 Stephen S. Roberts
Stephen S. Roberts Alexander J. Chou
Alexander J. Chou Nai-Kong V. Cheung
Nai-Kong V. Cheung- Department of Pediatrics, Memorial Sloan Kettering Cancer Center, New York, NY, USA
Pediatric sarcomas are a heterogeneous group of malignant tumors of bone and soft tissue origin. Although more than 100 different histologic subtypes have been described, the majority of pediatric cases belong to the Ewing’s family of tumors, rhabdomyosarcoma and osteosarcoma. Most patients that present with localized stage are curable with surgery and/or chemotherapy; however, those with metastatic disease at diagnosis or those who experience a relapse continue to have a very poor prognosis. New therapies for these patients are urgently needed. Immunotherapy is an established treatment modality for both liquid and solid tumors, and in pediatrics, most notably for neuroblastoma and osteosarcoma. In the past, immunomodulatory agents such as interferon, interleukin-2, and liposomal-muramyl tripeptide phosphatidyl-ethanolamine have been tried, with some activity seen in subsets of patients; additionally, various cancer vaccines have been studied with possible benefit. Monoclonal antibody therapies against tumor antigens such as disialoganglioside GD2 or immune checkpoint targets such as CTLA-4 and PD-1 are being actively explored in pediatric sarcomas. Building on the success of adoptive T cell therapy for EBV-related lymphoma, strategies to redirect T cells using chimeric antigen receptors and bispecific antibodies are rapidly evolving with potential for the treatment of sarcomas. This review will focus on recent preclinical and clinical developments in targeted agents for pediatric sarcomas with emphasis on the immunobiology of immune checkpoints, immunoediting, tumor microenvironment, antibody engineering, cell engineering, and tumor vaccines. The future integration of antibody-based and cell-based therapies into an overall treatment strategy of sarcoma will be discussed.
Introduction
Sarcomas are a heterogeneous group of malignant tumors arising from bone or soft tissues. More than 100 different subtypes of sarcoma have been described in adults and pediatrics; the majority of cases in children are rhabdomyosarcoma, Ewing’s family of tumors, osteosarcoma, and the non-rhabdomyosarcoma soft tissue sarcomas. Although these tumors are rare individually, as a group they account for 10–14% of all childhood cancers (1). While most patients who present with localized disease are highly curable with conventional therapies involving surgery and chemoradiotherapy, those who present with metastatic disease or who relapse post-therapy have an extremely poor prognosis, with little to no improvements in survival seen over the past 20 years. Furthermore, current therapies are highly toxic and associated with significant long-term morbidity in survivors; thus, new and effective therapies are urgently needed for these patients.
History of Immunotherapy for Sarcomas
That the immune system might be involved in cancer control was first observed in sarcoma patients when Wilhelm Busch in Germany reported in 1866 on tumor regressions in sarcoma patients who developed erysipelas infections (2). Immunotherapy for the treatment of sarcomas can be traced back at least as far as 1891, when William Coley, a prominent bone surgeon at Memorial Hospital in New York (now Memorial Sloan Kettering Cancer Center), published his report on the use of what came to be known as “Coley’s Toxin” to treat a series of sarcomas of the bone (3, 4). He found that injections with streptococcus organisms (originally live bacteria, later a heat-killed concoction that also included Serratia marcescens) could induce remissions in some patients with otherwise inoperable sarcomas. Though use of his toxins was highly controversial and eventually fell out of favor, they are considered by many to be the precursors of today’s modern anti-cancer immunotherapy (5). Perhaps the best conceptualization of what has become modern immunotherapy came from Paul Ehrlich in the early 1900s with his description of the “magischen kugeln” – the “Magic bullet” – specific medicines fashioned to attack and kill only the diseased cell while sparing the surrounding normal tissues (6). The increased frequency of lymphoid malignancies in patients with immunodeficiencies also suggests that the immune system plays an important role in carcinogenesis (7). In addition, development of sarcomas has been well described in allograft transplant recipients, with a risk more than double that of non-immunosuppressed patients (8).
Immune System in the Non-Malignant State
Our immune system is a complex organization of immune cells and mediators that interact with each other and with other accessory cells to protect against infections; simultaneously, this system must maintain tolerance toward self. The immune system consists of two layers of defense: the innate and adaptive spheres. The innate immune system includes dendritic cells, mast cells, and macrophages, as well as natural killer (NK) cells, neutrophils, basophils, and eosinophils. Innate immune cells serve as the initial defense against foreign antigens. Once activated, macrophages and mast cells release cytokines that engage additional immune cells and initiate an inflammatory response. Dendritic cells serve as antigen-presenting cells, taking in foreign antigens and subsequently presenting them for recognition by adaptive immune cells, thereby recruiting the second sphere of the immune system. NK cells can also interact with dendritic cells, either activating or eliminating them depending on context, thus they too can influence both the innate and adaptive immune systems.
The adaptive immune system includes B-lymphocytes, CD4+ T helper lymphocytes, and CD8+ cytotoxic T lymphocytes (CTLs). This arm of the immune system requires direct activation through antigen presentation by antigen-presenting cells. Upon antigen presentation and activation, antigen-specific T and B cells are generated. Together, the innate and adaptive pathways eliminate pathogens and remove damaged cells (7, 9). Unlike the innate system, the adaptive immune response requires training, but, once established, is antigen specific, has a memory, and can be recalled to rapid action in the future.
Immune Surveillance and Immunoediting
One of the basic principles of cancer immunosurveillance is that cancer cells possess antigens that distinguish them [or set them apart] from non-transformed cells. These so-called tumor “neoantigens” can be recognized by the endogenous immune system and targeted for destruction. These tumor antigens are generally products of mutated genes, abnormally expressed normal genes, or genes coding for viral proteins. Unfortunately, transformed cells, under the selective pressure of the normal host response, are sometimes able to evolve evasive or immune-suppressive mechanisms and thus avoid detection and/or eradication. This concept that the immune system, while protecting against cancer, influences tumor immunogenicity and ultimately tumor escape was proposed as the framework for cancer immunoediting (10). This process can be divided into three phases: elimination, equilibrium, and escape. During the elimination phase both the innate and adaptive immune systems work to identify a developing neoplasm and eliminate it, through various mechanisms including activation of innate immune effector cells such as NK cells, and secretion of interferons (IFNs) and subsequent activation of dendritic cells, which in turn promote adaptive anti-tumor immune responses. However, a subset of cancer cells may develop the ability to survive this elimination phase, and thus the developing neoplasm enters the equilibrium phase. Here, the immune system prevents tumor escape, yet fails to eradicate it completely and thus participates in influencing the immunogenicity of these remaining cells. Finally, in the escape phase, those tumor cells that evolved the ability to evade the immune system during the equilibrium phase progressively proliferate and present as clinically apparent tumors. Mechanisms by which this escape may occur include loss of tumor antigens, down regulation of histocompatibility locus antigens (HLA) from the tumor cell surface; altered tumor microenvironment that is immunosuppressive due to the recruitment of regulatory T cells (Tregs); myeloid-derived suppressor cells, tumor-associated M2 macrophages, and others (11–13); upregulation of inhibitory receptors (e.g., PD-1) on T cells; or upregulation of inhibitory ligands (e.g., PD-L1 or B7-H3) on stromal cells or tumor cells.
Immunomodulatory Agents
A variety of immunomodulatory agents have been investigated for the treatment of sarcomas, including cytokines such as interleukin-2 (IL-2) and IFN. The majority of sarcoma studies have been conducted in adult patients with advances fueling interest in the pediatric patient population.
Cytokines
Stimulation of the immune system has been attempted using various cytokines. Cytokines are involved in a wide array of immune functions including modulation of antigen presentation and T cell activation (14, 15). The list of cytokines continues to expand (15). Although the most widely studied clinically are IFN and IL-2, several other cytokines are also moving into the clinic.
Interleukin 2
Interleukin 2 stimulates T cells proliferation, induces generation of CTLs, and facilitates the maintenance of NK cells (16–18). IL-2 is FDA approved for the treatment of metastatic renal cell carcinoma and melanoma, and responses to IL-2 have been reported in several other cancers including lung and breast cancers (19, 20). In pediatrics, IL-2 has been used most notably for the treatment of high-risk neuroblastoma in combination with an anti-GD2 monoclonal antibody (mAb) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (21). A study of high-dose IL-2 in relapsed pediatric patients included four patients with osteosarcoma and two patients with Ewing sarcoma. Two of the four osteosarcoma patients had complete responses, while the other two and both Ewing sarcoma patients had progressive disease (20). However, use of high-dose infusional IL-2 is greatly hampered by significant toxicity, including capillary leak syndrome; continued use in pediatric sarcoma as a single agent seems unlikely. Several studies are ongoing in pediatrics combining IL-2 given in a variety of different routes and dosages with antibody therapy, vaccines, and adoptive cell therapy.
