 Tene Aneka Cage1
Tene Aneka Cage1 Yvan Chanthery2,3
Yvan Chanthery2,3 Louis Chesler4
Louis Chesler4 Matthew Grimmer1,2,5
Matthew Grimmer1,2,5 Zachary Knight6Kevan Shokat7William A. Weiss1,2,8,9,10
Zachary Knight6Kevan Shokat7William A. Weiss1,2,8,9,10 W. Clay Gustafson8,9,10*
W. Clay Gustafson8,9,10*- 1Department of Neurological Surgery, Brain Tumor Research Center, University of California San Francisco, San Francisco, CA, USA
- 2Department of Neurology, University of California San Francisco, San Francisco, CA, USA
- 3Department of Bioengineering, Santa Clara University, Santa Clara, CA, USA
- 4Division of Paediatric Solid Tumour Biology and Therapeutics, The Institute of Cancer Research, London, UK
- 5Department of Biochemistry, University of Southern California, Los Angeles, CA, USA
- 6Department of Physiology, University of California San Francisco, San Francisco, CA, USA
- 7Department of Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, CA, USA
- 8Department of Pediatrics, University of California San Francisco, San Francisco, CA, USA
- 9Department of Pediatric Hematology and Oncology, University of California San Francisco, San Francisco, CA, USA
- 10Hellen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, CA, USA
The MYCN proto-oncogene is associated with poor outcome across a broad range of pediatric tumors. While amplification of MYCN drives subsets of high-risk neuroblastoma and medulloblastoma, dysregulation of MYCN in medulloblastoma (in the absence of amplification) also contributes to pathogenesis. Since PI3K stabilizes MYCN, we have used inhibitors of PI3K to drive degradation. In this study, we show PI3K inhibitors by themselves induce cell cycle arrest, with modest induction of apoptosis. In screening inhibitors of PI3K against MYCN, we identified PIK-75 and its derivative, PW-12, inhibitors of both PI3K and of protein kinases, to be highly effective in destabilizing MYCN. To determine the effects of PW-12 treatment in vivo, we analyzed a genetically engineered mouse model for MYCN-driven neuroblastoma and a model of MYCN-driven medulloblastoma. PW-12 showed significant activity in both models, inducing vascular collapse and regression of medulloblastoma with prominent apoptosis in both models. These results demonstrate that inhibitors of lipid and protein kinases can drive apoptosis in MYCN-driven cancers and support the importance of MYCN as a therapeutic target.
Introduction
The MYC family of proto-oncogenes (MYC, MYCN, and MYCL) has been broadly implicated in cancer. MYC proteins are helix–loop–helix transcription factors, which share a large degree of homology and function. MYC transcriptionally regulates ~500 core targets with as many as 2000 additional cancer and/or cell type-specific targets, making pharmacologic targeting downstream of MYC proteins challenging. However, MYC proteins are tightly regulated at the level of protein stability through several upstream signaling events (1). The MYCN gene is amplified or overexpressed in a number of pediatric cancers including both medulloblastoma and neuroblastoma and is a negative prognostic factor in both diseases (2, 3).
Neuroblastoma is the most common extracranial solid tumor of childhood. High-risk neuroblastoma represents about 40% of all patients diagnosed. Despite significant progress in the treatment of low- and intermediate-risk patients, survival among children with high-risk disease remains poor even after significant escalations in the intensity of therapy (4, 5). Amplification of MYCN occurs in ~25% of patients and is the clearest genetic risk factor for high-risk disease, making MYCN a prominent candidate for targeted therapies (6, 7). Targeted expression of MYCN to the peripheral neural crest of mice results in neuroblastoma tumors with high penetrance. We have previously shown that indirect methods of blocking MYCN in these animals have led to decreased tumor size and improved survival (8, 9).
