 Jonathan Noujaim
Jonathan Noujaim Khin Thway1
Khin Thway1 Zia Bajwa
Zia Bajwa Charles Keller
Charles Keller- 1Royal Marsden Hospital, London, UK
- 2Children’s Cancer Therapy Development Institute, Fort Collins, CO, USA
- 3Adult and Paediatric Sarcoma Program, Tisch Cancer Institute, Mount Sinai School of Medicine, New York, NY, USA
Epithelioid sarcoma (ES) is a soft tissue sarcoma of children and young adults for which the preferred treatment for localized disease is wide surgical resection. Medical management is to a great extent undefined, and therefore for patients with regional and distal metastases, the development of targeted therapies is greatly desired. In this review, we will summarize clinically relevant biomarkers (e.g., SMARCB1, CA125, dysadherin, and others) with respect to targeted therapeutic opportunities. We will also examine the role of EGFR, mTOR, and polykinase inhibitors (e.g., sunitinib) in the management of local and disseminated disease. Toward building a consortium of pharmaceutical, academic, and non-profit collaborators, we will discuss the state of resources for investigating ES with respect to cell line resources, tissue banks, and registries so that a roadmap can be developed toward effective biology-driven therapies.
Introduction
Epithelioid sarcoma (ES), first described by Enzinger over half a century ago (1), is a rare neoplasm accounting for <1% of adult soft tissue sarcomas and between 4 and 8% of pediatric non-rhabdomyosarcomatous sarcomas (2, 3). ES is presumed to be a mesenchymal malignancy, but ES characteristically exhibits both mesenchymal and epithelial markers. The cell of origin and molecular drivers are still a matter of debate. ES is divided into two recognizable clinicopathological entities, classic ES (also called distal-type ES), and proximal-type ES (Figures 1A–E). These two subtypes are thought be a continuum of disease rather than distinct entities (4). Distal-type ES is histologically identifiable by tumor nodules with central necrosis surrounded by large polygonal cells and spindle cells merging in the periphery (5) (Figures 1A,B). Described variants include angiomatoid variant, fibroma-like variant, and myxoid variant. Proximal-type ES is characterized by a multinodular pattern and sheet-like growth of large polygonal cells, often accompanied by a focal or predominant rhabdoid morphology (6) (Figures 1C,D). A specific marker has not yet been identified in ES. On immunohistochemistry (IHC), virtually all cases are positive for cytokeratin (CK) and epithelial membrane antigen (EMA) and most cases co-express vimentin. The marker CD34 is expressed in 60–70% of cases. IHC studies are typically negative for S-100, neurofilament protein, carcinoembryonic antigen, factor VIII-related antigen and CD-31, and INI-1 (SMARCB1) whose expression is lost in tumor nuclei (7). Establishing a diagnosis of ES can be difficult as tumors can present with a wide range of appearances and immunophenotypes. The differential diagnosis include fibrous histiocytoma, nodular fasciitis, other reactive proliferations, fibromatosis, giant cell tumor of tendon sheath, sclerosing epithelioid fibrosarcoma, and even some carcinomas and melanomas (7). IHC is helpful in differentiating these entities. Epithelioid vascular tumors can resemble ES and efforts must be made to exclude a diagnosis of epithelioid hemangioendothelioma. In epithelioid hemangioendothelioma, the unique translocation t(1;3)(p36;q25), resulting in the fusion of WWTR1 with CAMTA1I establishes a firm diagnosis (8).
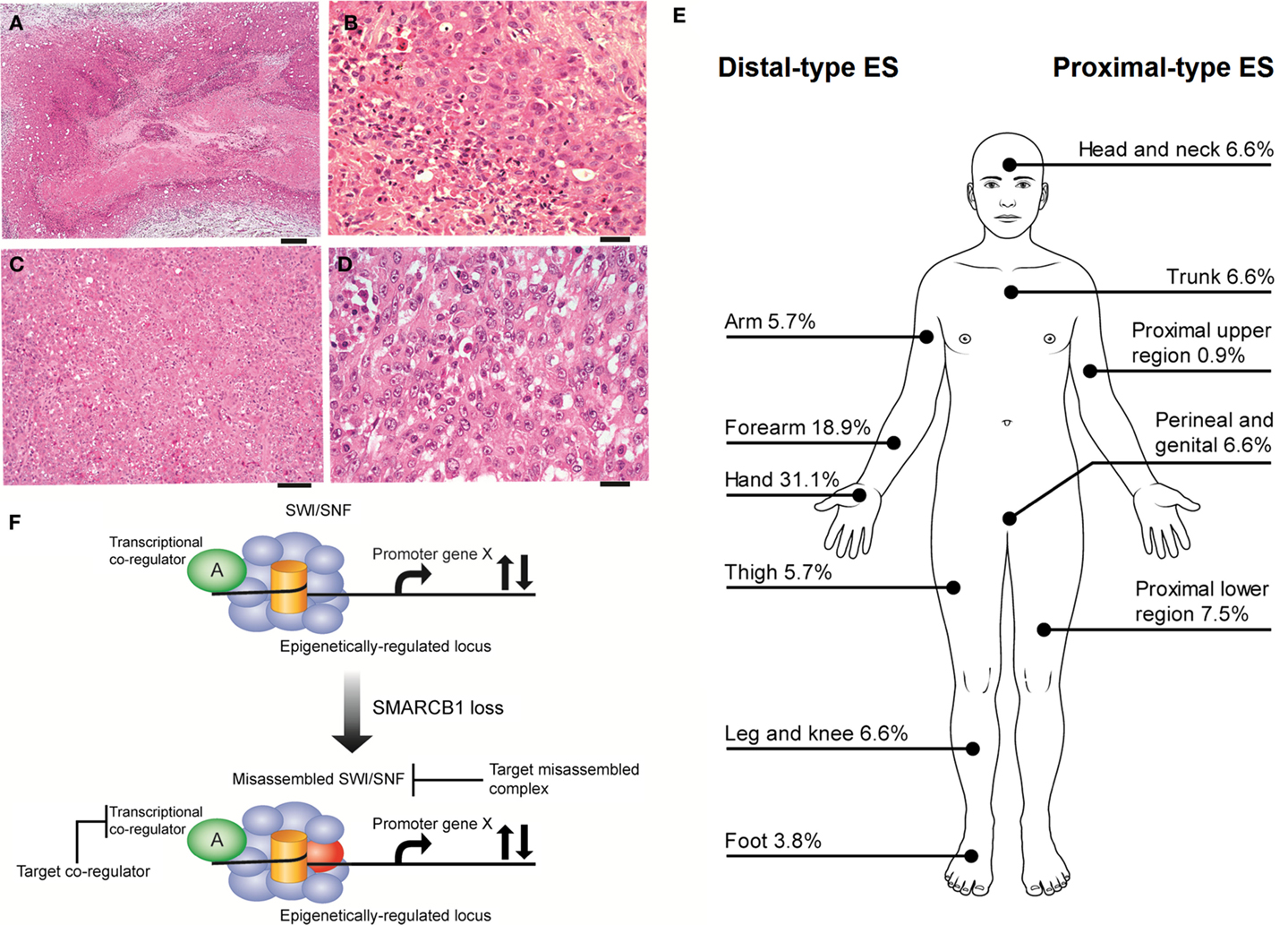
Figure 1. (A,B) Distal-type ES. (A) Low power histology shows a nodule of tumor present in the dermis and subcutis, comprising a large area of central geographic necrosis, surrounded by sheets of relatively uniform polygonal neoplastic cells (hematoxylin and eosin, ×40). Scale bar, 500 μM. (B) At higher power, these are medium-sized, rounded cells, with ovoid vesicular nuclei with even chromatin, and small nucleoli. This example is cellular, but more sparsely cellular neoplasms can appear subtle, and the neoplastic cells may be confused with inflammatory cells. The characteristic necrosis is seen abutting the tumor cells (bottom left of field) (hematoxylin and eosin, ×200). Scale bar, 50 μM. (C,D) Proximal-type ES. (C) At low power, proximal-type ES comprises sheets or lobules of medium-sized to large round cells, and is seen to lack the more defined architecture and geographic central necrosis of the distal-type variant (hematoxylin and eosin, ×40). Scale bar, 20 μM. (D) At higher power, this is characterized by a sheet-like growth of large polygonal cells, often with focal rhabdoid morphology, and which have ovoid vesicular nuclei, prominent large nucleoli, and relatively abundant eosinophilic cytoplasm. The cells are often more pleomorphic than those of the distal-type variant. On morphology alone, these cells are difficult to distinguish from other malignant epithelioid cells, such as those of carcinoma, melanoma, rhabdomyosarcoma, or epithelioid angiosarcoma, and therefore immunohistochemistry is crucial for establishing a correct diagnosis (hematoxylin and eosin, ×200). Scale bar, 50 μM. (E) Distributions of ES subytpes, adapted from the largest series reported by the French Sarcoma Group (9). (F) Vulnerabilities in the misassembled SWI\SNF complex when SMARCB1 is absent. Using epithelioid sarcoma as well as rhabdoid tumor as a basis for this model of SMARCB1 null tumors, the misassembled SWI/SNF complex has the potential to dysregulate target loci that may be co-regulated by other transcription factors (36, 38–40, 43) and thereby present indirect ways to drug target the misassembled complex.
