 Michael D. Deel
Michael D. Deel Jenny J. Li
Jenny J. Li Lisa E. S. Crose
Lisa E. S. Crose Corinne M. Linardic
Corinne M. Linardic- 1Division of Hematology-Oncology, Department of Pediatrics, Duke University School of Medicine, Durham, NC, USA
- 2Duke University School of Medicine, Durham, NC, USA
- 3Department of Pharmacology and Cancer Biology, Duke University School of Medicine, Durham, NC, USA
The Hippo signaling pathway is an evolutionarily conserved developmental network vital for the regulation of organ size, tissue homeostasis, repair and regeneration, and cell fate. The Hippo pathway has also been shown to have tumor suppressor properties. Hippo transduction involves a series of kinases and scaffolding proteins that are intricately connected to proteins in developmental cascades and in the tissue microenvironment. This network governs the downstream Hippo transcriptional co-activators, YAP and TAZ, which bind to and activate the output of TEADs, as well as other transcription factors responsible for cellular proliferation, self-renewal, differentiation, and survival. Surprisingly, there are few oncogenic mutations within the core components of the Hippo pathway. Instead, dysregulated Hippo signaling is a versatile accomplice to commonly mutated cancer pathways. For example, YAP and TAZ can be activated by oncogenic signaling from other pathways, or serve as co-activators for classical oncogenes. Emerging evidence suggests that Hippo signaling couples cell density and cytoskeletal structural changes to morphogenic signals and conveys a mesenchymal phenotype. While much of Hippo biology has been described in epithelial cell systems, it is clear that dysregulated Hippo signaling also contributes to malignancies of mesenchymal origin. This review will summarize the known molecular alterations within the Hippo pathway in sarcomas and highlight how several pharmacologic compounds have shown activity in modulating Hippo components, providing proof-of-principle that Hippo signaling may be harnessed for therapeutic application in sarcomas.
Introduction
Overview of Pediatric Sarcomas
Sarcomas account for ~1% of all malignancies, but occur with higher frequency in children compared to adults, comprising ~15% of all childhood malignancies (1). The mainstay of treatment includes combining primary tumor control with surgery and/or radiation and systemic chemotherapy. While survival rates for localized sarcomas have improved to >70%, children with metastatic or recurrent disease continue to have dismal outcomes (2, 3).
Malignant bone and soft-tissue sarcomas arise in connective tissues (including bone, fat, muscle, blood vessels, deep skin tissues, nerves, and cartilage) and represent a histologically and molecularly heterogeneous group of tumors. Although the precise cell of origin of most of these tumors is not known, sarcomas are thought to develop as a result of genetic alterations in mesenchymal progenitor cells. While older adult patients often develop sarcomas with complex genetic karyotypes, there are relatively few genetic mutations driving tumorigenesis for the majority of childhood sarcomas, with the exception of some characteristic chromosomal translocations. In cases where the underlying molecular pathogenesis has been identified, this has not translated into improvements in survival rates for those patients with advanced or aggressive tumors, as many of the molecular drivers have not been able to pharmacologically modulated (2, 3). Discovering therapeutically targetable proteins that may be collaborating with such tumorigenic drivers is a promising new frontier for molecular oncology.
Overview of Hippo Signaling
The delineation of the Hippo pathway began in 2003 with identification of the Drosophila hippo gene. Hippo loss-of-function phenotypes were described concurrently by the Pan and Hariharan laboratories while screening for genes that negatively regulate tissue growth (4, 5). Subsequent studies unveiled Hippo signaling as an evolutionarily conserved cascade consisting of adaptor proteins and inhibitory kinases that regulate Yorkie, a pro-growth transcriptional regulator (6–8). Hippo signaling is highly conserved between Drosophila and mammals, and homologous pathway components across species are well described (9, 10). For this review, focus will be on mammalian Hippo signaling.
As shown in Figure 1, the mammalian Hippo pathway relays plasma membrane and cytoplasmic signals into the nucleus, where it regulates the expression of a diverse group of target genes that control essential cellular processes, including proliferation, differentiation, and apoptosis. Canonical Hippo transduction involves serine/threonine kinases mammalian STE20-like protein kinase 1/2 (MST1/2, which are homologs of Drosophila Hippo) (4, 5, 11, 12) and large tumor suppressor homolog 1/2 (LATS1/2) (7, 13, 14), which, in conjunction with adaptor proteins Salvador homolog 1 (SAV1) (12) and Mob kinase activator 1 (MOB1) (15), phosphorylate and inhibit the transcriptional co-activators Yes-associated protein 1 (YAP, a homolog of Yorkie) and transcriptional co-activator with PDZ-binding motif (TAZ) [also known as WW domain-containing transcription regulator 1, WWTR1] (16). The Hippo pathway is “ON” when MST1/2 and LATS1/2 kinases are active. Through an interaction between the PPxY (PY) motifs of LATS1/2 and the WW domains of YAP and TAZ, activated LATS1/2 lead to phosphorylation of YAP and TAZ, which results in YAP/TAZ cytoplasmic retention and β-TRCP (β-transducin repeat-containing E3 ubiquitin protein ligase)-dependent proteasomal degradation (9, 10). When Hippo signaling is inactive or “OFF”, YAP and TAZ are localized to the nucleus, where they serve as transcriptional co-activators for TEA domain-containing sequence-specific transcription factors (TEADs) (17–21) as well as other transcription factors (16).
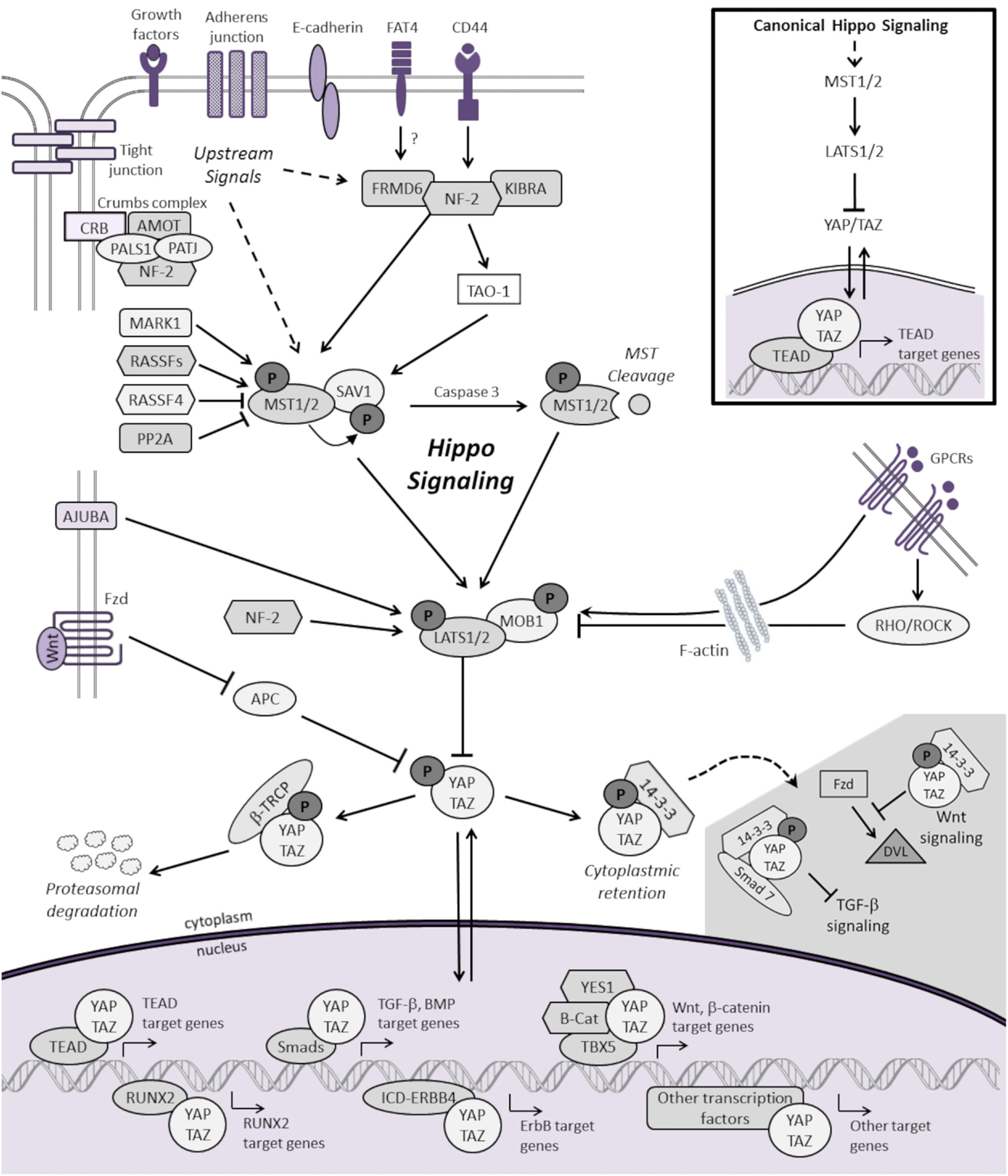
Figure 1. Schematic representation of the mammalian Hippo signaling cascade. Canonical Hippo transduction involves MST1/2 and LATS1/2 kinases, which, in conjunction with SAV1 and MOB1, phosphorylate, and inhibit the transcriptional co-activators YAP and TAZ. Regulation of YAP and TAZ are governed by plasma membrane proteins, cytoskeletal adaptor proteins, regulatory cross-talk from other signaling pathways, and intrinsic and extrinsic mechanical cues with the actin cytoskeleton. For simplicity, not all the known protein–protein interactions and regulators of Hippo signaling are represented. When Hippo signaling is “OFF”, YAP/TAZ translocate to the nucleus to serve as transcriptional co-activators for TEADs as well as other transcription factors (only a few of which are represented here) involved in cellular proliferation, differentiation, self-renewal, and apoptosis. See text for additional details.
Regulation of the Hippo Pathway
Much of our understanding of Hippo regulation comes from studies performed in epithelial tissue. In this context, the transcriptional activities of YAP and TAZ are regulated by four interconnected inputs: (1) plasma membrane proteins, which complex with YAP and TAZ directly to sequester them at cell–cell junctions; (2) upstream adaptor proteins, which activate core Hippo kinases to ultimately phosphorylate and repress YAP and TAZ; (3) regulatory cross-talk from other signaling pathways; and (4) intrinsic and extrinsic mechanical forces within the cell, which exert local control over YAP and TAZ localization. An overview of Hippo regulation is summarized below. For more detail, see the review by Grusche and colleagues (22), as well as three recent proteomic analyses that identified key protein–protein interactions with Hippo kinases, and YAP and TAZ within the global signaling network (23–25).
Regulation through Plasma Membrane Proteins
Growth control is signaled through plasma membrane proteins to upstream Hippo proteins, often in response to increased cell density. The Crumbs polarity complex, other polarity proteins, and adherens junctions, which all modulate each other, contribute inputs to various Hippo components (22, 26). E-cadherin and the junction-associated Ajuba protein family modulate MST and LATS kinases, respectively. The Crumbs complex involves transmembrane proteins that recruit scaffold proteins that localize to apical junctions and mediate cell polarity (27, 28). G-protein-coupled receptor (GPCR) ligands have been identified as regulators of Hippo signaling (29). Depending on the coupled G-protein, LATS1/2 kinases can either be activated or inhibited. YAP and TAZ directly influence the GPCR transcriptional activity, as YAP/TAZ are required for the expression of many GPCR-mediated target genes (29). The transmembrane hyaluronate receptor CD44 interacts with neurofibromin 2 (NF2, also known as Merlin) and other scaffold proteins to recruit LATS to the cell membrane, where it is phosphorylated (9, 30–32). Finally, the atypical cadherin protein Fat (Drosophila) is required for localization of Expanded (FRMD6 in mammals) to apical junctions, which results in activation of Hippo (MST1/2) (33). In avian cells, FAT4 has been shown to inhibit YAP1-mediated neuroprogenitor cell proliferation and differentiation (34).