Medulloblastoma is the most common malignant brain tumor in children and is divided into four molecular subgroups. MYC family oncogenes are amplified in ~10% of medulloblastoma tumors and amplification correlates with poor survival (10–12). MYCN is highly expressed in two of the medulloblastoma subgroups, the Sonic Hedgehog (SHH) group and the Group 4 tumors (13). Using a genetically engineered model (GEM), we have previously targeted expression of MYCN to the cerebellum of transgenic mice. These GTML mice are predisposed to SHH-independent, MYCN-amplified medulloblastoma (14). We have shown that transient downregulation of MYCN in these mice resulted in tumor cell senescence and improved survival (14, 15). In addition, medulloblastoma tumor cell lines with constitutive activation of the SHH pathway derived from Ptc1+/−p53−/− mice (SmoWT) (16–18), demonstrate robust tumorigenesis (17). Loss of the Ptch receptor in these mice leads to constitutive activation of the downstream smoothened transmembrane protein, resulting in medulloblastomas driven by MYCN and GLI. This model provides an excellent platform to explore MYCN-directed pre-clinical therapeutics in neuroblastoma and in medulloblastoma.
As transcription factors with no surfaces amenable to small molecule binding, MYC proteins are widely considered undruggable directly. However, the stability of MYCN protein is tightly regulated by a sequential series of key phosphorylation events downstream of CDK1/CyclinB and the PI3K/mTOR pathway, suggesting inhibitors of relevant kinases as an approach to target MYC and MYCN (1). Further, these phosphorylation events are known to be downstream of the activating ALKF1174L mutation, which plays a role in both sporadic and familial neuroblastomas (19). We have previously shown that the tool PI3K inhibitor LY294002 and the dual PI3K/mTOR inhibitor NVP-BEZ235 cause downregulation of MYCN protein and, in the case of NVP-BEZ235, decreased angiogenesis through a paracrine signaling mechanism (9, 20). These studies emphasize the importance of mouse models for MYCN-driven pediatric cancers and implicate further exploration of the interplay between PI3K and MYCN protein regulation through the use of alternate pharmacologic inhibitors of PI3K.
Several specific inhibitors of PI3K have been developed and characterized in other systems including PI-103, PIK-75, PW-12, and BEZ235 (21, 22). Resistance to PI3K inhibitors is scaffold-dependent and often mediated by point mutations (23). If PI3K inhibition is to be a viable strategy in cancer, demonstration of efficacy across an array of PI3K inhibitor scaffolds will likely be necessary. We have previously shown efficacy for the PI3K/mTOR inhibitor BEZ235 in neuroblastoma (9). In the current study, we show that the PI3K alpha inhibitor PIK-75 and its derivative, PW-12, destabilize MYCN protein and induce cell cycle arrest and apoptosis in MYCN-amplified cell lines. We further demonstrate that these drugs have pre-therapeutic efficacy in models of MYCN-driven neuroblastoma (24) and MYCN-driven medulloblastoma (16, 25). These studies provide evidence for PI3K inhibition with subsequent blockade of MYCN and vascular collapse as a therapeutic strategy across chemical scaffolds and an impetus for the continued development of PI3K inhibitors in MYCN-driven pediatric cancers, including both neuroblastoma and medulloblastoma.
Materials and Methods
Immunoblotting
Human Kelly neuroblastoma cell lines were obtained from the University of California at San Francisco. Cells were grown in RPMI medium with 10% FBS at 37°C. In most experiments, cells were conditioned in 2% FBS for 5 h and replaced with full medium and recombinant human insulin-like growth factor-I (20 ng/ml) (Invitrogen, Waltham, MA, USA) for 1 h before harvesting. Lysates were collected and sonicated. Primary antibodies were as follows: anti-MYCN, anti-phosphorylated Akt, anti-p-rpS6, anti-Akt, anti-rpS6 (Cell Signaling Technology, Danvers, MA, USA), and β-tubulin (Upstate, Buffalo, NY, USA). Immunoblots were developed with horseradish peroxidase-conjugated secondary antibodies (Calbiochem, San Diego, CA, USA) and Enhanced Chemiluminescence Plus reagents (Amersham, Piscataway, NJ, USA).
Cell Proliferation Assay and Flow Cytometry
Kelly neuroblastoma cells were suspended in basal RPMI medium with 10% FBS and plated in 96-well tissue culture plates (Corning/Sigma-Aldrich, Tewksbury, MA, USA). To measure viable cells, Kelly cells were incubated at 37°C for 16 h and then changed into fresh medium containing LY-294002 (Sigma, St. Louis, MO, USA), PI-103 (Calbiochem, San Diego, CA, USA), PIK-75, PW-12 (Kevan Shokat, Ph.D. Lab, University of California San Francisco, CA, USA), or DMSO vehicle. We analyzed DNA content at 48 h after plating using Water Soluable Tetrazolium (WST-1) Cell Proliferation Assay Kit (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s protocol. In this assay, A450 nm − A800 nm is proportional to secreted WST-1 activity and proliferation. In addition, cell cycle effects were measured using flow cytometry as previously described in our lab (26). For these experiments, Kelly neuroblastoma cells were treated for 48 h with 1 μM of PI-103 or PW-12.
