 Alessandro Rimessi
Alessandro Rimessi Simone Patergnani
Simone Patergnani Massimo Bonora
Massimo Bonora Mariusz R. Wieckowski
Mariusz R. Wieckowski Paolo Pinton
Paolo Pinton- 1Section of Pathology, Oncology and Experimental Biology, Laboratory for Technologies of Advanced Therapies (LTTA), Department of Morphology, Surgery and Experimental Medicine, University of Ferrara, Ferrara, Italy
- 2Department of Biochemistry, Nencki Institute of Experimental Biology, Warsaw, Poland
Cancer is sustained by defects in the mechanisms underlying cell proliferation, mitochondrial metabolism, and cell death. Mitochondrial Ca2+ ions are central to all these processes, serving as signaling molecules with specific spatial localization, magnitude, and temporal characteristics. Mutations in mtDNA, aberrant expression and/or regulation of Ca2+-handling/transport proteins and abnormal Ca2+-dependent relationships among the cytosol, endoplasmic reticulum, and mitochondria can cause the deregulation of mitochondrial Ca2+-dependent pathways that are related to these processes, thus determining oncogenic behavior. In this review, we propose that mitochondrial Ca2+ remodeling plays a pivotal role in shaping the oncogenic signaling cascade, which is a required step for cancer formation and maintenance. We will describe recent studies that highlight the importance of mitochondria in inducing pivotal “cancer hallmarks” and discuss possible tools to manipulate mitochondrial Ca2+ to modulate cancer survival.
Introduction
The mitochondrion is an endosymbiotic organelle that characterizes any eukaryotic cell, participating in many aspects of cell physiology, such as ATP production (1), intracellular Ca2+ signaling (2), lipid metabolism (3), reactive oxygen species (ROS) production (4), inflammasome activation (5), and cell death regulation (6).
Mitochondria can fulfill a large energy demand due to their biosynthetic capacities; during mitochondrial respiration, it can rapidly accumulate Ca2+ through an electrogenic pathway (7) and via their close apposition with the Ca2+-storing endoplasmic reticulum (ER) (8). Ca2+ released from the ER generates microdomains consisting of high [Ca2+] that are sequestered by the mitochondrial Ca2+ uniporter (MCU) (9, 10).
Because of the important role of mitochondria in cell fate, many pathological conditions are associated with mitochondrial failure, including metabolic and neurodegenerative diseases (11), inflammation, and cancer (12). Mitochondrial dysfunction is associated with cancer development and progression. During carcinogenesis in some malignant cells, the mitochondrial Ca2+ signaling was significantly remodeled, compromising mitochondrial functions in ways that caused them to overwhelm normal cells. Uncontrolled growth, programed cell death evasion, metabolic reprograming, invasion, and metastasis are “cancer hallmarks” that are linked to mitochondrial Ca2+ remodeling, which could be the prime reason for these transformations (Figure 1).
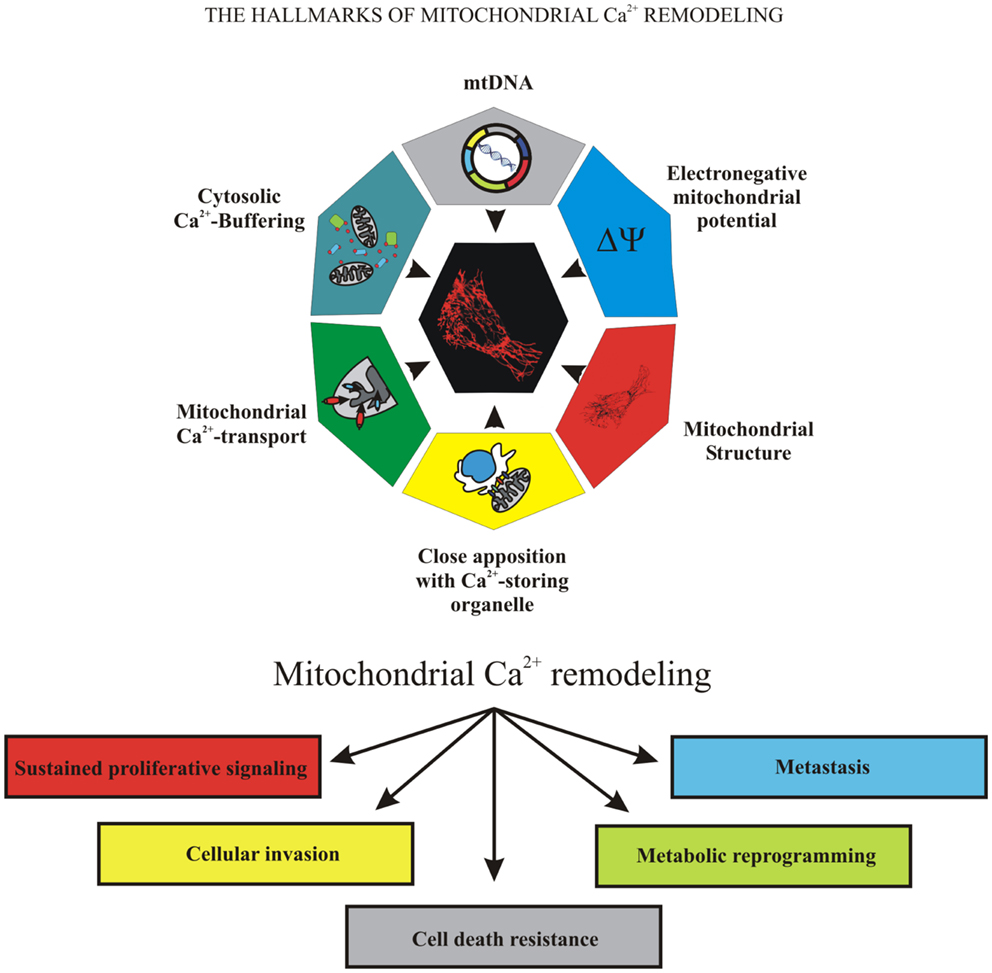
Figure 1. The hallmarks of mitochondrial Ca2+ remodeling. This illustration encompasses the six hallmarks of remodeled mitochondrial Ca2+ signaling that are involved in pathogenesis of some and perhaps all cancers. Uncontrolled growth, programed cell death evasion, metabolic reprograming, invasion, and metastasis are the “cancer hallmarks” that are linked to mitochondrial Ca2+ remodeling, the prime reason of oncogenic behavior.