Regulation through Upstream Intracellular Adaptor Proteins
The core Hippo pathway is controlled by a complex upstream regulatory network. MST and LATS kinase activity are regulated by several upstream proteins, including Ras-association domain-containing family proteins (RASSFs1-10) (35, 36), kidney and brain protein (KIBRA) (37–39), thousand and one amino acid protein 1 (TAO-1) (40), MAP/microtubule affinity-regulating kinase 1 (MARK1) (41), and NF2. Via their interaction through a homologous SARAH (SAlvador–RAssf–Hpo) binding domain, RASSFs and SAV1 regulate MST activity (42). MST1/2 complexes with SAV1 to directly phosphorylate LATS1/2. MST1/2 bound to SAV1 can also bind to and phosphorylate MOB1, which binds LATS1/2 to promote autophosphorylation. While a growing inventory of functional interactions between upstream proteins and Hippo kinases are well described, the degree to which their binding is dependent on tissue type or cellular context, as well as their reliance on canonical Hippo signaling, requires further investigation. Several of the aforementioned proteins can also directly alter YAP activity in a manner independent of MST and/or LATS kinases (31, 43).
The Hippo pathway plays a major role in arbitrating cell contact inhibition, cell proliferation, and promoting apoptosis (44). As cells increase in confluence, the tumor suppressor NF2 localizes near cell junctions to activate Hippo signaling (45, 46). YAP suppression has been shown to rescue the hyperproliferative phenotypes caused by NF2 inactivation in both mesothelioma (47) and meningioma (48). Furthermore, overexpression of a dominant-negative TEAD suppressed the tumor growth resulting from liver-specific NF2 deletion in mice (49). A negative feedback loop between YAP/TAZ and LATS2 has also been described. YAP and TAZ stimulation and TEAD binding induces LATS2 expression, both directly and by inducing NF2 (50). In addition, YAP and TAZ may negatively regulate each other. For example, Taz accumulates in the livers of Yap knockout mice, while either in vitro suppression or overexpression of Yap results in inverse changes to Taz protein expression (50).
Regulation through Cross-Talk with Other Pathways
Cell status and function, as well as overall tissue and organismal growth, is governed by an integrated network of morphogenic signals. Hippo transduction is proving to be a hub for such integration (51–53). Although studies are needed to clarify intra-pathway cross-talk in sarcomas, many of these pathways have been individually implicated in sarcomagenesis. YAP and TAZ are well recognized as co-activators for transcription factors of numerous signaling cascades. The specific ways in which signaling networks synergize or antagonize Hippo to coordinate biologic activity is only beginning to be understood. We highlight a few examples of regulatory cross-talk and refer to the studies referenced in Table 1 for additional details.
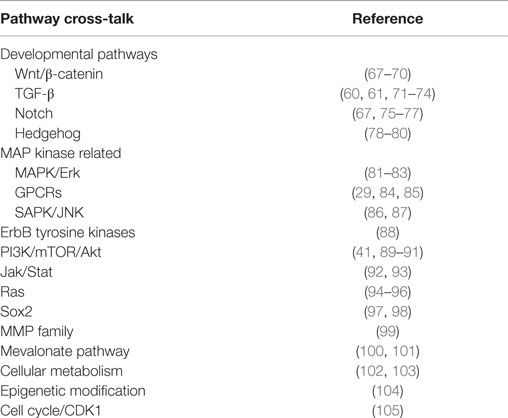
Table 1. Pathway cross-talk with Hippo signaling.
One example is illustrated by the relationship between the WNT and Hippo pathways. WNT activity is critical in myogenesis (54) and osteogenesis (55), and has recently been shown to be important in sarcomagenesis as well (56, 57). Rosenbluh et al. performed genome-scale loss-of-functions screens on 85 cancer cell lines (including osteosarcoma) and determined that WNT-active cancers are dependent upon β-catenin forming a complex with YAP and the transcription factor TBX5 to promote transcription of anti-apoptotic genes that are essential for cancer cell transformation and survival (58). This relationship was validated in a β-catenin-derived orthotopic colon cancer murine model, where Yap was required for tumor formation (58). In another study using murine cardiac muscle, knockdown of Hippo components Sav1, Mst1/2, or Lats2 results in increased Yap activity and cardiomyocyte proliferation with phenotypic cardiomegaly. Gene profiling from these mice reveal an elevated WNT signature, and the phenotypic effects could be offset by conditional loss of one β-catenin allele (59).
TGFβ and Hippo signaling also collaborate to direct cell behavior. YAP and TAZ associate with SMADs to promote transcription of TGFβ and BMP target genes (60–62). TGFβ signaling alters YAP/TAZ expression to drive mesenchymal stem cell (MSC) fate. For example, treatment of MSCs with BMP2 leads to increased TAZ expression and enhanced interaction with RUNX2 to promote osteoblast differentiation (63). Notch and Hippo signaling provide another example of coordinated cross-talk. Notch has been shown to be a driver of both bone and soft-tissue sarcomas (64–66). While no studies have examined the interplay of Notch and Hippo in sarcomas, overexpression of Yap1 in mouse intestinal epithelia stimulates Notch signaling and the expansion of undifferentiated progenitor cells. However, treatment with γ-secretase inhibitors to block Notch signaling prevents the intestinal dysplasia caused by YAP (67). Together, these insights provide a deeper appreciation for the complex molecular circuitry that regulates Hippo activity in cell biology and malignancy.
Cytoskeletal Regulation through Mechanical Influences
To sustain proper function, from facilitating organ development during embryogenesis to maintain homeostasis postnatally, cells must perceive their microenvironment and respond appropriately to stimuli. In addition to transmitting biochemical signals, cells also extract information from mechanical cues. Mechanotransduction is the ability to perceive and translate physical stimuli [elasticity of the extracellular matrix (ECM) and forces exerted by cell–cell or cell–matrix interactions] into biochemical signals on a cellular level. Cells adapt to changes in tension through rapid cytoskeletal remodeling (106–108). YAP and TAZ have emerged as dynamic factors linking remodeling to nuclear transcriptional outputs that control cell behavior. Thus, by modulating YAP/TAZ activity, mechanical stimuli can direct cell fate and guide stem cell maintenance, proliferation, and differentiation (107, 109–111). For example, in Drosophila, the tension modulated within the cytoskeleton causes proportionate changes in wing growth through an Ajuba-Warts (homolog of LATS) complex (112).
In situations of high mechanical stress and low cell confluence, YAP and TAZ are transcriptionally active, resulting in proliferation and tissue growth. However, with increasing cell contact, adhesion molecules stimulate LATS activity, resulting in YAP/TAZ phosphorylation and nuclear exclusion (44). Both F-actin polymerization and stress fiber formation lead to the nuclear localization and activation of YAP/TAZ, whereas disrupting F-actin inhibits YAP/TAZ transcriptional activity (113–116). As shown in Figure 2, ECM stiffness and cell shape/spreading can also regulate YAP/TAZ localization by regulating the activity of Rho-GTPases and the formation of stress fibers and actin bundles (106, 110, 113). In MSCs, YAP and TAZ act as both sensors of mechanotransduction and mediators of cellular responses to mechanical signals (117, 118). YAP and TAZ remain inactive in the cytoplasm and direct MSCs to differentiate into adipocytes when human MSCs are exposed to low ECM stiffness, are cultured on a soft matrix, or are manipulated into a small round shape. However, YAP and TAZ are active in the nucleus and MSCs differentiate into osteoblasts when they are subjected to high ECM stiffness, are grown on a stiff matrix, or are stretched and manipulated into a “spread-out” morphology (119, 120). This mechanical control over YAP/TAZ activity supersedes density cues from cell–cell or cell–matrix contact (113, 115).
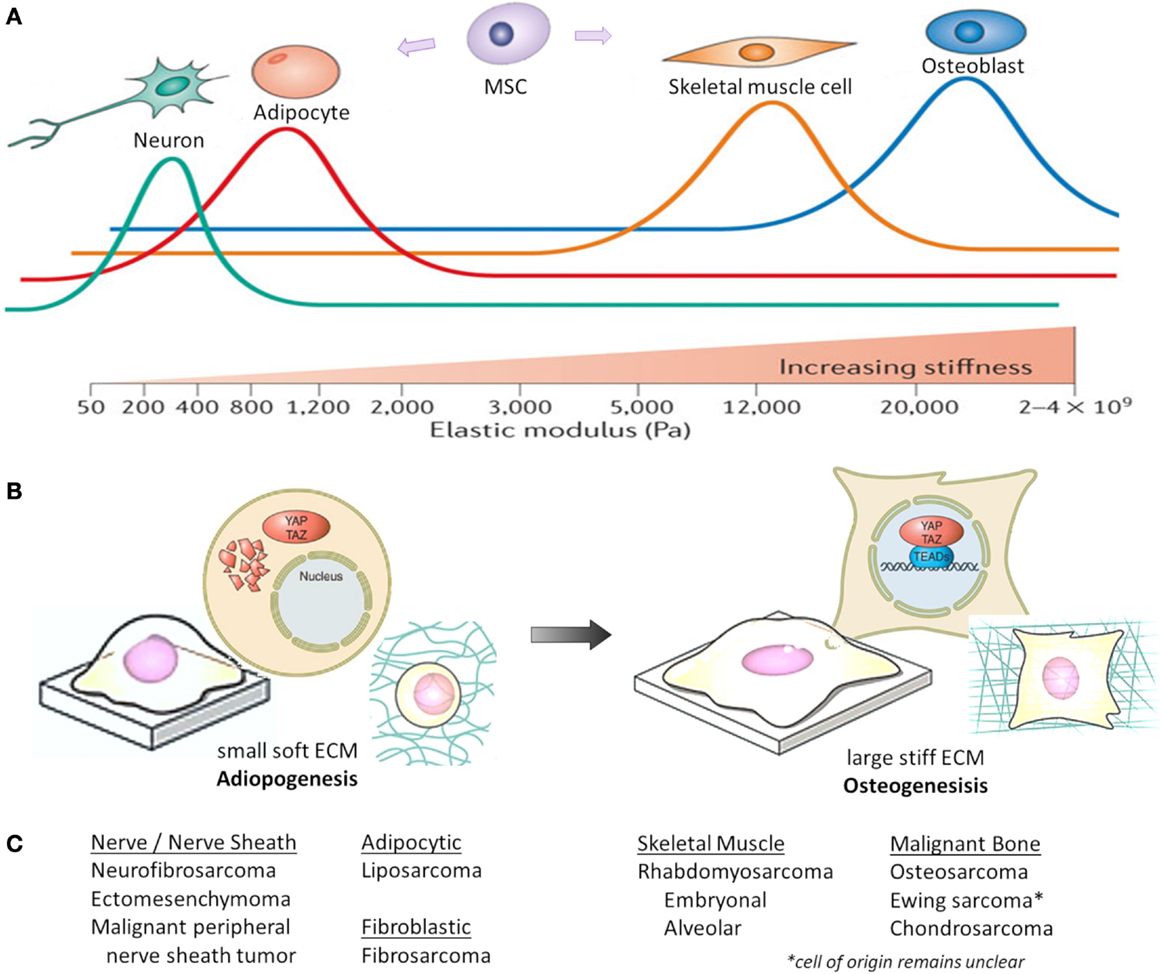
Figure 2. Mechanical and physical influences on MSC cell fate. Cell geometry and ECM stiffness regulate MSC lineage commitment into neurons, adipocytes, skeletal muscle cells, or osteoblasts. (A) Increasing ECM stiffness in vitro (by increasing type I collagen concentration and crosslinking) compromises tissue organization, inhibits apoptosis and lumen formation, and destabilizes adherens junctions. Through modeling different ECM elasticities in vitro, MSCs differentiate into the varying lineages at elasticities that recapitulate the physiological ECM stiffness of their corresponding natural niche (shown as colored lines, with peaks indicating maximal differentiation). Pa, Pascal. (B) When MSCs are either cultured on a soft matrix or are manipulated into a small round shape, YAP/TAZ remain inhibited in the cytoplasm and MSCs differentiate into adipocytes. However, when MSCs are either grown on a stiff matrix or stretched and manipulated into a “spread-out” morphology, YAP/TAZ localize to the nucleus as MSCs differentiate into osteoblasts. (C) Corresponding histologic sarcoma subtype [2013 WHO classification (230)], which may reflect varying lineage differentiation from mesenchymal progenitor cells. This represents only a theoretical link between mechanotransduction influencing mesenchymal progenitors and sarcoma, and not all sarcoma subtypes are represented. Figures (A,B) are modified with permission from Halder et al. (108) and Piccolo et al. (117).