Neuroblastoma Genetically Engineered Mouse Model
TH-MYCN transgenic mice were maintained in hemizygotic matings as previously described (24). All animals were housed and treated following University of California at San Francisco Institute on Animal Care and Use Committee (IACUC) guidelines. TH-MYCN mice with neuroblastoma tumors were treated either with 25 mg/kg PW-12 in 10% DMSO/water (n = 3) or vehicle alone [10% DMSO/water (n = 3)], once daily by IP injection, for 14 days. For Western blot analysis, tumors were snap-frozen at the time of harvesting. For immunohistochemistry, tumors were fixed in 10% formalin for 24 h and processed as described below. For tumor weight assessment, tumors were weighed on the final day of treatment, 14 days. Tumor diameters were measured on day 14 and volumes were calculated (mm3 = width2 × length/2).
SHH Medulloblastoma Subgroup Tumor Model
All athymic nude mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA), and were housed and treated following University of California at San Francisco Institute on Animal Care and Use Committee (IACUC) guidelines. About 6 × 106 SmoWT medulloblastoma cells derived from medulloblastoma tumors generated from Ptc1+/−p53+/− mice (16–18) were injected subcutaneously into the flank of athymic nude mice. A total of 10 mice were used for these experiments. Starting 8 h after implantation of cells, five animals were treated with 25 mg/kg PW-12 in 10% DMSO/water and five were treated with vehicle alone [(10% DMSO/water)], once daily by IP injection for 7 days. Tumor diameters were measured with calipers daily and volumes were calculated (mm3 = width2 × length/2).
Histology
For H&E analysis, 10-μm sections were cut paraffin blocks. Hematoxylin (Vector Labs, Burlingame, CA, USA) and bluing agent was added. Slides were washed three times with water and once with 70% ethanol (EtOH) for 5 min each. Eosin was added followed by 70, 95, and 100% EtOH, and dipped into xylene. Slides were mounted with Vectashield (Vector Labs) and analyzed with light microscopy. For immunohistochemistry, samples were prepared as described previously (20) using the following antibodies: Ki67 (1:200; M7249; Dako, Carpinteria, CA, USA), Ki67 (1:100; NB600-1252; Novus Biologicals, Littleton, CO, USA), cleaved-Caspase-3 (1:200; 9661; Cell Signaling Technology, Danvers, MA, USA), MYCN (1:50; 10159-2-AP; Proteintech, Chicago, IL, USA), and CD-31 (1:50; 55024; BD Bioschences, San Diego, CA, USA).
Statistical Analysis
To evaluate in vivo data, averages were calculated and error bars were generated using SD. Student’s t-test was used to calculate statistical significance. A p-value <0.05 was considered statistically significant.
Results
Both PIK-75 and PW-12 Potently Downregulate MYCN Protein
In order to assess the effects of pharmacologic inhibition of PI3K on MYCN-driven cells, human Kelly MYCN-amplified neuroblastoma cells were treated with various PI3K inhibitors: PI-103, LY-294002, PIK-75, and PW-12. PI-103 causes proliferative arrest but not apoptosis during in vivo glioma treatment (26). It acts by blocking class I PI3Ks and mTOR, and subsequent downstream stabilization of MYCN protein. LY-294002 also causes proliferative arrest but not apoptosis. It is also a pan-selective, broad-spectrum PI3K/mTOR inhibitor that blocks both p-Akt and the downstream target of mTOR p-rpS6 (22). LY-294002 is associated with significant cellular toxicity and therefore has limited effect as viable human clinical in vivo agent (26, 27). However, in xenograft models of neuroblastoma and glioma, it has been shown to effectively block progression of tumorigenesis though not apoptosis (26, 28, 29). PIK-75 is a PI3 kinase alpha selective inhibitor, specifically, it is an imidazopyridine inhibitor that induces apoptosis, not growth arrest, in glioma cells (29). However, this inhibitor carries significant cellular toxicity with broad off-target effects and therefore is not a viable clinical therapeutic option. Because of this, PIK-75 is an effective agent to study in vitro, but is toxic in vivo. PW-12, a derivative of PIK-75, induces growth arrest as well as some apoptosis. It has fewer off-target effects than its counterpart and therefore is a viable option for in vivo animal and human clinical therapeutics.