Interferon
Interferons are a complex family of molecules that bind to IFN receptors; IFNα and IFNβ activate type I receptors, while IFNγ activates type II receptors (15, 22). Both IFNα and IFNβ activate immune cells and increase antigen presentation to T cells. INFα is approved for use in melanoma and has also been studied in sarcomas. Most recently, the large EURAMOS study reported three-year follow-up data on 715 pediatric and adult osteosarcoma patients up to 40 years of age randomized to postoperative chemotherapy ± IFN; there was no survival benefit from IFNα when added to standard three-drug chemotherapy in osteosarcoma patients (74% chemotherapy alone vs. 77% chemotherapy + IFNα; EFS, p = 0.21) (23). Further development of IFN as a single agent in pediatric sarcoma seems unlikely; its role in pediatric sarcoma immunotherapy as an adjuvant combined with other immunotherapies such as adoptive cell therapy to increase antigen presentation remains to be defined.
Interleukin 15
Interleukin 15 (IL-15) (24, 25) is a 14–15 kDa glycoprotein that binds to a heterotrimeric receptor that shares the IL-2R/IL-15Rβ (CD122) and the common gamma (γc) chain (CD132) with the IL-2 receptor (26), as well as a unique α subunit (IL-15Rα) that confers receptor specificity. However, unlike IL-2, IL-15 is not required for the maintenance of Tregs (27); it does not induce activation-induced cell death (AICD) of CD8+ effector T cells (28); is required for the differentiation of NK, effector CD8+ and memory phenotype CD8+ T cells; and does not cause capillary leak syndrome (29). IL-15Rα binds to IL-15 with high affinity (Kd < 10–11 M) and retains IL-15 on the cell surface. IL-15Rα trans-presents IL-15 to IL-2R/IL-15Rβ-γc on neighboring NK and T cells through immunological synapses (30, 31). IL-15 has diverse immunologic effects (26). It stimulates the proliferation of activated CD4−CD8−, CD4+CD8+) CD4+, CD8+ T cells, induces cytotoxic CTLs, and stimulates the generation, proliferation, and activation of NK cells. Though not essential for the generation of memory CD8+ T cells, IL-15 is required for their homeostatic proliferation over long periods of time (32). IL-15 protects neutrophils from apoptosis, modulates phagocytosis, stimulates mast cell growth, induces B cell proliferation and differentiation partially independent of T cell help, and increases their immunoglobulin secretion, while stimulating secondary cytokine release from macrophages and maturing dendritic cells. When given as the IL15/IL15Rα complex, it is more effective and should be less toxic than the soluble IL15 (33–35). Several preclinical studies have shown that IL-15 may potentiate anti-sarcoma immunotherapy in Ewing and osteosarcoma models (36–38). A clinical trial combining recombinant human IL15 with NK cells for relapsed and refractory pediatric solid tumors, including sarcomas, is currently underway at the U.S. National Cancer Institute (NCT01875601). Although no clinical trial of IL-15 has been conducted specifically for sarcomas, this cytokine will likely play a major role in future immunotherapy strategies.
Liposomal-Muramyl Tripeptide Phosphatidyl-Ethanolamine
The immune modulator liposomal-muramyl tripeptide phosphatidyl-ethanolamine (L-MTP or mifamurtide) has been extensively studied, primarily in osteosarcoma. This compound is a non-specific modulator of innate immunity and is a synthetic analog of muramyl dipeptide derived from bacterial cell walls. It activates monocytes and macrophages leading to an increase of a wide variety of immunomodulatory molecules including: tumor necrosis factor-alpha (TNF-a), interleukin (IL)-1, IL-6, IL-8, IL-12, nitric oxide, prostaglandin E2, lymphocyte function-associated antigen 1 (LFA-1), and intercellular adhesion molecule 1 (ICAM1) (39). Preclinical studies suggested that this inflammatory response triggered by L-MTP could potentially eliminate minimal residual disease. A small study conducted by the EORTC Soft Tissue and Bone Sarcoma Group in the 1990s treated 20 adult patients with soft tissue sarcomas with MTP; there were no responses in that study (40). The largest clinical experience with combination chemotherapy and L-MTP derives from the Intergroup (INT-) 0133 osteosarcoma study. This prospective, double randomization, phase III trial tested first the utility of adding ifosfamide to the standard three-drug chemotherapy regimen (doxorubicin, cisplatin, and high-dose methotrexate); and second the impact on survival with the addition of L-MTP to either assigned chemotherapy arm. No difference in survival was found for patients who received ifosfamide in addition to the standard three-drug chemotherapy. The study did suggest that L-MTP had a beneficial impact on survival, improving the 5-year overall survival rate from 70 to 78% (p = 0.03) (41). However, when the 91 patients who had metastatic disease were analyzed separately, the difference in survival between those who did versus those who did not receive L-MTP, though suggesting improvement, did not reach statistical significance. The overall survival at 5 years was 53% for those randomized to receive L-MTP versus 40% for those who did not (p = 0.27) (42). Based, in part, on the updated results of the non-metastatic cohort of INT-0133, the European Medicines Agency granted L-MTP an indication for the treatment of non-metastatic osteosarcoma in 2009; the American Food and Drug Administration (FDA) did not. L-MTP is also approved for use in Turkey, Mexico, and Israel.
Antibody-Based Immunotherapy
Monoclonal Antibodies
Unmodified antibodies specific for tumor-associated surface antigens can engage tumor cells while activating innate immune effector cells, primarily macrophages and NK cells via their Fc receptors (FcγR). Once activated, the effector cell releases cytotoxic granules to kill the target cell, a process known as antibody-dependent cellular cytotoxicity (ADCC). It is important to note that T cells do not possess FcγR and have no affinity for conventional antibodies, and hence cannot be activated by these tumor selective antibodies.
Many mAbs have been developed for various cancer types. While there have been notable successes (for example, anti-CD20 for hematologic malignancies, anti-human epidermal growth factor receptor 2 (HER2) for breast cancer, and anti-GD2 for neuroblastoma), most mAbs have failed to improve outcomes despite their initial promise, especially in pediatric sarcomas.
Approximately 50% of osteosarcomas overexpress HER2, and HER2 expression was shown to correlate with a poorer prognosis (43); a phase II study was conducted by the Children’s Oncology Group (COG) to evaluate if the addition of trastuzumab (anti-HER2, Herceptin) to standard chemotherapy would improve survival in metastatic osteosarcoma patients. Ninety-six patients were enrolled, and 41 were found to have HER2 overexpression. Unfortunately, no significant difference in survival was seen in patients who received trastuzumab + chemotherapy compared to those who received chemotherapy alone {EFS of 32% in both arms, OS of 50% for chemotherapy alone compared to 59% for chemotherapy + trastuzumab, [p = 0.54 for EFS; p = 0.58 for OS] (44)}.
Instead of binding directly to tumors, antibodies can neutralize growth factors (e.g., insulin-like growth factor 1 (IGF1) or IGF2) or their receptors (e.g., IGF-1R, -A12). A large body of preclinical and early clinical data suggested that IGF1 and 2 might play an important role in the initiation and progression of a variety of cancers, including pediatric sarcomas (45–47). Several phase I and II studies were conducted evaluating anti-IGF1 mAbs in relapsed and refractory solid tumors including sarcomas, the largest being a phase II study by the COG that enrolled 116 patients, including 20 with rhabdomyosarcoma, 11 with osteosarcoma, and 10 with synovial sarcoma; there were no objective responses in any of the sarcoma patients (48, 49). Finally, a randomized phase II study of standard chemotherapy ± the anti-IGF-1R mAb ganitumab is ongoing within COG for Ewing sarcoma (NCT02306161). However, this agent failed to show improved outcomes in a large randomized phase III trial of adult pancreatic cancer patients (50) and the manufacturer has announced that they will not be pursuing development of this agent. Thus, regardless of the results of the ongoing trial, its future for pediatric sarcoma is unclear.
Although both IGF-1 and IGF-2 activate IGF-1R, the latter shares a similar tetrameric α2β2 structure with insulin receptor (IR). The IR can be expressed in two isoforms (IR-A and IR-B). IR-A binds to IGF-2 with the same affinity as it binds to insulin. In addition, insulin and IGF-1 receptor subunits can form hybrid heterodimeric receptors (51). Antibodies against IGF-1R only partially inhibit IR-A activity by disrupting the IR-A/IGF-1R hybrid, but completely fail to inhibit IR-A homodimers. Failure of IGF-1R inhibition results from two compensatory mechanisms: (1) IGF-2 is increased during treatment with IGF-1R mAb (52) which signals through IR-A, which is known to promote cancer survival (53). (2) Compensatory activation of the epidermal growth factor receptor (EGFR) allowing the cancer to continue to progress despite blockade of the IGF pathway (54). One novel approach to overcome these limitations is to reduce the serum and tissue levels of the IGF ligands, using neutralizing mAbs specific for both IGF-1 and IGF-2. By removing IGF-2, the escape mechanism of IGF-2-mediated IR-A activation can be aborted, suggesting that newer mAbs that target both IGF-1 and IGF-2 may have more success than the first-generation mAbs tested (55, 56).
