response to various stresses, including amino acid starvation, viral infection, and haem deficiencies, in addition to ER stress. PERK also directly phosphorylates transcription factor NF-E2-related factor 2 (Nrf2), which is known to have the dual role in cancer, tumor suppressor function and oncogenic function (54). Under unstressed condition, Nrf2 maintains the inactive state in the cytoplasm through interaction with kelch-like ECH-associated protein 1. Phosphorylated Nrf2 by PERK dissociates from kelch-like ECH-associated protein 1, resulting in translocation into the nucleus and expression of antioxidant genes (55, 56). However, artificial activation of PERK mainly induces ISR signaling target genes in an eIF2α phosphorylation-dependent manner (57). Although PERK is activated by not only ER stress but also other stress (e.g., glucose starvation and oxidative stress), PERK-mediated Nrf2 activation may be limited to the ER stress condition.
Most cancers are constitutively exposed to various stresses, including hypoxia and nutrient deprivation, even when glycolysis and angiogenesis are promoted in cancer cells (42–45). Activation of PERK signaling and the ISR are considered necessary for tumor survival during hypoxia and nutrient starvation (43). Hypoxia and oxidative stress increase the generation of reactive oxygen species (ROS). In addition, ER oxidoreductase 1α, which regulates ER redox status, is upregulated through PERK signaling in the ISR (58–60). Consistent with the enhanced production of ROS in tumors, the expression of ER oxidoreductase 1α is substantially increased in various types of cancers (61). The ablation of PERK signaling or the ISR leads to ROS production and thereby impairs tumor growth through oxidative DNA damage (62). Furthermore, the ISR, which induces ATF4 expression, is also required for tumor survival and growth. Suppression of ATF4 expression inhibits tumor survival and proliferation in response to both amino acid deprivation and glucose deprivation (63, 64). Taken together, cancer cells adapt to hypoxia, oxidative stress, and nutrient starvation by improving their PERK- and ISR-mediated redox homeostasis and metabolic homeostasis, respectively. Transcription factor CHOP, a downstream target of ATF4, induces cell death in response to the intense and/or prolonged ER stress. Deletion of CHOP or suppression DNA damage-inducible 34, a downstream of CHOP, promotes tumor dedifferentiation and survival. Thus, it is possible that this dual role of the PERK and ISR signaling pathways might be therapeutic targets for cancer. Indeed, it has been reported that PERK specific inhibitor GSK2656157 impairs angiogenesis and amino acid metabolism, resulting in prevention of tumorigenesis in vivo (65). Furthermore, in leukemic cells, salubrinal, a selective inhibitor of eIF2α dephosphorylation induces synergistic apoptosis under proteasome inhibitor-induced ER stress condition (66). These observations strongly suggest that the compounds, which regulate PERK pathway, may shed light on the treatment of cancer.
The IRE1α Branch
Inositol-requiring enzyme 1α is an ER type I transmembrane endoribonuclease/kinase protein that contains a kinase domain and an endoribonuclease domain in its cytosolic C-terminal region. During ER stress, activated IRE1α catalyzes the splicing of a 26-nt intron from the mRNA of X-box binding protein 1 (XBP1), resulting in the production of active XBP1 (XBP1s) transcription factor and the consequent promotion of ER-associated degradation (8, 15). IRE1α also cleaves non-specific mRNAs through a process termed regulated IRE1α-dependent mRNA decay (RIDD) (8, 15, 67, 68). Several reports have indicated that the IRE1α signaling, especially via the IRE1α-XBP1 pathway, is required for tumor growth and survival during hypoxia. For example, XBP1-deficient cells are vulnerable to hypoxia-induced apoptosis, leading to the impairment of hypoxic growth in tumor xenografts (18). A recent report suggests that expression of the spliced form of XBP1s is essential for tumor angiogenesis independently of vascular endothelial growth factor, a well-known angiogenic factor (69). XBP1s is constitutively activated through the hypoxia-inducible factor-1α pathway in response to hypoxia, resulting in tumor development (70). In addition, a recent analysis indicated that the IRE1α-RIDD pathway contributes to tumor growth, infiltration, and invasion under tumor microenvironment conditions (71).
Inositol-requiring enzyme 1α also induces activation of c-Jun N-terminal kinase (JNK) through interaction with tumor necrosis associated factor 2 and apoptosis signaling-regulating kinase 1 (72, 73). IRE1α-JNK pathway contributes to not only apoptotic cell death (72) but also cell survival via c-Jun activation (74, 75), suggesting that IRE1α-JNK pathway has binary effects on cell fate. In neuroblastoma, IRE1α-JNK pathway induced by severe ER stress triggers apoptosis (76). On the other hand, IRE1α-JNK pathway involves in induction of autophagy, which protect cancer cell death (77). Thus, it is considered that IRE1α signaling contributes to both tumor survival and cell death in some situations of chronic stress. Since activities of kinase and endoribonuclease of IRE1α can be selectively controlled by distinct classes of adenosine triphosphate (ATP)-competitive inhibitors (78), it is expected that regulation of pathway might be a therapeutic strategy for cancer.
Endoplasmic reticulum stress activates T cells in the adaptive immune system in tumor (79). IRE1α signaling phosphorylates IκB kinase and activates NF-κB pathway, leading to induction of inflammatory response (80). Pro-inflammatory cytokines interleukin (IL)-6 and tumor necrosis factor-α are induced by XBP1s in response to ER stress (81, 82). In macrophages, activation of UPR is triggered by culture media conditioned by ER stressor-treated cancer cells via toll-like receptor, suggesting that cancer cells non-cell-autonomously induce tumor inflammation through the transmission of UPR signaling to macrophages (83). Recent study has shown that loss of ER chaperone calreticulin results in the prevention of phagocytosis and antigen presentation in dendritic cells, and translocation of calreticulin to the plasma membrane induced by anti-neoplastic drugs anthracycline triggers tumor immunogenicity (84). Although anti-tumor agent cisplatin has no effect on induction of an anti-cancer immune response via the UPR activation, the combination of ER stress inducer (e.g., thapsigargin or tunicamycin) with cisplatin triggers translocation of calreticulin to the plasma membrane and trigger tumor immunogenicity (85). Thus, it is possibility that combination of anti-tumor drugs and ER stress inducers might become therapeutic strategies for various cancers.
The ATF6 Branch
Activating transcription factor 6 is an ER type II transmembrane protein that possesses a transcriptional activation domain in its cytosolic region. Upon ER stress, ATF6 is transported from the ER membrane to the Golgi, where it is sequentially cleaved by the Golgi-resident site-1 and site-2 proteases. The cleaved ATF6 is translocated into the nucleus and binds to ER stress elements and UPR elements, leading to the expression of UPR target genes such as ER chaperones (8, 15). Expression of GRP78, which is upregulated by the ATF6 signaling during ER stress, has been shown to be essential for tumor growth, survival, progression, and metastasis (86–88). Conversely, overexpression of GRP78 suppresses non-steroidal anti-inflammatory drug-induced apoptosis in cancer cells (86). Therefore, both ATF6 signaling and GRP78 appear to contribute to tumor adaptation to microenvironmental challenges by promoting protein folding.
Calcium Signaling from the ER
The ER is a major intracellular calcium store. Calcium contributes to a wide variety of intracellular signaling pathways as a second messenger (48, 89). However, depletion of calcium in the ER disturbs the function of ER chaperones and induces ER stress, leading to activation of the UPR (11, 13, 14). In addition, increasing intracellular calcium concentrations triggers cytotoxicity under certain conditions such as increased ROS generation (48). Thus, both cytosolic calcium and the concentration of calcium in the ER must be tightly regulated. Import of calcium into the ER is maintained by sarcoplasmic/ER calcium ATPase (SERCA) pumps, which function against a calcium concentration gradient (90). Conversely, ER-resident calcium is released through two tetrameric calcium channels, ryanodine receptors and inositol 1,4,5-trisphosphate receptors (IP3Rs) (91–93). These mediators are involved in protein synthesis, gene expression, secretion, the cell cycle, cell proliferation, and differentiation through the regulation of calcium signaling (48, 89, 94). Surprisingly, in most cancers, the calcium concentration in the ER is decreased, whereas the cytosolic calcium concentration is increased (47, 48). This indicates that various cancers alter the activity or expression of SERCA pumps and IP3Rs, resulting in the remodeling of calcium signaling. Because abnormal intracellular calcium homeostasis occurs via altered activity or expression of ER-resident calcium pumps and channels in cancer, regulation of the ER-resident calcium status may be a candidate for the treatment of cancer.