Human Kelly MYCN-amplified neuroblastoma cells were treated with increasing concentrations of PIK-75 or PW-12 and blotted for markers of PI3K activity and MYCN protein (Figure 1A). These two drugs were chosen from the PI3K panel because PIK-75 is a highly effective in vitro inhibitor but due to known toxicity is not a viable in vivo option. PW-12, however, is a less toxic derivative of PIK-75. There is clear downregulation of MYCN in response to both PIK-75 and PW-12 (Figure 1A). This corresponds directly with concomitant downregulation of AKT phosphorylation downstream of PI3K. These data indicate that the stability of MYCN protein is sensitive to inhibition of PI3K alpha in vitro as well as some mTOR inhibition as evidenced by blockade of p-rpS6. Further, blockade of PI3K by PIK-75 results in a significant decrease in viability in MYCN-amplified cells (Figure 1B) when PW-12 and PIK-75 are compared to LY294002 (a tool compound PI3K/mTOR inhibitor with lower affinity for these kinases) or PI-103, a PI3K/mTOR inhibitor with intermediate activity.
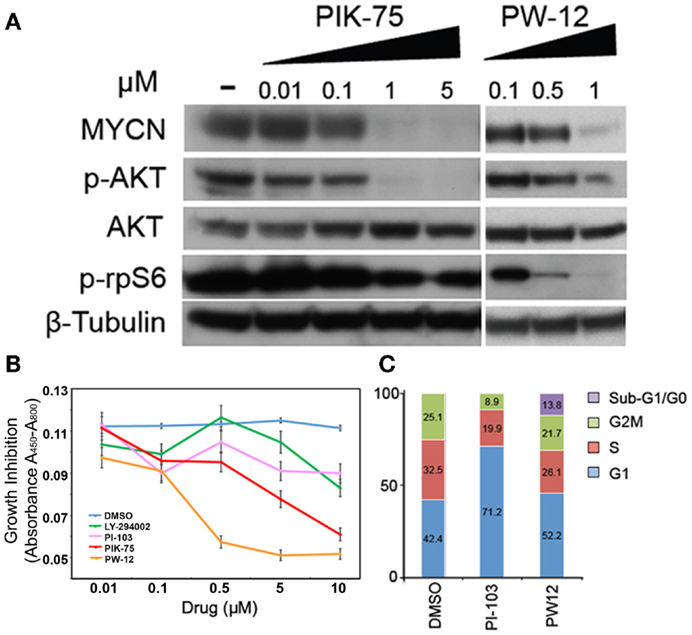
Figure 1. Inhibitors of PI3K downregulate MYCN protein, increase apoptosis, and decrease viability in MYCN-amplified tumor cells. (A) Kelly MYCN-amplified neuroblastoma cells were treated for 24 h with the indicated concentrations of inhibitor (PIK-75 or PW-12) and blotted for MYCN and markers downstream of PI3K (pAKT) and mTOR (prpS6). (B) Kelly neuroblastoma cells were treated with inhibitor (LY-294002, PI-103, PIK-75, PW-12, or DMSO) for 48 h and cell proliferation was measured by WST1 assay. (C) Kelly neuroblastoma cells were treated with 1 μM of inhibitor (PI-103, PW-12) for 48 h and cell cycle effects were analyzed by flow cytometry.
Next, we explored two PI3K inhibitors to evaluate effects on cell cycle, PI-103 and the PIK-75 derivative PW-12. These were chosen because their low-toxicity profiles make them more viable therapeutic options than their counterparts. When compared to the control, PW-12 demonstrated significant apoptosis, as measured by flow cytometry and measurement of the sub-G1/G0 fraction (Figure 1C), suggesting increased apoptosis as one contributor to the increase in activity when comparing PW-12 to PI-103 and to vehicle. This loss of cells to apoptosis could explain the differences in cell cycle profiles between PI-103 and PW-12. Notably, MYCN is critical for S-phase entry in MYCN-driven disease and both PW-12 and PIK-75 result in a loss of cells in S-phase and accumulation of cells in G1, perhaps because of increased potency against PI3K by PW-12 in these cells (Figure 1A). In addition, the smaller percentage of Kelly neuroblastoma cells treated with PW-12 are in the S and M phases of the cell cycle suggesting decreased proliferation.