Several trials of mAbs against the EGFR and the VEGFR (57) alone and in combination with chemotherapy have been conducted in children and young adults with sarcomas. The COG conducted a randomized trial of bevacizumab (anti-VEGFR) combined with vincristine, topotecan and cyclophosphamide in patients with recurrent Ewing sarcoma, as well as a randomized trial of bevacizumab and temsirolimus in combination with vinorelbine and cyclophosphamide in recurrent/refractory rhabdomyosarcoma patients. In the rhabdomyosarcoma trial, the bevacizumab arm was significantly worse than the temsirolimus arm and the study was stopped early (58); results for the Ewing sarcoma trial have not yet been published. Despite preclinical rationale for these targets (59–61), overall, these studies have not shown many significant responses in sarcomas, though some studies are ongoing.
A phase I trial of the anti-tumor necrosis factor-related apoptosis-inducing ligand receptor 2 (TRAIL-2) mAb lexatumumab was conducted by the U.S. National Cancer Institute (NCI) in pediatric solid tumors. This study enrolled 24 patients, including 21 with various sarcomas. No objective responses were seen and this mAb is no longer under clinical development (62).
Given the success of anti-GD2 mAb therapy in neuroblastoma (21, 63) and the expression of GD2 by many sarcomas (64, 65), studies exploring the use of these mAbs in sarcomas, particularly in osteosarcoma, are underway. Current trials include the anti-GD2 mAbs humanized3F8 (NCT01419834 and NCT01662804) and hu14.18K322A (NCT00743496).
Engineered Antibodies Including Bispecific Antibodies
Bispecific antibodies are engineered antibodies linking a tumor antigen recognition domain to a second domain that activates a receptor on immune effector cells, typically T cells (Figure 1). The anti-CD19/anti-CD3 bispecific antibody blinatumomab was approved by the FDA for the treatment of precursor B cell acute lymphoblastic leukemia in 2014, making it the first in its class to be approved in the US. Recently published preclinical data of an anti-GD2 T cell retargeting bispecific antibody showed excellent in vivo activity against GD2 expressing neuroblastomas and melanomas (66). Currently, there are limited clinical data on bispecific antibodies in pediatric sarcomas; there is one study that recently began enrolling OS patients (Activated T Cells Armed with GD2 Bispecific Antibody in Children and Young Adults With Neuroblastoma and Osteosarcoma, NCT02173093).
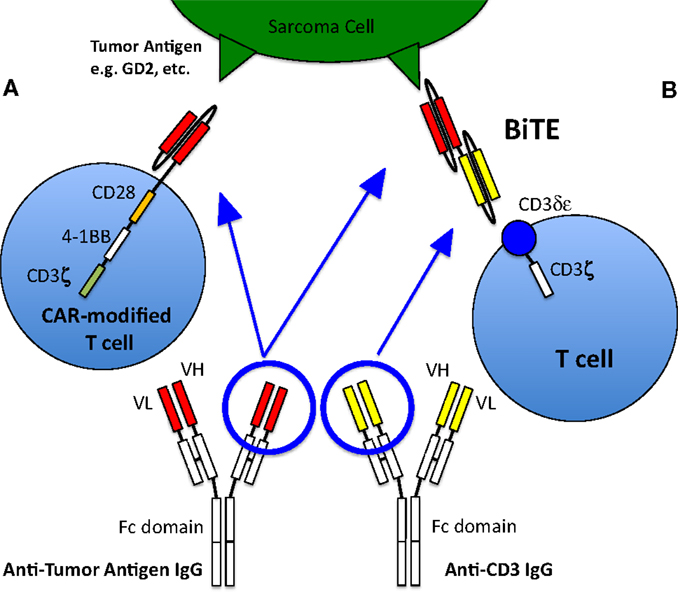
Figure 1. T cell activation and recruitment to tumor cells. Normally, T cells will only target cells via antigenic sequences presented to the T cell receptor via MHC. However, this rarely occurs, especially in pediatric sarcomas, thus native T cells generally are not active in killing tumor cells and other strategies to recruit T cells to the tumor are therefore required. (A) T cell with a chimeric antigen receptor to a sarcoma tumor-associated antigen is able to recognize the antigen and activate the T cell, allowing it to kill the targeted cell. The CAR-T cell depicted here is a so-called third generation CAR, because it contains three co-stimulatory domains (CD28, 4-1BB, and CD3ζ). (B) Bispecific antibody binding to a sarcoma tumor-associated antigen as well as to the CD3 receptor of a T cell, thus activating the T cell and allowing tumor cell killing. Figure adapted from Suzuki et al. (67).
Immunologic Checkpoint Blockade or Inhibitors
Recently, there has been much excitement about the potential of the immune checkpoint inhibitors in solid tumors including pediatric sarcomas following their clinical successes and approvals for treatment of metastatic melanoma and metastatic squamous non-small cell lung cancer.
CTLA-4 Blockade
Ipilimumab is a human IgG4 monoclonal antibody that blocks the anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and was the first of the new generation of checkpoint inhibitors to gain FDA approval (68). CTLA-4 is a member of the immunoglobulin superfamily; after T cell activation, CTLA-4 is expressed on the plasma membrane of cells where it acts to inhibit T cell function through a variety of mechanisms, allowing tumor cells to escape immune surveillance (69, 70). The experience of ipilimumab in pediatric patients is limited; GI toxicity was the major concern. A small phase II study in adults with synovial sarcoma had no clinical or immunological responses (71).
PD-1 Blockade
Antibodies targeting the programed cell death protein 1 pathway (PD-1/PD-L1) (nivolumab, pembrolizumab) function in a similar manner to ipilimumab by removing the brakes on T cells which then can perform active anti-tumor immune surveillance (69, 70). Preclinical studies have demonstrated expression of PD-1L in OS and suggest that high expression levels may correlate with worse clinical outcomes (72); In vivo studies using murine sarcoma models with anti-CTLA-4 antibodies have also shown promise for these agents (73). Currently, however, these agents have limited pediatric clinical data available; several trials with these agents for relapsed or refractory pediatric solid tumors are currently ongoing.
Despite the overall successes of checkpoint inhibitors, only subsets of patients with melanoma, lung cancer, ovarian cancer, NHL, and Hodgkin lymphoma have responded. Two important studies have examined the tumors of responders versus non-responders, one in melanoma and one in non-small-cell lung cancer (74–76). In both cases, treatment efficacy was associated with a higher number of mutations in the tumors. In melanoma patients treated with ipilimumab, the investigators carefully examined the tumors of those who responded versus those who did not, and found that the responders had tumors with higher mutation rates and tumor antigens and in particular, those whose tumor neoantigens shared tetrapeptide sequences with viral antigens were most likely to be responders to checkpoint inhibition (75). To improve on the quality of response to immune checkpoint blockade, CTLA-4 and PD-1/PD-L1 antibodies are being tested in combination or when added to other anti-cancer agents such as chemotherapy, targeted therapy, radiotherapy, and other immunotherapy (19, 69, 77). Currently, the COG is conducting a phase I/II study (NCT02304458) of nivolimab alone or in combination with ipilimumab for relapsed and refractory solid tumors including sarcomas.
Although there is much excitement currently surrounding these new agents, caution seems warranted in pediatric sarcomas. In contrast to melanoma and lung cancer, pediatric cancers in general and pediatric sarcomas in particular have an extremely low rate of recurrent mutations (<1 mutation per Mb for pediatric cancers compared to 15 per Mb for melanomas) (78, 79). Furthermore, many sarcomas do not express major histocompatibility complex (MHC) which is required for both the afferent and efferent arms of T cell response (80). Taken together, it seems probable that checkpoint inhibitors may have less efficacy in pediatric sarcomas (especially as single agents) than in melanoma and lung cancer; careful consideration of ideal clinical trial design using these agents will be critical for defining their potential role in the immunotherapy of pediatric sarcomas.
Tumor Vaccines
Vaccines directed against specific tumor antigens were some of the earliest targeted immunotherapies tested. The aim of tumor vaccines is to induce an anti-tumor response through exposure to tumor antigens. Of the high value tumor targets among the 75 candidates derived from the NCI consensus panel, only a few are directly adaptable to sarcoma (81). The most notable are the gangliosides GD2 and GD3, polysialic acid, and translocation breakpoints. In animal models and human clinical trials (9, 82), vaccines have shown efficacy in preventing tumor development or delaying progression, but have generally failed to mediate regressions of established tumors. Single-arm trials have investigated vaccines targeting whole cells, lysates, proteins, and peptides in both adult and pediatric patients with sarcomas. Results from most studies in sarcomas (adult and pediatric) have been disappointing, though some have shown potential benefit with either laboratory evidence of the development of an immune response, or prolonged stable disease or disease-free intervals (83–88). Several additional pediatric sarcoma studies remain ongoing (NCT01241162, NCT01803152, NCT01061840). Promising results of a recent phase I study of a bivalent GD2-GD3 gangliosides vaccine in combination with β-glucan in neuroblastoma patients in second remission (89) suggest that vaccines for sarcomas may be beneficial if given in the setting of minimal residual disease.