Role of SERCA Pumps in the Remodeling of Calcium Signaling
Sarcoplasmic/ER calcium ATPase pumps, which are type P ATPase pumps, import calcium into the ER from the cytosol to regulate the cytosolic calcium level. They have diverged into three different genes and are further composed of at least five isoforms (SERCA1a, 1b, 2a, 2b, and 3). Among the SERCA pumps, SERCA2b is ubiquitously expressed and has the highest calcium affinity (29, 90, 95–97). Phospholamban and sarcolipin have been reported to be endogenous negative regulators that suppress the affinity of SERCA pumps for calcium (98). Furthermore, the activity of SERCA pumps is also regulated by post-translational modifications including SUMOylation, glutathiolation, and nitration (99–101). Several reports have shown that the expression of SERCA pumps, especially SERCA2b and SERCA3, is frequently decreased in cancer cells. Korosec et al. and Endo et al. reported that in colon, lung, and oral cancer, SERCA2b expression was reduced by alterations of the promoter region or by an epigenetic mechanism as an early event in tumorigenesis (102, 103). Although the mechanism remains unclear, the ATPase activity of SERCA2b is also reduced in advanced tumorigenic thyroid cells, consistent with the reductions at the mRNA and protein levels (104). Furthermore, germline alterations in SERCA2b have been indicated as closely involved in the early phase of carcinogenesis in the colon and lung (102). Similar to SERCA2b, SERCA3 is also reduced in colon and gastric cancer cells (105, 106). In addition, SERCA3 expression is involved in the remodeling of intracellular calcium homeostasis during cell differentiation (105).
Role of IP3Rs in the Remodeling of Calcium Signaling
Inositol triphosphate receptors are ER-resident calcium channels that form tetrameric assemblies and thereby release calcium into the cytosol from the ER. IP3Rs play an important role in calcium signaling and homeostasis. IP3Rs consist of three subtypes (IP3R1, IP3R2, and IP3R3) that are differentially expressed in specific cell types (91, 107, 108). IP3, which is generated via the phospholipase C pathway and is an agonist of IP3Rs, directly binds to the cytosolic region of all three IP3R subtypes and enhances calcium release (109, 110). The activity of IP3Rs is also positively or negatively regulated by endogenous factors including calcium, ATP, and pH (111, 112). Interestingly, some reports have demonstrated that kinase and cytosolic proteins can regulate the activity of IP3Rs (113–115). It is reported that serine/threonine kinase Akt/protein kinase B phosphorylates IP3Rs, resulting in inhibition of calcium release activity of IP3Rs (113, 114). In addition, Bcl-2 family protein BAX and BAK positively regulates calcium release from the ER through a decrease of IP3Rs phosphorylation (115).
Recent studies have shown alterations in the activity and expression of IP3Rs during various processes of tumor cells including survival, growth, proliferation, invasion, and metastasis (116–127). In breast cancer cells, activation of IP3Rs, especially IP3R3, is enhanced by ATP, thereby promoting cell growth through regulation of the spatiotemporal pattern of intracellular calcium (116–119). Moreover, enhanced expression of IP3R1 and IP3R3 is also involved in the epithelial-mesenchymal transition (EMT) of breast cancer (117, 120). In addition to breast cancer, IP3R3 is specifically overexpressed in gastric cancer cells (121). Inhibition of IP3R3 by an antagonist of IP3Rs attenuates their proliferation and induces apoptosis (121). Additionally, although the IP3R subtype remains unclear, colorectal cancer (122), lung cancer (123, 124), melanoma (125), and insulinoma (126, 127) also show enhanced IP3R activity and expression, leading to the remodeling of calcium signaling. Therefore, IP3Rs might be therapeutic targets for treatment of cancer.
Lipid Signaling from the ER
The ER can be classically divided into two types, rough ER (rER) and smooth ER (sER), which have different functions and morphology (128). The rER is studded with ribosomes on the membrane surface; it forms a membranous sheet and plays an important role as a major site of protein synthesis. By contrast, the sER is ribosome-free and exists as a tubular or reticular network throughout the cytoplasm (128). A large number of reports have demonstrated that sER is the site of lipid biogenesis. Among various lipids and phospholipids, glycerophospholipids and sphingolipids are essential as major components of biological membranes, and they are also important as signaling molecules (129, 130). These signaling lipids play important roles in cellular processes such as cell proliferation, migration, metabolism, inflammation, and apoptosis (Figure 3). In addition, both rER and sER are composed mainly of glycerophospholipids [i.e., phosphatidylcholine, phosphatidylethanolamine, phosphatidylinositol (PI), and phosphatidyleserine], and sphingolipids [i.e., sphingosine-1-phosphate (S1P) and sphingosine] (130). In these phospholipids, including both saturated and unsaturated fatty acids are usually disturbed in cancer cells (36–38). Recent studies have demonstrated that changing the balance between membrane phospholipid saturation and unsaturation activates UPR signaling (10, 12). Furthermore, alterations in fatty acid unsaturation in membranes are implicated in cancer (131). Based on these findings, it is believed that the UPR-regulated signaling lipids or their precursors may have important roles in tumorigenesis and in the maintenance of cancer cell metabolism.
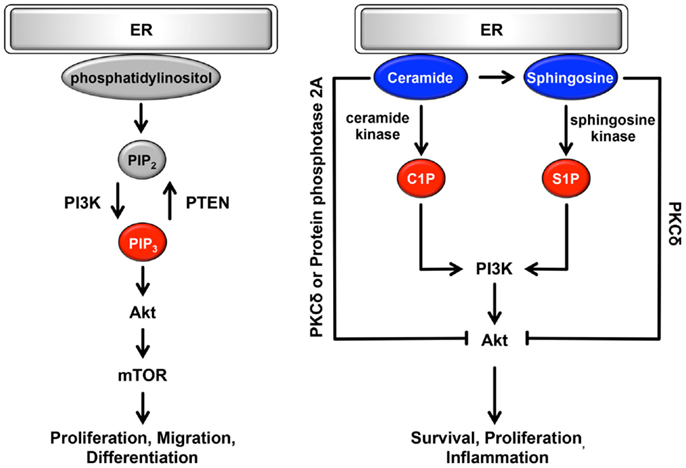
Figure 3. Role of the ER lipid signaling pathway. In the ER, glycerophospholipids and phosphatidylinositol are synthesized. Phosphatidylinositol is converted to highly phosphorylated forms. Conversion of PIP2 to PIP3 is promoted by PI3K, resulting in the regulation of cell proliferation, cell migration, and cell differentiation through the induction of PI3K-Akt-mTOR signaling. Sphingosine is also synthesized on the cytosolic surface of the ER through the de novo biosynthesis of ceramide. Both ceramide and sphingosine inhibit Akt activity via the activation of PKCδ and protein phosphatase 2A. Conversely, C1P and S1P antagonize the effects of ceramide and sphingosine and activate Akt, consequently contributing to cell survival, cell proliferation, and inflammation. Pro-oncogenesis-related lipids are indicated in red. Anti-oncogenesis-related lipids are indicated in blue.
Role of Lipid Signaling via Glycerophospholipids
Glycerophospholipids are glycerol-based phospholipids consisting of a polar group and hydrophobic chains. In general, glycerophospholipids are classified by differences in polar group (37, 130, 132). Among the glycerophospholipids, phosphatidylcholine, phosphatidylethanolamine, and PI are synthesized in the ER. Glycerol and fatty acids are transformed into phospholipid precursors, including triglycerides and diacylglycerol phosphate, by ER-resident enzymes. During the next synthesis step, on leaflets of the ER membrane, these phospholipid precursors are dephosphorylated and then converted to phosphatidylcholine or phosphatidylethanolamine. PI is also synthesized from l-myo-inositol and CDP-diacylglycerol through PI synthase in the ER. Synthesized PI can be converted to several highly phosphorylated forms called phosphoinositides. They are localized mainly in the plasma membrane and play important roles in various signaling pathways and vesicle trafficking (49, 130, 132). Among the various types of phosphoinositides, the plasma membrane-resident phosphoinositides PI-4,5-bisphosphate [PI(4,5)P2] and PI-3,4,5-bisphosphate [PI(3,4,5)P3] are specifically involved in cell growth- and survival-related intracellular signaling (37). In response to stimulation by growth factors, conversion of PIP2 to PIP3 is promoted through phosphorylation by the class I phosphoinositide 3-kinase (PI3K) (37, 133). This conversion is transient and dramatic, and it enhances the binding of Akt to PI(3,4,5)P3, and thereby to phosphorylated Akt, resulting in activation of important cascades, including mammalian target of rapamycin (mTOR) signaling (133, 134) (Figure 3). mTOR signaling is an oncogenic PI3K pathway that regulates protein synthesis, cell proliferation, migration, differentiation, and the cell cycle (134–137). Conversion of PI(3,4,5)P3 to PI(4,5)P2 occurs through dephosphorylation via the phospholipid phosphatase PTEN and is antagonized by PI3K pathway (138, 139). Thus, cellular functions are maintained through a physiological balance of phosphorylation and dephosphorylation by PI3K and PTEN, respectively. Interestingly, several cancers frequently show constitutively active PI3K mutations or loss-of-function mutations in PTEN, resulting in an increase in PI(3,4,5)P3 that in turn contributes to oncogenesis (37, 140, 141). These mutations are particularly observed in ovarian, colon, breast, and gastric cancer (142). Although the amount of additional PI, a precursor of PI(4,5)P2 and PI(3,4,5)3, remains poorly understood, PI3K, Akt, and mTOR may be therapeutic targets for various cancers.