PI3K Inhibition by PW-12 has Efficacy in a TH-MYCN Model of Neuroblastoma
Using the TH-MYCN neuroblastoma mouse model in which human MYCN is expressed under control of the mouse tyrosine hydroxylase promoter, we are able to generate mouse neuroblastoma tumors with high penetrance, which are genetically, histologically, and morphologically similar to human neuroblastomas (24). Abdominal tumors were detected in TH-MYCN mice by palpation and either PW-12 (25 mg/kg) or vehicle alone was administered by IP injection daily for 14 days. PW-12 treated tumors demonstrate a significant decrease in MYCN protein expression and in tumor mass (p < 0.05) when compared to vehicle treated controls (Figures 2A,B), consistent with direct blockade of MYCN and subsequent anti-tumor effects. They also showed decreased cellularity, decreased proliferation measured by Ki-67 staining, and increased apoptosis as measured by cleaved caspase 3 staining (Figure 2C). These results support the role of PW-12 in neuroblastoma tumor regression, thereby positioning it to be a pre-clinical therapeutic for MYCN-driven neuroblastoma.
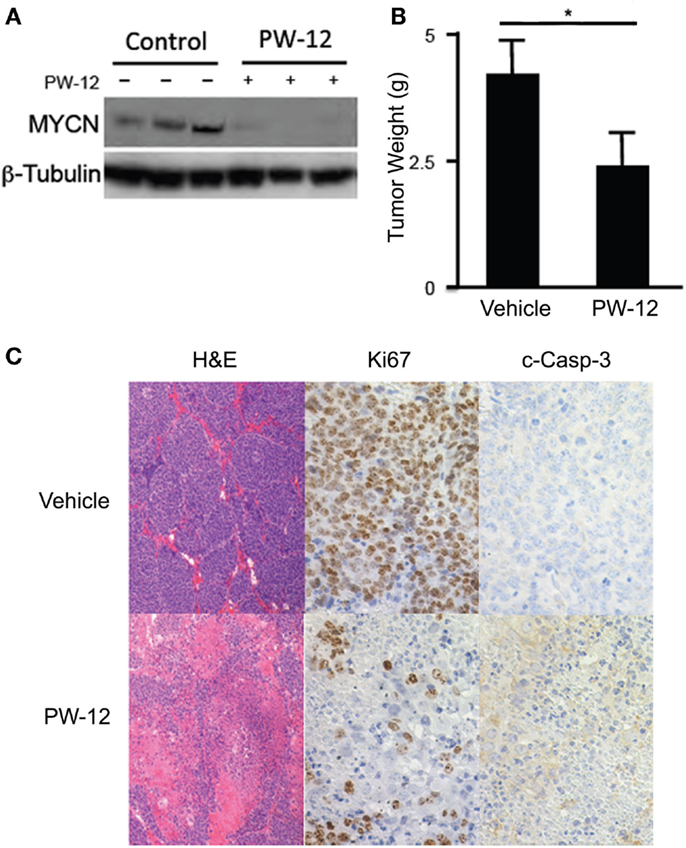
Figure 2. PW-12 is efficacious in a GEM model of MYCN-amplified neuroblastoma. Tumor-bearing TH-MYCN mice were treated for 14 days with either 25 mg/kg of PW-12 (n = 3) or vehicle alone (n = 3). (A) Western blot for MYCN protein from treated tumors confirm that PW-12 is effective at downregulating MYCN protein production. (B) Tumor size was significantly reduced in treated animals (* represents p < 0.05). (C) Histology for Ki67 and c-Caspase 3 shows increase in apoptosis and decrease in proliferation in treated tumors.