Adoptive Cell Therapy
Adoptive cell therapy is the term coined to describe the concept of giving a patient immune cells with cytolytic properties in sufficient numbers to cause an anti-tumor response. There are various strategies to accomplish this, including use of ex vivo expanded autologous cells and infusion of donor-derived allogeneic immune effector cells.
Natural Killer Cells
Natural Killer cells are lymphocytes of the innate immune system with both cytotoxic and regulatory functions and are important mediators of immune responses against infections and cancer. Unlike T and B cells, NK cells recognize their targets without prior sensitization and generally do not have the same memory system [with some exceptions (90)] as T or B cells. NK cells are activated through various receptors that recognize proteins that are upregulated by cell stress or are foreign. In turn, NK cells are negatively regulated by inhibitory receptors that primarily bind HLA as a means of preventing self-recognition, thus preventing autoimmunity. NK cell target cytotoxicity is triggered when the overall balance between the various activating and inhibitory signals is weighted toward activation (91). NK cells were initially identified through their ability to kill tumor cells (92); the anti-tumor actions of NK cells have subsequently been documented in many human and animal models. NK cells are neither HLA-restricted nor do they require activation via the adaptive immune system (93). These facts plus their ability to target and kill a wide variety of tumors has led to strong interest in their therapeutic potential (94). The first application of NK-cell-enriched cellular products to treat cancer was performed at the NCI using autologous cells in 1980 (95). Subsequently, clinical trials, primarily in acute myeloid leukemia, confirmed that haploidentical donor-derived NK cells can be expanded in vivo and can induce remissions (96). Preclinical data suggest that NK cell strategies may be of benefit in pediatric sarcomas (97). Specifically, various studies have shown that Ewing sarcoma, osteosarcoma, and rhabdomyosarcoma cell lines, including highly chemoresistant lines, are all sensitive to NK cell killing and that cytokine activation greatly enhances this killing ability both in vitro and in in vivo models (36, 38, 98). Several current NK-cell-based studies are open to pediatric sarcomas and apply various strategies, including post-allogeneic transplant and ex vivo expansion and/or cytokine stimulation; however, no results have yet been reported from these trials.
Cytotoxic T Lymphocytes
Cytotoxic T Lymphocytes are highly efficient at targeting and killing specific cells; thus, there has long been much interest in harnessing this ability for cancer immunotherapy. De novo T cells are generally of low frequency and incapacitated by the tumor microenvironment. Initial efforts to use T cells for cancer therapy involved ex vivo expansion of the so-called tumor infiltrating lymphocytes (TILs) freed from excised tumors. This approach is limited, however, by the fact that they cannot be reliably extracted or be expanded to sufficient numbers from most tumors. To date, there are no studies in pediatric cancer patients (9). Despite their limitations, TILs are an important proof-of-concept of the potential value of T-cell-based immunotherapy as they were the first immunotherapy to induce regressions of bulky tumors (99). To overcome these limitations, polyclonal T cells can be genetically modified to express T cell receptors (TCRs) that recognize tumor peptide antigens in the context of MHC. These transgenic TCRs function like their natural counterparts, but remain restricted by MHC, thus limiting the use of these cells to the patient’s specific individual HLA alleles. As approximately 50% of the Caucasian population in the U.S. express HLA A*0201, many studies have focused on associated antigens, particularly the cancer testis antigens. Among these, NY-ESO-1 is one of the most studied with expression found in 70–80% of synovial sarcomas, but only sporadically in other sarcomas (100, 101); in a pilot feasibility study, four of six patients with synovial sarcoma had an objective response (101, 102). Further studies using NY-ESO-1 CTLs in synovial sarcoma are ongoing (NCT01343043).
Chimeric Antigen Receptor-Modified T Cells
Because T cells do not carry Fcγ-receptors, these potent effector cells cannot recognize tumor-bound antibodies, and have therefore traditionally not been recruited by such antibodies to tumor sites. Furthermore, T cells need to recognize tumor peptides in the context of their own MHC antigens to be effective killers. However, many tumors down regulate or lose their HLA, or even tumor peptides, making them transparent to even the primed T cells. To overcome these issues of HLA and to broaden the selection of targets (e.g., to carbohydrates or lipids), chimeric antigen receptors (CARs) can be engineered into T cells. These receptors are not classic TCRs, but derived from conventional antibodies specific for any target. A chimeric molecule consisting of an antibody in the form of single chain Fv (scFv) as the ectodomain, and T cell signaling machinery as the intracellular domain, forms this artificial receptor through which T cells are activated when they come into contact with the specific antigen, without the necessity of MHC. These CARs are inserted into T cells using viral vectors, DNA transposons, or RNA transfection. In the early versions (so-called First generation) of CAR-modified T cells (CAR T cells), signaling was done through a single activation domain (either the CD3-ζ chain or FcϵRIγ). Second- and third-generation CAR T cells contain one or two additional co-stimulatory signaling domains such as CD28, 4-1BB, and OX40 (67) (Figure 1). The first-generation CAR T cells did not show significant activity in clinical trials presumably because many tumor cells lack co-stimulatory ligands (103), and because of poor persistence of the T cells, although a phase I study of anti-GD2 CAR T cells in relapsed neuroblastoma patients saw some objective clinical responses including complete remission in three patients (104, 105). Second-generation CAR T cells have shown improvements in T cell proliferation and survival (106) and have shown promising results in hematologic malignancies (107). Several studies with CAR T cells are underway that include pediatric sarcoma patients. Two of the open trials target HER2 expressing sarcomas (NCT00902044, NCT00889954), while two more target GD2 expression (NCT01953900, NCT02107963); it remains to be seen whether similar successes seen in hematologic malignancies can be achieved in solid tumors. The death of a patient receiving third-generation anti-HER2 CAR T cells has raised concerns regarding the safety of highly activatable T cells even when the expression of the antigen in normal tissues was low (NCT00924287) (108).
Challenges
Toxicity
In general, although immunotherapy may have less long-term toxicity than chemotherapy or radiation therapy, which is particular appealing for pediatric cancer, major short-term toxicities can be daunting. These include immediate infusion-related allergic reactions with mAbs, and autoimmune reactions to the checkpoint inhibitors, some of which were life threatening (109). In a recently completed phase I study of ipilimumab (NCT01445379) in pediatric patients with refractory solid tumors including sarcomas, no objective responses were seen but significant autoimmune toxicity was observed, with up to 50% of patients experiencing symptoms (Personal communication, Dr. L. Wexler, 2015); however, no pediatric safety data for these agents are yet published. In adults, enterocolitis, hepatitis, and dermatitis were the most commonly seen toxicities, but autoimmune-related toxicities due to unregulated T cell activity have been reported in nearly every organ system (109). Adoptive cell transfer also carries the real potential for serious adverse events. T cell therapy is highly potent such that even normal tissues with low target antigen expression can become innocent bystanders. These unintended and unexpected toxicities to critical organs can be life threatening (110) and have limited the choice of certain targets for redirected T-cell-based therapy (111). Additionally, T cells have been associated with severe, sometimes fatal, cytokine release. Cytokine release syndrome (CRS) occurs when extremely high levels of immune cells are activated thereby stimulating release of large amounts of inflammatory cytokines, leading to organ dysfunction and death. CRS is particularly seen with second- and third-generation CARs as well as bispecific antibodies (112), but can occur after antibody infusion as well as with other adoptive lymphocyte therapies. Corticosteroids are the mainstay of treatment, while anti-IL6R antibody can also be helpful (113).