Interestingly, manipulation of YAP/TAZ expression can overrule mechanical influences to direct differentiation. When YAP/TAZ is suppressed, MSCs grown on a stiff ECM will undergo adipogenic differentiation. However, when activated YAP is overexpressed, MSCs grown on a soft ECM will undergo osteogenic differentiation (113). Knockdown of LATS1/2 has almost no effect on YAP/TAZ regulation by mechanical cues, and LATS-insensitive TAZ still responds to mechanical cues (113). Therefore, cellular mechanical stress can directly impact proliferation and tissue growth through YAP/TAZ, independent from Hippo signaling. Together, these studies emphasize the importance of cytoskeletal regulation of YAP and TAZ transcriptional activity, and demonstrate that YAP and TAZ are required for mechanical signals to direct MSC fate.
Summary of Hippo Regulation
In summary, while the mechanistic and functional interactions between Hippo signaling and other regulatory pathways and cellular processes are not entirely understood, it is apparent that Hippo transduction links cell density and cell contact cues to morphogenic signals that regulate cell behavior. During development and tissue regeneration, the tumor suppressor function of Hippo signaling serves to offset the proliferative effects of other pathways. However, during malignant transformation, Hippo transduction is suppressed as cells evade contact inhibition, allowing the downstream effectors, YAP and TAZ, to co-activate TEADs as well as other transcription factors, to promote pro-proliferative and anti-apoptotic properties.
Hippo Signaling in Mesenchymal Stem Cell Fate
While the precise cellular origin for most sarcomas remains uncertain, they are presumed to arise from mesenchymal precursors that fail to undergo terminal differentiation. These precursors have stem-like characteristics, including high proliferative and self-renewal potential. Therefore, insight into MSC regulation, lineage commitment, and differentiation (121), may shed light on sarcoma biology. As shown in Figure 2C, sarcoma subtypes are histologically described by the features of their presumed mesenchymal lineage. Summarized below are the known roles of Hippo signaling in modulating normal bone (osteogenic), fat (adipogenic), and muscle (myogenic) development, which are the origins of the most common sarcomas. YAP/TAZ are also critical mediators of cancer stem cell biology, a topic reviewed by others (122).
Hippo Signaling in Osteogenic Differentiation
Osteogenic differentiation is coordinated by the transcription factor, RUNX2, and a host of co-regulators (123), which activate the expression of osteoblast-specific genes, including osteocalcin (63, 124, 125). Through direct binding of the TAZ WW domain to the PY motif on RUNX2, TAZ has been identified as a transcriptional co-activator of RUNX2. Expression of an active TAZ mutant enhances RUNX2-driven gene expression two to threefold (63, 126), while knockdown of TAZ in MSCs inhibits osteogenesis when the cells are cultured under conditions favoring osteoblast differentiation (63). Transgenic mice with osteoblast-specific overexpression of Taz have significantly higher whole body bone mineral density, increased bone formation, and higher expression of RUNX2, osteocalcin, ALP, and osterix (127). TAZ-mediated osteogenesis may also occur downstream of the WNT pathway, since WNT3A can cause PP1A-mediated TAZ dephosphorylation, leading to TAZ nuclear localization and induction of osteogenic differentiation (68).
While the role of TAZ in supporting osteogenesis is clear, the role of YAP is more complex. When an activated YAP mutant was overexpressed in MSCs, osteogenic differentiation was promoted over adipogenic differentiation, even under conditions favoring the latter (113). However, YAP can also act as repressor of RUNX2 when it is regulated by non-canonical pathways (128). For example, when Src/Yes tyrosine signaling is inhibited, Yap tyrosine phosphorylation is blocked, Yap dissociates from RUNX2, and osteocalcin is induced (128, 129). Last, there is evidence that YAP is a direct target of SOX2, a transcription factor important for MSC cell fate; in situations of high SOX2 or YAP expression, osteogenesis is blocked, while depletion of either SOX2 or YAP enhances osteogenesis (98).
In addition to YAP/TAZ, there is evidence that upstream scaffold proteins influence osteogenesis. Rassf2 knockout mice develop bone-remodeling defects, and in vitro studies show that ablation of RASSF2 suppresses osteoblastogenesis while promoting osteoclastogenesis (130).
Hippo Signaling in Adipogenic Differentiation
A key transcription factor orchestrating adipogenesis is peroxisome proliferator-activated receptor gamma (PPARγ), which contains a PY motif for binding the WW domains on YAP and TAZ (63). In this context, binding of TAZ has an inhibitory role, suppressing transcriptional activity. When cultured under conditions that promote adipogenic differentiation, knockdown of TAZ permits MSCs to differentiate toward this lineage (63). Similarly, treatment with the small molecule KR62980 (a ligand for PPARγ that antagonizes adipocyte differentiation) does so by promoting TAZ nuclear localization and enhanced interaction between TAZ and PPARγ (131).
Recent work has shed light on the role of YAP in adipogenesis. Similar to osteogenesis, YAP is downstream of SOX2. However, YAP levels must be fine-tuned; both over or under-expression of YAP inhibits adipogenesis. Mechanistically, YAP induces the Wnt antagonist Dkk1 to diminish osteogenic signaling in favor of adipogenesis. In addition to YAP and TAZ, upstream Hippo regulators have been implicated. The Hippo adaptor protein SAV1 contains WW domains that can interact with the PY motif within PPARγ (132). MST1/2 stimulated SAV1 to bind PPARγ, which stabilizes and increases PPARγ levels, ultimately leading to adipogenic differentiation. In addition, knockdown of MST1/2 or SAV1 results in the inhibition of adipogenesis (132), though it is not known whether this effect is through canonical Hippo transduction or an alternate pathway.
Hippo Signaling in Myogenic Differentiation
Myogenic differentiation is driven by the myogenic regulatory factor family [MRFs: MyoD, myogenin, MRF4, and myogenic factor 5 (Myf5)]) (133–135) in coordination with myocyte-specific MEF2 enhancer factors (136, 137). In murine C2C12 skeletal muscle myoblasts, YAP supports an undifferentiated phenotype and promotes myoblast proliferation (138–140). Upon differentiation, nuclear YAP is translocated to the cytoplasm, with a 20-fold increase in YAP phosphorylation. Overexpression of YAP S127A, a mutant that cannot be phosphorylated at the LATS-regulated site, impedes myotube formation, and alters the expression of MRFs (139). Activation of YAP causes upregulation of Myf5, which promotes myoblast proliferation. Activated YAP also leads to down-regulation of MyoD and MEF2, which are important in cell-cycle exit and differentiation, as well as upregulation of inhibitors of MyoD and MEF2, such as ID2, Twist1, and Snai1/2 (133, 138). In activated satellite cells, which are resident stem cells of skeletal muscle, high YAP activity prevents differentiation and promotes proliferation (138, 140). YAP suppression dramatically reduces satellite cell-derived myoblast proliferation (140). Additionally, muscle CAT (MCAT) elements, which are TEAD-binding sites, are found in the promoters of genes that are selectively expressed in terminally differentiated skeletal muscle (140, 141).
Interestingly, while YAP inhibits myogenic differentiation, some studies suggest TAZ may enhance myogenesis. TAZ physically binds MyoD to enhance binding to the myogenin gene promoter to activate MyoD-dependent gene transcription (142, 143). Ectopic overexpression of TAZ in C2C12 myoblasts results in accelerated myofiber formation, whereas TAZ loss lessened myogenic differentiation (142).
Evidence of upstream Hippo pathway regulators in muscle differentiation is limited. However, MST was found to have a pro-differentiation role during an investigation of caspase 3 in myogenesis (144). While caspases are classically known for their role in apoptosis, non-apoptotic functions have been reported. This appears to be the case in myogenesis, as caspase 3 was robustly activated in differentiating myoblasts without inducing apoptosis. Caspase 3-deficient myoblasts or C2C12 cells treated with caspase inhibitors are less able to differentiate, in part due to caspase 3-mediated regulation of MST1. Additionally, MST1 is a substrate for caspase 3, and cleaved MST1 was enriched in myoblasts undergoing differentiation. In caspase 3-deficient myoblasts, introduction of the cleaved MST1 induced myogenic differentiation, proving a link between these two pathways. However, MST1 activation must be tightly controlled, as MST1 activation in wild-type myoblasts ultimately led to cell death (144). While this study suggests a role for MST1 in myogenic differentiation, connections between MST1 activation by caspase 3 and the canonical Hippo pathway in muscle remain to be determined.
The Molecular Basis for Hippo Signaling in Sarcomas
Sarcomas comprise a group of clinically and histologically diverse tumors of mesenchymal origin. They can develop anywhere in the body, with about half arising in bone and half in soft tissues. In children and adolescents, osteosarcoma (OS) and Ewing sarcoma (EWS) are the two most common malignant bone sarcomas, while rhabdomyosarcoma (RMS) and non-rhabdomyosarcoma soft-tissue sarcomas (NRSTSs) are the major classes of malignant soft-tissue sarcomas (145).
As reviewed earlier, Hippo signaling is essential for proper organ growth, amplification of tissue-specific progenitor cells during tissue regeneration, and cellular proliferation (10, 146). In 2007, Dong and colleagues generated a liver-specific conditional Yap1 transgenic mouse model that develops hepatocellular carcinoma (10). This led to the understanding that YAP is important in cancer and identified Hippo signaling as a tumor suppressor pathway in mammals. In other genetically engineered mouse models (GEMMs), mutations or altered expression of Hippo pathway genes gives rise to sarcomas, substantiating Hippo pathway deregulation in sarcomagenesis (138, 147–149). The next section will review the molecular basis of dysregulated Hippo signaling in bone and soft-tissue sarcomas. Each subsection will highlight the pro-tumorigenic role of YAP/TAZ, with subsequent cataloging of other Hippo pathway member involvement. Table 2 summarizes these alterations.