The reported overall peak incidence of ES is around 35 years of age (9, 10). Distal-type ES is more frequently diagnosed and tends to affect a younger (20–40 years of age) and more predominantly male population compared to proximal-type ES, which is usually found in an older population (20–65 years of age) (9, 11). Distal-type ES can present itself as superficial, slow growing painless firm nodules leading to chronic non-healing ulcers affecting mostly the hands and arms. Distal-type ES can also arise as deep-seated slow growing tumors in the extremities or in the tenosynovial tissues. Proximal-type ES is more often diagnosed as deep infiltrating soft tissue masses affecting axial proximal regions and is thought to be associated with a more aggressive course (6). Figure 1E illustrates the sites of involvement of disease. In the largest reported cohort, a majority of ES patients (47%) had localized disease at presentation (2). ES is one of the rare sarcomas that regularly spread to lymph nodes (2, 12, 13). The course of disease is characterized by multiple local recurrences and eventual metastatic spread in 30–50% of cases with the lungs being the primary site of involvement (11). It might be said that local recurrence is the gateway to metastasis.
Management and Prognosis
Optimal management of this rare sarcoma remains to be defined. The cornerstone of treatment of localized disease is wide surgical resection (14). Neo-adjuvant or adjuvant radiation therapy is often administered in an attempt to reduce local relapses (15, 16). The role of adjuvant chemotherapy is unclear (13, 14, 17, 18). Despite multimodal management, the relapse rate remains high and recurrences tend to occur many years later following initial therapy. Reported local relapse rates are ~35% (11, 18, 19). Patients with localized disease fare better compared to regional disease [5-year overall survival (OS) of 75 vs. 49%]. Pediatric patients seem to have a favorable prognosis [5-year OS of 92.4%] as they are more likely to be diagnosed with localized distal-type ES and are less likely to have nodal or metastatic involvement at presentation (3).
Even though reasonable control of localized disease is possible, metastatic spread is seen in approximately half of patients (2). The available literature on palliative chemotherapy in ES is limited to case reports and small retrospectives studies. The most commonly administered chemotherapy regimens are single-agent anthracycline therapy or the combination an anthracycline with ifosfamide (20). A single group reported activity of a regimen combining gemcitabine with docetaxel, but the experience is limited to a small number of patients (21). The activity of navelbine was raised in a case report and may warrant further investigation (22). Partial responses are rare. Most patients achieve stable disease at best with palliative chemotherapy. In one study, tumor regression was only seen in distal-type disease (20). However, another group reported high-response rates in proximal-type ES using doxorubicin-ifosfamide combination (23). With the medical evidence being limited to small retrospectives studies, it is difficult to draw definitive conclusions regarding the chemosensitivity of this histological subtype.
Despite the administration of palliative chemotherapy, patients with metastasis have a poor prognosis. The reported median survival is ~52 weeks and the 1- and 5-year survival rates are 46 and 0%, respectively (2, 20). Therefore, a substantial unmet need exists to improve the medical management of ES patients by establishing novel systemic regimens and exploring novel targeted therapy. In this review, we will summarize our current understanding of the underlying biology of this rare disease by highlighting implicated signaling pathways and potential actionable biomarkers (Table 1). In order to establish a roadmap that can be developed toward effective biology-driven therapies, we will highlight therapeutics opportunities and drugs with promising activity.
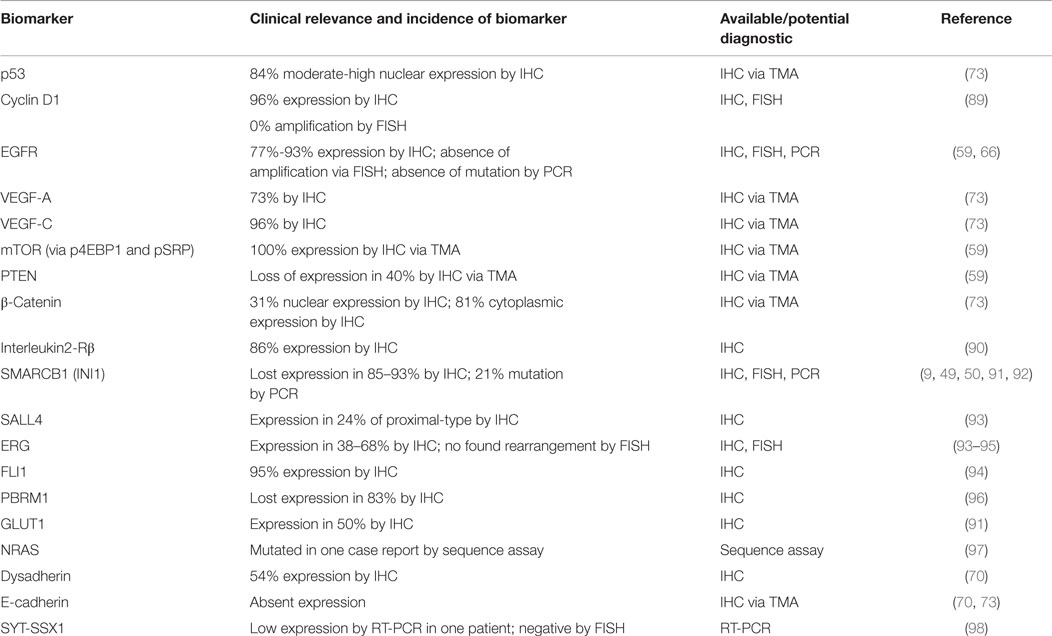
Table 1. Potential actionable biomarkers in clinical epithelioid sarcoma samples.
Cytogenetic Analyses
Cytogenetic analyses were first attempted to better understand the biology of ES (24–29). The karyotype analysis on clinical samples or cells lines varied greatly and was mostly done in adult cases. A minority of samples were diploid, some polypoid, while a great majority had complex patterns consisting of multiple numerical and structural rearrangements (see Table 3). Pediatric cytogenetic analyses seem to indicate less complex genetic alterations compared to adults and may therefore offer an explanation of their more favorable prognosis (30, 31). Translocations t(8;22)(q22;q11) in distal-type ES and t(10;22) in proximal-type ES were found (24, 32). However, compared to other translocation-driven sarcomas, there is no unique identifiable reoccurring cytogenetic pattern in ES. The only identified recurrent breakpoints have been structural rearrangements involving 18q11 and 22q11. The observation that a substantial number of ES had either rearrangements or deletions of 22q led to the hypothesis that this region may contain a tumor suppressor gene (32, 33). Further studies identified SMARCB1 as being involved in the tumorigenesis of ES (34).