PI3K Inhibition by PW-12 has Efficacy in a MYCN-Driven SHH Medulloblastoma
To study MYCN-driven medulloblastoma, we used a subcutaneous flank allograft model where SmoWT cells were injected in to the flank of athymic nude mice. In this model, SmoWT cells from tumors of Ptc1+/−p53+/− mice undergo tumorigenesis via activation of the Shh pathway. Allografted animals were treated with either PW-12 (25 mg/kg) or vehicle by IP injection daily for 7 days. In response to therapy, treated tumors showed a statistically significant (p < 0.05) decrease in tumor volume (Figure 3A) with preserved medulloblastoma histology features on H&E (Figure 3B). The small amount of tumor mass in the treated tumors precluded us from immunoblotting these specimens. However, immunohistochemistry revealed uniform and profoundly decreased levels of MYCN across tumors in all five mice treated with PW-12 as well as decreased vascularity in treated tumors as measured by CD-31 compared to untreated tumors (Figure 3B). In addition, treated tumors showed a clear decrease in cell proliferation by Ki67 and marked induction of apoptosis, cleaved caspase (Figures 3B,C). These results establish the flank SmoWT tumor cell implantation as a useful model for pre-clinical therapeutics in medulloblastoma and suggest a potential for inhibitors of PI3K in children with MYCN-driven disease.
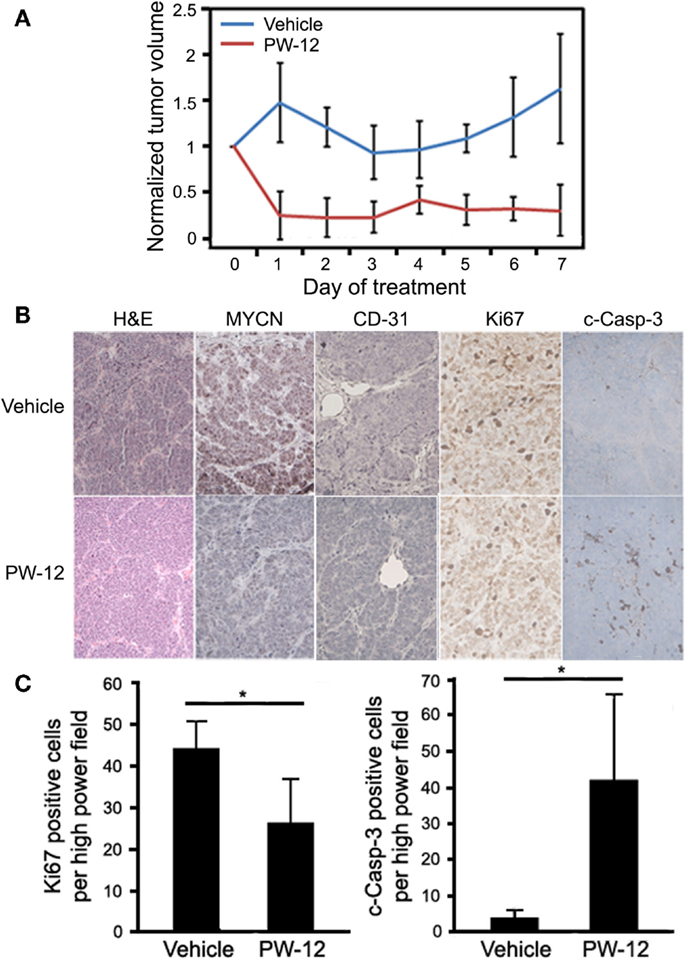
Figure 3. PW-12 is efficacious in an orthotopic flank tumor model of MYCN-amplified medulloblastoma. Mice were implanted with 6 × 106 SmoWT medulloblastoma cells in the hindflank. Animals were treated for 7 days with 25 mg/kg of PW-12 (n = 5) or vehicle alone (n = 5). (A) Tumor size was significantly reduced in treated animals (all time points days 1–7 resulted in statistically significant differences between the treatment and control groups). (B) Histology on H&E stain is consistent with medulloblastoma (H&E, 20×). Tumor cell proliferation (Ki67, 40×) was decreased, apoptosis (cleaved-caspase-3, 40×) increased, MYCN (20×) expression downregulated, and vascularity decreased in PW-12 treated tumors as confirmed by histology (B) and cell quantification for apoptosis and cell proliferation (C). (* representsp < 0.05).