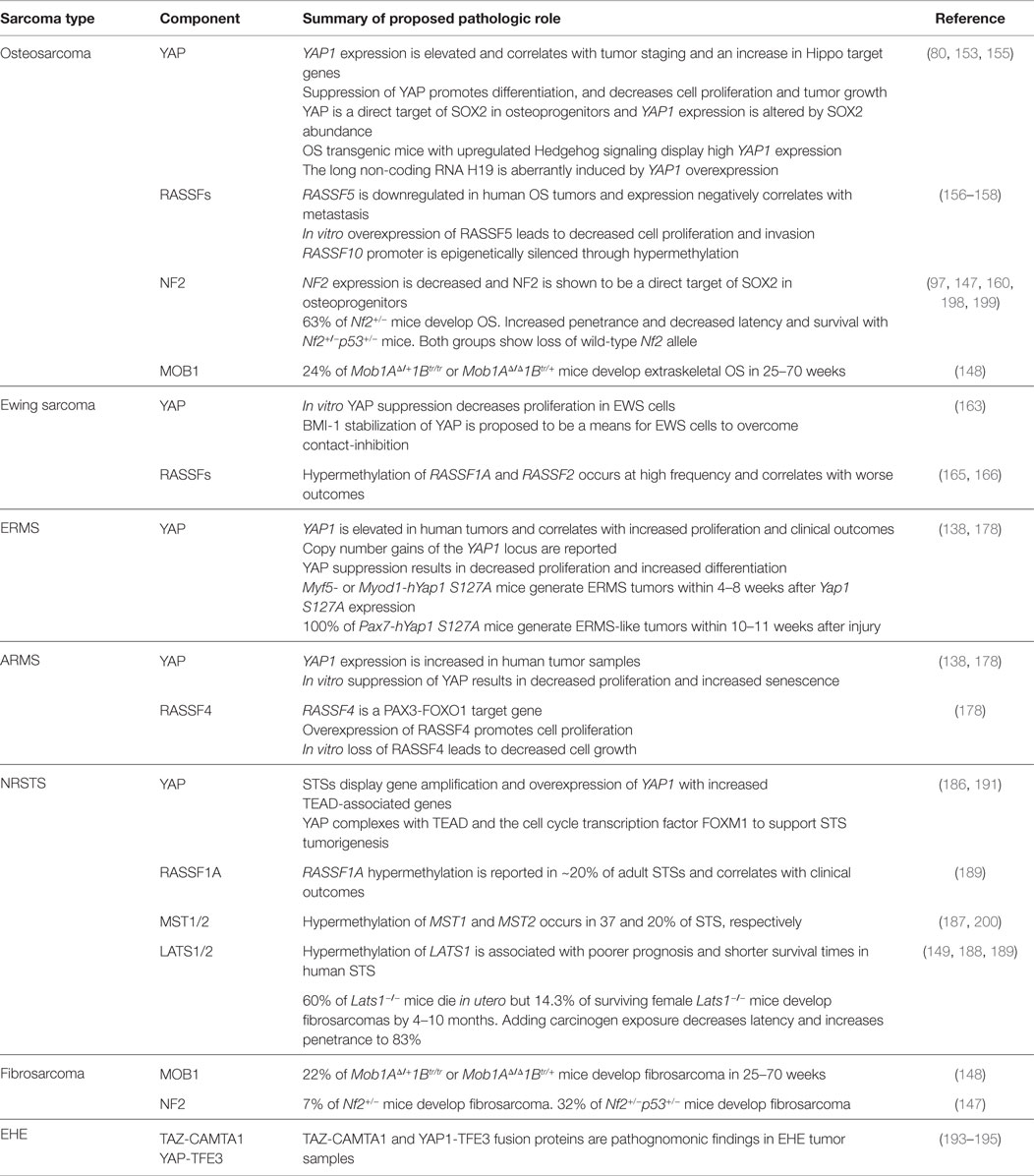
Table 2. Proposed involvement of Hippo pathway components in sarcoma biology.
Hippo Signaling in Osteosarcoma
Osteosarcoma is the most common primary malignancy of bone, with a 5-year overall survival of 60–70% (150). Given its decreased radiosensitivity compared to other sarcomas, surgical resection with chemotherapy is the mainstay of treatment. OS tumors are characterized by complex genomic rearrangements as well as copy number variations (151, 152). Mutations or loss-of-function of tumor suppressors RB1 and TP53 are two of the most common genetic alterations and are reported in ~50 and ~30% of tumors, respectively (151). Aberrations in Hippo signaling are proving to be important in the biology of OS.
YAP
Human tissue microarray analyses have revealed high YAP1 protein expression in OS compared to surrounding non-cancerous tissue, and expression correlates with staging (153). These findings corroborate other studies which showed high YAP1 expression in 78% of human OS samples and an increase in Hippo pathway target genes (80, 97, 154). Nuclear localization of Yap was found in Kios-5 murine OS cell lines, and Yap (and Taz, to a lesser extent) protein expression was also increased. In vitro suppression of Yap was associated with decreased cell proliferation and invasion, as well as decreased expression of Runx2, CyclinD1, and MMP-9. Decreased tumor growth was observed with in vivo Yap suppression in murine xenografts (155), as well as transgenic mouse models (80).
The mechanism of YAP upregulation in OS is complex but appears to be due in part to the stem cell transcription factor SOX2. In murine OS cell lines, Sox2 was found to directly repress the Hippo pathway activators, Nf2 and Kibra, leading to increased YAP. When grown as osteospheres, where stem cells are enriched, YAP expression was higher (and Nf2 lower) compared to adherent cells. In cells depleted of Sox2, either Yap overexpression or Nf2 suppression restored osteosphere formation. Conversely, suppressing Yap or overexpressing Nf2 promoted osteogenic differentiation and prevented osteosphere formation. The differentiated phenotype of OS cells induced by Nf2 could be overcome by either overexpressing wild-type or constitutively active mutant Yap, but not mutant Yap with a deficient TEAD-binding site. This regulation of Yap by Sox2 occurs through canonical Hippo signaling, as suppression of either Mst1/2 or Lats1/2 abolished Nf2-induced osteogenic differentiation as well as changes in Yap expression and function (97).
YAP can also be upregulated by Hedgehog (Hh) pathway activation. Malignant OS occurs with high penetrance in Ptch1c/+;p53+/−;HOC-Cre mutant mice, in which Hh signaling is partially upregulated in a p53 heterozygous background. Resultant tumors have high Yap1 expression, which is significantly reduced with Hh inhibition, and suppression of Yap1 blocks tumor progression. This same study showed that the Hh-Yap axis may regulate the expression of H19, a maternally imprinted long non-coding RNA implicated in tumorigenesis (80).
RASSFs
Two RASSFs (RASSF5 and RASSF10) have been implicated as tumor suppressors in OS. Similar to other RASSF family members, RASSF5 and RASSF10 are seen downregulated in human tumors (including OS) by CpG island promoter hypermethylation (156). In a human tissue microarray representing 45 OS samples, RASSF5 was significantly downregulated and expression negatively correlated with distant metastasis (157). In human U2OS cells, in vitro suppression of RASSF5 conveyed resistance to TNF-α-induced apoptosis, which is thought to occur through interaction and inactivation of the pro-apoptotic function of MST1 (158). Conversely, overexpression of RASSF5 in human OS cell lines decreases cell proliferation, increases apoptosis, and inhibits invasion.
NF2
In humans, germline or somatic mutations in one allele of NF2 result in the disease neurofibromatosis type 2, which is associated with schwannomas, meningiomas, and ependymomas. However, mice heterozygous for Nf2 develop a variety of malignant tumors at high frequency, including OS (63%). Somatic mutations of the wild-type Nf2 allele were found in almost all of these tumors, implying that loss of heterozygosity of Nf2 may be required for sarcomagenesis (147).
CD44
CD44 is a cell-surface glycoprotein that transmits extracellular signals to the ERK, AKT, and Hippo pathways (82, 159). CD44 was found to be suppressed by NF2, leading to decreased migration and invasion in OS cell lines in vitro, although an enhanced OS malignant phenotype was observed with knockdown of CD44 in mice xenografts (160). Others have shown that NF2 mediates contact growth inhibition through ECM signals by complexing with CD44 (32).
MOB1
In vitro overexpression of MOB1A impairs cellular proliferation, while suppression of MOB1A leads to aberrant mitosis (15). In double-mutant mice lacking both Mob1A and Mob1B, complete loss of both alleles (Mob1AΔ/Δ1Btr/tr, null mutation of Mob1A, gene trap of Mob1B) is embryonically lethal. However, double-mutant mice retaining one allele of either (Mob1AΔ/+1Btr/tr or Mob1AΔ/Δ1Btr/+) survive and spontaneously develop tumors with 100% penetrance within 70 weeks. Extraskeletal OS arose in 24% (9/37) of mice, while benign exostosis occurred in 92% (34/37). All the tumors examined from either single heterozygote (Mob1AΔ/+1Btr/tr or Mob1AΔ/Δ1Btr/+) group revealed loss of the wild-type Mob1 allele, suggesting loss of heterozygosity may be necessary for tumor growth (148).
Hippo Signaling in Ewing Sarcoma
Ewing sarcoma is the second most common malignant bone tumor in children and young adults. Although the 5-year overall survival is about 70%, 30–40% of patients either present with metastatic disease or develop recurrence, where outcomes are worse (161). EWS is characterized by a t(11;22) chromosomal translocation, which generates a fusion gene encoding the EWS-FLI1 chimeric protein that is thought to be the predominant driver of EWS tumorigenesis (162). The molecular basis for dysregulated Hippo signaling in EWS is beginning to be studied, as summarized below.
YAP
YAP suppression in human EWS cell lines decreases proliferation and anchorage-independent colony formation (163). A relationship between YAP and BMI-1, a Polycomb complex protein involved in chromatin remodeling (164), has been proposed. In studies examining the effect of cell density in cultured EWS cells, loss of BMI-1 had no effect in low-density, while it caused cell-cycle arrest and death under conditions of confluence. These findings may be due in part to the role of BMI-1 in stabilizing YAP expression and activity, and may serve as a means for BMI-1-driven EWS cells to overcome contact inhibition (163).
RASSFs
Hypermethylation of the promoter regions of RASSF1A and RASSF2 has been described in EWS and is correlated with worse clinical outcome (165, 166). One study of 55 human EWS tumors reported methylation rates for RASSF1A and RASSF2 of ~52 and ~42%, respectively (165). In in vitro studies, overexpression of either RASSF1A or RASSF2 in EWS cells reduced their ability to form colonies (165). In a separate study, methylation of RASSF1A was observed in 75% (3/4) of EWS cell lines and 68% (21/31) of human tumors (166), though these studies are contradicted by other reports that did not demonstrate increased RASSF1A hypermethylation (167, 168). The EWS-FLI1 fusion protein has recently been shown to provoke widespread epigenetic changes, including altered DNA methylation, although it is not known whether there is a direct effect on RASSF expression (168, 169).
Hippo Signaling in Rhabdomyosarcoma
Rhabdomyosarcomas are soft-tissue sarcomas and account for approximately 8% of all pediatric solid tumors (170). The two major histological subtypes are termed embryonal (ERMS) and alveolar (ARMS) rhabdomyosarcoma. ERMS, which is more common, typically arises in the head and neck or retroperitoneum of younger children and conveys a better prognosis (localized tumors have >70% 5-year overall survival) (171, 172). ERMS tumors demonstrate numerous chromosomal aberrations, including genomic amplifications, loss of heterozygosity of specific chromosomal regions, frequent chromosomal gains in 2, 8, 12, and 13, and loss-of-imprinting (171–174). ARMS make up about 25–30% of cases and usually arise in the extremities or trunk and occur more frequently in adolescents. ARMS is characterized by recurrent chromosomal translocations, principally t(2;13) and t(1;13), which result in the expression of PAX3-FOXO1 and PAX7-FOXO1 fusion proteins, respectively (175). These aberrant chimeric proteins are oncogenic transcription factors that confer a poor prognosis (5-year overall survival <15% for metastatic or recurrent tumors) (173, 175–177). Interestingly, fusion-negative histologic ARMS have a cytogenetic and molecular profile similar to ERMS, and correspondingly similar clinical behavior (177).
YAP
YAP protein is upregulated in both ERMS and ARMS tumors (138, 178). In ERMS and fusion-negative ARMS, this is due in part to increased YAP1 locus copy number. The importance of YAP in ERMS was confirmed by the remarkable finding that expression of YAP S127A is sufficient for ERMS tumorigenesis in a GEMM (138). This finding was particularly surprising given prior work showing YAP1 S127A expression in adult mouse muscle caused atrophy (179). Similar to this study, limb stiffness and gait defects were the initial phenotypes observed in Myf5/MyoD-YAP1 S127A mice (138). However, analysis of their muscle beds found that within the muscle damage were sites of active muscle regeneration and expansion of mononucleated cells. These were confirmed to be ERMS lesions, as they stained positive for ERMS histological markers. Tumor cells from these mice were transplantable, leading to secondary ERMS tumors with short latency. Given the high proportion of mononucleated cells in the primary tumor, Tremblay and colleagues hypothesized that satellite cells could serve as an ERMS cell of origin in this model. While expression of YAP1 S127A in the Pax7 (satellite) cell lineage did not induce ERMS formation, YAP1 S127A did transform satellite cells in the context of muscle injury. This suggests that hyperactive YAP signaling in activated satellite cells has transformative properties.