In this review, we will describe the current knowledge regarding the role of mitochondrial Ca2+ signaling in cancer and the rationale for targeting cancer cells with mitochondrial Ca2+ modulators as a pharmacological therapeutic strategy.
Mitochondrial Ca2+ Homeostasis
Due to their strong electronegative potential (ΔΨ) and by topological organization, mitochondria are able to accumulate Ca2+ in the mitochondrial matrix. This organelle can generate contact sites with the ER (13), where the channels responsible for Ca2+-release (inositol trisphosphate receptor, IP3R) are juxtaposed to mitochondrial Ca2+ handling/transport (14). Voltage-dependent anion selective channels (VDACs) are permeable to Ca2+ and are in the outer mitochondrial membrane (OMM), toward the matrix-specific Ca2+ transporter, where these channels participate in structures devoted functionally to facilitating Ca2+-flow toward the mitochondrion (15). This flow allows the generation of a high [Ca2+] microdomain that is sufficient to trigger the opening of mitochondrial uniporter complex (16). MCU is a two-transmembrane domain protein that spans across the mitochondrial inner membrane, oligomerizing it to form the Ca2+-channel portion of the complex (9, 10), and where it can accommodate the MCU paralog MCUb. This protein does not exhibit any channel-forming activity and may behave as a negative regulator of MCU complex (17). MCU/MCUb oligomers physically interact with two regulators located in the intermembrane space, MICU1 and MICU2. Silencing studies have indicated that both regulators participate in setting the Ca2+-threshold for mitochondrial Ca2+-uptake (18–20). Finally, the MCU regulator EMRE mediates the interaction between MCU oligomers and MICU1/MICU2 (16).
The accumulation of Ca2+ requires efficient mechanisms to turn off the signal to prevent the mitochondrial permeability transition pore (mPTP) opening. The most well-characterized Ca2+-efflux mechanism involves the mitochondrial Na+/Ca2+ exchanger (21). This protein forms dimers that transport either Na+ or Li+ in exchange for Ca2+. Then, accumulated Na+ is extruded via mitochondrial Na+/H+ exchange. This activity induces the dissipation of ΔΨ, causing mitochondrial Ca2+-efflux (22). In contrast, a non-electrogenic Ca2+/2H+ exchange has been proposed to be responsible for Ca2+-efflux in non-excitable cells. Its molecular identity has been proposed to be leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) (23). Ongoing discussions have proposed that mPTP could be a component of the Ca2+-efflux system due to the involvement of a non-specific channel that has been identified as the C subunit of mitochondrial F1/FO ATP synthase (24). The manipulation of C subunit expression is able to promote or inhibit mPTP opening without affecting the physiological mitochondrial Ca2+ homeostasis (25).
Mitochondrial Ca2+ Homeostasis during Proliferation and Cell Death
Cancer is sustained by defects in the mechanisms underlying cell proliferation and cell death, and mitochondrial Ca2+ ions are central to both events (Figure 1).
To meet the increased energy demand of cells during proliferation, mitochondria utilize fundamental pathways for energy metabolism, including the citric acid cycle (TCA) and oxidative phosphorylation. An increase in mitochondrial ATP levels was shown to occur in parallel with an evoked increase in mitochondrial [Ca2+] (26). The first results were obtained by Denton and McCormack, who showed that mitochondrial Ca2+-activated dehydrogenase enzymes led to increased NADH and thus ATP production (27, 28). The involvement of different mitochondrial targets in the regulation of oxidative phosphorylation by Ca2+, including dehydrogenase activity, F1-F0-ATPase, and mitochondrial substrate-transport has been proposed (29).
Three are the mitochondrial dehydrogenase sensitive to Ca2+ of TCA: Pyruvate dehydrogenase (PDH), isocitrate dehydrogenase and α-ketoglutarate dehydrogenase. All of these molecules exhibit different Ca2+-dependent mechanisms, suggesting that the origins or specific activation signals are quite dissimilar. Although the deregulation of mitochondrial metabolism is intimately linked to oncogenic behavior, the role of cellular metabolism in oncogene-induced transformation is unclear. Recently, the mitochondrial gatekeeper PDH was shown to be a crucial mediator of oncogene-induced senescence promoted by BRAF (V600E), which is the oncogene that is commonly mutated in tumors (30).