Target Selection
Perhaps the most critical first step in designing cancer immunotherapy is identifying appropriate immunologic targets. A good immunotherapy target must be highly expressed on tumor tissues but not on normal tissues. Ideally, a good target will play a role in the underlying oncogenesis of the tumor, though this is not always required. Targets that meet these attributes are rare (81). An alternative approach has been to target markers that are highly expressed on cancers and expressed in the so-called non-vital tissues, such that targeting and loss of these normal cells are tolerated by the patient. Monoclonal antibody targeting of CD20, and CAR T cells and bispecific antibodies targeting CD19 are examples of this approach. Adding further difficulty to target selection is that they by necessity must be present on the surface of the cell for immune recognition, which limits the potential target list. In fact, of the 75 NCI consensus high value targets, two-thirds are internal antigens (81). The only way to target these internal antigens is through their peptides presented on the HLA; hence the description of such antibodies as TCR like. Less than 100 publications have been published on the discovery of such antibodies, but the best characterized are those against the RMFPNAPYL peptide of the Wilm’s tumor-1 (WT1) antigen presented on HLA0201 (114). However, this approach is limited by the restriction to specific HLA subtypes. Most pediatric sarcomas lack HLA expression (80), and among those that have it, only individuals with the specific restricted subtype would be sensitive to the immunotherapy. Efforts to mine gene expression databases for potential new antibody targets are promising but still in early stages; validation of these mRNA level exploratory analyses at the protein level will be critical (115, 116). Tables 1 and 2 list some of the pediatric sarcoma-specific targets, both MHC non-restricted (Table 1), and MHC restricted (Table 2), currently in preclinical and/or clinical development.
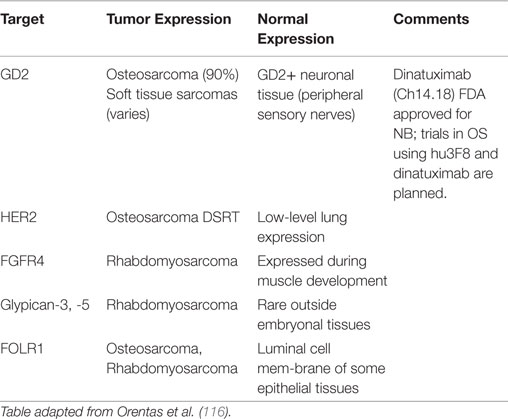
Table 1. Cell surface targets for MHC non-restricted immunotherapy of pediatric sarcomas.

Table 2. MHC-restricted immunotherapy targets for pediatric sarcomas.
Future Directions
Sarcoma immunotherapy remains in its infancy. To date, while we have not seen the successes seen in other malignancies, there are glimpses of activity which suggest that immunotherapy could be an effective treatment modality. However, to fully realize that potential we believe that the following four areas must be carefully considered:
Target Discovery and Validation
Given the narrow mutation landscape in sarcomas, and especially so among those with translocations, neoantigens derived from gene mutations are predicted to be rare. Translocation fusion sequences have remained difficult to target with T cells, or to be used as vaccines. Without neoantigens, even checkpoint blockades used at recommended dosage levels might not be effective. By default, differentiation antigens and tissue antigens deserve to be more carefully explored. These include the gangliosides GD2 (124) and GD3 (65), ROR2 (125), HER2 (126, 127), B7-H3 (128), CSPG4 (129, 130), polysialic acid (131), and glypican 3 (132). All of these antigens have established antibodies ready for construction of CAR T cells or bispecific antibodies. Importantly, most of these antibodies have already been tested in humans with acceptable toxicities. Considerations should also be given to novel engineered forms such as bispecific antibodies to retarget T cells (66) or bispecific antibodies for multistep targeting to greatly improve therapeutic index (133). Given the early glimpses of response to IGF1R antibodies and a better understanding as to why tumors escape, the new generation of dual-specific antibodies for IGF1 and IGF2 should be considered (56, 134).
Careful Patient Selection
The majority of clinical trials to date have shown that immunotherapy is generally not effective against large, bulky disease. Thus, it is imperative that the proper patient population is selected for clinical trials moving forward. For example, we are developing a phase II anti-GD2 immunotherapy protocol for osteosarcoma patients in second or greater remission, with the goal of targeting pulmonary minimal residual disease. This is based on our experience in OS patients treated on our phase I protocol where we found that patients with visible metastatic lesions progressed rapidly while those with minimal residual disease have shown increased time to progression compared to historical controls. It would appear that the clinical efficacy of immunotherapy for pediatric sarcoma can best be tested in clinical trials designed to treat patients after their overt disease burden has been reduced as much as possible.
Development of Combined Modality Regimens
To date, the majority of studies using single immunotherapy modalities have not demonstrated significant activity in solid tumors in general and in pediatric sarcomas in particular. However, rational combinations of new immunotherapies are being developed and will need to be carefully explored. Antibodies combined with immunomodulatory agents are the most mature of these combinatorial approaches. Anti-GD2 mAbs combined with GM-CSF or GM-CSF and IL-2 are effective against neuroblastoma (21, 63) with studies planned in osteosarcoma. While checkpoint inhibitors, for reasons described above, are unlikely to be of significant benefit when used alone in pediatric sarcomas, their combination with adoptive cell therapy or bispecific antibodies has the potential to enhance the efficacy of these T cell-based strategies. Additionally, preclinical studies suggest that prior radiotherapy can induce tumor neoantigen expression and increased effectiveness of checkpoint blockade, echoing the abscopal effect in the clinic (135). Studies exploring this strategy in adults are underway and may be warranted in children. T cells could also be combined with NK cells: MHC down regulation by the tumor cells as a means of escape from T cell killing should render these cells more susceptible to NK cell killing, which does not require MHC, but is instead inhibited by high MHC expression (91).
Tolerance of Increased Toxicity
This last point is perhaps the most controversial. However, historical precedent suggests that learning to manage toxicities associated with therapies can allow otherwise effective treatments to be developed. Anti-GD2 immunotherapy is associated with significant infusional toxicities including severe pain; this pain side effect was completely unexpected when these mAbs were first used (136). Fortunately, rather than halting the development of these antibody treatments, ways to overcome the toxicities were developed and as a result, anti-GD2 immunotherapy is now proven effective in neuroblastoma and is in active trials in sarcoma patients. Similarly, it seems likely that newer immunotherapy treatments, especially combination therapies as suggested above, will have both predictable, as well as unexpected, and potentially severe side effects. However, an unwillingness to carefully explore and manage novel toxicities may limit the adoption of some potentially beneficial treatments. With checkpoint blockade, the autoimmune toxicity seen shows that children do, in fact, have autoreactive T cells that will react with self if the “brakes” are sufficiently released. Since many tumors overexpress normal self antigens, it is plausible that “releasing the brakes” enough (by combining ipilimumab with nivolumab while pushing the dose of both) could allow an autoreactive T cell to target a protein on the tumor that would otherwise be tolerated by the immune system. The currently approved dose of ipilimumab for patients with melanoma, however, achieves the target trough concentration of 20 mcg/mL, the level at which ipilimumab attains maximum CTLA-4 blockade, in only 30% of patients (68), suggesting that increasing the dose could yield improved clinical benefit, if toxicities can be managed. Several clinical trials testing this hypothesis in adults are underway. Similarly, combination therapy with adoptive T cells and checkpoint blockade could have significantly increased toxicity, especially for on-target, off-tumor effects, such that appropriate target selection and clinical trial design to minimize these risks are critical.
Conclusion
Pediatric cancer immunotherapy continues to advance; we believe these advances will improve outcomes in patients who have not benefited from conventional therapy alone. Late toxicities remain a major challenge for those patients who underwent life saving chemotherapy and radiation therapy. Immunotherapy offers an opportunity to consolidate remission while reducing genotoxic therapy. We are cautiously optimistic that immunotherapy will improve not just survival but also the quality of life in children with sarcomas.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Pizzo PA, Poplack DG. Principles and Practice of Pediatric Oncology. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins Health (2011).
3. Coley WB II. Contribution to the knowledge of sarcoma. Ann Surg (1891) 14(3):199–220. doi: 10.1097/00000658-189112000-00015
4. McCarthy EF. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop J (2006) 26:154–8.
5. Society AC. Coleys Toxins (2008). Available from: http://www.cancer.org/treatment/treatmentsandsideeffects/complementaryandalternativemedicine/pharmacologicalandbiologicaltreatment/coley-toxins%3E
6. Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer (2008) 8(6):473–80. doi:10.1038/nrc2394
7. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer (2006) 6(1):24–37. doi:10.1038/nrc1782
8. Penn I. Sarcomas in organ allograft recipients. Transplantation (1995) 60(12):1485–91. doi:10.1097/00007890-199560120-00020
9. Mackall CL, Merchant MS, Fry TJ. Immune-based therapies for childhood cancer. Nat Rev Clin Oncol (2014) 11(12):693–703. doi:10.1038/nrclinonc.2014.177
10. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science (2011) 331(6024):1565–70. doi:10.1126/science.1203486
11. Diaz-Montero CM, Finke J, Montero AJ. Myeloid-derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Semin Oncol (2014) 41(2):174–84. doi:10.1053/j.seminoncol.2014.02.003
12. Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front Immunol (2014) 5:489. doi:10.3389/fimmu.2014.00489
13. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol (2014) 27:1–7. doi:10.1016/j.coi.2013.12.005
14. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol (2014) 192(12):5451–8. doi:10.4049/jimmunol.1490019
15. Margolin KLM, Kaufman HL. Cytokines in the treatment of cancer. In: Curiel TJ, editor. Cancer Immunotherapy: Paradigms, Practice, and Promise. New York, NY: Springer (2012). p. 173–210.
16. Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol (2013) 14(6):e218–28. doi:10.1016/S1470-2045(12)70582-X
17. Nelson BH. IL-2, regulatory T cells, and tolerance. J Immunol (2004) 172(7):3983–8. doi:10.4049/jimmunol.172.7.3983
18. Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer Immunol Res (2015) 3(3):219–27. doi:10.1158/2326-6066.CIR-15-0009
19. D’Angelo SP, Tap WD, Schwartz GK, Carvajal RD. Sarcoma immunotherapy: past approaches and future directions. Sarcoma (2014) 2014:391967. doi:10.1155/2014/391967
20. Schwinger W, Klass V, Benesch M, Lackner H, Dornbusch HJ, Sovinz P, et al. Feasibility of high-dose interleukin-2 in heavily pretreated pediatric cancer patients. Ann Oncol (2005) 16(7):1199–206. doi:10.1093/annonc/mdi226
21. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med (2010) 363(14):1324–34. doi:10.1056/NEJMoa0911123
22. Pestka S. The interferons: 50 years after their discovery, there is much more to learn. J Biol Chem (2007) 282(28):20047–51. doi:10.1074/jbc.R700004200
23. Bielack SS, Smeland S, Whelan JS, Marina N, Jovic G, Hook JM, et al. MAP plus maintenance pegylated interferon α-2b (MAPIfn) versus MAP alone in patients with resectable high-grade osteosarcoma and good histologic response to preoperative MAP: first results of the EURAMOS-1 “good response” randomization. J Clin Oncol (2013) 31:LBA10504. doi:10.1200/JCO.2014.60.0734
24. Burton JD, Bamford RN, Peters C, Grant AJ, Kurys G, Goldman CK, et al. A lymphokine, provisionally designated interleukin T and produced by a human adult T-cell leukemia line, stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc Natl Acad Sci U S A (1994) 91(11):4935–9. doi:10.1073/pnas.91.11.4935
25. Grabstein KH, Eisenman J, Shanebeck K, Rauch C, Srinivasan S, Fung V, et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science (1994) 264(5161):965–8. doi:10.1126/science.8178155
26. Steel JC, Waldmann TA, Morris JC. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol Sci (2012) 33(1):35–41. doi:10.1016/j.tips.2011.09.004
27. Berger C, Berger M, Hackman RC, Gough M, Elliott C, Jensen MC, et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood (2009) 114(12):2417–26. doi:10.1182/blood-2008-12-189266
28. Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, et al. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A (2000) 97(21):11445–50. doi:10.1073/pnas.200363097
29. Munger W, DeJoy SQ, Jeyaseelan R Sr., Torley LW, Grabstein KH, Eisenmann J, et al. Studies evaluating the antitumor activity and toxicity of interleukin-15, a new T cell growth factor: comparison with interleukin-2. Cell Immunol (1995) 165(2):289–93. doi:10.1006/cimm.1995.1216
30. Dubois S, Mariner J, Waldmann TA, Tagaya Y. IL-15Ralpha recycles and presents IL-15 In trans to neighboring cells. Immunity (2002) 17(5):537–47. doi:10.1016/S1074-7613(02)00429-6
31. Kobayashi H, Dubois S, Sato N, Sabzevari H, Sakai Y, Waldmann TA, et al. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood (2005) 105(2):721–7. doi:10.1182/blood-2003-12-4187
32. Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med (2002) 195(12):1541–8. doi:10.1084/jem.20020369
33. Wong RL, Liu B, Zhu X, You L, Kong L, Han KP, et al. Interleukin-15:Interleukin-15 receptor alpha scaffold for creation of multivalent targeted immune molecules. Protein Eng Des Sel (2011) 24(4):373–83. doi:10.1093/protein/gzq116
34. Han KP, Zhu X, Liu B, Jeng E, Kong L, Yovandich JL, et al. IL-15:IL-15 receptor alpha superagonist complex: high-level co-expression in recombinant mammalian cells, purification and characterization. Cytokine (2011) 56(3):804–10. doi:10.1016/j.cyto.2011.09.028
35. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol (2015) 33(1):74–82. doi:10.1200/JCO.2014.57.3329
36. Verhoeven DH, de Hooge AS, Mooiman EC, Santos SJ, ten Dam MM, Gelderblom H, et al. NK cells recognize and lyse Ewing sarcoma cells through NKG2D and DNAM-1 receptor dependent pathways. Mol Immunol (2008) 45(15):3917–25. doi:10.1016/j.molimm.2008.06.016
37. Pahl JH, Ruslan SE, Buddingh EP, Santos SJ, Szuhai K, Serra M, et al. Anti-EGFR antibody cetuximab enhances the cytolytic activity of natural killer cells toward osteosarcoma. Clin Cancer Res (2012) 18(2):432–41. doi:10.1158/1078-0432.CCR-11-2277
38. Buddingh EP, Schilham MW, Ruslan SE, Berghuis D, Szuhai K, Suurmond J, et al. Chemotherapy-resistant osteosarcoma is highly susceptible to IL-15-activated allogeneic and autologous NK cells. Cancer Immunol Immunother (2011) 60(4):575–86. doi:10.1007/s00262-010-0965-3
39. Kleinerman ES, Jia SF, Griffin J, Seibel NL, Benjamin RS, Jaffe N. Phase II study of liposomal muramyl tripeptide in osteosarcoma: the cytokine cascade and monocyte activation following administration. J Clin Oncol (1992) 10(8):1310–6.
40. Verweij J, Judson I, Steward W, Coleman R, Woll P, van Pottelsberghe C, et al. Phase II study of liposomal muramyl tripeptide phosphatidylethanolamine (MTP/PE) in advanced soft tissue sarcomas of the adult. An EORTC soft tissue and bone sarcoma group study. Eur J Cancer (1994) 30A(6):842–3. doi:10.1016/0959-8049(94)90303-4
41. Meyers PA, Schwartz CL, Krailo MD, Healey JH, Bernstein ML, Betcher D, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival – a report from the children’s oncology group. J Clin Oncol (2008) 26(4):633–8. doi:10.1200/JCO.2008.14.0095
42. Chou AJ, Kleinerman ES, Krailo MD, Chen Z, Betcher DL, Healey JH, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the children’s oncology group. Cancer (2009) 115(22):5339–48. doi:10.1002/cncr.24566
43. Gorlick R, Huvos AG, Heller G, Aledo A, Beardsley GP, Healey JH, et al. Expression of HER2/erbB-2 correlates with survival in osteosarcoma. J Clin Oncol (1999) 17(9):2781–8.
44. Ebb D, Meyers P, Grier H, Bernstein M, Gorlick R, Lipshultz SE, et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: a report from the children’s oncology group. J Clin Oncol (2012) 30(20):2545–51. doi:10.1200/JCO.2011.37.4546
45. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8(12):915–28. doi:10.1038/nrc2536
46. Pollak M. Targeting insulin and insulin-like growth factor signalling in oncology. Curr Opin Pharmacol (2008) 8(4):384–92. doi:10.1016/j.coph.2008.07.004
47. Benini S, Manara MC, Baldini N, Cerisano V, Massimo S, Mercuri M, et al. Inhibition of insulin-like growth factor I receptor increases the antitumor activity of doxorubicin and vincristine against Ewing’s sarcoma cells. Clin Cancer Res (2001) 7(6):1790–7.
48. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, et al. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the children’s oncology group. Pediatr Blood Cancer (2014) 61(3):452–6. doi:10.1002/pbc.24605
49. Malempati S, Weigel B, Ingle AM, Ahern CH, Carroll JM, Roberts CT, et al. Phase I/II trial and pharmacokinetic study of cixutumumab in pediatric patients with refractory solid tumors and Ewing sarcoma: a report from the children’s oncology group. J Clin Oncol (2012) 30(3):256–62. doi:10.1200/JCO.2011.37.4355
50. Fuchs CS, Azevedo S, Okusaka T, Van Laethem JL, Lipton LR, Riess H, et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: the GAMMA trialdagger. Ann Oncol (2015) 26(5):921–7. doi:10.1093/annonc/mdv027
51. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev (2007) 28(1):20–47. doi:10.1210/er.2006-0001
52. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther (2010) 9(10):2652–64. doi:10.1158/1535-7163.MCT-10-0318
53. Belfiore A, Malaguarnera R. Insulin receptor and cancer. Endocr Relat Cancer (2011) 18(4):R125–47. doi:10.1530/ERC-11-0074
54. Fidler MJ, Shersher DD, Borgia JA, Bonomi P. Targeting the insulin-like growth factor receptor pathway in lung cancer: problems and pitfalls. Ther Adv Med Oncol (2012) 4(2):51–60. doi:10.1177/1758834011427576
55. Gao J, Chesebrough JW, Cartlidge SA, Ricketts SA, Incognito L, Veldman-Jones M, et al. Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res (2011) 71(3):1029–40. doi:10.1158/0008-5472.CAN-10-2274
56. Zhao Q, Tran H, Dimitrov DS, Cheung NK. A dual-specific anti-IGF-1/IGF-2 human monoclonal antibody alone and in combination with temsirolimus for therapy of neuroblastoma. Int J Cancer (2015). doi:10.1002/ijc.29588
57. Okada K, Yamasaki K, Tanaka C, Fujisaki H, Osugi Y, Hara J. Phase I study of bevacizumab plus irinotecan in pediatric patients with recurrent/refractory solid tumors. Jpn J Clin Oncol (2013) 43(11):1073–9. doi:10.1093/jjco/hyt124
58. Mascarenhas L, Meyer WH, Lyden E, Rodeberg DA, Indelicato DJ, Linardic CM, et al. Randomized phase II trial of bevacizumab and temsirolimus in combination with vinorelbine (V) and cyclophosphamide (C) for first relapse/disease progression of rhabdomyosarcoma (RMS): A report from the children’s oncology group (COG). J Clin Oncol (2014) 32(suppl; abstr 10003):5s.