Using this GEMM model, hyperactive YAP signaling in ERMS tumors was found to induce a myogenic differentiation block. When YAP S127A expression was reduced, tumors rapidly regressed, and tumor cells spontaneously expressed markers of terminal muscle differentiation. Similarly, endogenous YAP suppression in ERMS RD cell xenografts caused myogenic differentiation (138). These findings are in line with earlier work implicating a role for YAP signaling in regulating myogenic differentiation. In proliferating C2C12 and satellite cells, YAP levels are high and localized in the nucleus. Upon differentiation stimulus, YAP mRNA expression is reduced and YAP becomes cytoplasmic (139, 140). This suggests an important role for YAP signaling in maintaining a high proliferative and anti-differentiation state. Similarly, YAP S127A can block C2C12 and satellite cell in vitro differentiation. This differentiation block is believed to be due to transcriptional changes induced by YAP-TEAD, particularly through upregulation of pro-proliferative genes and repression of MYOD1 and MEF2 regulation of terminal differentiation genes (138).
Additional studies have supported a role for YAP in RMS. A subset of ERMS tumors harbor mutations in the PKN1 gene (encoding a kinase of the protein kinase C superfamily), which imparts a gene expression signature associated with activated YAP (180). In ARMS cells, in vitro genetic suppression of YAP induces growth arrest and senescence (178).
RASSF4
A role for the Hippo pathway in ARMS began with the identification of RASSF4 as a PAX3-FOXO1 target gene (178). Using transcriptional profiling studies, PAX3-FOXO1-expressing myoblasts were found to upregulate RASSF4 expression. Further, PAX3-FOXO1-positive ARMS cell lines and human tumors had elevated RASSF4 levels, and high RASSF4 expression was associated with worse RMS clinical prognosis. Loss-of-function studies demonstrated that RASSF4 was promoting cell proliferation and senescence evasion in ARMS cells. These RASSF4 functions were due to inhibition of MST1 signaling to MOB1. While no changes in signaling to LATS1 were observed, RASSF4-deficient ARMS cells did express lower levels of YAP1 protein. However, cells expressing a hyperactive YAP1 (YAP S127A) could not reverse the phenotypes associated with RASSF4 loss, suggesting an indirect connection between RASSF4 and YAP signaling (178). Altogether, these studies suggest that suppression of MST1-MOB1 signaling is an important oncogenic function of RASSF4 in ARMS.
TEAD-NCOA2 Fusions
NCOA2 is a transcriptional co-activator for steroid and nuclear hormone receptors. Fusion of TEAD to NCOA2 was found in tumor tissue removed from a 4-week-old child with spindle cell RMS (181), a rare variant of ERMS (182). While NCOA2 gene rearrangements with other gene partners are seen in high frequency in congenital spindle cell RMS and mesenchymal chondrosarcomas (181, 183), the clinical and molecular significance of TEAD as a binding partner in this case is not known.
Hippo Signaling in Non-Rhabdomyosarcoma Soft-Tissue Sarcomas
Non-rhabdomyosarcoma soft-tissue sarcomas comprise the fifth most common group of solid tumors in children, accounting for 8–9% of childhood malignancies. These are histologically heterogeneous tumors that share some biologic characteristics. Surgical resection results in remission for about 80% of patients presenting with localized disease, though survival for those with unresected or metastatic disease remains poor (184). Many NRSTS, particularly those common in children, are characterized by disease-defining chromosomal translocations. Examples include synovial sarcoma t(X;18) and alveolar soft part sarcoma t(X;17), which result in the SYT-SSX and ASPL-TFE3 oncogenic fusion proteins, respectively (145). Other NRSTSs that are more common in adults, such as leiomyosarcoma or undifferentiated sarcoma, display multiple complex karyotypic abnormalities with frequent mutations in the TP53 and RB tumor suppressor pathways (185).
YAP
Nuclear staining for YAP is increased in a subset of human STS samples, compared to corresponding normal connective tissue (186). KRAS-based [LSLKrasG12D/+;Tp53fl/fl (KP)] GEMMs were used to further investigate the role of YAP in STS. Yap suppression in allograft tumors generated from KP cells results in decreased cell proliferation and tumor growth, and treatment with verteporfin to block the YAP–TEAD interaction decreased transcription of Yap1 target genes. Many of the downregulated mRNAs in this model were noted to also be targets of Foxm1, a transcription factor involved in cell-cycle progression. FOXM1 is ordinarily inhibited by direct interaction with members of the TP53 and RB tumor suppressor pathways, and it is often overexpressed in malignancies where these tumor suppressor functions have been lost (186). FOXM1 expression was found to be increased in a variety of human sarcoma samples. In xenograft studies, FOXM1 suppression inhibited sarcoma growth. Co-immunoprecipitation and ChIP-seq experiments reveal that FOXM1 physically associates with a YAP/TEAD complex (186). YAP suppression in human sarcoma cell lines resulted in decreased proliferation and decreased FOXM1 expression, suggesting a novel role for YAP in co-activating FOXM1-mediated transcription in STS.
MST1/2
Hypermethylation of MST1 and MST2 promoters occurs in 37 and 20% of all STS (including RMS), respectively (187). In leiomyosarcoma samples, hypermethylation of RASSF1A and MST2 were mutually exclusive, implying a common signaling pathway may exist for both genes. Surprisingly, methylation of the MST1 promoter appears to correlate with a decreased risk of tumor-related mortality (187), albeit from a retrospective cohort with a small sample size.
LATS
Reduced LATS gene expression was observed in 14% (7/50) of human adult STS tumors (188). These findings correlate with subtype, as three of four myxoid liposarcomas, three of seven leiomyosarcomas, and one of nine malignant fibrous histiocytomas showed reduced or no expression of LATS1. In one of those samples, an allelic loss of the LATS1 locus in chromosome 6q23-25.1, resulting from a missense point mutation, was observed. The other six samples showed aberrant hypermethylation of the putative LATS1 promoter (188), corroborating another study showing hypermethylation of the LATS1 promoter in 7% (3/43) of human STS samples (187). Hypermethylation of LATS1 in STSs is associated with a worse prognosis and shorter survival times (189). It is not known whether epigenetic regulation of Hippo pathway kinases alters the expression of YAP and TAZ.
In transgenic mouse models, most mice (60/101) homozygous for a null mutation in Lats1 died in utero or within post-natal day 1. However, ~14% of surviving female Lats1−/− mice developed large NRSTS by 4–10 months of age consistent with fibrosarcomas. After exposure to the carcinogen DMBA and repeated exposure to UVB, 83% (10/12) of Lats1−/− mice developed STSs, whereas no wild-type or heterozygous Lats1+/− mice developed tumors (149).
RASSF1A
Epigenetic silencing of RASSF1A via hypermethylation of its promoter occurs in 20% (17/84) of adult STSs (189). (This study included six cases of RMS, which did not reveal RASSF1A hypermethylation.) RASSF1A silencing was especially common in leiomyosarcomas, and overall was associated with an increase in tumor-related death.
VGLL3
Like YAP, VGLL3 is a TEAD co-activator and has been identified as an inhibitor of terminal adipogenic differentiation, suggesting that it has a core role in mesenchymal cell fate (190). In a study of 404 adult STSs, recurrent amplifications of chromosomes 11q22 and 3p12, which contain genes for YAP1 and Vestigial-like 3 (VGLL3), respectively, were identified in 10% of cases. Genomic amplification corresponded to overexpression of YAP1 and VGLL3 at the message level, and an increase in TEAD-associated genes. In vitro suppression of YAP1 or VGLL3 decreased cell proliferation and in the case of VGLL3, decreased migration (191). In a smaller study, analysis of eight NRSTS tumors identified 3p11-12 as a commonly amplified region of a ring chromosome 3 that was associated with high expression of VGLL3 (192).
TAZ-CAMTA1 and YAP-TFE3 Fusions
Fusions between the WWTR1 (gene name for TAZ protein) and CAMTA1 genes were first noted in a NRSTS subtype termed epithelioid hemangioendothelioma (EHE) (193). EHEs are vascular sarcomas that can develop in bone, soft tissue, or visceral organs, and they demonstrate a clinical behavior intermediate between benign hemangiomas and high-grade angiosarcomas. Sequencing of two tumors identified the t(1;3) translocation between WWTR1 and CAMTA1, and showed the fusion product to be under transcriptional control of the TAZ promoter. A larger study investigating 17 EHE tumors confirmed the translocation in all samples. The translocation and resulting transcript were not seen in epithelioid hemangioma and epithelioid angiosarcoma, morphologic mimics of EHE (194).
Subsequently, a YAP1-TFE3 fusion product was identified in nine EHE samples that were morphologically different from the WWTR1-CAMTA1 fusion-positive tumors (195). These findings were corroborated by two additional studies, the largest of which included 35 tumors and used a combination of IHC, FISH, and RT-PCR to validate WWTR1-CAMTA1 fusion events in 33 cases and YAP1-TFE3 protein in two cases (196, 197). The oncogenic role of these signature fusions in EHE, or the role of Hippo signaling in vascular sarcomas, has not yet been established.
Targeting Hippo Signaling for Therapy
Recognition of the importance of Hippo signaling in malignancy has led to preclinical studies aimed at targeting components of this pathway for anti-cancer therapy. Modeled genetic manipulation of Hippo components exhibit profound effects on tumorigenicity, which provides optimism that modulators of Hippo components could be effective in patients. Indeed, the Hippo cascade involves many protein–protein interactions that could serve as novel targets. For details on each potential modulator, see recent reviews in Ref. (201, 202).
Small Molecule Modulators of the Hippo Pathway
As listed in Table 3 and highlighted in Figure 3, several pharmacologic compounds, that directly or indirectly modulate Hippo pathway activity, have been identified. However, a number of important challenges exist. First, while kinases are often excellent targets for small molecule inhibitors, the majority of kinases in the Hippo pathway are tumor suppressors, and restoring lost tumor suppressive function is not easily achieved. Moreover, and as highlighted here, aberrant hyperactivity of oncogenic YAP and TAZ is often seen in malignancy as a result of mutations in proteins from other signaling networks, even in the presence of intact upstream Hippo kinase activity. However, small molecules aimed at increasing YAP/TAZ phosphorylation-induced nuclear export and proteosomal degradation could be effective at reducing their activity.
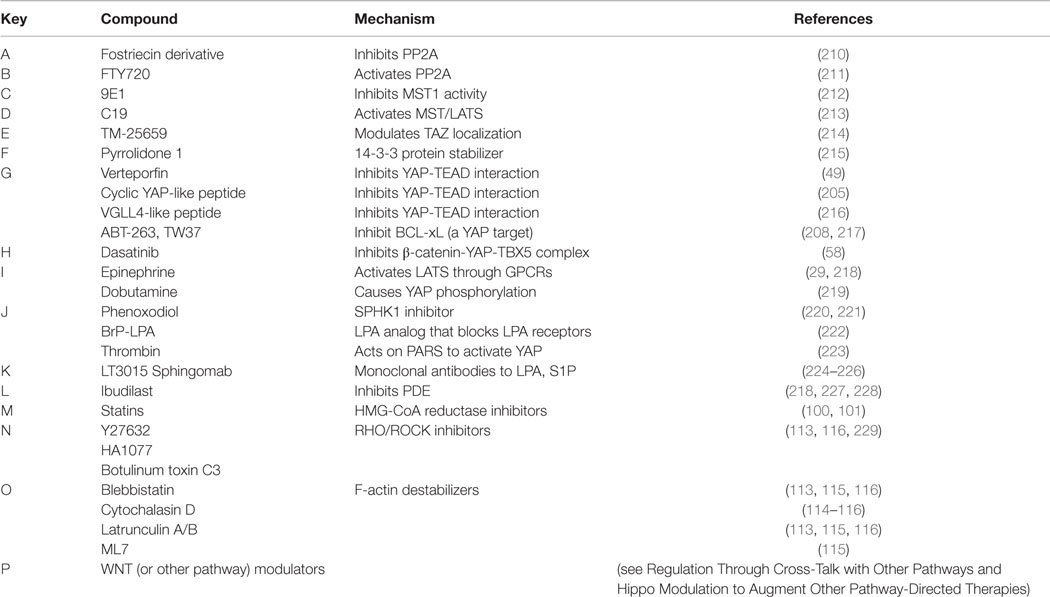
Table 3. Pharmacologic modulators of the Hippo pathway.