Role of Lipid Signaling via Sphingolipids
Sphingolipids are sphingoid-based phospholipids that are produced through the de novo biosynthesis of ceramide, which is generated by a condensation reaction involving serine and palmitoyl-CoA via serine palmitoyl transferase (37, 107, 109). This biosynthesis occurs on the cytosolic surface of the ER. Then, ceramide is coordinated by various enzymes, including ceramide kinase, glucosylceramide synthase, galactosyltransferase, sphingomyelin synthase, and ceramidase, consequently producing ceramide 1-phosphate (C1P), glycosphingolipids, sulphatide, sphingomyelin, sphingosine, and S1P. Conversely, ceramide is synthesized from these products via several metabolic pathways (37, 107, 109). Among sphingolipids, ceramide, C1P, sphingosine and S1P are potent signaling molecules and have important roles in cellular processes. Ceramide and sphingosine are involved in anti-oncogenic processes such as apoptosis, cell-cycle arrest, and cellular senescence (37, 143–146) (Figure 3). By contrast, C1P and S1P antagonize the pro-apoptotic effects of ceramide and sphingosine and contribute to survival, proliferation, and inflammation (37, 38, 143–145). Ceramide and sphingosine can attenuate Akt activity (37, 38); they directly activate PKCδ, which inhibits the translocation of Akt to the plasma membrane, leading to suppression of Akt activity. As another inhibitory mechanism, ceramide dephosphorylates Akt through the activation of cytosolic protein phosphatase 2A. By contrast, C1P and S1P are positive regulators of the Akt signaling pathway via activation of PI3K (37, 143, 146). Interestingly, S1P is linked to endothelial growth factor-, platelet-derived growth factor-, and transforming growth factor β-related signaling pathways (146–148). Consequently, Akt, ERK1/2, and PKC-β are activated by crosstalk between S1P and growth factors. Surprisingly, it has been reported that several sphingolipid synthases and metabolites are dysregulated in many types of cancer. Especially in leukemia, melanoma, breast, ovarian, and colon cancers, sphingosine kinase is substantially upregulated, resulting in increased generation of S1P (149–155). Conversely, the production of ceramide is suppressed by increased ceramide kinase in liver and breast cancer (156, 157). Furthermore, in the de novo biogenesis of ceramide, the expression of serine palmitoyl transferase, which is localized on the surface of the ER, is decreased (149). Based on these findings, it is possible that cancers evade the anti-oncogenic effects of sphingolipids via the dysregulation of sphingolipid metabolism including the upregulation of anti-apoptotic sphingolipids.
Signaling Pathways from the MAM
As noted above, in addition to the rER and sER, recent reports have defined a subdomain of the ER that is a site of direct communication between the ER and mitochondria, called the MAM (49). It is believed that this tether is important for lipid and calcium trafficking between the ER and mitochondria to regulate lipid metabolism and mitochondrial calcium homeostasis (25–30, 49). Several proteins involved in lipid synthesis and trafficking are enriched at the MAM. Acyl-CoA synthase 4, acyl-CoA cholesterol acyl transferase 1, phosphatidyl serine synthase, phosphatidylethanolamine N-methyltransferase 2, and acyl-CoA:diacylglycerol acyltransferase 2 have been identified as MAM-enriched enzymes (25, 32–35). Among these enzymes, acyl-CoA synthase 4 is well known as a marker protein of the MAM. Furthermore, it has also been reported that the MAM is important for the de novo biogenesis of phospholipids and for their transport between the ER and mitochondria (25, 32–35). At the MAM, IP3R binds to the outer-mitochondrial-membrane-localized calcium channel VDAC1 via the cytosolic chaperone GRP75, leading to the transfer of calcium to the mitochondrion (22). Furthermore, the ER chaperone sigma-1 receptor is also localized in the MAM and can interact with IP3R (158). This interaction is related to the stability of IP3R and sustains the calcium transfer from the ER to the mitochondria. SERCA2b is also localized at the MAM and regulates the import of calcium into the ER (159). More recently, in addition to proteins involved in lipid and calcium trafficking, a variety of MAM-resident proteins, including proteins related to oncogenic signaling, have been characterized (Figure 2; Table 1).
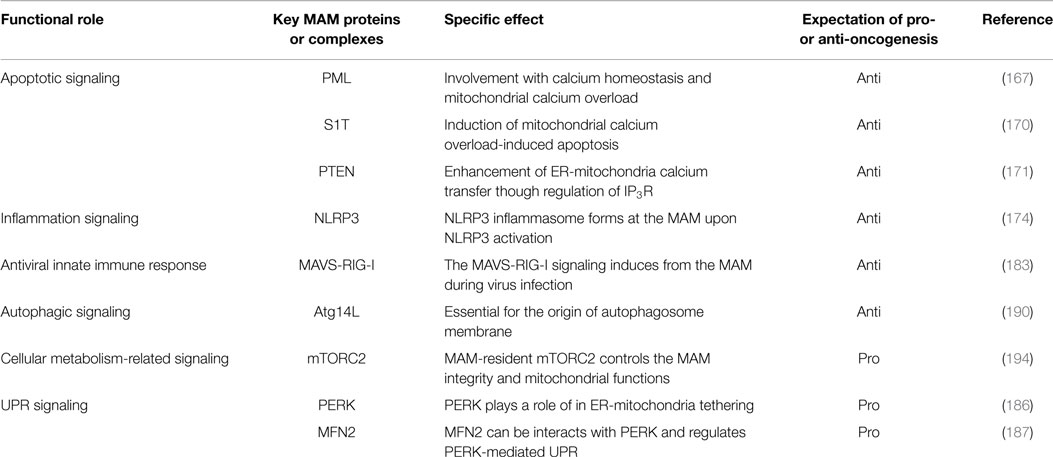
Table 1. MAM-resident proteins associated with a variety of signaling pathways and cancers.
Apoptotic Signaling Pathway at the MAM
Mitochondria are intracellular power plants that produce ATP and many biosynthetic intermediates while also contributing to the metabolism of amino acids and lipids and to the maintenance of intracellular calcium homeostasis (160–164). Mitochondria also have non-canonical functions such as the induction and amplification of apoptotic cell death (165, 166). Interestingly, recent reports have shown that the MAM functions in the apoptotic signaling pathway through dysregulation of calcium transfer from the ER to mitochondria. Mitochondrial calcium uptake is enhanced during apoptosis. Promyelocytic leukemia (PML) protein, which is involved in calcium homeostasis and apoptosis, is localized at the MAM and functions there (167). PML normally forms complexes with IP3Rs, Akt, and protein phosphatase 2, resulting in a reduction in the hyper-phosphorylation of IP3R3, which in turn results in mitochondrial calcium overload through an increase in calcium release from the ER (167). Interestingly, PML is often decreased or dysregulated in cancer cells, leading to an escape from mitochondrial calcium-mediated apoptosis (168, 169). Thus, it is considered that MAM-resident PML is involved in anti-oncogenesis and is a tumor suppressor.
Another MAM-resident protein involved in apoptosis, the truncated form of the SERCA (S1T), has been shown to induce pro-apoptotic mitochondrial calcium overload (170). High expression of S1T increases basal mitochondrial calcium and triggers abnormal mitochondrial structure and function, resulting in apoptotic cell death. S1T knockdown results in an impairment of mitochondrial calcium overload-induced apoptosis. Moreover, S1T has been shown to be involved in ER stress (170). Taken together, it is possible that S1T is important for cell fate through ER-mitochondrion calcium transfer-induced ER stress.
PTEN dephosphorylates PI(3,4,5)P3, converting it to PI(4,5)P2, thereby attenuating the oncogenic PI3K signaling cascade (138, 139). Thus, PTEN has been considered a tumor suppressor protein. Interestingly, some PTEN proteins are localized at the MAM and function in calcium transfer from the ER to mitochondria and in pro-apoptotic mitochondrial calcium overload (171). PTEN knockdown impairs calcium release from the ER, decreases the cytosolic and mitochondrial calcium levels, and thereby reduces cytochrome C release and caspase 3 cleavage. These results indicate that ablation of PTEN attenuates calcium-dependent apoptotic cell death. By contrast, overexpression of a specific chimeric PTEN that is localized at the cytoplasmic surface of the ER membrane enhances ER-mitochondrion calcium transfer and the vulnerability of cells to arachidonic acid-induced apoptosis. This effect of PTEN is dependent on IP3R function. PTEN interacts with IP3Rs, thereby inhibiting their hyper-phosphorylation and consequently increasing calcium release from the ER. Surprisingly, the protein phosphatase activity of MAM-resident PTEN is independent of its lipid phosphatase activity (171). Based on these findings, PTEN is thought to prevent oncogenic signaling through its canonical and non-canonical activities.