Discussion
Effective pharmacologic targeting of the MYC family of transcription factor oncogenes, particularly targeting MYCN in pediatric cancer, has been a long sought after goal (1). Pre-clinical testing of drugs across pharmacologic scaffolds is an indicator of robustness of a particular drug target and mechanism as well as a potential means of bypassing emergent resistance. PI3K inhibition has previously been implicated in stabilization of MYCN. Here, we show that PW-12, a bioavailable derivative of the PI3K inhibitor PIK-75, shows knockdown of MYCN in a neuroblastoma cell line with marked reduction in viability and changes in cell cycle consistent with a MYCN-specific effect. Using a transgenic model of MYCN-amplified neuroblastoma and a model of medulloblastoma where murine-derived allografted cells are implanted into mice, we demonstrate significant in vivo efficacy against both neuroblastoma and medulloblastoma tumors. We further show on-target efficacy against tumor MYCN protein, marked decrease in proliferation and increase in tumor apoptosis as well as apparent tumor vascular collapse.
The past decade of experience with targeted inhibitors has shown that emergent resistance is nearly inevitable. Effective blockade of a particular pharmacologic target requires an array of pharmacologic scaffolds to bypass resistance by point mutation within the target, a resistance mechanism that has been shown to be active for PI3K inhibitors (23). To our knowledge, this is the first in vivo pre-therapeutic trial of PW12 in neuroblastoma or medulloblastoma and the first implicating blockade of MYCN in its effect. By broadening in vivo and cell line studies across this new pharmacologic scaffold, these studies add to the growing evidence implicating PI3K/mTOR inhibition as a viable strategy in MYCN-driven pediatric cancers including both neuroblastoma and medulloblastoma.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by NIH K08NS079485 (WG); Alex’s Lemonade Stand (WG and WW); Frank A. Campini Foundation (WG); P01CA081403 (WW and KS); CureSearch Grand Challenge Award (WW); R01CA102321 (WW); R01CA159859 (WW); R01CA148699 (WW); R01NS089868 (WW); P30CA82103 (WW); Katie Dougherty Foundation (WW and WG); Howard Hughes Medical Institute (KS); The Samuel Waxman Cancer Research Foundation (KS and WW); and The Ross K. MacNeill Foundation (WW).
References
1. Gustafson W, Weiss W. Myc proteins as therapeutic targets. Oncogene (2010) 29:1249–59. doi: 10.1038/onc.2009.512
2. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Springs Harb Perspect Med (2013) 3:a014415. doi:10.1101/cshperspect.a014415
3. Roussel MF, Robinson GW. Role of MYC in medulloblastoma. Cold Springs Harb Perspect Med (2013) 3:a014308. doi:10.1101/cshperspect.a014308
4. Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, et al. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol (2009) 27:1007–13. doi:10.1200/JCO.2007.13.8925
5. Maris J. Recent advances in neuroblastoma. N Engl J Med (2010) 362:2202–11. doi:10.1056/NEJMra0804577
6. Brodeur G, Seeger R, Schwab M, Varmus H, Bishop J. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science (1985) 224:1121–4. doi:10.1126/science.6719137
7. Seeger R, Brodeur G, Sather H, Dalton A, Siegel S, Wong K, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med (1985) 313:1111–6. doi:10.1056/NEJM198510313131802
8. Chesler L, Goldenberg DD, Collins R, Grimmer M, Kim G, Tihan T, et al. Chemotherapy-induced apoptosis in a transgenic model of neuroblastoma proceeds through p53 induction. Neoplasia (2008) 10:1268–74. doi:10.1593/neo.08778
9. Chanthery Y, Gustafson W, Itsara M, Persson A, Hackett CS, Grimmer M, et al. Paracrine signaling through MYCN enhances tumor-vascular interactions in neuroblastoma. Sci Transl Med (2012) 4:115ra3. doi:10.1126/scitranslmed.3002977
10. Huse J, Holland E. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer (2010) 10:319–31. doi:10.1038/nrc2818