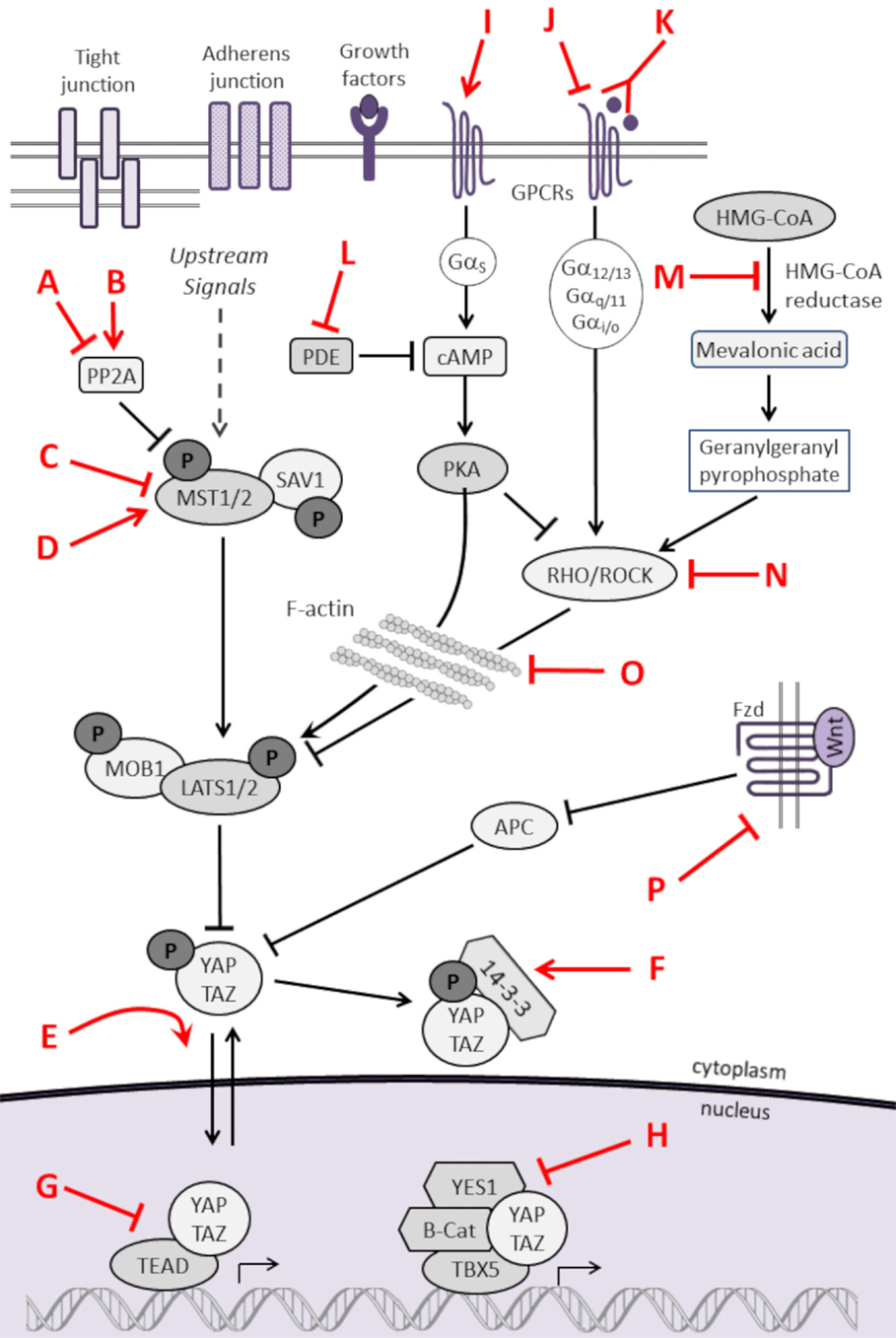
Figure 3. Pharmacologic modulators of the Hippo pathway. The Hippo cascade involves many protein–protein interactions that could serve as novel targets, and numerous pharmacologic compounds either directly or indirectly modulate Hippo activity. Some of the compounds activate Hippo components and others have an inhibitory role. While not all referenced studies have proven that modulation of upstream regulators result in concomitant changes in YAP or TAZ activity, these provide proof of principal that targeting Hippo signaling could be harnessed as a novel strategy to treat sarcomas. This is not an inclusive list, and other compounds are known to modulate Hippo components. Figure is modified with permission from Park et al. (202). Letters in Red correspond to the letters in the Key in Table 3.
As such, inhibiting the activity of YAP/TAZ is the most obvious and presumably the most potent anti-cancer approach. Three porphyrin-related compounds were identified as top hits in a small molecule library screen of potential modulators for inhibiting the transcriptional activity of YAP in vitro. Verteporfin is a photosensitizer used clinically to treat patients with macular degeneration (203). Verteporfin binding to YAP alters YAP conformation to prevent it from binding to TEAD transcription factors. In vivo experiments in murine systems show verteporfin inhibits YAP-induced liver overgrowth by decreasing cell proliferation (49). In vitro treatment of retinoblastoma cells with verteporfin caused decreased cell proliferation and down-regulation of the pluripotency marker OCT4 (204). Other small molecule inhibitors, such as cyclic YAP-like peptides and TM-25659, have been developed to interfere with YAP/TAZ–TEAD interactions (205).
Another challenge is that the Hippo pathway is ubiquitously expressed and thus, systemic treatment may cause detrimental side effects. This is particularly important in the pediatric population, where normal growth and development in most tissues likely rely on intact Hippo signaling. Similarly, GPCRs, although relatively accessible to inhibition, have broad physiological functions. However, intestine-specific conditional Yap1 knockout mice develop normally (206), implying that in some instances, YAP/TAZ may be dispensable for tissue development. YAP and TAZ are responsive to tissue-specific regulatory elements, presenting a theoretical possibility of targeting Hippo signaling in specific cells or tissues.
Hippo Modulation to Augment Other Pathway-Directed Therapies
Evidence suggests Hippo-directed therapies may synergize with other targeted modulators. By serving as a parallel means of cancer cell survival, YAP promotes resistance to RAF and MEK inhibitors in BRAF/RAS-mutated tumors. YAP overexpression was observed in tumors harboring a BRAF mutation from patients with melanoma or NSCLC, and YAP expression levels inversely correlated to the patients’ initial response to RAF and MEK inhibition. Furthermore, YAP suppression enhanced MEK inhibition in murine xenografts of human NSCLC, melanoma, and pancreatic adenocarcinoma with BRAF or KRAS mutations (207, 208). Similarly, YAP upregulation of EGFR through a YAP–TEAD complex at the EGFR promoter has been shown to partly explain the reduced translational impact of EGFR inhibitors in cancer. Inhibition of the YAP–TEAD interaction using verteporfin results in decreased EGFR expression and enhanced chemosensitivity to 5-fluorouracil and EGFR inhibitors in mouse xenografts of esophageal cancer (209). Finally, mTOR inhibition with rapamycin results in decreased TAZ expression in hepatocellular carcinoma (90).
Conclusion
The Hippo signaling pathway is an evolutionarily conserved tumor suppressor network important not only for proper cell, tissue and organ development, homeostasis, and repair, but it is also found dysregulated in many human cancers. While much of the early investigation on Hippo signaling in cancer was performed in epithelial malignancies, dysregulation of the Hippo pathway also occurs in sarcomas, cancers of mesenchymal origin. In a range of bone and soft-tissue sarcomas, Hippo signaling is commonly thwarted by upregulation of YAP or TAZ. However, genetic and epigenetic dysregulation of upstream core Hippo pathway members, and adaptor proteins has been noted. The role of Hippo signaling in mechanotransduction in both normal and cancerous mesenchymal cell behavior and fate provides additional insight into sarcoma biology. Further studies will be needed to clarify the underlying mechanisms of Hippo pathway dysregulation in specific sarcoma subtypes, providing a foundation upon which to develop successful therapeutic interventions.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Katrina Slemmons (Linardic laboratory) for manuscript review, and support received from the Norman & Bettina Roberts Foundation Inc. (to MD), a Derfner-Children’s Miracle Network Hospital Award (to MD), Alex’s Lemonade Stand Innovation (to CL) and “A” (to LC) Awards, and Hyundai Hope on Wheels (to CL).
Abbreviations
AMOT, angiomotin; APC, adenomatous polyposis coli; ARMS, alveolar rhabdomyosarcoma; β-TRCP, β-transducin repeat-containing E3 ubiquitin protein ligase; BMI-1, B-lymphoma Mo-MLV insertion region 1 homolog; BMP2, bone morphogenetic protein 2; BRAF, v-raf murine sarcoma viral oncogene homolog B; cAMP, adenylyl cyclase pathway; CD44, CD44 antigen; CDKN2A, cyclin-dependent kinase inhibitor 2A; ChIP-Seq, chromatin immunoprecipitation followed by high-throughput DNA sequencing; CRB, crumbs complex proteins; CTGF, connective tissue growth factor; dLats, large tumor suppressor (or warts); DMBA, 9,10-dimethyl-1,2-benzanthracene; DVL, disheveled; ECM, extracellular matrix; EGFR, epidermal growth factor receptor; EHE, epithelioid hemangioendothelioma; ERK, extracellular signal-regulated kinases; ERMS, embryonal rhabdomyosarcoma; EWS, Ewing sarcoma; FAT4, FAT tumor suppressor homolog 4; FOXM1, transcription factor forkhead box M1; GEMM, genetically engineered mouse model; GPCRs, G protein-coupled receptors; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; hTERT, telomerase reverse transcriptase; ID2, inhibitor of DNA binding 2; KIBRA, kidney and brain protein; LATS1/2, large tumor suppressor homolog 1/2; LLGL1, lethal giant larvae homolog 1; MAPK, mitogen-activated protein kinase; MARK1, MAP/microtubule affinity-regulating kinase 1; MCAT, muscle CAT elements; MDM2, mouse double minute 2; MEF2, myocyte enhancer factor 2; MEK, MAPK kinase; MMP-9, matrix metallopeptidase 9; MOB1, Mob kinase activator 1; MRFs, myogenic regulatory factor family; MSC, mesenchymal stem cell; MST1/2, serine/threonine kinases mammalian STE20-like protein kinase 1/2; mTOR, mechanistic target of rapamycin; MYCN, v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog; Myf5, myogenic factor 5; MyHC, myosin heavy chain; MyoD, myogenic differentiation 1; NF2, neurofibromin 2 (or Merlin); NRSTS, non-rhabdomyosarcoma soft-tissue sarcoma; NSCLC, non-small cell lung cancer; OCT4, octamer-binding transcription factor-4; OS, osteosarcoma; p16INK4A, prototypic INK4 protein; PCNA, proliferating cell nuclear antigen; PDE, phosphodiesterase; PKA, protein kinase A; PKN1, protein kinase N1; PP1A, protein phosphatase 1, catalytic subunit, alpha isozyme; PP2A, protein phosphatase 2, regulatory subunit B, delta1; PPARγ, peroxisome proliferator-activated receptor gamma; RAF, v-raf murine sarcoma viral oncogene homolog; RAS, rat sarcoma viral oncogene homolog; RASSFs, Ras-association domain-containing family of proteins; RB1, retinoblastoma 1; RHO, rhodopsin; RMS, rhabdomyosarcoma; ROCK1, Rho-associated, coiled-coil containing protein kinase 1; RUNX2, runt-related transcription factor 2; SARAH domain, Salvador-Rassf-Hpo binding domain; SAV1, salvador homolog 1; SMADs, mothers against decapentaplegic proteins; Snai1/2, snail family zinc finger 1/2; SOX2, SRY (sex determining region Y)-box 2; STS, soft-tissue sarcoma; TAO-1, thousand and one amino acid protein 1; TAZ, transcriptional co-activator with PDZ-binding motif (or WWTR1); TCGA, The Cancer Genome Atlas; TEADs, TEA domain-containing sequence-specific transcription factors; TGFβ, transforming growth factor beta; TNF-α, tumor necrosis factor alpha; TP53, tumor protein p53; TP73, tumor protein p73; Twist1, twist family bHLH transcription factor 1; UVB, ultraviolet radiation B; VGLL3, vestigial-like 3; WNT, wingless-type MMTV integration site family; YAP, yes-associated protein 1.