Inflammatory Signaling Pathway at the MAM
Inflammasomes are important large multiprotein complexes of the innate immune system that act as molecular platforms for immune defenses against microbial-, viral infection- and stress-mediated cellular danger signals via maturation or release of pro-inflammatory cytokines including IL-1β and IL-18 (172, 173). The components and mechanism of activation of the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome have been particularly well analyzed (172, 173). In response to microbial stress, NLRP3 becomes oligomerized prior to its activation. It interacts with apoptosis-associated speck-like protein containing a CARD (ASC) and recruits procaspase-1, which in turn is converted to active caspase-1, leading to the generation of mature IL-1β (172, 173). NLRP3 protein has been reported to be enriched both in the ER and at the MAM (174). In addition, NLRP3 is co-localized with ASC proteins at the MAM when NLRP3 is activated by stimulation with nigericin or monosodium urate, which are known as inflammasome inducers. Thus, the NLRP3 inflammasome may be formed at the MAM. The detailed mechanisms, such as how and why the NLRP3 inflammasome is localized at the MAM, remain unclear. However, it has been reported that ROS generation from damaged mitochondria is required for NLRP3 inflammasome activation (174). There is a possibility that NLRP3 at the MAM senses mitochondrial function. The role of inflammasomes includes not only immune responses but also cancer pathogenesis. In various cancers, the NLRP3 inflammasome has been shown to be involved in the prevention of tumorigenesis, malignancy, and growth (175). NLRP3−/− mice exhibit tumors of increased mass, number, and size. The deletion of the NLRP3 inflammasome components ASC and caspase-1 produces a similar phenotype. These findings indicate that the NLRP3 inflammasome has anti-tumor functions (175). Furthermore, it has been reported that NLRP3 inflammasome components, including NLRP3, ASC, and procaspase-1, are downregulated in hepatocellular carcinoma (HCC) (176). This downregulation is dependent on a decrease in their mRNA levels. Interestingly, the expression pattern of NLRP3 inflammasome components changes dynamically in the development stage of hepatocarcinogenesis. Although the components of the NLRP3 inflammasome components are expressed at low levels in the healthy liver, they are upregulated in hepatitis and hepatic cirrhosis. However, their expression levels substantially decrease during hepatocarcinoma development. From these findings, this report concluded that dynamic regulation of the expression of NLRP3 inflammasome components is required for HCC development and progression. The MAM may be a central platform for NLRP3 inflammasome-mediated anti-tumorigenic functions.
Antiviral Innate Immune Response at the MAM
Retinoic acid-inducible gene I (RIG-I) is a cytosolic pathogen recognition receptor that contributes to the innate immune response against viral infection via its adaptor protein mitochondrial antiviral signaling (MAVS) (177, 178). RIG-I-MAVS signaling triggers the production of type I IFN and pro-inflammatory cytokines and results in interference with viral replication (177, 178). Viral RNA genome fragments trigger apoptotic cell death in cancer cells through the pro-apoptotic protein, TNF-related apoptosis-including ligand and NOXA, which are induced by RIG-I-MAVS signaling (179). The RIG-MAVS signaling pathway is believed to function in the cellular antitumor immune response (180). Some groups have reported that MAVS can be localized to the mitochondria or peroxisomes (181, 182). The main localization site of MAVS has been shown to be the MAM (183). Moreover, RIG-1 is recruited to the MAM-resident MAVS upon virus infection, leading to the activation of RIG-I-MAVS signaling (183). When the formation of the MAM is inhibited by MFN2 knockdown, MAVS localization increases at the peroxisomes, resulting in the enhancement of IFN-β promoter activity via RIG-I-MAVS signaling. These results suggest that the MAM can regulate the localization and signaling of MAVS. Surprisingly, when cells are infected with hepatitis C virus, NS3/4A protease selectively cleaves the MAM-resident MAVS to avoid the antiviral response. Because cleaved MAVS does not have antiviral activity, it suppresses IFN-β promoter activity. The MAM may function as a platform for innate immune signaling.
Role of UPR Proteins at the MAM
Several UPR-related proteins, including ER chaperones, calcium channels, and ER-resident oxidoreductase, have been shown to reside at the MAM (159, 184, 185). MAM-resident PERK has been shown to possess heterogeneous roles. For example, PERK is important for the maintenance of the MAM and triggers ROS-mediated mitochondrial apoptosis (186). PERK deficiency leads to ER fragmentation and aberrant calcium release. This functional change of the ER requires defective MAM formation due to PERK deletion. Interestingly, transient transfection with a kinase-dead PERK mutant can reconstitute the formation of the MAM, indicating that the role of PERK in MAM formation is independent of its kinase activity. Furthermore, this PERK function is involved in mitochondrial sensitization during ROS-mediated stress and induces ROS-mediated apoptosis. PERK has been shown to interact directly with MFN2, which forms a molecular bridge between the ER and mitochondria (187). MFN2 deletion induces ER stress and activates the three branches of UPR signaling; the PERK, ATF6, and IRE1α pathways (187). Although UPR signaling mediates the pro-apoptotic pathway under prolonged ER stress conditions, MFN2 deletion results in the inhibition of ER stress-induced apoptosis. This MFN2-mediated apoptosis requires PERK. Furthermore, when PERK is deleted in MFN2-deleted cells, ROS production and mitochondrial calcium overload are reduced, and mitochondrial morphology is improved. Therefore, in addition to its role in the UPR, PERK is involved in the regulation of mitochondrial morphology and function. Based on these reports, it is believed that PERK has multiple functions through its canonical and non-canonical activities both in the ER and at the MAM. Although the role of MAM-resident PERK in oncogenesis remains unclear, it is known that PERK is involved in the adaptation of cancer cells to tumor microenvironmental challenges. Thus, it is possible that MAM-resident PERK also has a pathological function and might be a therapeutic target in cancer.
Role of the MAM in the Autophagic Signaling Pathway
Autophagy is one of the proteolytic systems that contribute to the lysosome-mediated degradation pathway required for maintaining intracellular homeostasis (188). Defects in autophagy result in enhanced tumorigenesis, suggesting that this system plays an important role in tumor suppression (189). Autophagy is tightly regulated by a set of autophagy-related proteins (Atgs) that mediate the formation of autophagosomes (188). Autophagy requires two ubiquitin-like conjugation systems mediated by the covalent binding of Atg12 to Atg5 and a linkage between LC3 and phosphatidylethanolamine. The Atg12–Atg5 conjugate interacts with Atg16 and organizes a non-covalent multimeric complex. It localizes in nascent autophagosomes and functions as an E3 ligase of LC3, resulting in autophagosome formation. More recently, the MAM has been discovered to be the origin of autophagosome formation (190). Among the many Atg proteins, Atg14L, which is well known as a pre-autophagosome marker, re-localizes to the MAM during starvation, and Atg5 then localizes to the same site during autophagosome formation. Furthermore, in terms of the mechanism of autophagosome formation at the MAM, it has been reported that MAM-resident Atg14L interacts with the ER-resident SNARE protein syntaxin 17. Although the detailed mechanism of autophagosomal membrane generation is poorly understood, this event may be regulated via the coordination of MAM-resident proteins.
Cellular Metabolism-Related Signaling Pathway at the MAM
Mammalian target of rapamycin plays an important role in the regulation of various cellular processes (134). It forms two functionally distinct multi-protein complexes, mTOR complex 1 (mTORC1) and mTORC2 (135, 191). mTORC1 is composed of mTOR, Raptor, mLST8, and DEPTOR, and it is sensitive to rapamycin. This complex contributes to the regulation of protein synthesis and cell growth (134, 135, 191, 192). mTORC2 is composed of mTOR, Rictor, Sin1 mLST8, and DEPTOR, and it regulates AGC subfamily kinases such as Akt and consequently controls cell growth, cell proliferation, cell spreading, and organization of the actin cytoskeleton (134, 135, 191, 193). Furthermore, in addition to these roles, mTORC2 contributes to cell survival, cell metabolism, and cell migration in cancer (193). Thus, mTORC2 is considered a key regulator of cancer metabolic reprograming, including glycolytic metabolism, lipid metabolism, glutamine metabolism, and ROS metabolism, for protection against microenvironmental challenges (193). Interestingly, a recent report has shown that mTORC2 is localized to the MAM during growth factor stimulation (194).