59. Dalal S, Berry AM, Cullinane CJ, Mangham DC, Grimer R, Lewis IJ, et al. Vascular endothelial growth factor: a therapeutic target for tumors of the Ewing’s sarcoma family. Clin Cancer Res (2005) 11(6):2364–78. doi:10.1158/1078-0432.CCR-04-1201
60. Maris JM, Courtright J, Houghton PJ, Morton CL, Gorlick R, Kolb EA, et al. Initial testing of the VEGFR inhibitor AZD2171 by the pediatric preclinical testing program. Pediatr Blood Cancer (2008) 50(3):581–7. doi:10.1002/pbc.21232
61. Hughes DP, Thomas DG, Giordano TJ, Baker LH, McDonagh KT. Cell surface expression of epidermal growth factor receptor and Her-2 with nuclear expression of Her-4 in primary osteosarcoma. Cancer Res (2004) 64(6):2047–53. doi:10.1158/0008-5472.CAN-03-3096
62. Merchant MS, Geller JI, Baird K, Chou AJ, Galli S, Charles A, et al. Phase I trial and pharmacokinetic study of lexatumumab in pediatric patients with solid tumors. J Clin Oncol (2012) 30(33):4141–7. doi:10.1200/JCO.2012.44.1055
63. Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, et al. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. J Clin Oncol (2012) 30(26):3264–70. doi:10.1200/JCO.2011.41.3807
64. Roth M, Linkowski M, Tarim J, Piperdi S, Sowers R, Geller D, et al. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer (2014) 120(4):548–54. doi:10.1002/cncr.28461
65. Chang HR, Cordon-Cardo C, Houghton AN, Cheung NK, Brennan MF. Expression of disialogangliosides GD2 and GD3 on human soft tissue sarcomas. Cancer (1992) 70(3):633–8. doi:10.1002/1097-0142(19920801)70:3<633::AID-CNCR2820700315>3.0.CO;2-F
66. Xu H, Cheng M, Guo H, Chen Y, Huse M, Cheung NK. Retargeting T cells to GD2 pentasaccharide on human tumors using bispecific humanized antibody. Cancer Immunol Res (2015) 3(3):266–77. doi:10.1158/2326-6066.CIR-14-0230-T
67. Suzuki M, Curran KJ, Cheung NK. Chimeric antigen receptors and bispecific antibodies to retarget T cells in pediatric oncology. Pediatr Blood Cancer (2015) 62(8):1326–36. doi:10.1002/pbc.25513
68. Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol (2010) 11(2):155–64. doi:10.1016/S1470-2045(09)70334-1
69. Callahan MK, Postow MA, Wolchok JD. CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol (2014) 4:385. doi:10.3389/fonc.2014.00385
70. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol (2015) 33(17):1974–82. doi:10.1200/JCO.2014.59.4358
71. Maki RG, Jungbluth AA, Gnjatic S, Schwartz GK, D’Adamo DR, Keohan ML, et al. A pilot study of anti-CTLA4 antibody ipilimumab in patients with synovial sarcoma. Sarcoma (2013) 2013:168145. doi:10.1155/2013/168145
72. Shen JK, Cote GM, Choy E, Yang P, Harmon D, Schwab J, et al. Programmed cell death ligand 1 expression in osteosarcoma. Cancer Immunol Res (2014) 2(7):690–8. doi:10.1158/2326-6066.CIR-13-0224
73. Kawano M, Itonaga I, Iwasaki T, Tsumura H. Enhancement of antitumor immunity by combining anti-cytotoxic T lymphocyte antigen-4 antibodies and cryotreated tumor lysate-pulsed dendritic cells in murine osteosarcoma. Oncol Rep (2013) 29(3):1001–6. doi:10.3892/or.2013.2224
74. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi:10.1126/science.aaa1348
75. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi:10.1056/NEJMoa1406498
76. Snyder A, Wolchok JD, Chan TA. Genetic basis for clinical response to CTLA-4 blockade. N Engl J Med (2015) 372(8):783. doi:10.1056/NEJMc1415938
77. Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med (2013) 369(2):122–33. doi:10.1056/NEJMoa1302369
78. Watson IR, Takahashi K, Futreal PA, Chin L. Emerging patterns of somatic mutations in cancer. Nat Rev Genet (2013) 14(10):703–18. doi:10.1038/nrg3539
79. Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, et al. A landscape of driver mutations in melanoma. Cell (2012) 150(2):251–63. doi:10.1016/j.cell.2012.06.024
80. Haworth KB, Leddon JL, Chen CY, Horwitz EM, Mackall CL, Cripe TP. Going back to class I: MHC and immunotherapies for childhood cancer. Pediatr Blood Cancer (2015) 62(4):571–6. doi:10.1002/pbc.25359
81. Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res (2009) 15(17):5323–37. doi:10.1158/1078-0432.CCR-09-0737
82. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med (2004) 10(9):909–15. doi:10.1038/nm1100
83. Dillman RO, Wiemann M, Nayak SK, DeLeon C, Hood K, DePriest C. Interferon-gamma or granulocyte-macrophage colony-stimulating factor administered as adjuvants with a vaccine of irradiated autologous tumor cells from short-term cell line cultures: a randomized phase 2 trial of the cancer biotherapy research group. J Immunother (2003) 26(4):367–73. doi:10.1097/00002371-200307000-00009
84. Dillman R, Barth N, Selvan S, Beutel L, de Leon C, DePriest C, et al. Phase I/II trial of autologous tumor cell line-derived vaccines for recurrent or metastatic sarcomas. Cancer Biother Radiopharm (2004) 19(5):581–8. doi:10.1089/1084978042484812
85. Geiger JD, Hutchinson RJ, Hohenkirk LF, McKenna EA, Yanik GA, Levine JE, et al. Vaccination of pediatric solid tumor patients with tumor lysate-pulsed dendritic cells can expand specific T cells and mediate tumor regression. Cancer Res (2001) 61(23):8513–9.