References
1. Burningham Z, Hashibe M, Spector L, Schiffman JD. The epidemiology of sarcoma. Clin Sarcoma Res (2012) 2(1):14. doi:10.1186/2045-3329-2-14
2. Gorlick R, Janeway K, Lessnick S, Randall RL, Marina N. Children’s Oncology Group’s 2013 blueprint for research: bone tumors. Pediatr Blood Cancer (2013) 60(6):1009–15. doi:10.1002/pbc.24429
3. Hawkins DS, Spunt SL, Skapek SX. Children’s Oncology Group’s 2013 blueprint for research: soft tissue sarcomas. Pediatr Blood Cancer (2013) 60(6):1001–8. doi:10.1002/pbc.24435
4. Wu S, Huang JB, Dong JX, Pan DJ. Hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell (2003) 114(4):445–56. doi:10.1016/S0092-8674(03)00549-X
5. Harvey KF, Pfleger CM, Hariharan IK. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell (2003) 114(4):457–67. doi:10.1016/S0092-8674(03)00557-9
6. Huang JB, Wu S, Barrera J, Matthews K, Pan DJ. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell (2005) 122(3):421–34. doi:10.1016/j.cell.2005.06.007
7. Tapon N, Harvey KF, Bell DW, Wahrer DCR, Schiripo TA, Haber DA, et al. Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell (2002) 110(4):467–78. doi:10.1016/S0092-8674(02)00824-3
8. Pan D. The hippo signaling pathway in development and cancer. Dev Cell (2010) 19(4):491–505. doi:10.1016/j.devcel.2010.09.011
9. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Gene Dev (2007) 21(21):2747–61. doi:10.1101/gad.1602907
10. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell (2007) 130(6):1120–33. doi:10.1016/j.cell.2007.07.019
11. Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat Cell Biol (2003) 5(10):921–7. doi:10.1038/ncb1051
12. Udan RS, Kango-Singh M, Nolo R, Tao CY, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat Cell Biol (2003) 5(10):914–20. doi:10.1038/ncb1050
13. Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. The Drosophila tumor-suppressor gene warts encodes a homolog of human myotonic-dystrophy kinase and is required for the control of cell-shape and proliferation. Gene Dev (1995) 9(5):534–46. doi:10.1101/gad.9.5.534
14. Xu T, Wang W, Zhang S, Stewart RA, Yu W. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development (1995) 121(4):1053–63.
15. Hergovich A. MOB control: reviewing a conserved family of kinase regulators. Cell Signal (2011) 23(9):1433–40. doi:10.1016/j.cellsig.2011.04.007
16. Hong W, Guan K-L. The YAP and TAZ transcription co-activators: key downstream effectors of the mammalian Hippo pathway. Semin Cell Dev Biol (2012) 23(7):785–93. doi:10.1016/j.semcdb.2012.05.004
17. Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev (2008) 22(14):1962–71. doi:10.1101/gad.1664408
18. Zhao B, Kim J, Ye X, Lai ZC, Guan KL. Both TEAD-binding and WW domains are required for the growth stimulation and oncogenic transformation activity of yes-associated protein. Cancer Res (2009) 69(3):1089–98. doi:10.1158/0008-5472.CAN-08-2997
19. Zhang H, Liu CY, Zha ZY, Zhao B, Yao J, Zhao SM, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem (2009) 284(20):13355–62. doi:10.1074/jbc.M900843200
20. Chan SW, Lim CJ, Loo LS, Chong YF, Huang CX, Hong WJ. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J Biol Chem (2009) 284(21):14347–58. doi:10.1074/jbc.M901568200
21. Han D, Byun SH, Park S, Kim J, Kim I, Ha S, et al. YAP/TAZ enhance mammalian embryonic neural stem cell characteristics in a Tead-dependent manner. Biochem Biophys Res Commun (2015) 458(1):110–6. doi:10.1016/j.bbrc.2015.01.077
22. Grusche FA, Richardson HE, Harvey KF. Upstream regulation of the Hippo size control pathway. Curr Biol (2010) 20(13):R574–82. doi:10.1016/j.cub.2010.05.023
23. Wang WQ, Li X, Huang J, Feng L, Dolinta KG, Chen JJ. Defining the protein–protein interaction network of the human Hippo pathway. Mol Cell Proteomics (2014) 13(1):119–31. doi:10.1074/mcp.M113.030049
24. Kwon Y, Vinayagam A, Sun X, Dephoure N, Gygi SP, Hong P, et al. The Hippo signaling pathway interactome. Science (2013) 342(6159):737–40. doi:10.1126/science.1243971
25. Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal (2013) 6(302):rs15. doi:10.1126/scisignal.2004712
26. Schroeder MC, Halder G. Regulation of the Hippo pathway by cell architecture and mechanical signals. Semin Cell Dev Biol (2012) 23(7):803–11. doi:10.1016/j.semcdb.2012.06.001
27. Zhou D, Zhang Y, Wu H, Barry E, Yin Y, Lawrence E, et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc Natl Acad Sci U S A (2011) 108(49):E1312–20. doi:10.1073/pnas.1110428108
28. Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC, et al. The p130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Sci Signal (2013) 6(291):ra77. doi:10.1126/scisignal.2004060
29. Yu F-X, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell (2012) 150(4):780–91. doi:10.1016/j.cell.2012.06.037
30. Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, et al. The tumour-suppressor genes NF2/Merlin and expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol (2006) 8(1):27–36. doi:10.1038/ncb1339
31. Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell (2013) 154(6):1342–55. doi:10.1016/j.cell.2013.08.025
32. Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev (2001) 15(8):968–80. doi:10.1101/gad.189601
33. Silva E, Tsatskis Y, Gardano L, Tapon N, McNeill H. The tumor-suppressor gene fat controls tissue growth upstream of expanded in the hippo signaling pathway. Curr Biol (2006) 16(21):2081–9. doi:10.1016/j.cub.2006.09.004
34. Van Hateren NJ, Das RM, Hautbergue GM, Borycki AG, Placzek M, Wilson SA. FatJ acts via the Hippo mediator Yap1 to restrict the size of neural progenitor cell pools. Development (2011) 138(10):1893–902. doi:10.1242/dev.064204
35. Avruch J, Praskova M, Ortiz-Vega S, Liu M, Zhang XF. Nore1 and RASSF1 regulation of cell proliferation and of the MST1/2 kinases. Methods Enzymol (2006) 407:290–310. doi:10.1016/S0076-6879(05)07025-4
36. Polesello C, Huelsmann S, Brown NH, Tapon N. The Drosophila RASSF homolog antagonizes the hippo pathway. Curr Biol (2006) 16(24):2459–65. doi:10.1016/j.cub.2006.10.060
37. Yu J, Zheng Y, Dong J, Klusza S, Deng WM, Pan D. Kibra functions as a tumor suppressor protein that regulates Hippo signaling in conjunction with Merlin and Expanded. Dev Cell (2010) 18(2):288–99. doi:10.1016/j.devcel.2009.12.012
38. Baumgartner R, Poernbacher I, Buser N, Hafen E, Stocker H. The WW domain protein Kibra acts upstream of Hippo in Drosophila. Dev Cell (2010) 18(2):309–16. doi:10.1016/j.devcel.2009.12.013
39. Genevet A, Wehr MC, Brain R, Thompson BJ, Tapon N. Kibra is a regulator of the Salvador/Warts/Hippo signaling network. Dev Cell (2010) 18(2):300–8. doi:10.1016/j.devcel.2009.12.011
40. Poon CL, Lin JI, Zhang X, Harvey KF. The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Dev Cell (2011) 21(5):896–906. doi:10.1016/j.devcel.2011.09.012
41. Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, et al. A genetic screen identifies an LKB1–MARK signalling axis controlling the Hippo–YAP pathway. Nat Cell Biol (2014) 16(1):108–17. doi:10.1038/ncb2884
42. Scheel H, Hofmann K. A novel interaction motif, SARAH, connects three classes of tumor suppressor. Curr Biol (2003) 13(23):R899–900. doi:10.1016/j.cub.2003.11.007
43. Moleirinho S, Chang N, Sims AH, Tilston-Lunel AM, Angus L, Steele A, et al. KIBRA exhibits MST-independent functional regulation of the Hippo signaling pathway in mammals. Oncogene (2013) 32(14):1821–30. doi:10.1038/onc.2012.196
44. Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell (2008) 13(3):188–92. doi:10.1016/j.ccr.2008.02.011
45. Lallemand D, Curto M, Saotome I, Giovannini M, McClatchey AI. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Gene Dev (2003) 17(9):1090–100. doi:10.1101/gad.1054603
46. Gladden AB, Hebert AM, Schneeberger EE, McClatchey AI. The NF2 tumor suppressor, Merlin, regulates epidermal development through the establishment of a junctional polarity complex. Dev Cell (2010) 19(5):727–39. doi:10.1016/j.devcel.2010.10.008
47. Yokoyama T, Osada H, Murakami H, Tatematsu Y, Taniguchi T, Kondo Y, et al. YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation. Carcinogenesis (2008) 29(11):2139–46. doi:10.1093/carcin/bgn200
48. Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia (2008) 10(11):1204–12. doi:10.1593/neo.08642
49. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee S-J, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Gene Dev (2012) 26(12):1300–5. doi:10.1101/gad.192856.112
50. Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev (2015) 29(12):1271–84. doi:10.1101/gad.262816.115
51. Mauviel A, Nallet-Staub F, Varelas X. Integrating developmental signals: a Hippo in the (path)way. Oncogene (2012) 31(14):1743–56. doi:10.1038/onc.2011.363
52. Yin MX, Zhang L. Hippo signaling: a hub of growth control, tumor suppression and pluripotency maintenance. J Genet Genomics (2011) 38(10):471–81. doi:10.1016/j.jgg.2011.09.009
53. Irvine KD. Integration of intercellular signaling through the Hippo pathway. Semin Cell Dev Biol (2012) 23(7):812–7. doi:10.1016/j.semcdb.2012.04.006
54. Brack AS, Conboy IM, Conboy MJ, Shen J, Rando TA. A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell (2008) 2(1):50–9. doi:10.1016/j.stem.2007.10.006
55. Gaur T, Lengner CJ, Hovhannisyan H, Bhat RA, Bodine PV, Komm BS, et al. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J Biol Chem (2005) 280(39):33132–40. doi:10.1074/jbc.M500608200
56. Zhao S, Kurenbekova L, Gao Y, Roos A, Creighton CJ, Rao P, et al. NKD2, a negative regulator of Wnt signaling, suppresses tumor growth and metastasis in osteosarcoma. Oncogene (2015). doi:10.1038/onc.2014.429
57. Kephart JJ, Tiller RG, Crose L, Slemmons KK, Chen PH, Hinson AR, et al. Secreted frizzled related protein 3 (SFRP3) is required for tumorigenesis of PAX3-FOXO1-positive alveolar rhabdomyosarcoma. Clin Cancer Res (2015). doi:10.1158/1078-0432.CCR-14-1797
58. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell (2012) 151(7):1457–73. doi:10.1016/j.cell.2012.11.026
59. Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science (2011) 332(6028):458–61. doi:10.1126/science.1199010
60. Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S, et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell (2009) 139(4):757–69. doi:10.1016/j.cell.2009.09.035
61. Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, et al. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev Cell (2010) 19(6):831–44. doi:10.1016/j.devcel.2010.11.012
62. Varelas X, Miller BW, Sopko R, Song S, Gregorieff A, Fellouse FA, et al. The hippo pathway regulates Wnt/beta-catenin signaling. Dev Cell (2010) 18(4):579–91. doi:10.1016/j.devcel.2010.03.007
63. Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science (2005) 309(5737):1074–8. doi:10.1126/science.1110955
64. Tao J, Jiang MM, Jiang L, Salvo JS, Zeng HC, Dawson B, et al. Notch activation as a driver of osteogenic sarcoma. Cancer Cell (2014) 26(3):390–401. doi:10.1016/j.ccr.2014.07.023
65. Rota R, Ciarapica R, Miele L, Locatelli F. Notch signaling in pediatric soft tissue sarcomas. BMC Med (2012) 10:141. doi:10.1186/1741-7015-10-141
66. Belyea BC, Naini S, Bentley RC, Linardic CM. Inhibition of the Notch-Hey1 axis blocks embryonal rhabdomyosarcoma tumorigenesis. Clin Cancer Res (2011) 17(23):7324–36. doi:10.1158/1078-0432.CCR-11-1004
67. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol (2007) 17(23):2054–60. doi:10.1016/j.cub.2007.10.039
68. Byun MR, Hwang JH, Kim AR, Kim KM, Hwang ES, Yaffe MB, et al. Canonical Wnt signalling activates TAZ through PP1A during osteogenic differentiation. Cell Death Differ (2014) 21(6):854–63. doi:10.1038/cdd.2014.8
69. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, et al. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell (2014) 158(1):157–70. doi:10.1016/j.cell.2014.06.013
70. Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, et al. Role of TAZ as mediator of Wnt signaling. Cell (2012) 151(7):1443–56. doi:10.1016/j.cell.2012.11.027
71. Hiemer SE, Szymaniak AD, Varelas X. The transcriptional regulators TAZ and YAP direct transforming growth factor beta-induced tumorigenic phenotypes in breast cancer cells. J Biol Chem (2014) 289(19):13461–74. doi:10.1074/jbc.M113.529115
72. Mullen AC. Hippo tips the TGF-beta scale in favor of pluripotency. Cell Stem Cell (2014) 14(1):6–8. doi:10.1016/j.stem.2013.12.009
73. Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol (2008) 10(7):837–48. doi:10.1038/ncb1748
74. Lian I, Kim J, Okazawa H, Zhao J, Zhao B, Yu J, et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev (2010) 24(11):1106–18. doi:10.1101/gad.1903310
75. Genevet A, Polesello C, Blight K, Robertson F, Collinson LM, Pichaud F, et al. The Hippo pathway regulates apical-domain size independently of its growth-control function. J Cell Sci (2009) 122(Pt 14):2360–70. doi:10.1242/jcs.041806
76. Polesello C, Tapon N. Salvador-warts-hippo signaling promotes Drosophila posterior follicle cell maturation downstream of notch. Curr Biol (2007) 17(21):1864–70. doi:10.1016/j.cub.2007.11.005
77. Yu J, Poulton J, Huang YC, Deng WM. The hippo pathway promotes Notch signaling in regulation of cell differentiation, proliferation, and oocyte polarity. PLoS One (2008) 3(3):e1761. doi:10.1371/journal.pone.0001761
78. Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, et al. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev (2009) 23(23):2729–41. doi:10.1101/gad.1824509
79. Huang J, Kalderon D. Coupling of Hedgehog and Hippo pathways promotes stem cell maintenance by stimulating proliferation. J Cell Biol (2014) 205(3):325–38. doi:10.1083/jcb.201309141
80. Chan LH, Wang W, Yeung W, Deng Y, Yuan P, Mak KK. Hedgehog signaling induces osteosarcoma development through Yap1 and H19 overexpression. Oncogene (2014) 33(40):4857–66. doi:10.1038/onc.2013.433
81. Reddy BV, Irvine KD. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev Cell (2013) 24(5):459–71. doi:10.1016/j.devcel.2013.01.020
82. Yu S, Cai X, Wu C, Wu L, Wang Y, Liu Y, et al. Adhesion glycoprotein CD44 functions as an upstream regulator of a network connecting ERK, AKT and Hippo-YAP pathways in cancer progression. Oncotarget (2015) 6(5):2951–65.