Mitochondria-associated ER membrane-resident mTORC2 can interact with the IP3R-GRP75-VDAC1 complex, which is a molecular bridge at the MAM. Defective mTORC2 results in a decrease of the ER-mitochondrion contact site, producing mitochondrial dysfunction including abnormal ATP production and mitochondrial calcium overload. Mechanistically, mTORC2 phosphorylates IP3Rs, hexokinase 2, and phosphofurin acidic cluster sorting protein 2 through the phosphorylation of Akt. The phosphorylation of these molecules mediates mitochondrial functions and the integrity of the MAM. Furthermore, activation of the mTORC2-Akt-HK2 pathway at the MAM is involved in energy metabolism and cell survival. Thus, in addition to the canonical roles of mTORC2, MAM-resident mTORC2 may control mitochondrial function and cellular metabolism.
Conclusion
The ER extends throughout the cytoplasm and has heterogeneous morphology and various functions. In addition to newly synthesized protein folding, calcium homeostasis, and phospholipid synthesis, the ER is a platform for various intracellular signaling pathways. The current understanding of these signaling pathways suggests that multiple functions of the ER are relevant to cancer pathogenesis. The signaling pathways from the ER contribute not only to oncogenesis but also to tumor suppression. The UPR is well known to contribute to the adaptation to tumor microenvironmental challenges through its constitutive activation (Figure 4). Furthermore, the maintenance of calcium homeostasis in cancer cells is critical for tumorigenesis and tumor cell survival. Similarly, lipid metabolism is also affected, inducing abnormal glycerophospholipid- and sphingolipid-related signaling, thereby leading to tumor cell survival. More recently, it has been reported that the MAM is a platform for various signaling pathways, including apoptotic signaling, inflammatory signaling, antiviral innate immune responses, autophagic signaling, and metabolism-related signaling pathways. Interestingly, these signaling pathways are closely related to both pro- and anti-oncogenic processes. Although research into the relationship between the MAM and cancer has only recently begun, it is expected that further characterization of the signaling pathways from the ER, especially from the MAM, will reveal new insight into novel cancer therapies (Figure 4).
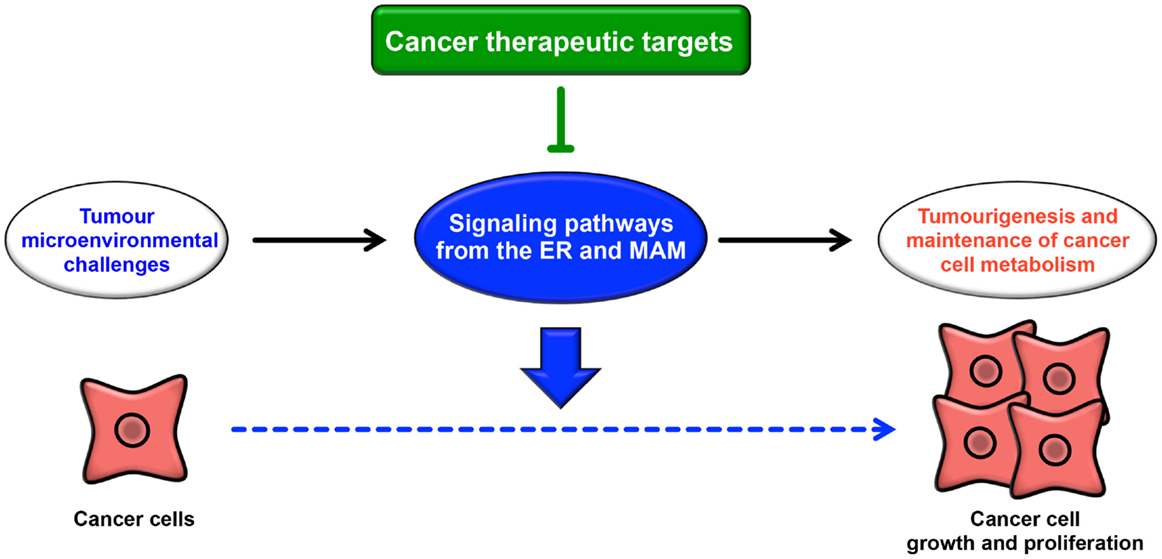
Figure 4. Roles of signaling pathways from the ER and MAM in cancer. Cancer cells are constitutively exposed to microenvironmental challenges, resulting in the remodeling of signaling pathways from the ER and MAM. Dysregulation of these signaling pathways directly or indirectly contributes to tumorigenesis and the maintenance of cancer cell metabolism. Thus, these pathways may lead to novel therapeutic strategies for various cancers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all members of the Laboratory of Biochemistry and Molecular Biology for their valuable discussion. This work was supported by Grant-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (No. 24390418 to HN; No. 26860641 to HK).
References
1. Tehlivets O, Scheuringer K, Kohlwein SD. Fatty acid synthesis and elongation in yeast. Biochim Biophys Acta (2007) 1771:255–70. doi: 10.1016/j.bbalip.2006.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Kleizen B, Braakman I. Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol (2004) 16:343–9. doi:10.1016/j.ceb.2004.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Vance JE, Vance DE. Phospholipid biosynthesis in mammalian cells. Biochem Cell Biol (2004) 82:113–28. doi:10.1139/o03-073
4. Brostrom MA, Brostrom CO. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell Calcium (2003) 34:345–63. doi:10.1016/S0143-4160(03)00127-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium (2002) 32:235–49. doi:10.1016/S0143416002001823
6. Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, et al. Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell (2002) 110:119–31. doi:10.1016/S0092-8674(02)00797-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol (2012) 197:857–67. doi:10.1083/jcb.201110131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol (2007) 8:519–29. doi:10.1038/nrm2199
9. Volmer R, Van Der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci U S A (2013) 110:4628–33. doi:10.1073/pnas.1217611110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Kitai Y, Ariyama H, Kono N, Oikawa D, Iwawaki T, Arai H. Membrane lipid saturation activates IRE1alpha without inducing clustering. Genes Cells (2013) 18:798–809. doi:10.1111/gtc.12074
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Mekahli D, Bultynck G, Parys JB, De Smedt H, Missiaen L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb Perspect Biol (2011) 3:a004317. doi:10.1101/cshperspect.a004317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Ariyama H, Kono N, Matsuda S, Inoue T, Arai H. Decrease in membrane phospholipid unsaturation induces unfolded protein response. J Biol Chem (2010) 285:22027–35. doi:10.1074/jbc.M110.126870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell (2010) 140:900–17. doi:10.1016/j.cell.2010.02.034
14. Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci U S A (1990) 87:2466–70. doi:10.1073/pnas.87.7.2466
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol (2004) 14:20–8. doi:10.1016/j.tcb.2003.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Lee AS. GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res (2007) 67:3496–9. doi:10.1158/0008-5472.CAN-07-0325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Moenner M, Pluquet O, Bouchecareilh M, Chevet E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res (2007) 67:10631–4. doi:10.1158/0008-5472.CAN-07-1705
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res (2004) 64:5943–7. doi:10.1158/0008-5472.CAN-04-1606
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol (2006) 174:915–21. doi:10.1083/jcb.200604016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science (1998) 280:1763–6. doi:10.1126/science.280.5370.1763
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. De Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature (2008) 456:605–10. doi:10.1038/nature07534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Szabadkai G, Bianchi K, Varnai P, De Stefani D, Wieckowski MR, Cavagna D, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol (2006) 175:901–11. doi:10.1083/jcb.200608073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Iwasawa R, Mahul-Mellier AL, Datler C, Pazarentzos E, Grimm S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J (2011) 30:556–68. doi:10.1038/emboj.2010.346
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. De Vos KJ, Morotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet (2012) 21:1299–311. doi:10.1093/hmg/ddr559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Rusinol AE, Cui Z, Chen MH, Vance JE. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem (1994) 269:27494–502.
26. Vance JE. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J Biol Chem (1991) 266:89–97.
27. Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem (1990) 265:7248–56.
28. Filippin L, Magalhaes PJ, Di Benedetto G, Colella M, Pozzan T. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J Biol Chem (2003) 278:39224–34. doi:10.1074/jbc.M302301200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science (1993) 262:744–7. doi:10.1126/science.8235595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142:270–83. doi:10.1016/j.cell.2010.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Sano R, Annunziata I, Patterson A, Moshiach S, Gomero E, Opferman J, et al. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol Cell (2009) 36:500–11. doi:10.1016/j.molcel.2009.10.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Browman DT, Resek ME, Zajchowski LD, Robbins SM. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci (2006) 119:3149–60. doi:10.1242/jcs.03060
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Hayashi T, Su TP. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: roles in endoplasmic reticulum lipid compartmentalization and export. J Pharmacol Exp Ther (2003) 306:718–25. doi:10.1124/jpet.103.051284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Lewin TM, Kim JH, Granger DA, Vance JE, Coleman RA. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J Biol Chem (2001) 276:24674–9. doi:10.1074/jbc.M102036200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Stone SJ, Vance JE. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem (2000) 275:34534–40. doi:10.1074/jbc.M002865200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J (2012) 279:2610–23. doi:10.1111/j.1742-4658.2012.08644.x
37. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol (2008) 9:162–76. doi:10.1038/nrm2335
38. Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer (2004) 4:604–16. doi:10.1038/nrc1411
39. Van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta (2014) 1843:2253–62. doi:10.1016/j.bbamcr.2014.03.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta (2013) 1833:213–24. doi:10.1016/j.bbamcr.2012.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Simmen T, Lynes EM, Gesson K, Thomas G. Oxidative protein folding in the endoplasmic reticulum: tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta (2010) 1798:1465–73. doi:10.1016/j.bbamem.2010.04.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta (2014) 1843:2143–9. doi:10.1016/j.bbamcr.2014.01.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell (2014) 25:563–73. doi:10.1016/j.ccr.2014.03.015
44. Park HR, Tomida A, Sato S, Tsukumo Y, Yun J, Yamori T, et al. Effect on tumor cells of blocking survival response to glucose deprivation. J Natl Cancer Inst (2004) 96:1300–10. doi:10.1093/jnci/djh243
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res (1998) 58:1408–16.