SMARCB1
The SMARCB1 gene, located at 22q11, codes for BAF47, a core subunit of the SWI/SNF ATP-dependent chromatin remodeling complex and acts as a tumor suppressor gene (35). Components of the SWI/SNF complex are mutated in 20% of cancers, most notably rhabdoid tumor (36). This complex regulates genes by enabling the nucleosome to reposition itself in relation to the DNA sequence (37). Inactivation of SMARCB1 leads to neoplastic transformation by transcriptional deregulation of target genes implicated in regulating genomic stability, cell-cycle progression, and other signaling pathways in cooperation with transcriptional co-regulators (e.g., MyoD, Olig2) (36, 38, 39). SMARCB1 was shown to transcriptionally regulate p16INK4a and/or p21 and repress cyclin D1, thereby suppressing E2F activity and its target genes (40–42). SMARCB1-deficient cells have been implicated to have aberrant Hedgehog signaling pathway activation (40, 43). Brenca et al. demonstrated that loss of SMARCB1 expression in the ES cell line VAESBJ was caused by homozygous deletion of SMARCB1 through mutations of exon 1. They also identified equally prevalent homozygous deletion of CDKN2A and CDKN2B loci, responsible for encoding p16, p14, and p15 proteins. Restoration of SMARCB1 led to a reduction of cell proliferation and cell migration and to an increase in sensitivity to genotoxic stress, thereby providing evidence to support SMARCB1 inactivation in the tumorigenesis of ES (44). For rhabdoid tumor, SWI/SNF disruption is sufficient to cause neoplastic transformation (45). However, in the context of ES, loss of SMARCB1 by itself is not sufficient. Interestingly, knockout of SMARCB1 in primary fibroblast cells causes rapid growth arrest and p53-mediated programed cell death (46). However, when mutations of TP53 co-exist, tumor proliferation is dramatically increased (47). Brenca et al. demonstrated that the VAESBJ cell line retains wild-type TP53, but hypothesized that the homozygous loss of CDKN2A which leads to impaired p16/RB and p14/TP53 responses likely contributes to the genomic instability seen in this cell line (44). Hence, other signaling pathways may contribute to tumor progression in ES as witnessed by the complex genetic landscape reported in cytogenetic studies. Whether the SMARCB1-deficient SWI/SNF complex exists in a misassembled state as it does in rhabdoid tumor (48), and to what extent the misassembled complex aberrantly deregulates loci that are not normally associated with the SWI/SNF complex remains to be investigated. Most certainly, the milieu of transcriptional co-regulators in ES will be different than in rhabdoid tumor.
Targeting SMARCB1 is complicated by the different mechanisms of loss of expression. IHC studies demonstrated that the loss of expression of SMARCB1 ranges from 85 to 93% of cases (9, 34, 49, 50). Allelic homozygous deletions varied from 5 to 71%; however, the true value may be ~10% (51–53). Papp et al. identified different mechanisms to explain the loss of expression of SMARCB1: 13% of cases had biallelic deletions, 33% showed single-allelic deletion, and 4% had point mutations (52). In 59% of cases, both alleles were intact and no cases had promoter hypermethylation nor post-translational modification. The authors went on to show that loss of SMARCB1 protein expression in those cases is due to epigenetic gene silencing by oncomiRs. Three of the overexpressed miRNAs (miR-206, miR-381, and miR-671-5p) could silence the SMARCB1 mRNA expression in cell cultures (54). The role of oncomiRs was also validated by another group where miR193a-5p could equally inhibit the mRNA expression of SMARCB1 (55). Beyond targeting the misassembled SWI/SNF complex, transcriptional co-regulators are also theoretical targets (Figure 1F). In summary, loss of SMARCB1 has a crucial role in the pathogenesis of ES (along with other signaling pathways) and therefore is an interesting target to pursue for the development of new therapies. Acknowledging that the restoration of SMARCB1 function is likely the primary therapeutic opportunity in ES, in the paragraphs to follow we discuss other therapeutic opportunities related to consistent alterations in other signaling pathways that may also contribute to the pathogenesis of ES.
PI3K–AKT–mTOR Signaling Pathway
The phosphatidylinositol 3-kinase/protein kinase-B/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling pathway has been studied extensively and is activated in a myriad of cancers. This signaling pathway’s signaling regulates cell proliferation, differentiation, cellular metabolism, and cytoskeletal reorganization leading to apoptosis and cancer cell survival (56). A previous study done on SMARCB1-deficient tumor cells revealed persistent AKT activation (57). This finding led Imura et al. to further investigate the importance of this signaling pathway in ES (58). By studying two SMARCB1-deficient cell lines (VAESBJ and Asra-EPS), this group has shown that AKT/mTOR pathway is constitutively hyperactivated. Results demonstrated that silencing mTOR by transfecting cell lines with anti-mTOR-specific siRNAs suppressed cell proliferation. However, inhibition of mTOR with everolimus caused tumor growth delay without shrinkage. Blocking the mTOR signaling pathway with everolimus caused an increase in AKT and ERK activity, which was subsequently shown to be dependent of c-MET activation. Blocking c-MET activation had a variable effect on growth inhibition on studied cell lines. This variability could be partially explained by the degree of loss of PTEN, which is thought to contribute to resistance to c-MET inhibitors through sustained AKT activation upon mTOR blockade. Combining agents to block both AKT and c-MET were more effective in inducing tumor arrest compared to using either one alone. The importance of AKT and c-MET/HGF pathways was also highlighted through immunohistochemical analysis of random clinical samples. The variability of AKT activation and loss of PTEN expression in different cell lines were also confirmed by another group and thought to correlate with sensitivity of rapamycin (59). This heterogeneity could highlight the complex genetics of the disease as well as the variable importance of PI3K/AKT/mTOR signaling pathway in the tumorigenesis of ES. In vitro sensitivity to mTOR inhibitors is likely an imperfect surrogate for clinical activity. Nonetheless, these preclinical data are interesting and may warrant additional studies before pursuing clinical trials. Resistance to single-agent mTOR inhibitors can not only be a potential issue but can also possibly be overcome by simultaneously targeting other pathways. These findings are consistent with the shortcomings of targeting mTOR signaling pathway in general and highlight the importance of patient selection and identification of putative biomarkers (60).
EGF Pathway
The human epidermal growth factor signaling pathway regroups four distinct receptor tyrosine kinases, HER1 (ErbB-1, EGFR), HER2 (ErbB-2), HER3 (ErbB-3), and HER4 (ErbB-4) and is implicated in cell proliferation, apoptosis, and angiogenesis (61). The role of EGFR in malignant transformation of carcinomas has been extensively studied. Recently, EGFR expression was revealed to be present in soft tissue and bone sarcomas (62, 63). However, subsets of disease demonstrating tyrosine kinase domain mutations were rare (64, 65). These findings sparked an interest in studying EGFR in ES. Cascio et al. showed that 93% of clinical samples (including distal and proximal-type ES) expressed EGFR by IHC (66). This high level of expression of EGFR was also corroborated by Xie et al. (59). Furthermore, Cascio et al. went on to show an absence of EGFR amplification via FISH studies. Sequencing of the EFGR gene tyrosine kinase domain revealed no point mutations, insertions, or deletions. Xie et al. further investigated the role of EGFR pathway in the tumorigenesis of ES. EGF-induction contributes to cell-cycle progression partly through upregulation of cyclin D1. EGFR activation also causes an increase in migration and invasion of ES cells where high levels of expression of MMP2 and MMP9 were found. Next, this group tested whether EGFR inhibition with erlotinib would be a viable therapy. Exposure to erlotinib caused tumor growth delay without causing tumor arrest. An explanation for this incomplete response is given by the cooperation of HGFR/MET pathway with EGFR in sustaining AKT and ERK phosphorylation. Dual inhibition of both those pathways had a synergistic effect in decreasing cell proliferation (44). Combining inhibition of EGFR pathway with erlotinib and mTOR pathway with rapamycin also proved to be synergistic causing cell-cycle arrest as well as an increase in apoptosis (59). Targeting solely the EGFR pathway may not translate to a possible clinical benefit. However, combined inhibition of EFGR with either mTOR or HGFR\MET may worth investigating further through preclinical animal studies.
Other Possible Actionable Pathways and Targets
Dysadherin is a cancer-associated cell membrane glycoprotein shown to downregulate E-cadherin cell-mediated adhesion and to promote metastasis (67). Dysadherin contributes to metastatic progression through autocrine activation of CCL2 expression in part through activation of the nuclear factor-kappaB pathway (68). Dysadherin also has the ability to attribute stem-cell like properties to cancer cells (68, 69). Higher mRNA expression levels of dysadherin were documented in cell lines derived from proximal-type ES compared to distal-type ES (70). This difference in expression in levels of dysadherin may offer a possible explanation to the poor prognosis associated with proximal-type ES. Interestingly, in breast cancer cell lines, dysadherin overexpression was shown to possibly enhance AKT activation. Subsequently, inhibiting AKT reduced dysadherin’s ability to promote cell mobility and tumor cell invasion (71). Targeting dysadherin could be potentially exploited to treat ES, but further work is needed. Agonists of the CCL2 receptor, CCR2, such as PF-04634817, may be one area to begin investigation.