The efficient mitochondrial respiration and maintenance of normal cell bioenergetics require basal constitutive low-level Ca2+ signaling to sustain the mitochondrial Ca2+-uptake provided by IP3R (31). The enhanced resistance to apoptosis of cancer cells may involve its downregulated expression or activation, compromising IP3R-mediated Ca2+ release. Indeed, in bladder cancer cells, the acquisition of cisplatin resistance was sustained by the drug-induced downregulation of IP3R1 expression (32). Additionally, the disruption of IP3R-mediated ER–mitochondria crosstalk compromised the production of mitochondrial ATP, increasing the AMP/ATP ratio, which subsequently activated the energy sensor AMP-activated protein kinase, inducing autophagy (33, 34). This cascade of events occurs in advanced cancer under hypoxic and nutrient-deficient conditions that activate autophagy as a pro-survival mechanism (35).
The failure of mitochondria to take-up Ca2+ results in a failed intracellular Ca2+ buffering with consequent aberrant activation of cytosolic Ca2+-dependent enzymes, such as calpain proteases (36) and calmodulin-dependent kinases (37). In turn, these enzymes alter cellular signaling cascades with consequent effects on cell growth (38) or lead to increased glycolysis and tumor cell invasion (39). However, the activation of calcineurin protein phosphatase by increased cytosolic Ca2+ leads to the dephosphorylation of IκBβ and successive NF-κB activation (40), which promote apoptosis resistance and cell migration (41). The migratory capacity of tumor cells is linked with mitochondria morphology (42, 43). In breast cancer, higher fragmented mitochondria content correlates with metastatic potential and the cell’s ability to migrate and invade is significantly reduced when the Dynamin-related protein 1 is silenced. The relationship between mitochondrial shaping and Ca2+ is well represented by the involvement of Rho-GTPase Miro and by Drp1 [as reviewed in Ref. (44, 45)].
Endoplasmic reticulum–mitochondria contact sites allow the accumulation of Ca2+ in response to not only physiological agonists but also in response to apoptotic stimuli that induce ER Ca2+-release (46). While moderate Ca2+ levels are essential for normal mitochondrial activities, mitochondrial Ca2+ overload is detrimental to mitochondrial morphology, causing mitochondrial permeabilization and organelle swelling, with the consequent release of pro-apoptotic factors (6). Recently, mitochondrial Ca2+ overload has also been implicated in mitophagy, the selective degradation of damaged mitochondria (45).
A critical link between Ca2+ and apoptosis was established by the oncoprotein Bcl-2. Evidence show that it reduces the filling state of intracellular Ca2+-stores, reducing the Ca2+ signaling evoked by physiological and pathological stimuli, but this observation is still debated (47, 48). Bcl-2 is also a critical regulator of IP3R and interacts directly with this molecule to inhibit channel opening and ER Ca2+-release, thereby assuring Ca2+-dependent cell proliferation and apoptosis protection (49, 50). The mechanism by which Bcl-2 family members control apoptosis remains unclear; however, their activity involves controlling OMM permeability, which also requires interactions with OMM proteins such as VDACs (51). Pro-apoptotic proteins of the Bcl-2 family (such as Bax) exert opposite effects (i.e., a potentiation of Ca2+-mediated signals) counteracting the effects of Bcl-2 on ER Ca2+ filling (52).
Voltage-dependent anion-selective channels are readily permeable to Ca2+ and can interact with IP3Rs at the ER (53). VDACs are expressed in three isoforms that have common channeling properties but different roles in cell survival (54, 55). Although all three VDAC isoforms are equivalent in facilitating mitochondrial Ca2+-uptake upon agonist stimulation, VDAC1 is preferentially involved in the transmission of low-amplitude apoptotic Ca2+ signals to mitochondria. VDAC1 gene expression levels are predictors of poor outcome in NSCLC and in other cancers (56). VDACs are subjected to multiple levels of regulation, including expression levels, post-translational modification, and protein–protein interactions, all of which can limit Ca2+-uptake. For example, VDAC may inhibit apoptosis, promoting tumorigenesis and glycolysis through specific interactions with hexokinase-2 (HK2). The upregulation of HK expression in tumor cells and its binding to VDAC provide both metabolic benefits and apoptosis-suppressive capacity, offering advantages in terms of growth and resistance to chemotherapy (57, 58). Mitochondrial HK2 interferes with the ability of Bax to bind to mitochondria and release cytochrome c, thus antagonizing cell death (59).
Mitochondria are also the critical sites for “apoptotic” Ca2+ signaling due to MCU, which is responsible for low-affinity Ca2+-uptake, influencing a myriad of Ca2+-dependent processes, including cell death (60). MCU-overexpressing cells treated with apoptotic stimuli, such as H2O2 and ceramide, exhibited more pronounced apoptotic responses (10), while the overexpression of a MCU-targeting microRNA miR-25 in colon cancer cells impaired mitochondrial Ca2+-uptake and increased apoptosis resistance (61). Thus, MCU appears to be a crucial protein for tumorigenesis, and its specific pharmacological activators, if identified, could become a useful tool.
Much remains to be understood regarding the additional signals that converge on mitochondria and switch their function to apoptotic inducers. These organelles can handle large Ca2+ loads under physiological conditions (e.g., in cardiac myocytes, significant amounts of Ca2+ accumulate in mitochondria with every heartbeat) with no deleterious effects. Oxidative stress is considered an important factor for additional “apoptotic signal” (62, 63). ROS is involved in the regulation of physiological processes but may also be harmful if produced excessively by oxidative phosphorylation. A feature of transformed cells is increased mitochondrial ROS levels, which are attributed to inefficiencies in electron transport at the respiratory chain, increased metabolic demand, reduced ROS scavenging, oncogene-induced replicative stress, and altered mitochondrial dynamics (64, 65). Indeed, ROS promote tumorigenesis in numerous ways, including the stabilization of hypoxia-inducible factor HIF-α, the induction of oxidative base damage to mtDNA, the activation of both NRF2 and NF-kB transcription factors, and the alteration of Ca2+ flux (66).