46. Papp B, Brouland JP, Arbabian A, Gelebart P, Kovacs T, Bobe R, et al. Endoplasmic reticulum calcium pumps and cancer cell differentiation. Biomolecules (2012) 2:165–86. doi:10.3390/biom2010165
47. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer (2011) 11:609–18. doi:10.1038/nrc3105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer (2008) 8:361–75. doi:10.1038/nrc2374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Bravo R, Parra V, Gatica D, Rodriguez AE, Torrealba N, Paredes F, et al. Endoplasmic reticulum and the unfolded protein response: dynamics and metabolic integration. Int Rev Cell Mol Biol (2013) 301:215–90. doi:10.1016/B978-0-12-407704-1.00005-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell (2000) 6:1099–108. doi:10.1016/S1097-2765(00)00108-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Trinh MA, Klann E. Translational control by eIF2alpha kinases in long-lasting synaptic plasticity and long-term memory. Neurobiol Learn Mem (2013) 105:93–9. doi:10.1016/j.nlm.2013.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Wek RC, Cavener DR. Translational control and the unfolded protein response. Antioxid Redox Signal (2007) 9:2357–71. doi:10.1089/ars.2007.1764
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc Trans (2006) 34:7–11. doi:10.1042/BST0340007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev (2013) 27:2179–91. doi:10.1101/gad.225680.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem (2004) 279:20108–17. doi:10.1074/jbc.M314219200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol (2003) 23:7198–209. doi:10.1128/MCB.23.20.7198-7209.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, et al. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J (2004) 23:169–79. doi:10.1038/sj.emboj.7600030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol (2013) 15:481–90. doi:10.1038/ncb2738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest (2008) 118:3378–89. doi:10.1172/JCI34587
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev (2004) 18:3066–77. doi:10.1101/gad.1250704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Kutomi G, Tamura Y, Tanaka T, Kajiwara T, Kukita K, Ohmura T, et al. Human endoplasmic reticulum oxidoreductin 1-alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci (2013) 104:1091–6. doi:10.1111/cas.12177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene (2010) 29:3881–95. doi:10.1038/onc.2010.153
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Wang Y, Alam GN, Ning Y, Visioli F, Dong Z, Nor JE, et al. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res (2012) 72:5396–406. doi:10.1158/0008-5472.CAN-12-0474
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J (2010) 29:2082–96. doi:10.1038/emboj.2010.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res (2013) 73:1993–2002. doi:10.1158/0008-5472.CAN-12-3109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Drexler HC. Synergistic apoptosis induction in leukemic cells by the phosphatase inhibitor salubrinal and proteasome inhibitors. PLoS One (2009) 4:e4161. doi:10.1371/journal.pone.0004161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol (2009) 186:323–31. doi:10.1083/jcb.200903014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell (2009) 35:551–61. doi:10.1016/j.molcel.2009.08.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature (1992) 359:845–8. doi:10.1038/359845a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Chen X, Iliopoulos D, Zhang Q, Tang Q, Greenblatt MB, Hatziapostolou M, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature (2014) 508:103–7. doi:10.1038/nature13119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Dejeans N, Pluquet O, Lhomond S, Grise F, Bouchecareilh M, Juin A, et al. Autocrine control of glioma cells adhesion and migration through IRE1alpha-mediated cleavage of SPARC mRNA. J Cell Sci (2012) 125:4278–87. doi:10.1242/jcs.099291
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev (2002) 16:1345–55. doi:10.1101/gad.992302
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science (2000) 287:664–6. doi:10.1126/science.287.5453.664
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Fuest M, Willim K, Macnelly S, Fellner N, Resch GP, Blum HE, et al. The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology (2012) 55:408–18. doi:10.1002/hep.24699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Zhao P, Xiao X, Kim AS, Leite MF, Xu J, Zhu X, et al. c-Jun inhibits thapsigargin-induced ER stress through up-regulation of DSCR1/Adapt78. Exp Biol Med (Maywood) (2008) 233:1289–300. doi:10.3181/0803-RM-84
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Yang W, Tiffany-Castiglioni E, Koh HC, Son IH. Paraquat activates the IRE1/ASK1/JNK cascade associated with apoptosis in human neuroblastoma SH-SY5Y cells. Toxicol Lett (2009) 191:203–10. doi:10.1016/j.toxlet.2009.08.024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, et al. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol (2007) 171:513–24. doi:10.2353/ajpath.2007.070188
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Wang L, Perera BG, Hari SB, Bhhatarai B, Backes BJ, Seeliger MA, et al. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat Chem Biol (2012) 8:982–9. doi:10.1038/nchembio.1094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Wheeler MC, Rizzi M, Sasik R, Almanza G, Hardiman G, Zanetti M. KDEL-retained antigen in B lymphocytes induces a proinflammatory response: a possible role for endoplasmic reticulum stress in adaptive T cell immunity. J Immunol (2008) 181:256–64. doi:10.4049/jimmunol.181.1.256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell (2000) 103:239–52. doi:10.1016/S0092-8674(00)00116-1
81. Mahadevan NR, Fernandez A, Rodvold JJ, Almanza G, Zanetti M. Prostate cancer cells undergoing ER stress in vitro and in vivo activate transcription of pro-inflammatory cytokines. J Inflamm Res (2010) 3:99–103. doi:10.2147/JIR.S11190
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol (2010) 11:411–8. doi:10.1038/ni.1857
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M. Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells. Proc Natl Acad Sci U S A (2011) 108:6561–6. doi:10.1073/pnas.1008942108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13:54–61. doi:10.1038/nm1523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Martins I, Kepp O, Schlemmer F, Adjemian S, Tailler M, Shen S, et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene (2011) 30:1147–58. doi:10.1038/onc.2010.500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Zhang J, Jiang Y, Jia Z, Li Q, Gong W, Wang L, et al. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin Exp Metastasis (2006) 23:401–10. doi:10.1007/s10585-006-9051-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Fu Y, Lee AS. Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol Ther (2006) 5:741–4. doi:10.4161/cbt.5.7.2970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Jamora C, Dennert G, Lee AS. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc Natl Acad Sci U S A (1996) 93:7690–4. doi:10.1073/pnas.93.15.7690
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Coe H, Michalak M. Calcium binding chaperones of the endoplasmic reticulum. Gen Physiol Biophys (2009) 28:F96–103.