The role of Wnt/β-catenin signaling pathway in cancer is well documented. APC deficiency or β-catenin mutations preventing its degradation lead to constitutive activation of β-catenin signaling, which in turn contribute to stem-cell renewal and proliferation (72). In ES, IHC studies revealed low expressions of nuclear β-catenin (73). Furthermore, no β-catenin gene mutations were found (74). Therefore, the proliferative abilities of ES cells are probably related to other mechanisms than Wnt/β-catenin signaling pathway. This finding is in contradistinction to the β-catenin activation seen in rhabdoid tumor (39). Interestingly, IHC studies identified a complete loss of E-cadherin (70, 73). E-cadherin is a calcium-dependent glycoprotein responsible for cell–cell adhesion (75). E-cadherin/β-catenin protein complexes have an active role in epithelial-to-mesenchymal transition (EMT), an important mechanism for the subsequent development of metastasis (76, 77). Further studies are needed to elucidate the importance of loss of adhesion molecules in tumor progression in ES.
CA125 was first identified and used as a serum marker for epithelial ovarian carcinoma (78). IHC studies revealed high positivity and specificity of CA125 in ES compared to other sarcomas (79). High expressions of the MUC16 gene were identified by RT-PCR in ES cell lines. Serum levels of CA125 also seem to correlate with disease progression (80). Measuring CA125 serum levels is well-established and routinely available and could potentially be useful in monitoring disease status and evaluating response to therapy. Targeted immune strategies toward CA125 and MUC16 are active areas of research in ovarian cancer and any potential breakthroughs could possibly be applicable in treating ES (81, 82).
Tyrosine Kinase Inhibitors
As stated previously, chemotherapy has a limited role in the management of ES. Early studies explored the reasons underlying chemotherapy resistance. A study of the SARCCR2 cell line showed overexpression of P-glycoprotein, an ATP-binding cassette (ABC) chemotherapy export pump. Using verapamil and cyclosporine A to reverse multidrug resistance, the authors showed increased sensitivity to doxorubicin and vincristine (83). For the GRU cell line, expression of P-glycoprotein and MRP could also be observed (84). However, one study identified an absence of expression of P-glycoprotein and MRP in the ES-OMC-MN and SFT-8606 cell lines (85). In contradistinction, these studies demonstrated the presence of lung resistant protein (LRP), which mediates multidrug resistance (MDR). Results showed that reversing MDR with cyclosporin A increased sensitivity to actinomycin D, vincristine, and adriamycin. The use of tyrosine kinase inhibitors, the newest ABC inhibitors, to reverse multidrug resistance remains unexplored and may potentially enhance the efficacy of chemotherapy in ES.
The medical evidence for the utility of tyrosine kinase inhibitors impacting ES is scarce. To our knowledge, only one case was reported in the medical literature. Sunitinib showed reasonable disease stabilization in a patient with metastatic ES (86). The underlying reasons why sunitinib was active in this patient are unknown and cannot be explained with what is currently known about the biology of this disease. Pazopanib, a recent oral tyrosine kinase inhibitor approved for the treatment of soft tissue sarcoma (87), is worth investigating prospectively as its activity is similar to sunitinib. Work is still needed in mapping out active signaling pathways and identifying actionable tyrosine kinase domain mutations. Polykinase inhibitors remain therefore greatly unexplored in the management of ES and may one day improve outcome.
Future Perspectives
Researching and developing new treatment strategies in rare cancers is a challenge, but possible with technology and resources available today and regulatory agency incentives (88). ES is a perfect model to envision what personalized medicine promises for the future. The intent of this review was to draw a roadmap to develop efficient biology-driven therapy. Achieving this will start with the selection of representative cell lines and mouse models of ES (Available cell lines and potential actionable targets are summarized in Tables 2 and 3). Many of the potential targets highlighted in this article were based on IHC-expression or reverse-transcriptase PCR studies. DNA deep-sequencing projects may demonstrate underlying genomic amplification and mutations that can be targeted. Partnership with pharmaceutical companies would allow screening of thousands of compounds on selected cell lines presenting mutations or other actionable targets. Active drugs may then undergo preclinical testing. Those most promising can be prioritized for clinical trials. Drugs being developed in other cancers that share common signaling pathways aberrations with ES may also prove to be useful. It is possible to perform basket trials in rare cancers, and this could be a way of evaluating novel agents in this extremely rare disease. On the way of developing new therapies, possible pitfalls can be expected. As demonstrated on ES cell line models, targeting a single signaling pathway may be insufficient. The complexity of the genetic landscape and the crosstalk between multiple signaling pathways contribute to resistance. This can be overcome by targeting multiple signaling pathways simultaneously. Only through international collaboration between pediatric and adult units, we can remain hopeful that targeted and immune therapy will have a major impact in the management of ES in the near future.
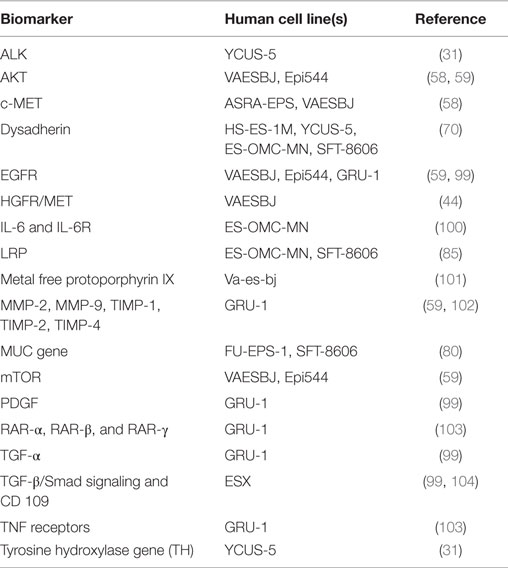
Table 2. Epithelioid sarcoma potential targets and corresponding experimental model systems.
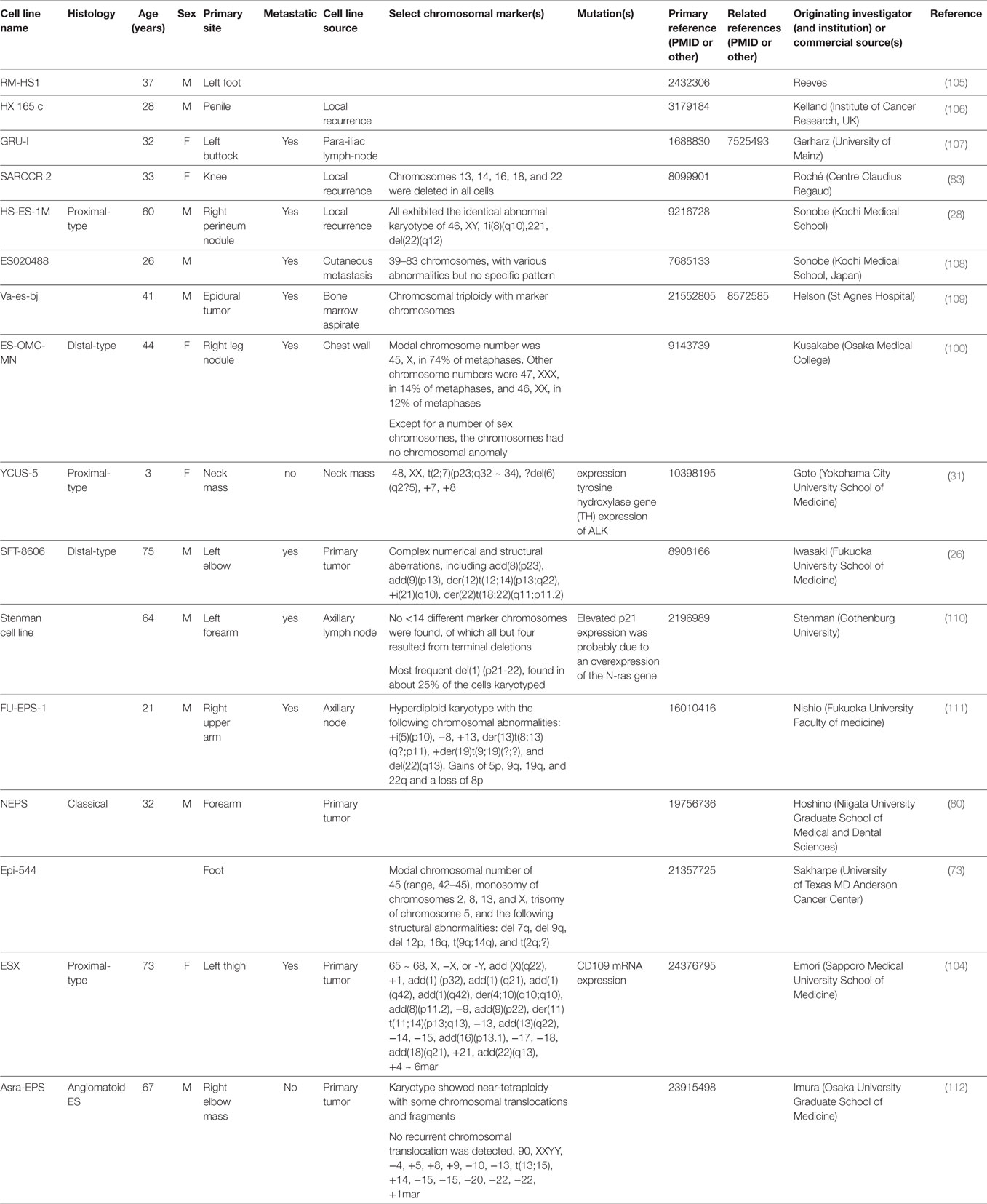
Table 3. Demographic and biological features of human epithelioid sarcoma cell lines.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Anne Wadmore for assistance with figure graphics. This work was supported in part by the support from the Scott Carter Foundation.