Alterations to Mitochondrial Ca2+ Homeostasis in Cancer Cells
A growing number of tumor suppressor genes and oncogenes have been investigated due to their ability to regulate mitochondrial function, influencing the physiological processes of this organelle and its capacity to uptake Ca2+.
Among these genes, increasing evidence has highlighted the role of the oncogene Ras in the growth and maintenance of the tumor environment (67). The first link between mitochondria and Ras is represented by the direct translocation of Ras into the organelle, where the phosphorylated form exerts apoptotic activity (68). Although some evidence has suggested a possible role of mitochondria in the regulation of Ras-driven modifications, a clear molecular mechanism underlying this relationship has not yet been addressed. The oncogenic function of Ras facilitates Stat3 translocation to mitochondria, which promotes mtDNA transcription (69), and Ras-dependent mitochondrial dysfunction causes metabolic changes and ROS, which promote tumor development (70). A direct link between Ca2+ regulation and oncogenic Ras has been shown by Rimessi et al., in which H-Ras affects the correct transmission of Ca2+ waves from the ER to mitochondria (71). As a result, mitochondrial metabolism and apoptosis are deeply compromised, and a neoplastic phenotype is induced. Recently, cancer stem cells in pancreatic cancer were shown to survive oncogenic K-Ras ablation by relying on oxidative phosphorylation (72) and by exhibiting high sensitivity to oxidative phosphorylation inhibitors to prevent tumors. This finding is consistent with other recent reports referring to leukemia cancer cells (73–75).
Additionally, the oncogene AKT regulates cell growth and apoptosis, and upon PI3K stimulation, it rapidly accumulates in mitochondria (76), where it inhibits apoptosis in an HK-dependent manner (77, 78). AKT phosphorylates several pro-apoptotic proteins, such as Bad and Bax, inhibiting their cell death functions. AKT also phosphorylates HK2, promoting binding with VDAC1, and preventing a Ca2+-dependent apoptotic response (79). In addition, AKT phosphorylates the IP3R isoforms that inhibit ER Ca2+ release and apoptosis, avoiding mitochondrial Ca2+ overload (80), preferentially phosphorylating the IP3R3 (81). AKT is intimately regulated by the tumor suppressor protein phosphatase and tensin homolog (PTEN), commonly lost or mutated in human cancers (82). PTEN was demonstrated to perform its growth-attenuating activity by regulating ER Ca2+ release via IP3R3. In this study, PTEN was shown to directly interact with IP3R, counteracting the reduced IP3R-dependent Ca2+ release mediated by AKT phosphorylation in a phosphatase-dependent manner (83). Another tumor suppressor that found to downregulate the anti-apoptotic features of AKT is PML. This tumor suppressor, where its tumoral behavior is regulated by PIAS1 and SUMOylation machinery (84), promotes the formation of a multiprotein complex containing IP3R3, AKT, and the protein phosphatase PP2a, orchestrating ER–mitochondria Ca2+ flux (85). An other example is the tumor suppressor BRCA1, recruited to the ER during apoptosis in an IP3R-dependent manner, sensitizing the IP3R to its ligand (86).
Additionally, the tumor suppressor p53 can regulate tumorigenesis in a Ca2+-dependent pathway. Different reports have documented the ability of p53 to regulate cell fate via post-translational modifications (87, 88); however, p53 was recently shown to function in modulating Ca2+ transfer from ER to mitochondria. An extranuclear pool of p53 interacts with the Ca2+-ATPase SERCA, modulating Ca2+-dependent ER–mitochondria crosstalk, mitochondrial swelling and apoptosis induction (89, 90).
The tumor suppressor FHIT functions in a different manner. This hydrolase, which acts as a tumor suppressor in vivo and in vitro, enhances apoptosis susceptibility by acting directly on the mitochondrial compartment. Specifically, FHIT increases the affinity of the mitochondrial machinery for physiological agonist- and apoptotic challenge-triggered Ca2+-uptake into mitochondria (91).
Anti-Cancer Drugs Act on Mitochondrial Ca2+ Homeostasis
The Ca2+ ion regulates various cellular processes, some of which are involved in tumorigenesis and which are thus considered as attractive drug targets for cancer therapy. Mitochondrial Ca2+ homeostasis may be affected by anti-cancer drugs that directly influence mitochondria or targets that indirectly regulate mitochondrial Ca2+-uptake. Some of these drugs may affect mitochondrial Ca2+ uptake due to the depolarization of ΔΨ. The compounds included in this category are as follows: pancratistatin (a natural alkaloid exhibiting anti-cancer effects against human colorectal adenocarcinoma xenografts (92) and colorectal carcinoma cell lines, rhodamine-123 [the anti-cancer effects of rhodamine-123 can be explained by its higher retention in kidney and breast cancer cells compared to non-tumorigenic cells (93, 94)], a naphthyridine derivative [4-phenyl-2,7-di(piperazin-1-yl)-1,8-naphthyridine (95)], PMT7 [redox-active quinone phloroglucinol derivative (96)], edelfosine [1-O-octadecyl-2-O-methyl-racglycero-3-phosphocholine; its redistribution to the mitochondria in HeLa cells causes mitochondrial depolarization and apoptosis (97)], and many others that inhibit the mitochondrial respiratory chain that have been reviewed in detail by Wen et al. (98).