11. Kool M, Korshunov A, Remke M, Schlanstein M, Northcott P, Cho Y, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, group 3, and group 4 medulloblastomas. Acta Neuropathol (2012) 123:473–84. doi:10.1007/s00401-012-0958-8
12. Korshunov A, Remke M, Kool M, Hielscher T, Northcott P, Williamson D, et al. Biological and clinical heterogeneity of MYCN-amplified medulloblastoma. Acta Neuropathol (2012) 123:515–27. doi:10.1007/s00401-011-0918-8
13. Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol (2012) 123:465–72. doi:10.1007/s00401-011-0922-z
14. Swartling F, Grimmer M, Hackett C, Northcott P, Fan Q-W, Goldenberg D, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev (2010) 24:1059–72. doi:10.1101/gad.1907510
15. Swartling F, Savov V, Persson A, Chen J, Hackett C, Northcott P, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell (2012) 21:601–13. doi:10.1016/j.ccr.2012.04.012
16. Romer J, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(±)p53(−/−) mice. Cancer Cell (2004) 6:229–40. doi:10.1016/j.ccr.2004.08.019
17. Yauch R, Dijkgraaf G, Alicke B, Januario T, Ahn C, Holcomb T, et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science (2009) 326:572–4. doi:10.1126/science.1179386
18. Tang Y, Gholamin S, Schubert S, Willardson M, Lee A, Bandopadhayay P, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat Med (2014) 20:732–40. doi:10.1038/nm.3613
19. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell (2012) 22:117–30. doi:10.1016/j.ccr.2012.06.001
20. Chesler L, Schelieve C, Goldenberg D, Kenney A, Kim G, Mcmillan A, et al. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer Res (2006) 66:8139–46. doi:10.1158/0008-5472.CAN-05-2769
21. Guillard S, Clarke P, Poele T, Mohri Z, Bejerke L, Valenti M, et al. Molecular pharmacology of phosphatidylinositol 3-kinase inhibition in human glioma. Cell Cycle (1009) 8:443–53. doi:10.4161/cc.8.3.7643
22. Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, et al. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell (2006) 125:733–47. doi:10.1016/j.cell.2006.03.035
23. Zunder E, Knight Z, Houseman B, Apsel B, Shokat K. Discovery of drug-resistant and drug-sensitizing mutations in teh oncogenic PI3K isoform p110alpha. Cancer Cell (2008) 14(2):180–92. doi:10.1016/j.ccr.2008.06.014
24. Weiss W, Aldape K, Mohapatra G, Feuerstein B, Bishop J. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J (1997) 16:2985–95. doi:10.1093/emboj/16.11.2985
25. Kim KW, Paul P, Qiao J, Lee S, Chung DH. Enhanced autophagy blocks angiogenesis via degradation of gastrin-releasing peptide in neuroblastoma cells. Autophagy (2013) 9:1579–90. doi:10.4161/auto.25987
26. Fan Q-W, Knight Z, Goldenberg D, Yu W, Mostov K, Stokoe D, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell (2006) 9:341–9. doi:10.1016/j.ccr.2006.03.029
27. Vivanco I, Sawyers C. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev Cancer (2002) 2:489–501. doi:10.1038/nrc839
28. Fan Q, Specht K, Kzhang C, Goldenberg D, Shokat K, Weiss W. Combinatorial efficacy achieved through two-point blockade within a signaling pathway-a chemical genetic approach. Cancer Res (2003) 63:8930–8.
Keywords: pediatric, cancer, neuroblastoma, medulloblastoma, MYCN, PI3 kinase, cell signaling, mTOR
Citation: Cage TA, Chanthery Y, Chesler L, Grimmer M, Knight Z, Shokat K, Weiss WA and Gustafson WC (2015) Downregulation of MYCN through PI3K inhibition in mouse models of pediatric neural cancer. Front. Oncol. 5:111. doi: 10.3389/fonc.2015.00111
Received: 02 February 2015; Accepted: 27 April 2015;
Published: 12 May 2015
Edited by:
Arturo Sala, Brunel University, UK; University College London, UKReviewed by:
Stephen L. Hart, University College London, UKVincenzo De Laurenzi, University of Chieti, Italy
Copyright: © 2015 Cage, Chanthery, Chesler, Grimmer, Knight, Shokat, Weiss and Gustafson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: W. Clay Gustafson, Departments of Pediatrics, Pediatric Hematology, and Oncology, Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, 1450 3rd Street, San Francisco, CA 94158, USA,Z3VzdGFmc29uY0BwZWRzLnVjc2YuZWR1