References
1. Enzinger FM. Epitheloid sarcoma. A sarcoma simulating a granuloma or a carcinoma. Cancer (1970) 26(5):1029–41. doi: 10.1002/1097-0142(197011)26:5<1029::AID-CNCR2820260510>3.0.CO;2-R
2. Jawad M, Extein J, Min E, Scully S. Prognostic factors for survival in patients with epithelioid sarcoma: 441 cases from the SEER database. Clin Orthop Relat Res (2009) 467(11):2939–48. doi:10.1007/s11999-009-0749-2
3. Casanova M, Ferrari A, Collini P, Bisogno G, Alaggio R, Cecchetto G, et al. Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer (2006) 106(3):708–17. doi:10.1002/cncr.21630
4. Rakheja D, Wilson K, Meehan J, Schultz R, Gomez A. “Proximal-type” and classic epithelioid sarcomas represent a clinicopathologic continuum: case report. Pediatr Dev Pathol (2005) 8(1):105–14. doi:10.1007/s10024-005-0148-y
5. Armah H, Parwani A. Epithelioid sarcoma. Arch Pathol Lab Med (2009) 133(5):814–9. doi:10.1043/1543-2165-133.5.814
6. Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol (1997) 21(2):130–46. doi:10.1097/00000478-199702000-00002
7. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol (2006) 13(3):114–21. doi:10.1097/00125480-200605000-00002
8. Errani C, Zhang L, Sung Y, Hajdu M, Singer S, Maki R, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer (2011) 50(8):644–53. doi:10.1002/gcc.20886
9. Chbani L, Guillou L, Terrier P, Decouvelaere A, Grégoire F, Terrier Lacombe M, et al. Epithelioid sarcoma: a clinicopathologic and immunohistochemical analysis of 106 cases from the French sarcoma group. Am J Clin Pathol (2009) 131(2):222–7. doi:10.1309/AJCPU98ABIPVJAIV
10. Brennan MF, Antonescu CR, Maki RG. Management of Soft Tissue Sarcoma. New York: Springer (2013).
11. Chase DR, Enzinger FM. Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am J Surg Pathol (1985) 9(4):241–63. doi:10.1097/00000478-198504000-00001
12. Ross HM, Lewis JJ, Woodruff JM, Brennan MF. Epithelioid sarcoma: clinical behavior and prognostic factors of survival. Ann Surg Oncol (1997) 4(6):491–5. doi:10.1007/BF02303673
13. Callister MD, Ballo MT, Pisters PW, Patel SR, Feig BW, Pollock RE, et al. Epithelioid sarcoma: results of conservative surgery and radiotherapy. Int J Radiat Oncol Biol Phys (2001) 51(2):384–91. doi:10.1016/S0360-3016(01)01646-7
14. Wolf P, Flum D, Tanas M, Rubin B, Mann G. Epithelioid sarcoma: the University of Washington experience. Am J Surg (2008) 196(3):407–12. doi:10.1016/j.amjsurg.2007.07.029
15. Shimm DS, Suit HD. Radiation therapy of epithelioid sarcoma. Cancer (1983) 52(6):1022–5. doi:10.1002/1097-0142(19830915)52:6<1022::AID-CNCR2820520616>3.0.CO;2-I
16. Livi L, Shah N, Paiar F, Fisher C, Judson I, Moskovic E, et al. Treatment of epithelioid sarcoma at the Royal Marsden Hospital. Sarcoma (2003) 7(3–4):149–52. doi:10.1080/13577140310001644760
17. Hasegawa T, Matsuno Y, Shimoda T, Umeda T, Yokoyama R, Hirohashi S. Proximal-type epithelioid sarcoma: a clinicopathologic study of 20 cases. Mod Pathol (2001) 14(7):655–63. doi:10.1038/modpathol.3880368
18. Baratti D, Pennacchioli E, Casali P, Bertulli R, Lozza L, Olmi P, et al. Epithelioid sarcoma: prognostic factors and survival in a series of patients treated at a single institution. Ann Surg Oncol (2007) 14(12):3542–51. doi:10.1245/s10434-007-9628-9
19. Levy A, Le Péchoux C, Terrier P, Bouaita R, Domont J, Mir O, et al. Epithelioid sarcoma: need for a multimodal approach to maximize the chances of curative conservative treatment. Ann Surg Oncol (2014) 21(1):269–76. doi:10.1245/s10434-013-3247-4
20. Jones R, Constantinidou A, Olmos D, Thway K, Fisher C, Al Muderis O, et al. Role of palliative chemotherapy in advanced epithelioid sarcoma. Am J Clin Oncol (2012) 35(4):351–7. doi:10.1097/COC.0b013e3182118cf7
21. Pink D, Richter S, Gerdes S, Andreou D, Tunn P-U, Busemann C, et al. Gemcitabine and docetaxel for epithelioid sarcoma: results from a retrospective, multi-institutional analysis. Oncology (2014) 87(2):95–103. doi:10.1159/000362602
22. Tariq Z, Ghose A, Bawany M, Saeed B, Mohamed I, Harmon D. A case report of complete remission of pulmonary metastases from epithelioid sarcoma to navelbine chemotherapy. Am J Ther (2012) 19(2):e95–7. doi:10.1097/MJT.0b013e3181e70a50
23. Piovesan C, Dileo P, Puma E, et al. Proximal-type epithelioid sarcomas: a retrospective analyses of 31 patients. Poster session presented at the 15th Annual Meeting of the Connective Tissue Oncology Society. Miami, FL (2009).
24. Cordoba JC, Parham DM, Meyer WH, Douglass EC. A new cytogenetic finding in an epithelioid sarcoma, t(8;22)(q22;q11). Cancer Genet Cytogenet (1994) 72(2):151–4. doi:10.1016/0165-4608(94)90132-5
25. Dal Cin P, Van den Berghe H, Pauwels P. Epithelioid sarcoma of the proximal type with complex karyotype including i(8q). Cancer Genet Cytogenet (1999) 114(1):80–2.
26. Iwasaki H, Ohjimi Y, Ishiguro M, Isayama T, Kaneko Y, Yoh S, et al. Epithelioid sarcoma with an 18q aberration. Cancer Genet Cytogenet (1996) 91(1):46–52. doi:10.1016/S0165-4608(95)00315-0
27. Molenaar WM, DeJong B, Dam Meiring A, Postma A, DeVries J, Hoekstra HJ. Epithelioid sarcoma or malignant rhabdoid tumor of soft tissue? Epithelioid immunophenotype and rhabdoid karyotype. Hum Pathol (1989) 20(4):347–51. doi:10.1016/0046-8177(89)90044-0
28. Sonobe H, Ohtsuki Y, Sugimoto T, Shimizu K. Involvement of 8q, 22q, and monosomy 21 in an epithelioid sarcoma. Cancer Genet Cytogenet (1997) 96(2):178–80.