90. Guerrero-Hernandez A, Dagnino-Acosta A, Verkhratsky A. An intelligent sarco-endoplasmic reticulum Ca2+ store: release and leak channels have differential access to a concealed Ca2+ pool. Cell Calcium (2010) 48:143–9. doi:10.1016/j.ceca.2010.08.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Taylor CW, Tovey SC. IP(3) receptors: toward understanding their activation. Cold Spring Harb Perspect Biol (2010) 2:a004010. doi:10.1101/cshperspect.a004010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem (2007) 76:367–85. doi:10.1146/annurev.biochem.76.053105.094237
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Marks AR. Ryanodine receptors/calcium release channels in heart failure and sudden cardiac death. J Mol Cell Cardiol (2001) 33:615–24. doi:10.1006/jmcc.2000.1343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Clapham DE. Calcium signaling. Cell (2007) 131:1047–58. doi:10.1016/j.cell.2007.11.028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Vandecaetsbeek I, Trekels M, De Maeyer M, Ceulemans H, Lescrinier E, Raeymaekers L, et al. Structural basis for the high Ca2+ affinity of the ubiquitous SERCA2b Ca2+ pump. Proc Natl Acad Sci U S A (2009) 106:18533–8. doi:10.1073/pnas.0906797106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Brini M, Carafoli E. Calcium pumps in health and disease. Physiol Rev (2009) 89:1341–78. doi:10.1152/physrev.00032.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Periasamy M, Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve (2007) 35:430–42. doi:10.1002/mus.20745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Bhupathy P, Babu GJ, Periasamy M. Sarcolipin and phospholamban as regulators of cardiac sarcoplasmic reticulum Ca2+ ATPase. J Mol Cell Cardiol (2007) 42:903–11. doi:10.1016/j.yjmcc.2007.03.738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Kho C, Lee A, Jeong D, Oh JG, Chaanine AH, Kizana E, et al. SUMO1-dependent modulation of SERCA2a in heart failure. Nature (2011) 477:601–5. doi:10.1038/nature10407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Knyushko TV, Sharov VS, Williams TD, Schoneich C, Bigelow DJ. 3-Nitrotyrosine modification of SERCA2a in the aging heart: a distinct signature of the cellular redox environment. Biochemistry (2005) 44:13071–81. doi:10.1021/bi051226n
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, et al. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med (2004) 10:1200–7. doi:10.1038/nm1119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Korosec B, Glavac D, Rott T, Ravnik-Glavac M. Alterations in the ATP2A2 gene in correlation with colon and lung cancer. Cancer Genet Cytogenet (2006) 171:105–11. doi:10.1016/j.cancergencyto.2006.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Endo Y, Uzawa K, Mochida Y, Shiiba M, Bukawa H, Yokoe H, et al. Sarcoendoplasmic reticulum Ca(2+) ATPase type 2 downregulated in human oral squamous cell carcinoma. Int J Cancer (2004) 110:225–31. doi:10.1002/ijc.20118
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Pacifico F, Ulianich L, De Micheli S, Treglia S, Leonardi A, Vito P, et al. The expression of the sarco/endoplasmic reticulum Ca2+-ATPases in thyroid and its down-regulation following neoplastic transformation. J Mol Endocrinol (2003) 30:399–409. doi:10.1677/jme.0.0300399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Gelebart P, Kovacs T, Brouland JP, Van Gorp R, Grossmann J, Rivard N, et al. Expression of endomembrane calcium pumps in colon and gastric cancer cells. Induction of SERCA3 expression during differentiation. J Biol Chem (2002) 277:26310–20. doi:10.1074/jbc.M201747200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Brouland JP, Gelebart P, Kovacs T, Enouf J, Grossmann J, Papp B. The loss of sarco/endoplasmic reticulum calcium transport ATPase 3 expression is an early event during the multistep process of colon carcinogenesis. Am J Pathol (2005) 167:233–42. doi:10.1016/S0002-9440(10)62968-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Sorrentino V, Barone V, Rossi D. Intracellular Ca(2+) release channels in evolution. Curr Opin Genet Dev (2000) 10:662–7. doi:10.1016/S0959-437X(00)00139-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium (1999) 26:237–51. doi:10.1054/ceca.1999.0090
109. Kiselyov K, Shin DM, Muallem S. Signalling specificity in GPCR-dependent Ca2+ signalling. Cell Signal (2003) 15:243–53. doi:10.1016/S0898-6568(02)00074-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem (2001) 70:281–312. doi:10.1146/annurev.biochem.70.1.281
111. Narayanan D, Adebiyi A, Jaggar JH. Inositol trisphosphate receptors in smooth muscle cells. Am J Physiol Heart Circ Physiol (2012) 302:H2190–210. doi:10.1152/ajpheart.01146.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Patterson RL, Boehning D, Snyder SH. Inositol 1,4,5-trisphosphate receptors as signal integrators. Annu Rev Biochem (2004) 73:437–65. doi:10.1146/annurev.biochem.73.071403.161303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci U S A (2008) 105:2427–32. doi:10.1073/pnas.0711324105
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Khan MT, Wagner L II, Yule DI, Bhanumathy C, Joseph SK. Akt kinase phosphorylation of inositol 1,4,5-trisphosphate receptors. J Biol Chem (2006) 281:3731–7. doi:10.1074/jbc.M509262200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, et al. Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A (2005) 102:105–10. doi:10.1073/pnas.0408352102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Azimi I, Roberts-Thomson SJ, Monteith GR. Calcium influx pathways in breast cancer: opportunities for pharmacological intervention. Br J Pharmacol (2014) 171:945–60. doi:10.1111/bph.12486
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Mound A, Rodat-Despoix L, Bougarn S, Ouadid-Ahidouch H, Matifat F. Molecular interaction and functional coupling between type 3 inositol 1,4,5-trisphosphate receptor and BKCa channel stimulate breast cancer cell proliferation. Eur J Cancer (2013) 49:3738–51. doi:10.1016/j.ejca.2013.07.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Szatkowski C, Parys JB, Ouadid-Ahidouch H, Matifat F. Inositol 1,4,5-trisphosphate-induced Ca2+ signalling is involved in estradiol-induced breast cancer epithelial cell growth. Mol Cancer (2010) 9:156. doi:10.1186/1476-4598-9-156
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. De Mattia F, Gubser C, Van Dommelen MM, Visch HJ, Distelmaier F, Postigo A, et al. Human Golgi antiapoptotic protein modulates intracellular calcium fluxes. Mol Biol Cell (2009) 20:3638–45. doi:10.1091/mbc.E09-05-0385
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Davis FM, Parsonage MT, Cabot PJ, Parat MO, Thompson EW, Roberts-Thomson SJ, et al. Assessment of gene expression of intracellular calcium channels, pumps and exchangers with epidermal growth factor-induced epithelial-mesenchymal transition in a breast cancer cell line. Cancer Cell Int (2013) 13:76. doi:10.1186/1475-2867-13-76
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Sakakura C, Hagiwara A, Fukuda K, Shimomura K, Takagi T, Kin S, et al. Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res (2003) 23:3691–7.
122. Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. J Cell Sci (2014) 127:1607–19. doi:10.1242/jcs.141408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Bergner A, Kellner J, Tufman A, Huber RM. Endoplasmic reticulum Ca2+-homeostasis is altered in small and non-small cell lung cancer cell lines. J Exp Clin Cancer Res (2009) 28:25. doi:10.1186/1756-9966-28-25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Choe CU, Ehrlich BE. The inositol 1,4,5-trisphosphate receptor (IP3R) and its regulators: sometimes good and sometimes bad teamwork. Sci STKE (2006) 2006:re15. doi:10.1126/stke.3632006re15
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Baljinnyam E, De Lorenzo MS, Xie LH, Iwatsubo M, Chen S, Goydos JS, et al. Exchange protein directly activated by cyclic AMP increases melanoma cell migration by a Ca2+-dependent mechanism. Cancer Res (2010) 70:5607–17. doi:10.1158/0008-5472.CAN-10-0056
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Somers G, Devis G, Malaisse WJ. Calcium antagonists and islet function. IX. Is extracellular calcium required for insulin release? Acta Diabetol Lat (1979) 16:9–18. doi:10.1007/BF02590758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Rasschaert J, Malaisse WJ. Expression of the calcium-sensing receptor in pancreatic islet B-cells. Biochem Biophys Res Commun (1999) 264:615–8. doi:10.1006/bbrc.1999.1577
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. English AR, Zurek N, Voeltz GK. Peripheral ER structure and function. Curr Opin Cell Biol (2009) 21:596–602. doi:10.1016/j.ceb.2009.04.004
129. Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol (2009) 19:R1046–52. doi:10.1016/j.cub.2009.09.058
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Fagone P, Jackowski S. Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res (2009) 50(Suppl):S311–6. doi:10.1194/jlr.R800049-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Ntambi JM. The regulation of stearoyl-CoA desaturase (SCD). Prog Lipid Res (1995) 34:139–50. doi:10.1016/0163-7827(94)00010-J
132. Van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol (2008) 9:112–24. doi:10.1038/nrm2330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol (2012) 4:a011189. doi:10.1101/cshperspect.a011189
134. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci (2009) 122:3589–94. doi:10.1242/jcs.051011
135. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell (2006) 124:471–84. doi:10.1016/j.cell.2006.01.016
136. Yecies JL, Manning BD. mTOR links oncogenic signaling to tumor cell metabolism. J Mol Med (Berl) (2011) 89:221–8. doi:10.1007/s00109-011-0726-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Howell JJ, Manning BD. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab (2011) 22:94–102. doi:10.1016/j.tem.2010.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Maehama T, Taylor GS, Dixon JE. PTEN and myotubularin: novel phosphoinositide phosphatases. Annu Rev Biochem (2001) 70:247–79. doi:10.1146/annurev.biochem.70.1.247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem (1998) 273:13375–8. doi:10.1074/jbc.273.22.13375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Georgescu MM. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes Cancer (2010) 1:1170–7. doi:10.1177/1947601911407325