Anti-cancer drugs can also influence the mitochondrial Ca2+-uptake indirectly through the induction of ER stress. For example, tunicamycin induces the accumulation of unfolded proteins in the ER, which is known to enhance antitumor effects of different chemotherapy drugs in vitro and in vivo (99). The cannabinoids induce apoptosis of pancreatic tumor cells via the ER-stress-related genes activating transcription factor 4 and TRB3 (100), or by the photo-oxidative treatment with hypericin that causes ER stress, induction of UPR target genes, and depletion of ER Ca2+-stores (101). Brefeldin A blocks protein transport from the ER to the Golgi apparatus (102), causes the deregulation of ER Ca2+ homeostasis, or affects IP3R activity. Bortezomib, a selective proteasome inhibitor approved for use in patients with multiple myeloma, requires the mitochondrial uniporter as critical regulatory factor for its cytotoxicity (103).
The anti-apoptotic Ca2+ effects of Bcl-2 are attributable to direct interactions with IP3R through the BH4 domain (50, 104). A peptide covering this residue was used to disrupt the IP3R/Bcl-2-protein complex, enhancing the Ca2+-dependent apoptotic response in a variety of tumoral cells (105–108).
The HK–VDAC complex is also an oncological target of erastin, which is an antitumor agent that is selective for tumor cells bearing oncogenic Ras (109), and of the chemo-potentiator 3-bromopyruvate, which is an inhibitor of energy metabolism in tumor cells (109, 110). Clotrimazole and bifonazole are also effective (111, 112), as is the anti-cancer agent furano naphthoquinone, which induces a caspase-dependent apoptosis pathway through ROS production (113).
The most direct effects of potential anti-cancer drugs on mitochondrial Ca2+-uptake were obtained by potentiating MCU complex expression/activity. Unfortunately, recent molecular discoveries have impeded the identification of an MCU-specific inducer. Alternative approaches could include MCU-targeting anti-miRNA-based therapy and anti-miRNA 25 oligonucleotides that could be used as potential agents against cancer (114).
Conclusion
“Uncontrolled growth, programed cell death evasion, metabolic reprograming, invasion, and metastasis are ‘cancer hallmarks’ that are linked to mitochondrial Ca2+ remodeling, which could be the prime reason for these transformations.” This speculation in the Section “Introduction” is sustained by the knowledge described thus far and by the most recent discoveries that highlight the role of oxidative phosphorylation in tumor progression and maintenance. Previously, mitochondrial dysfunction has been considered a consequence rather than a key player in tumor development. Recently, cancer stem cells were shown to survive oncogenic ablation by relying on oxidative phosphorylation (72), and only mtDNA-depleted cancer cells were capable of recovering mtDNA from host-formed metastasizing cancers in vivo (115), thus highlighting the malicious capacity of tumor cells to acquire mtDNA from normal bystander cells. Considering that the driving force for mitochondrial Ca2+ internalization is ΔΨ, which is generated by respiratory chain, mitochondrial Ca2+ signaling has become a prime factor that is responsible for oncogenic behavior. Although mitochondrial Ca2+ remains a novel area of research in oncology, considerable further work is required to clarify the molecular mechanisms by which it contributes to cancer formation and maintenance, and greater knowledge of this target may be of importance in the development of new therapeutic strategies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was supported by the Italian Ministry of Health to AR (GR-2009-1594541 and GR-2011-02346964); the Italian Association for Cancer Research (AIRC: IG 14442), local funds from the University of Ferrara, the Italian Ministry of Education, University and Research (COFIN: 20129JLHSY_002, FIRB: RBAP11FXBC_002, Futuro in Ricerca: RBFR10EGVP_001). SP was supported by FISM (Fondazione Italiana Sclerosi Multipla) research fellowship (2012/B/11). MW was supported by the grants W100/HFSC/2011 and HFSP RGP0027/2011.
References
1. Tarasov AI, Griffiths EJ, Rutter GA. Regulation of ATP production by mitochondrial Ca(2+). Cell Calcium (2012) 52:28–35. doi: 10.1016/j.ceca.2012.03.003
2. Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, et al. Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta (2009) 1787:1342–51. doi:10.1016/j.bbabio.2009.03.015
3. Scharwey M, Tatsuta T, Langer T. Mitochondrial lipid transport at a glance. J Cell Sci (2013) 126:5317–23. doi:10.1242/jcs.134130
4. Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab (2014) 2:17. doi:10.1186/2049-3002-2-17
5. Rimessi A, Bezzerri V, Patergnani S, Marchi S, Cabrini G, Pinton P. Mitochondrial Ca(2+)-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat Commun (2015) 6:6201. doi:10.1038/ncomms7201
6. Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial Ca(2+) and apoptosis. Cell Calcium (2012) 52:36–43. doi:10.1016/j.ceca.2012.02.008
7. Rimessi A, Giorgi C, Pinton P, Rizzuto R. The versatility of mitochondrial calcium signals: from stimulation of cell metabolism to induction of cell death. Biochim Biophys Acta (2008) 1777:808–16. doi:10.1016/j.bbabio.2008.05.449
8. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science (1998) 280:1763–6. doi:10.1126/science.280.5370.1763
9. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. doi:10.1038/nature10234
10. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. doi:10.1038/nature10230
12. Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion (2012) 12:77–85. doi:10.1016/j.mito.2011.07.004
13. Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid Redox Signal (2015) 22:995–1019. doi:10.1089/ars.2014.6223
14. Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta (2014) 1843:2164–83. doi:10.1016/j.bbamcr.2014.03.007
15. Messina A, Reina S, Guarino F, De Pinto V. VDAC isoforms in mammals. Biochim Biophys Acta (2012) 1818:1466–76. doi:10.1016/j.bbamem.2011.10.005
16. Sancak Y, Markhard AL, Kitami T, Kovacs-Bogdan E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (2013) 342:1379–82. doi:10.1126/science.1242993
17. Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J (2013) 32:2362–76. doi:10.1038/emboj.2013.157
18. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One (2013) 8:e55785. doi:10.1371/journal.pone.0055785
19. Kamer KJ, Mootha VK. MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep (2014) 15:299–307. doi:10.1002/embr.201337946
20. Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, et al. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell (2014) 53:726–37. doi:10.1016/j.molcel.2014.01.013
21. Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A (2010) 107:436–41. doi:10.1073/pnas.0908099107
22. Nicholls DG, Crompton M. Mitochondrial calcium transport. FEBS Lett (1980) 111:261–8. doi:10.1016/0014-5793(80)80806-4
23. Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci U S A (2013) 110:E2249–54. doi:10.1073/pnas.1308558110
24. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, et al. Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle (2013) 12:674–83. doi:10.4161/cc.23599
25. De Marchi E, Bonora M, Giorgi C, Pinton P. The mitochondrial permeability transition pore is a dispensable element for mitochondrial calcium efflux. Cell Calcium (2014) 56:1–13. doi:10.1016/j.ceca.2014.03.004
26. Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci U S A (1999) 96:13807–12. doi:10.1073/pnas.96.24.13807