29. Debiec Rychter M, Sciot R, Hagemeijer A. Common chromosome aberrations in the proximal type of epithelioid sarcoma. Cancer Genet Cytogenet (2000) 123(2):133–6. doi:10.1016/S0165-4608(00)00320-4
30. de Vries J, Hoekstra HJ, Oosterhuis JW, Postma A, Schraffordt Koops H. Epithelioid sarcoma in children and adolescents: a report of four cases. J Pediatr Surg (1989) 24(2):186–8. doi:10.1016/S0022-3468(89)80246-5
31. Goto H, Takahashi H, Funabiki T, Ikuta K, Sasaki H, Nagashima Y. Brief report: neural differentiation of a novel cell line, YCUS-5, established from proximal-type epithelioid sarcoma of a child. Med Pediatr Oncol (1999) 33(2):137–8. doi:10.1002/(SICI)1096-911X(199908)33:2<137::AID-MPO18>3.0.CO;2-N
32. Lualdi E, Modena P, Debiec Rychter M, Pedeutour F, Teixeira M, Facchinetti F, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer (2004) 41(3):283–90. doi:10.1002/gcc.20086
33. Quezado MM, Middleton LP, Bryant B, Lane K, Weiss SW, Merino MJ. Allelic loss on chromosome 22q in epithelioid sarcomas. Hum Pathol (1998) 29(6):604–8. doi:10.1016/S0046-8177(98)80010-5
34. Modena P, Lualdi E, Facchinetti F, Galli L, Teixeira M, Pilotti S, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res (2005) 65(10):4012–9. doi:10.1158/0008-5472.CAN-04-3050
35. Kalpana GV, Marmon S, Wang W, Crabtree GR, Goff SP. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science (1994) 266(5193):2002–6. doi:10.1126/science.7801128
36. Wang X, Haswell J, Roberts CWM. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer – mechanisms and potential therapeutic insights. Clin Cancer Res (2014) 20(1):21–7. doi:10.1158/1078-0432.CCR-13-0280
37. Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene (2009) 28(14):1653–68. doi:10.1038/onc.2009.4
38. Wöhrle S, Weiss A, Ito M, Kauffmann A, Murakami M, Jagani Z, et al. Fibroblast growth factor receptors as novel therapeutic targets in SNF5-deleted malignant rhabdoid tumors. PLoS One (2013) 8(10):e77652. doi:10.1371/journal.pone.0077652
39. Mora Blanco EL, Mishina Y, Tillman EJ, Cho YJ, Thom CS, Pomeroy SL, et al. Activation of β-catenin/TCF targets following loss of the tumor suppressor SNF5. Oncogene (2014) 33(7):933–8. doi:10.1038/onc.2013.37
40. Medjkane S, Novikov E, Versteege I, Delattre O. The tumor suppressor hSNF5/INI1 modulates cell growth and actin cytoskeleton organization. Cancer Res (2004) 64(10):3406–13. doi:10.1158/0008-5472.CAN-03-3004
41. Oruetxebarria I, Venturini F, Kekarainen T, Houweling A, Zuijderduijn LMP, Mohd Sarip A, et al. P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J Biol Chem (2004) 279(5):3807–16. doi:10.1074/jbc.M309333200
42. Zhang Z-K, Davies K, Allen J, Zhu L, Pestell R, Zagzag D, et al. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol (2002) 22(16):5975–88. doi:10.1128/MCB.22.16.5975-5988.2002
43. Jagani Z, Mora Blanco EL, Sansam C, McKenna E, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med (2010) 16(12):1429–33. doi:10.1038/nm.2251
44. Brenca M, Rossi S, Lorenzetto E, Piccinin E, Piccinin S, Rossi F, et al. SMARCB1/INI1 genetic inactivation is responsible for tumorigenic properties of epithelioid sarcoma cell line VAESBJ. Mol Cancer Ther (2013) 12(6):1060–72. doi:10.1158/1535-7163.MCT-13-0005
45. Lee R, Stewart C, Carter S, Ambrogio L, Cibulskis K, Sougnez C, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest (2012) 122(8):2983–8. doi:10.1172/JCI64400
46. Klochendler Yeivin A, Picarsky E, Yaniv M. Increased DNA damage sensitivity and apoptosis in cells lacking the Snf5/Ini1 subunit of the SWI/SNF chromatin remodeling complex. Mol Cell Biol (2006) 26(7):2661–74. doi:10.1128/MCB.26.7.2661-2674.2006
47. Isakoff M, Sansam C, Tamayo P, Subramanian A, Evans J, Fillmore C, et al. Inactivation of the Snf5 tumor suppressor stimulates cell cycle progression and cooperates with p53 loss in oncogenic transformation. Proc Natl Acad Sci U S A (2005) 102(49):17745–50. doi:10.1073/pnas.0509014102
48. Wang X, Sansam C, Thom C, Metzger D, Evans J, Nguyen PTL, et al. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res (2009) 69(20):8094–101. doi:10.1158/0008-5472.CAN-09-0733
49. Hornick J, Dal Cin P, Fletcher CDM. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol (2009) 33(4):542–50. doi:10.1097/PAS.0b013e3181882c54
50. Kohashi K, Izumi T, Oda Y, Yamamoto H, Tamiya S, Taguchi T, et al. Infrequent SMARCB1/INI1 gene alteration in epithelioid sarcoma: a useful tool in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol (2009) 40(3):349–55. doi:10.1016/j.humpath.2008.08.007
51. Le Loarer F, Zhang L, Fletcher C, Ribeiro A, Singer S, Italiano A, et al. Consistent SMARCB1 homozygous deletions in epithelioid sarcoma and in a subset of myoepithelial carcinomas can be reliably detected by FISH in archival material. Genes Chromosomes Cancer (2014) 53(6):475–86. doi:10.1002/gcc.22159
52. Papp G, Changchien Y-C, Péterfia B, Pecsenka L, Krausz T, Stricker T, et al. SMARCB1 protein and mRNA loss is not caused by promoter and histone hypermethylation in epithelioid sarcoma. Mod Pathol (2013) 26(3):393–403. doi:10.1038/modpathol.2012.190
53. Sullivan L, Folpe A, Pawel B, Judkins A, Biegel J. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol (2013) 26(3):385–92. doi:10.1038/modpathol.2012.175
54. Papp G, Krausz T, Stricker T, Szendrői M, Sápi Z. SMARCB1 expression in epithelioid sarcoma is regulated by miR-206, miR-381, and miR-671-5p on Both mRNA and protein levels. Genes Chromosomes Cancer (2014) 53(2):168–76. doi:10.1002/gcc.22128
55. Kohashi K, Yamamoto H, Kumagai R, Yamada Y, Hotokebuchi Y, Taguchi T, et al. Differential microRNA expression profiles between malignant rhabdoid tumor and epithelioid sarcoma: miR193a-5p is suggested to downregulate SMARCB1 mRNA expression. Mod Pathol (2014) 27(6):832–9. doi:10.1038/modpathol.2013.213
56. Vivanco I, Sawyers C. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer (2002) 2(7):489–501. doi:10.1038/nrc839
57. Darr J, Klochendler A, Isaac S, Eden A. Loss of IGFBP7 expression and persistent AKT activation contribute to SMARCB1/Snf5-mediated tumorigenesis. Oncogene (2014) 33(23):3024–32. doi:10.1038/onc.2013.261
58. Imura Y, Yasui H, Outani H, Wakamatsu T, Hamada K, Nakai T, et al. Combined targeting of mTOR and c-MET signaling pathways for effective management of epithelioid sarcoma. Mol Cancer (2014) 13:185. doi:10.1186/1476-4598-13-185
59. Xie X, Ghadimi MPH, Young E, Belousov R, Zhu Q-S, Liu J, et al. Combining EGFR and mTOR blockade for the treatment of epithelioid sarcoma. Clin Cancer Res (2011) 17(18):5901–12. doi:10.1158/1078-0432.CCR-11-0660
60. Lim H, Crowe P, Yang J-L. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J Cancer Res Clin Oncol (2015) 141(4):671–89. doi:10.1007/s00432-014-1803-3
61. Sibilia M, Kroismayr R, Lichtenberger B, Natarajan A, Hecking M, Holcmann M. The epidermal growth factor receptor: from development to tumorigenesis. Differentiation (2007) 75(9):770–87. doi:10.1111/j.1432-0436.2007.00238.x
62. Dobashi Y, Takei N, Suzuki S, Yoneyama H, Hanawa M, Ooi A. Aberration of epidermal growth factor receptor expression in bone and soft-tissue tumors: protein overexpression, gene amplification and activation of downstream molecules. Mod Pathol (2004) 17(12):1497–505. doi:10.1038/modpathol.3800218