86. Mackall CL, Rhee EH, Read EJ, Khuu HM, Leitman SF, Bernstein D, et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin Cancer Res (2008) 14(15):4850–8. doi:10.1158/1078-0432.CCR-07-4065
87. Pritchard-Jones K, Spendlove I, Wilton C, Whelan J, Weeden S, Lewis I, et al. Immune responses to the 105AD7 human anti-idiotypic vaccine after intensive chemotherapy, for osteosarcoma. Br J Cancer (2005) 92(8):1358–65. doi:10.1038/sj.bjc.6602500
88. Suminoe A, Matsuzaki A, Hattori H, Koga Y, Hara T. Immunotherapy with autologous dendritic cells and tumor antigens for children with refractory malignant solid tumors. Pediatr Transplant (2009) 13(6):746–53. doi:10.1111/j.1399-3046.2008.01066.x
89. Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK. Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res (2014) 20(5):1375–82. doi:10.1158/1078-0432.CCR-13-1012
90. Sun JC, Ugolini S, Vivier E. Immunological memory within the innate immune system. EMBO J (2014) 33(12):1295–303. doi:10.1002/embj.201387651
91. Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol (2012) 12(4):239–52. doi:10.1038/nri3174
92. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol (1975) 5(2):117–21. doi:10.1002/eji.1830050208
93. Caligiuri MA. Human natural killer cells. Blood (2008) 112(3):461–9. doi:10.1182/blood-2007-09-077438
94. Bachanova V, Miller JS. NK cells in therapy of cancer. Crit Rev Oncog (2014) 19(1–2):133–41. doi:10.1615/CritRevOncog.2014011091
95. Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med (1987) 316(15):889–97. doi:10.1056/NEJM198704093161501
96. Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood (2005) 105(8):3051–7. doi:10.1182/blood-2004-07-2974
97. Tarek N, Lee DA. Natural killer cells for osteosarcoma. Adv Exp Med Biol (2014) 804:341–53. doi:10.1007/978-3-319-04843-7_19
98. Cho D, Shook DR, Shimasaki N, Chang YH, Fujisaki H, Campana D. Cytotoxicity of activated natural killer cells against pediatric solid tumors. Clin Cancer Res (2010) 16(15):3901–9. doi:10.1158/1078-0432.CCR-10-0735
99. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res (2011) 17(13):4550–7. doi:10.1158/1078-0432.CCR-11-0116
100. Lai JP, Rosenberg AZ, Miettinen MM, Lee CC. NY-ESO-1 expression in sarcomas: a diagnostic marker and immunotherapy target. Oncoimmunology (2012) 1(8):1409–10. doi:10.4161/onci.21059
101. Lai JP, Robbins PF, Raffeld M, Aung PP, Tsokos M, Rosenberg SA, et al. NY-ESO-1 expression in synovial sarcoma and other mesenchymal tumors: significance for NY-ESO-1-based targeted therapy and differential diagnosis. Mod Pathol (2012) 25(6):854–8. doi:10.1038/modpathol.2012.31
102. Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol (2011) 29(7):917–24. doi:10.1200/JCO.2010.32.2537
103. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20 Pt 1):6106–15. doi:10.1158/1078-0432.CCR-06-1183
104. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14(11):1264–70. doi:10.1038/nm.1882
105. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118(23):6050–6. doi:10.1182/blood-2011-05-354449
106. Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res (2006) 66(22):10995–1004. doi:10.1158/0008-5472.CAN-06-0160
107. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5(177):177ra138. doi:10.1126/scitranslmed.3005930
108. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther (2010) 18(4):843–51. doi:10.1038/mt.2010.24
109. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi:10.1056/NEJMoa1003466
110. Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother (2013) 36(2):133–51. doi:10.1097/CJI.0b013e3182829903
111. Corrigan-Curay J, Kiem HP, Baltimore D, O’Reilly M, Brentjens RJ, Cooper L, et al. T-cell immunotherapy: looking forward. Mol Ther (2014) 22(9):1564–74. doi:10.1038/mt.2014.148
112. Topp MS, Gökbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol (2015) 16(1):57–66. doi:10.1016/S1470-2045(14)71170-2
113. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124(2):188–95. doi:10.1182/blood-2014-05-552729
114. Dao T, Yan S, Veomett N, Pankov D, Zhou L, Korontsvit T, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med (2013) 5(176):176ra133. doi:10.1126/scitranslmed.3005661
115. Orentas RJ, Yang JJ, Wen X, Wei JS, Mackall CL, Khan J. Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front Oncol (2012) 2:194. doi:10.3389/fonc.2012.00194
116. Orentas RJ, Lee DW, Mackall C. Immunotherapy targets in pediatric cancer. Front Oncol (2012) 2:3. doi:10.3389/fonc.2012.00003
117. Jacobs JF, Brasseur F, Hulsbergen-van de Kaa CA, van de Rakt MW, Figdor CG, Adema GJ, et al. Cancer-germline gene expression in pediatric solid tumors using quantitative real-time PCR. Int J Cancer (2007) 120(1):67–74. doi:10.1002/ijc.22118
118. Hu-Lieskovan S, Zhang J, Wu L, Shimada H, Schofield DE, Triche TJ. EWS-FLI1 fusion protein up-regulates critical genes in neural crest development and is responsible for the observed phenotype of Ewing’s family of tumors. Cancer Res (2005) 65(11):4633–44. doi:10.1158/0008-5472.CAN-04-2857
119. Grunewald TG, Bach H, Cossarizza A, Matsumoto I. The STEAP protein family: versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol Cell (2012) 104(11):641–57. doi:10.1111/boc.201200027
120. Nakatsuka S, Oji Y, Horiuchi T, Kanda T, Kitagawa M, Takeuchi T, et al. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod Pathol (2006) 19(6):804–14. doi:10.1038/modpathol.3800588
121. Ohta H, Hashii Y, Yoneda A, Takizawa S, Kusuki S, Tokimasa S, et al. WT1 (Wilms tumor 1) peptide immunotherapy for childhood rhabdomyosarcoma: a case report. Pediatr Hematol Oncol (2009) 26(1):74–83. doi:10.1080/08880010802435500
122. van den Broeke LT, Pendleton CD, Mackall C, Helman LJ, Berzofsky JA. Identification and epitope enhancement of a PAX-FKHR fusion protein breakpoint epitope in alveolar rhabdomyosarcoma cells created by a tumorigenic chromosomal translocation inducing CTL capable of lysing human tumors. Cancer Res (2006) 66(3):1818–23. doi:10.1158/0008-5472.CAN-05-2549
123. Worley BS, van den Broeke LT, Goletz TJ, Pendleton CD, Daschbach EM, Thomas EK, et al. Antigenicity of fusion proteins from sarcoma-associated chromosomal translocations. Cancer Res (2001) 61(18):6868–75.
124. Suzuki M, Cheung NK. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert Opin Ther Targets (2015) 19(3):349–62. doi:10.1517/14728222.2014.986459
125. Rebagay G, Yan S, Liu C, Cheung NK. ROR1 and ROR2 in human malignancies: potentials for targeted therapy. Front Oncol (2012) 2:34. doi:10.3389/fonc.2012.00034
126. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther (2009) 17(10):1779–87. doi:10.1038/mt.2009.133
127. Rainusso N, Brawley VS, Ghazi A, Hicks MJ, Gottschalk S, Rosen JM, et al. Immunotherapy targeting HER2 with genetically modified T cells eliminates tumor-initiating cells in osteosarcoma. Cancer Gene Ther (2012) 19(3):212–7. doi:10.1038/cgt.2011.83
128. Wang L, Zhang Q, Chen W, Shan B, Ding Y, Zhang G, et al. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS One (2013) 8(8):e70689. doi:10.1371/journal.pone.0070689
129. Bruland OS, Høifødt H, Saeter G, Smeland S, Fodstad O. Hematogenous micrometastases in osteosarcoma patients. Clin Cancer Res (2005) 11(13):4666–73. doi:10.1158/1078-0432.CCR-05-0165
130. Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer (2014) 2:25. doi:10.1186/2051-1426-2-25
131. Glüer S, Schelp C, von Schweinitz D, Gerardy-Schahn R. Polysialylated neural cell adhesion molecule in childhood rhabdomyosarcoma. Pediatr Res (1998) 43(1):145–7. doi:10.1203/00006450-199801000-00022
132. Chan ES, Pawel BR, Corao DA, Venneti S, Russo P, Santi M, et al. Immunohistochemical expression of glypican-3 in pediatric tumors: an analysis of 414 cases. Pediatr Dev Pathol (2013) 16(4):272–7. doi:10.2350/12-06-1216-OA.1
133. Cheal SM, Xu H, Guo HF, Zanzonico PB, Larson SM, Cheung NK. Preclinical evaluation of multistep targeting of diasialoganglioside GD2 using an IgG-scFv bispecific antibody with high affinity for GD2 and DOTA metal complex. Mol Cancer Ther (2014) 13(7):1803–12. doi:10.1158/1535-7163.MCT-13-0933
134. Bid HK, London CA, Gao J, Zhong H, Hollingsworth RE, Fernandez S, et al. Dual targeting of the type 1 insulin-like growth factor receptor and its ligands as an effective antiangiogenic strategy. Clin Cancer Res (2013) 19(11):2984–94. doi:10.1158/1078-0432.CCR-12-2008
135. Postow MA, Callahan MK, Barker CA, Yamada Y, Yuan J, Kitano S, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med (2012) 366(10):925–31. doi:10.1056/NEJMoa1112824
Keywords: pediatric sarcoma, immunotherapy of cancer, antibodies, monoclonal, CAR T cells, tumor vaccines, natural killer cells, osteosarcoma
Citation: Roberts SS, Chou AJ and Cheung N-KV (2015) Immunotherapy of childhood Sarcomas. Front. Oncol. 5:181. doi: 10.3389/fonc.2015.00181
Received: 18 May 2015; Accepted: 23 July 2015;
Published: 07 August 2015
Edited by:
Thomas Grunewald, Ludwig Maximilian University of Munich, GermanyReviewed by:
Francis Jay Mussai, Birmingham Children’s Hospital, UKDavid Anthony Rodeberg, Brody School of Medicine at East Carolina University, USA
Stefan Burdach, Technische Universität München, Germany
Copyright: © 2015 Roberts, Chou and Cheung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephen S. Roberts, Pediatrics, Memorial Sloan Kettering Cancer Center, 1275 York Avenue, New York, NY 10065, USA,cm9iZXJ0c3NAbXNrY2Mub3Jn