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr Cancer Drug Targets (2008) 8:187–98. doi:10.2174/156800908784293659
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol (2013) 10:143–53. doi:10.1038/nrclinonc.2013.10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Tornquist K. Sphingosine 1-phosphate and cancer: lessons from thyroid cancer cells. Biomolecules (2013) 3:303–15. doi:10.3390/biom3020303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Ryland LK, Fox TE, Liu X, Loughran TP, Kester M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol Ther (2011) 11:138–49. doi:10.4161/cbt.11.2.14624
145. Modrak DE, Gold DV, Goldenberg DM. Sphingolipid targets in cancer therapy. Mol Cancer Ther (2006) 5:200–8. doi:10.1158/1535-7163.MCT-05-0420
146. Miller AV, Alvarez SE, Spiegel S, Lebman DA. Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol Cell Biol (2008) 28:4142–51. doi:10.1128/MCB.01465-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. El-Shewy HM, Johnson KR, Lee MH, Jaffa AA, Obeid LM, Luttrell LM. Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine 1-phosphate receptors. J Biol Chem (2006) 281:31399–407. doi:10.1074/jbc.M605339200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Waters C, Sambi B, Kong KC, Thompson D, Pitson SM, Pyne S, et al. Sphingosine 1-phosphate and platelet-derived growth factor (PDGF) act via PDGF beta receptor-sphingosine 1-phosphate receptor complexes in airway smooth muscle cells. J Biol Chem (2003) 278:6282–90. doi:10.1074/jbc.M208560200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Duan RD, Nilsson A. Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog Lipid Res (2009) 48:62–72. doi:10.1016/j.plipres.2008.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Ruckhaberle E, Rody A, Engels K, Gaetje R, Von Minckwitz G, Schiffmann S, et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat (2008) 112:41–52. doi:10.1007/s10549-007-9836-9
151. Bayerl MG, Bruggeman RD, Conroy EJ, Hengst JA, King TS, Jimenez M, et al. Sphingosine kinase 1 protein and mRNA are overexpressed in non-Hodgkin lymphomas and are attractive targets for novel pharmacological interventions. Leuk Lymphoma (2008) 49:948–54. doi:10.1080/10428190801911654
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Kawamori T, Osta W, Johnson KR, Pettus BJ, Bielawski J, Tanaka T, et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J (2006) 20:386–8. doi:10.1096/fj.05-4331fje
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Johnson KR, Johnson KY, Crellin HG, Ogretmen B, Boylan AM, Harley RA, et al. Immunohistochemical distribution of sphingosine kinase 1 in normal and tumor lung tissue. J Histochem Cytochem (2005) 53:1159–66. doi:10.1369/jhc.4A6606.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Van Brocklyn JR, Jackson CA, Pearl DK, Kotur MS, Snyder PJ, Prior TW. Sphingosine kinase-1 expression correlates with poor survival of patients with glioblastoma multiforme: roles of sphingosine kinase isoforms in growth of glioblastoma cell lines. J Neuropathol Exp Neurol (2005) 64:695–705. doi:10.1097/01.jnen.0000175329.59092.2c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res (2003) 63:5962–9.
156. Ruckhaberle E, Karn T, Rody A, Hanker L, Gatje R, Metzler D, et al. Gene expression of ceramide kinase, galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. J Cancer Res Clin Oncol (2009) 135:1005–13. doi:10.1007/s00432-008-0536-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Sugiura M, Kono K, Liu H, Shimizugawa T, Minekura H, Spiegel S, et al. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J Biol Chem (2002) 277:23294–300. doi:10.1074/jbc.M201535200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell (2007) 131:596–610. doi:10.1016/j.cell.2007.08.036
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Lynes EM, Bui M, Yap MC, Benson MD, Schneider B, Ellgaard L, et al. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J (2012) 31:457–70. doi:10.1038/emboj.2011.384
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Otera H, Mihara K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem (2011) 149:241–51. doi:10.1093/jb/mvr002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev (2009) 89:799–845. doi:10.1152/physrev.00030.2008
162. Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem (2007) 76:751–80. doi:10.1146/annurev.biochem.76.071905.090048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol (2006) 16:R551–60. doi:10.1016/j.cub.2006.06.054
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol (2004) 287:C817–33. doi:10.1152/ajpcell.00139.2004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol (2010) 11:872–84. doi:10.1038/nrm3013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Suen DF, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev (2008) 22:1577–90. doi:10.1101/gad.1658508
167. Pinton P, Giorgi C, Pandolfi PP. The role of PML in the control of apoptotic cell fate: a new key player at ER-mitochondria sites. Cell Death Differ (2011) 18:1450–6. doi:10.1038/cdd.2011.31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Reineke EL, Lam M, Liu Q, Liu Y, Stanya KJ, Chang KS, et al. Degradation of the tumor suppressor PML by Pin1 contributes to the cancer phenotype of breast cancer MDA-MB-231 cells. Mol Cell Biol (2008) 28:997–1006. doi:10.1128/MCB.01848-07
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst (2004) 96:269–79. doi:10.1093/jnci/djh043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Chami M, Oules B, Szabadkai G, Tacine R, Rizzuto R, Paterlini-Brechot P. Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell (2008) 32:641–51. doi:10.1016/j.molcel.2008.11.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ (2013) 20:1631–43. doi:10.1038/cdd.2013.77
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi:10.1016/j.cell.2010.01.040
173. Franchi L, Munoz-Planillo R, Reimer T, Eigenbrod T, Nunez G. Inflammasomes as microbial sensors. Eur J Immunol (2010) 40:611–5. doi:10.1002/eji.200940180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
174. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi:10.1038/nature09663
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Allen IC, Tekippe EM, Woodford RM, Uronis JM, Holl EK, Rogers AB, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med (2010) 207:1045–56. doi:10.1084/jem.20100050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest (2014) 94:52–62. doi:10.1038/labinvest.2013.126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
177. Sumpter R Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol (2005) 79:2689–99. doi:10.1128/JVI.79.5.2689-2699.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
178. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol (2004) 5:730–7. doi:10.1038/ni1087
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Matsushima-Miyagi T, Hatano K, Nomura M, Li-Wen L, Nishikawa T, Saga K, et al. TRAIL and NOXA are selectively upregulated in prostate cancer cells downstream of the RIG-I/MAVS signaling pathway by nonreplicating Sendai virus particles. Clin Cancer Res (2012) 18:6271–83. doi:10.1158/1078-0432.CCR-12-1595
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Kaneda Y. The RIG-I/MAVS signaling pathway in cancer cell-selective apoptosis. Oncoimmunology (2013) 2:e23566. doi:10.4161/onci.23566
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell (2010) 141:668–81. doi:10.1016/j.cell.2010.04.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell (2005) 122:669–82. doi:10.1016/j.cell.2005.08.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Horner SM, Liu HM, Park HS, Briley J, Gale M Jr. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci U S A (2011) 108:14590–5. doi:10.1073/pnas.1110133108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, et al. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones (2010) 15:619–29. doi:10.1007/s12192-010-0174-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Myhill N, Lynes EM, Nanji JA, Blagoveshchenskaya AD, Fei H, Carmine Simmen K, et al. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell (2008) 19:2777–88. doi:10.1091/mbc.E07-10-0995
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ (2012) 19:1880–91. doi:10.1038/cdd.2012.74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Munoz JP, Ivanova S, Sanchez-Wandelmer J, Martinez-Cristobal P, Noguera E, Sancho A, et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J (2013) 32:2348–61. doi:10.1038/emboj.2013.168
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell (2011) 147:728–41. doi:10.1016/j.cell.2011.10.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev (2011) 25:795–800. doi:10.1101/gad.2016211
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, et al. Autophagosomes form at ER-mitochondria contact sites. Nature (2013) 495:389–93. doi:10.1038/nature11910
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell (2009) 137:873–86. doi:10.1016/j.cell.2009.03.046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Chen J, Long F. mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development (2014) 141:2848–54. doi:10.1242/dev.108811
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Masui K, Cavenee WK, Mischel PS. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol Metab (2014) 25:364–73. doi:10.1016/j.tem.2014.04.002
194. Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. Feature article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci U S A (2013) 110:12526–34. doi:10.1073/pnas.1302455110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: endoplasmic reticulum, mitochondria-associated ER membrane, unfolded protein response, ER stress, cancer
Citation: Kato H and Nishitoh H (2015) Stress responses from the endoplasmic reticulum in cancer. Front. Oncol. 5:93. doi: 10.3389/fonc.2015.00093
Received: 30 January 2015; Accepted: 31 March 2015;
Published: 20 April 2015
Edited by:
Matiullah Khan, AIMST University, MalaysiaReviewed by:
Min Hee Kang, Texas Tech University Health Sciences Center School of Medicine, USAAbhishek D. Garg, KU Leuven – University of Leuven, Belgium
Copyright: © 2015 Kato and Nishitoh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hideki Nishitoh, Laboratory of Biochemistry and Molecular Biology, Department of Medical Sciences, University of Miyazaki, 5200 Kihara, Miyazaki 889-1692, JapanbmlzaGl0b2hAbWVkLm1peWF6YWtpLXUuYWMuanA=