63. Dobashi Y, Suzuki S, Sugawara H, Ooi A. Involvement of epidermal growth factor receptor and downstream molecules in bone and soft tissue tumors. Hum Pathol (2007) 38(6):914–25. doi:10.1016/j.humpath.2006.12.005
64. Bode B, Frigerio S, Behnke S, Senn B, Odermatt B, Zimmermann D, et al. Mutations in the tyrosine kinase domain of the EGFR gene are rare in synovial sarcoma. Mod Pathol (2006) 19(4):541–7. doi:10.1038/modpathol.3800560
65. Tawbi H, Thomas D, Lucas D, Biermann JS, Schuetze S, Hart A, et al. Epidermal growth factor receptor expression and mutational analysis in synovial sarcomas and malignant peripheral nerve sheath tumors. Oncologist (2008) 13(4):459–66. doi:10.1634/theoncologist.2007-0166
66. Cascio M, O’Donnell R, Horvai A. Epithelioid sarcoma expresses epidermal growth factor receptor but gene amplification and kinase domain mutations are rare. Mod Pathol (2010) 23(4):574–80. doi:10.1038/modpathol.2010.2
67. Ino Y, Gotoh M, Sakamoto M, Tsukagoshi K, Hirohashi S. Dysadherin, a cancer-associated cell membrane glycoprotein, down-regulates E-cadherin and promotes metastasis. Proc Natl Acad Sci U S A (2002) 99(1):365–70. doi:10.1073/pnas.012425299
68. Nam J-S, Kang M-J, Suchar A, Shimamura T, Kohn E, Michalowska A, et al. Chemokine (C-C motif) ligand 2 mediates the prometastatic effect of dysadherin in human breast cancer cells. Cancer Res (2006) 66(14):7176–84. doi:10.1158/0008-5472.CAN-06-0825
69. Park J-R, Kim R-J, Lee Y-K, Kim S-R, Roh K-J, Oh S, et al. Dysadherin can enhance tumorigenesis by conferring properties of stem-like cells to hepatocellular carcinoma cells. J Hepatol (2011) 54(1):122–31. doi:10.1016/j.jhep.2010.06.026
70. Izumi T, Oda Y, Hasegawa T, Nakanishi Y, Iwasaki H, Sonobe H, et al. Prognostic significance of dysadherin expression in epithelioid sarcoma and its diagnostic utility in distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Mod Pathol (2006) 19(6):820–31. doi:10.1038/modpathol.3800599
71. Lee Y-K, Lee S-Y, Park J-R, Kim R-J, Kim S-R, Roh K-J, et al. Dysadherin expression promotes the motility and survival of human breast cancer cells by AKT activation. Cancer Sci (2012) 103(7):1280–9. doi:10.1111/j.1349-7006.2012.02302.x
72. Behrens J. Control of beta-catenin signaling in tumor development. Ann N Y Acad Sci (2000) 910:21–33. doi:10.1111/j.1749-6632.2000.tb06698.x
73. Sakharpe A, Lahat G, Gulamhusein T, Liu P, Bolshakov S, Nguyen T, et al. Epithelioid sarcoma and unclassified sarcoma with epithelioid features: clinicopathological variables, molecular markers, and a new experimental model. Oncologist (2011) 16(4):512–22. doi:10.1634/theoncologist.2010-0174
74. Saito T, Oda Y, Itakura E, Shiratsuchi H, Kinoshita Y, Oshiro Y, et al. Expression of intercellular adhesion molecules in epithelioid sarcoma and malignant rhabdoid tumor. Pathol Int (2001) 51(7):532–42. doi:10.1046/j.1440-1827.2001.01232.x
75. van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci (2008) 65(23):3756–88. doi:10.1007/s00018-008-8281-1
76. Tian X, Liu Z, Niu B, Zhang J, Tan T, Lee S, et al. E-cadherin/β-catenin complex and the epithelial barrier. J Biomed Biotechnol (2011) 2011:567305.
77. Wu Y, Zhou B. New insights of epithelial-mesenchymal transition in cancer metastasis. Chin J Biochem Biophys (2008) 40(7):643–50. doi:10.1111/j.1745-7270.2008.00443.x
78. Bast RC, Klug TL, St John E, Jenison E, Niloff JM, Lazarus H, et al. A radioimmunoassay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N Engl J Med (1983) 309(15):883–7. doi:10.1056/NEJM198310133091503
79. Kato H, Hatori M, Kokubun S, Watanabe M, Smith R, Hotta T, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol (2004) 34(3):149–54. doi:10.1093/jjco/hyh027
80. Hoshino M, Kawashima H, Ogose A, Kudo N, Ariizumi T, Hotta T, et al. Serum CA 125 expression as a tumor marker for diagnosis and monitoring the clinical course of epithelioid sarcoma. J Cancer Res Clin Oncol (2010) 136(3):457–64. doi:10.1007/s00432-009-0678-1
81. Sabbatini P, Harter P, Scambia G, Sehouli J, Meier W, Wimberger P, et al. Abagovomab as maintenance therapy in patients with epithelial ovarian cancer: a phase III trial of the AGO OVAR, COGI, GINECO, and GEICO – the MIMOSA study. J Clin Oncol (2013) 31(12):1554–61. doi:10.1200/JCO.2012.46.4057
82. Buzzonetti A, Fossati M, Catzola V, Scambia G, Fattorossi A, Battaglia A. Immunological response induced by abagovomab as a maintenance therapy in patients with epithelial ovarian cancer: relationship with survival-a substudy of the MIMOSA trial. Cancer Immunol Immunother (2014) 63(10):1037–45. doi:10.1007/s00262-014-1569-0
83. Roché H, Julia AM, Jozan S, Muller C, Dastugue N, Arquier MA, et al. Characterization and chemosensitivity of a human epithelioid sarcoma cell line (SARCCR 2). Int J Cancer (1993) 54(4):663–8. doi:10.1002/ijc.2910540423
84. Reinecke P, Knopf C, Schmitz M, Schneider EM, Gabbert HE, Gerharz CD. Growth inhibitory effects of paclitaxel on human epithelioid sarcoma in vitro: heterogeneity of response and the multidrug resistance phenotype. Cancer (2000) 88(7):1614–22. doi:10.1002/(SICI)1097-0142(20000401)88:7<1614::AID-CNCR16>3.0.CO;2-X
85. Kusakabe H, Iwasaki H, Sano K, Kiyokane K. Expression of lung resistance protein in epithelioid sarcoma in vitro and in vivo. Arch Dermatol Res (2000) 292(6):292–300. doi:10.1007/s004030000127
86. Penot P, Bouaziz JD, Battistella M, Kerob D, Pagès C, Vilmer C, et al. Stabilization of multiple metastatic epithelioid sarcoma under treatment with sunitinib malate. Br J Dermatol (2013) 168(4):871–3. doi:10.1111/bjd.12038
87. van der Graaf WTA, Blay J-Y, Chawla S, Kim D-W, Bui Nguyen B, Casali P, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (2012) 379(9829):1879–86. doi:10.1016/S0140-6736(12)60651-5
88. Sokolowski E, Turina CB, Kikuchi K, Langenau DM, Keller C. Proof-of-concept rare cancers in drug development: the case for rhabdomyosarcoma. Oncogene (2014) 33(15):1877–89. doi:10.1038/onc.2013.129
89. Lin L, Hicks D, Xu B, Sigel J, Bergfeld W, Montgomery E, et al. Expression profile and molecular genetic regulation of cyclin D1 expression in epithelioid sarcoma. Mod Pathol (2005) 18(5):705–9. doi:10.1038/modpathol.3800349
90. Lee MW, Jee KJ, Han SS, Gong GY, Choi JH, Moon KC, et al. Comparative genomic hybridization in epithelioid sarcoma. Br J Dermatol (2004) 151(5):1054–9. doi:10.1111/j.1365-2133.2004.06246.x
91. Orrock J, Abbott J, Gibson L, Folpe A. INI1 and GLUT-1 expression in epithelioid sarcoma and its cutaneous neoplastic and nonneoplastic mimics. Am J Dermatopathol (2009) 31(2):152–6. doi:10.1097/DAD.0b013e31818a5c4f
92. Raoux D, Péoc’h M, Pedeutour F, Vaunois B, Decouvelaere A, Folpe A. Primary epithelioid sarcoma of bone: report of a unique case, with immunohistochemical and fluorescent in situ hybridization confirmation of INI1 deletion. Am J Surg Pathol (2009) 33(6):954–8. doi:10.1097/PAS.0b013e31819b92d5
93. Kohashi K, Yamada Y, Hotokebuchi Y, Yamamoto H, Taguchi T, Iwamoto Y, et al. ERG and SALL4 expressions in SMARCB1/INI1-deficient tumors: a useful tool for distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol (2014) 46(2):225–30.