27. Denton RM, Randle PJ, Martin BR. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J (1972) 128:161–3.
28. McCormack JG, Halestrap AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev (1990) 70:391–425.
29. Bonora M, Patergnani S, Rimessi A, De Marchi E, Suski JM, Bononi A, et al. ATP synthesis and storage. Purinergic Signal (2012) 8:343–57. doi:10.1007/s11302-012-9305-8
30. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, et al. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature (2013) 498:109–12. doi:10.1038/nature12154
31. Cardenas C, Foskett JK. Mitochondrial Ca(2+) signals in autophagy. Cell Calcium (2012) 52:44–51. doi:10.1016/j.ceca.2012.03.001
32. Tsunoda T, Koga H, Yokomizo A, Tatsugami K, Eto M, Inokuchi J, et al. Inositol 1,4,5-trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene (2005) 24:1396–402. doi:10.1038/sj.onc.1208313
33. Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142:270–83. doi:10.1016/j.cell.2010.06.007
34. Decuypere JP, Bultynck G, Parys JB. A dual role for Ca(2+) in autophagy regulation. Cell Calcium (2011) 50:242–50. doi:10.1016/j.ceca.2011.04.001
35. Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell (2014) 159:1263–76. doi:10.1016/j.cell.2014.11.006
36. Storr SJ, Carragher NO, Frame MC, Parr T, Martin SG. The calpain system and cancer. Nat Rev Cancer (2011) 11:364–74. doi:10.1038/nrc3050
37. Racioppi L, Means AR. Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem (2012) 287:31658–65. doi:10.1074/jbc.R112.356485
38. Prevarskaya N, Skryma R, Shuba Y. Calcium in tumour metastasis: new roles for known actors. Nat Rev Cancer (2011) 11:609–18. doi:10.1038/nrc3105
39. Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene (2002) 21:7839–49. doi:10.1038/sj.onc.1205983
40. Biswas G, Tang W, Sondheimer N, Guha M, Bansal S, Avadhani NG. A distinctive physiological role for IkappaBbeta in the propagation of mitochondrial respiratory stress signaling. J Biol Chem (2008) 283:12586–94. doi:10.1074/jbc.M710481200
41. Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol (2005) 5:749–59. doi:10.1038/nri1703
42. Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene (2012) 32:4814–24. doi:10.1038/onc.2012.494
43. Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J (2013) 104:2077–88. doi:10.1016/j.bpj.2013.03.025
44. Campello S, Scorrano L. Mitochondrial shape changes: orchestrating cell pathophysiology. EMBO Rep (2010) 11:678–84. doi:10.1038/embor.2010.115
45. Rimessi A, Bonora M, Marchi S, Patergnani S, Marobbio CM, Lasorsa FM, et al. Perturbed mitochondrial Ca2+ signals as causes or consequences of mitophagy induction. Autophagy (2013) 9:1677–86. doi:10.4161/auto.24795
46. Ferrari D, Pinton P, Campanella M, Callegari MG, Pizzirani C, Rimessi A, et al. Functional and structural alterations in the endoplasmic reticulum and mitochondria during apoptosis triggered by C2-ceramide and CD95/APO-1/FAS receptor stimulation. Biochem Biophys Res Commun (2010) 391:575–81. doi:10.1016/j.bbrc.2009.11.101
47. He H, Lam M, McCormick TS, Distelhorst CW. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J Cell Biol (1997) 138:1219–28. doi:10.1083/jcb.138.6.1219
48. Pinton P, Ferrari D, Rapizzi E, Di Virgilio F, Pozzan T, Rizzuto R. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J (2001) 20:2690–701. doi:10.1093/emboj/20.11.2690
49. Rong YP, Barr P, Yee VC, Distelhorst CW. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim Biophys Acta (2009) 1793:971–8. doi:10.1016/j.bbamcr.2008.10.015
50. Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, et al. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A (2009) 106:14397–402. doi:10.1073/pnas.0907555106
51. Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem (2010) 285:6053–62. doi:10.1074/jbc.M109.082990
52. Chami M, Prandini A, Campanella M, Pinton P, Szabadkai G, Reed JC, et al. Bcl-2 and Bax exert opposing effects on Ca2+ signaling, which do not depend on their putative pore-forming region. J Biol Chem (2004) 279:54581–9. doi:10.1074/jbc.M409663200
53. Rowland AA, Voeltz GK. Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat Rev Mol Cell Biol (2012) 13:607–25. doi:10.1038/nrm3440
54. Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol (2002) 159:613–24. doi:10.1083/jcb.200205091
55. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ (2012) 19:267–73. doi:10.1038/cdd.2011.92
56. Grills C, Jithesh PV, Blayney J, Zhang SD, Fennell DA. Gene expression meta-analysis identifies VDAC1 as a predictor of poor outcome in early stage non-small cell lung cancer. PLoS One (2011) 6:e14635. doi:10.1371/journal.pone.0014635
57. Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, et al. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J Exp Med (2011) 208:313–26. doi:10.1084/jem.20101470
58. Shoshan-Barmatz V, Golan M. Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr Med Chem (2012) 19:714–35. doi:10.2174/092986712798992110
59. Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem (2002) 277:7610–8. doi:10.1074/jbc.M109950200
60. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol (2014) 592:829–39. doi:10.1113/jphysiol.2013.268235
61. Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, et al. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol (2013) 23:58–63. doi:10.1016/j.cub.2012.11.026
62. Csordas G, Hajnoczky G. SR/ER-mitochondrial local communication: calcium and ROS. Biochim Biophys Acta (2009) 1787:1352–62. doi:10.1016/j.bbabio.2009.06.004
63. Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, et al. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct (2012) 2012:329635. doi:10.1155/2012/329635
64. Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell (2006) 10:175–6. doi:10.1016/j.ccr.2006.08.015
65. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell (2012) 48:158–67. doi:10.1016/j.molcel.2012.09.025
66. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci (2010) 35:505–13. doi:10.1016/j.tibs.2010.04.002
67. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer (2003) 3:459–65. doi:10.1038/nrc1097
68. Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell (2006) 21:481–93. doi:10.1016/j.molcel.2006.01.012
69. Gough DJ, Corlett A, Schlessinger K, Wegrzyn J, Larner AC, Levy DE. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science (2009) 324:1713–6. doi:10.1126/science.1171721
70. Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res (2012) 22:399–412. doi:10.1038/cr.2011.145
71. Rimessi A, Marchi S, Patergnani S, Pinton P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene (2014) 33:2329–40. doi:10.1038/onc.2013.192
72. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature (2014) 514:628–32. doi:10.1038/nature13611
73. Samudio I, Harmancey R, Fiegl M, Kantarjian H, Konopleva M, Korchin B, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest (2010) 120:142–56. doi:10.1172/JCI38942
74. Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell (2011) 20:674–88. doi:10.1016/j.ccr.2011.10.015
75. Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell (2013) 12:329–41. doi:10.1016/j.stem.2012.12.013
76. Bijur GN, Jope RS. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J Neurochem (2003) 87:1427–35. doi:10.1046/j.1471-4159.2003.02113.x
77. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev (2001) 15:1406–18. doi:10.1101/gad.889901
78. Majewski N, Nogueira V, Bhaskar P, Coy PE, Skeen JE, Gottlob K, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell (2004) 16:819–30. doi:10.1016/j.molcel.2004.11.014
79. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell (2007) 129:1261–74. doi:10.1016/j.cell.2007.06.009
80. Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, et al. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun (2008) 375:501–5. doi:10.1016/j.bbrc.2008.07.153
81. Marchi S, Marinello M, Bononi A, Bonora M, Giorgi C, Rimessi A, et al. Selective modulation of subtype III IP(3)R by Akt regulates ER Ca(2)(+) release and apoptosis. Cell Death Dis (2012) 3:e304. doi:10.1038/cddis.2012.45
82. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell (2008) 133:403–14. doi:10.1016/j.cell.2008.04.013
83. Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, et al. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death Differ (2013) 20:1631–43. doi:10.1038/cdd.2013.77
84. Rabellino A, Carter B, Konstantinidou G, Wu SY, Rimessi A, Byers LA, et al. The SUMO E3-ligase PIAS1 regulates the tumor suppressor PML and its oncogenic counterpart PML-RARA. Cancer Res (2012) 72:2275–84. doi:10.1158/0008-5472.CAN-11-3159
85. Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science (2010) 330:1247–51. doi:10.1126/science.1189157
86. Hedgepeth SC, Garcia MI, Wagner LE II, Rodriguez AM, Chintapalli SV, Snyder RR, et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J Biol Chem (2015) 290:7304–13. doi:10.1074/jbc.M114.611186
87. Moll UM, Wolff S, Speidel D, Deppert W. Transcription-independent pro-apoptotic functions of p53. Curr Opin Cell Biol (2005) 17:631–6. doi:10.1016/j.ceb.2005.09.007
88. Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature (2009) 458:1127–30. doi:10.1038/nature07986
89. Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, et al. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget (2015) 6:1435–45.
90. Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112:1779–84. doi:10.1073/pnas.1410723112
91. Rimessi A, Marchi S, Fotino C, Romagnoli A, Huebner K, Croce CM, et al. Intramitochondrial calcium regulation by the FHIT gene product sensitizes to apoptosis. Proc Natl Acad Sci U S A (2009) 106:12753–8. doi:10.1073/pnas.0906484106
92. Griffin C, Karnik A, McNulty J, Pandey S. Pancratistatin selectively targets cancer cell mitochondria and reduces growth of human colon tumor xenografts. Mol Cancer Ther (2011) 10:57–68. doi:10.1158/1535-7163.MCT-10-0735
93. Bernal SD, Lampidis TJ, Summerhayes IC, Chen LB. Rhodamine-123 selectively reduces clonal growth of carcinoma cells in vitro. Science (1982) 218:1117–9. doi:10.1126/science.7146897
94. Summerhayes IC, Lampidis TJ, Bernal SD, Nadakavukaren JJ, Nadakavukaren KK, Shepherd EL, et al. Unusual retention of rhodamine 123 by mitochondria in muscle and carcinoma cells. Proc Natl Acad Sci U S A (1982) 79:5292–6. doi:10.1073/pnas.79.17.5292
95. Capozzi A, Mantuano E, Matarrese P, Saccomanni G, Manera C, Mattei V, et al. A new 4-phenyl-1,8-naphthyridine derivative affects carcinoma cell proliferation by impairing cell cycle progression and inducing apoptosis. Anticancer Agents Med Chem (2012) 12:653–62. doi:10.2174/187152012800617731
96. Broadley K, Larsen L, Herst PM, Smith RA, Berridge MV, McConnell MJ. The novel phloroglucinol PMT7 kills glycolytic cancer cells by blocking autophagy and sensitizing to nutrient stress. J Cell Biochem (2011) 112:1869–79. doi:10.1002/jcb.23107
97. Mollinedo F, Fernandez M, Hornillos V, Delgado J, Amat-Guerri F, Acuna AU, et al. Involvement of lipid rafts in the localization and dysfunction effect of the antitumor ether phospholipid edelfosine in mitochondria. Cell Death Dis (2011) 2:e158. doi:10.1038/cddis.2011.41
98. Wen S, Zhu D, Huang P. Targeting cancer cell mitochondria as a therapeutic approach. Future Med Chem (2013) 5:53–67. doi:10.4155/fmc.12.190
99. Hou H, Sun H, Lu P, Ge C, Zhang L, Li H, et al. Tunicamycin potentiates cisplatin anticancer efficacy through the DPAGT1/Akt/ABCG2 pathway in mouse Xenograft models of human hepatocellular carcinoma. Mol Cancer Ther (2013) 12:2874–84. doi:10.1158/1535-7163.MCT-13-0201
100. Carracedo A, Gironella M, Lorente M, Garcia S, Guzman M, Velasco G, et al. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res (2006) 66:6748–55. doi:10.1158/0008-5472.CAN-06-0169
101. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ (2012) 19:1880–91. doi:10.1038/cdd.2012.74
102. Shao RG, Shimizu T, Pommier Y. Brefeldin A is a potent inducer of apoptosis in human cancer cells independently of p53. Exp Cell Res (1996) 227:190–6. doi:10.1006/excr.1996.0266
103. Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT. Mitochondrial-mediated dysregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res (2005) 65:3828–36. doi:10.1158/0008-5472.CAN-04-3684
104. Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol (2004) 166:193–203. doi:10.1083/jcb.200309146
105. Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, et al. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Mol Cell (2008) 31:255–65. doi:10.1016/j.molcel.2008.06.014
106. Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science (2011) 334:1129–33. doi:10.1126/science.1206727
107. Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, et al. Induction of Ca(2)+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood (2011) 117:2924–34. doi:10.1182/blood-2010-09-307405
108. Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis (2013) 4:e632. doi:10.1038/cddis.2013.140
109. Yagoda N, Von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature (2007) 447:864–8. doi:10.1038/nature05859
110. Cardaci S, Ciriolo MR. TCA cycle defects and cancer: when metabolism tunes redox state. Int J Cell Biol (2012) 2012:161837. doi:10.1155/2012/161837
111. Penso J, Beitner R. Clotrimazole and bifonazole detach hexokinase from mitochondria of melanoma cells. Eur J Pharmacol (1998) 342:113–7. doi:10.1016/S0014-2999(97)01507-0
112. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene (2006) 25:4777–86. doi:10.1038/sj.onc.1209603
113. Simamura E, Hirai K, Shimada H, Koyama J, Niwa Y, Shimizu S. Furanonaphthoquinones cause apoptosis of cancer cells by inducing the production of reactive oxygen species by the mitochondrial voltage-dependent anion channel. Cancer Biol Ther (2006) 5:1523–9. doi:10.4161/cbt.5.11.3302
114. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med (2014) 20:460–9. doi:10.1016/j.molmed.2014.06.005
Keywords: mitochondrial dysfunction, cancer, Ca2+ signaling, oncogene and oncosuppressor
Citation: Rimessi A, Patergnani S, Bonora M, Wieckowski MR and Pinton P (2015) Mitochondrial Ca2+ remodeling is a prime factor in oncogenic behavior. Front. Oncol. 5:143. doi: 10.3389/fonc.2015.00143
Received: 22 February 2015; Accepted: 11 June 2015;
Published: 25 June 2015
Edited by:
Gavin P. McStay, New York Institute of Technology, USAReviewed by:
Llewelyn Roderick, University of Leuven, BelgiumPatrizia Agostinis, University of Leuven, Belgium
Copyright: © 2015 Rimessi, Patergnani, Bonora, Wieckowski and Pinton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Pinton, Section of Pathology, Oncology and Experimental Biology, Department of Morphology, Surgery and Experimental Medicine, University of Ferrara, Via Borsari 46, Ferrara 44121, Italy,cG5wQHVuaWZlLml0