94. Stockman D, Hornick J, Deavers M, Lev D, Lazar A, Wang W-L. ERG and FLI1 protein expression in epithelioid sarcoma. Mod Pathol (2014) 27(4):496–501. doi:10.1038/modpathol.2013.161
95. Miettinen M, Wang Z, Sarlomo Rikala M, Abdullaev Z, Pack S, Fetsch J. ERG expression in epithelioid sarcoma: a diagnostic pitfall. Am J Surg Pathol (2013) 37(10):1580–5. doi:10.1097/PAS.0b013e31828de23a
96. Li L, Fan X-S, Xia Q-Y, Rao Q, Liu B, Yu B, et al. Concurrent loss of INI1, PBRM1, and BRM expression in epithelioid sarcoma: implications for the cocontributions of multiple SWI/SNF complex members to pathogenesis. Hum Pathol (2014) 45(11):2247–54. doi:10.1016/j.humpath.2014.06.027
97. Kusakabe H, Yonebayashi K, Sakatani S, Iki M, Nakasuji K, Kiyokane K, et al. Metastatic epithelioid sarcoma with an N-ras oncogene mutation. Am J Dermatopathol (1994) 16(3):294–300. doi:10.1097/00000372-199406000-00012
98. de Torres C, Cardesa T, Rodríguez Perales S, Cigudosa J, Mora J. Epithelioid sarcoma with SYT-SSX1 fusion gene expression: molecular and cytogenetic analysis. Cancer Genet Cytogenet (2005) 162(1):50–6. doi:10.1016/j.cancergencyto.2005.03.009
99. Gerharz CD, Ramp U, Reinecke P, Schardt C, Friebe U, Déjosez M, et al. Analysis of growth factor-dependent signalling in human epithelioid sarcoma cell lines: clues to the role of autocrine, juxtacrine and paracrine interactions in epithelioid sarcoma. Eur J Cancer (2000) 36(9):1171–9. doi:10.1016/S0959-8049(00)00053-8
100. Kusakabe H, Sakatani S, Yonebayashi K, Kiyokane K. Establishment and characterization of an epithelioid sarcoma cell line with an autocrine response to interleukin-6. Arch Dermatol Res (1997) 289(4):224–33. doi:10.1007/s004030050184
101. Braverman S, Helson C, Helson L. Hemin toxicity in a human epithelioid sarcoma cell line. Anticancer Res (1995) 15(5B):1963–7.
102. Engers R, Gerharz CD, Donner A, Mrzyk S, Krause Paulus R, Petek O, et al. In vitro invasiveness of human epithelioid-sarcoma cell lines: association with cell motility and inverse correlation with the expression of tissue inhibitor of metalloproteinases. Int J Cancer (1999) 80(3):406–12. doi:10.1002/(SICI)1097-0215(19990129)80:3<406::AID-IJC12>3.0.CO;2-L
103. Engers R, van Roy F, Heymer T, Ramp U, Moll R, Dienst M, et al. Growth inhibition in clonal subpopulations of a human epithelioid sarcoma cell line by retinoic acid and tumour necrosis factor alpha. Br J Cancer (1996) 73(4):491–8. doi:10.1038/bjc.1996.86
104. Emori M, Tsukahara T, Murase M, Kano M, Murata K, Takahashi A, et al. High expression of CD109 antigen regulates the phenotype of cancer stem-like cells/cancer-initiating cells in the novel epithelioid sarcoma cell line ESX and is related to poor prognosis of soft tissue sarcoma. PLoS One (2013) 8(12):e84187. doi:10.1371/journal.pone.0084187
105. Reeves BR, Fisher C, Smith S, Courtenay VD, Robertson D. Ultrastructural, immunocytochemical, and cytogenetic characterization of a human epithelioid sarcoma cell line (RM-HS1). J Natl Cancer Inst (1987) 78(1):7–18.
106. Kelland LR, Bingle L. Radiosensitivity and characterisation of a newly established cell line from an epithelioid sarcoma. Br J Cancer (1988) 58(3):322–5. doi:10.1038/bjc.1988.211
107. Gerharz CD, Moll R, Ramp U, Mellin W, Gabbert HE. Multidirectional differentiation in a newly established human epithelioid sarcoma cell line (GRU-1) with co-expression of vimentin, cytokeratins and neurofilament proteins. Int J Cancer (1990) 45(1):143–52. doi:10.1002/ijc.2910450126
108. Sonobe H, Furihata M, Iwata J, Oka T, Ohtsuki Y, Hamasato S, et al. Morphological characterization of a new human epithelioid sarcoma cell line, ES020488, in vitro and in vivo. Virchows Arch B Cell Pathol (1993) 63(4):219–25. doi:10.1007/BF02899265
109. Helson C, Melamed M, Braverman S, Traganos F, Preti R, Helson L. Va-es-bj – an epithelioid sarcoma cell-line. Int J Oncol (1995) 7(1):51–6.
110. Stenman G, Kindblom LG, Willems J, Angervall L. A cell culture, chromosomal and quantitative DNA analysis of a metastatic epithelioid sarcoma. Deletion 1p, a possible primary chromosomal abnormality in epithelioid sarcoma. Cancer (1990) 65(9):2006–13. doi:10.1002/1097-0142(19900501)65:9<2006::AID-CNCR2820650921>3.0.CO;2-Q
111. Nishio J, Iwasaki H, Nabeshima K, Ishiguro M, Naumann S, Isayama T, et al. Establishment of a new human epithelioid sarcoma cell line, FU-EPS-1: molecular cytogenetic characterization by use of spectral karyotyping and comparative genomic hybridization. Int J Oncol (2005) 27(2):361–9. doi:10.3892/ijo.27.2.361
Keywords: epithelioid sarcoma, SMARCB1, INI1, BAF47, SWI/SNF complex
Citation: Noujaim J, Thway K, Bajwa Z, Bajwa A, Maki RG, Jones RL and Keller C (2015) Epithelioid sarcoma: opportunities for biology-driven targeted therapy. Front. Oncol. 5:186. doi: 10.3389/fonc.2015.00186
Received: 14 May 2015; Accepted: 03 August 2015;
Published: 17 August 2015
Edited by:
Thomas Grunewald, Ludwig Maximilian University of Munich, GermanyReviewed by:
Jaume Mora, Hospital Sant Joan de Déu Barcelona, SpainStefan Burdach, Technische Universität München, Germany
Copyright: © 2015 Noujaim, Thway, Bajwa, Bajwa, Maki, Jones and Keller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Noujaim, Royal Marsden Hospital, Fulham Road, London SW3 6JJ, UK,am9uYXRoYW4ubm91amFpbUBybWgubmhzLnVr;
Charles Keller, Children’s Cancer Therapy Development Institute, 320 East Vine Drive, Fort Collins, CO 80524, USA,Y2hhcmxlc0BjYy10ZGkub3Jn