83. Byun MR, Kim AR, Hwang JH, Kim KM, Hwang ES, Hong JH. FGF2 stimulates osteogenic differentiation through ERK induced TAZ expression. Bone (2014) 58:72–80. doi:10.1016/j.bone.2013.09.024
84. O’Hayre M, Degese MS, Gutkind JS. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr Opin Cell Biol (2014) 27:126–35. doi:10.1016/j.ceb.2014.01.005
85. Zhou X, Wang Z, Huang W, Lei QY. G protein-coupled receptors: bridging the gap from the extracellular signals to the Hippo pathway. Acta Biochim Biophys Sin (Shanghai) (2015) 47(1):10–5. doi:10.1093/abbs/gmu108
86. Sun G, Irvine KD. Ajuba family proteins link JNK to Hippo signaling. Sci Signal (2013) 6(292):ra81. doi:10.1126/scisignal.2004324
87. Sun G, Irvine KD. Regulation of Hippo signaling by Jun kinase signaling during compensatory cell proliferation and regeneration, and in neoplastic tumors. Dev Biol (2011) 350(1):139–51. doi:10.1016/j.ydbio.2010.11.036
88. Haskins JW, Nguyen DX, Stern DF. Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci Signal (2014) 7(355):ra116. doi:10.1126/scisignal.2005770
89. Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, et al. YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat Cell Biol (2012) 14(12):1322–9. doi:10.1038/ncb2615
90. Chiang J, Martinez-Agosto JA. Effects of mTOR Inhibitors on Components of the Salvador-Warts-Hippo Pathway. Cells (2012) 1(4):886–904. doi:10.3390/cells1040886
91. Sciarretta S, Zhai P, Maejima Y, Del Re DP, Nagarajan N, Yee D, et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep (2015) 11(1):125–36. doi:10.1016/j.celrep.2015.03.010
92. Karpowicz P, Perez J, Perrimon N. The Hippo tumor suppressor pathway regulates intestinal stem cell regeneration. Development (2010) 137(24):4135–45. doi:10.1242/dev.060483
93. Staley BK, Irvine KD. Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr Biol (2010) 20(17):1580–7. doi:10.1016/j.cub.2010.07.041
94. Corbo V, Ponz-Sarvise M, Tuveson DA. The RAS and YAP1 dance, who is leading? EMBO J (2014) 33(21):2437–8. doi:10.15252/embj.201490014
95. Hong X, Nguyen HT, Chen Q, Zhang R, Hagman Z, Voorhoeve PM, et al. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. EMBO J (2014) 33(21):2447–57. doi:10.15252/embj.201489385
96. Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Sci Signal (2014) 7(324):ra42. doi:10.1126/scisignal.2005049
97. Basu-Roy U, Bayin NS, Rattanakorn K, Han E, Placantonakis DG, Mansukhani A, et al. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat Commun (2015) 6:6411. doi:10.1038/ncomms7411
98. Seo E, Basu-Roy U, Gunaratne Preethi H, Coarfa C, Lim D-S, Basilico C, et al. SOX2 regulates YAP1 to maintain stemness and determine cell fate in the osteo-adipo lineage. Cell Rep (2013) 3(6):2075–87. doi:10.1016/j.celrep.2013.05.029
99. Tang Y, Rowe RG, Botvinick EL, Kurup A, Putnam AJ, Seiki M, et al. MT1-MMP-dependent control of skeletal stem cell commitment via a beta1-integrin/YAP/TAZ signaling axis. Dev Cell (2013) 25(4):402–16. doi:10.1016/j.devcel.2013.04.011
100. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol (2014) 16(4):357–66. doi:10.1038/ncb2936
101. Wang Z, Wu Y, Wang H, Zhang Y, Mei L, Fang X, et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc Natl Acad Sci U S A (2014) 111(1):E89–98. doi:10.1073/pnas.1319190110
102. Enzo E, Santinon G, Pocaterra A, Aragona M, Bresolin S, Forcato M, et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. EMBO J (2015) 34(10):1349–70. doi:10.15252/embj.201490379
103. Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol (2015) 17(4):490–9. doi:10.1038/ncb3113
104. Oudhoff MJ, Freeman SA, Couzens AL, Antignano F, Kuznetsova E, Min PH, et al. Control of the hippo pathway by Set7-dependent methylation of Yap. Dev Cell (2013) 26(2):188–94. doi:10.1016/j.devcel.2013.05.025
105. Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, et al. CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res (2013) 73(22):6722–33. doi:10.1158/0008-5472.CAN-13-2049
106. Schwartz MA. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harbor Perspect Biol (2010) 2(12):a005066. doi:10.1101/cshperspect.a005066
107. Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol (2006) 7(4):265–75. doi:10.1038/nrm1890
108. Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol (2012) 13(9):591–600. doi:10.1038/nrm3416
109. Discher DE, Mooney DJ, Zandstra PW. Growth factors, matrices, and forces combine and control stem cells. Science (2009) 324(5935):1673–7. doi:10.1126/science.1171643
110. Jaalouk DE, Lammerding J. Mechanotransduction gone awry. Nat Rev Mol Cell Biol (2009) 10(1):63–73. doi:10.1038/nrm2597
111. Mammoto A, Ingber DE. Cytoskeletal control of growth and cell fate switching. Curr Opin Cell Biol (2009) 21(6):864–70. doi:10.1016/j.ceb.2009.08.001
112. Rauskolb C, Sun S, Sun G, Pan Y, Irvine KD. Cytoskeletal tension inhibits Hippo signaling through an Ajuba-Warts complex. Cell (2014) 158(1):143–56. doi:10.1016/j.cell.2014.05.035
113. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature (2011) 474(7350):179–83. doi:10.1038/nature10137
114. Sansores-Garcia L, Bossuyt W, Wada K-I, Yonemura S, Tao C, Sasaki H, et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J (2011) 30(12):2325–35. doi:10.1038/emboj.2011.157
115. Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development (2011) 138(18):3907–14. doi:10.1242/dev.070987
116. Zhao B, Li L, Wang L, Wang C-Y, Yu J, Guan K-L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Gene Dev (2012) 26(1):54–68. doi:10.1101/gad.173435.111
117. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev (2014) 94(4):1287–312. doi:10.1152/physrev.00005.2014
118. Tschumperlin DJ, Liu F, Tager AM. Biomechanical regulation of mesenchymal cell function. Curr Opin Rheumatol (2013) 25(1):92–100. doi:10.1097/BOR.0b013e32835b13cd
119. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell (2006) 126(4):677–89. doi:10.1016/j.cell.2006.06.044
120. McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell (2004) 6(4):483–95. doi:10.1016/S1534-5807(04)00075-9
121. Kolf CM, Cho E, Tuan RS. Mesenchymal stromal cells. Biology of adult mesenchymal stem cells: regulation of niche, self-renewal and differentiation. Arthritis Res Ther (2007) 9(1):204. doi:10.1186/ar2116
122. Mo JS, Park HW, Guan KL. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep (2014) 15(6):642–56. doi:10.15252/embr.201438638
123. Hakelien AM, Bryne JC, Harstad KG, Lorenz S, Paulsen J, Sun J, et al. The regulatory landscape of osteogenic differentiation. Stem Cells (2014) 32(10):2780–93. doi:10.1002/stem.1759
124. Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J (1999) 18(9):2551–62. doi:10.1093/emboj/18.9.2551
125. Cui CB, Cooper LF, Yang X, Karsenty G, Aukhil I. Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol Cell Biol (2003) 23(3):1004–13. doi:10.1128/MCB.23.3.1004-1013.2003
126. Hong JH, Yaffe MB. TAZ: a beta-catenin-like molecule that regulates mesenchymal stem cell differentiation. Cell Cycle (2006) 5(2):176–9. doi:10.4161/cc.5.2.2362
127. Yang JY, Cho SW, An JH, Jung JY, Kim SW, Kim SY, et al. Osteoblast-targeted overexpression of TAZ increases bone mass in vivo. PLoS One (2013) 8(2):e56585. doi:10.1371/journal.pone.0056585
128. Zaidi SK, Sullivan AJ, Medina R, Ito Y, van Wijnen AJ, Stein JL, et al. Tyrosine phosphorylation controls Runx2-mediated subnuclear targeting of YAP to repress transcription. EMBO J (2004) 23(4):790–9. doi:10.1038/sj.emboj.7600073
129. Id Boufker H, Lagneaux L, Najar M, Piccart M, Ghanem G, Body JJ, et al. The Src inhibitor dasatinib accelerates the differentiation of human bone marrow-derived mesenchymal stromal cells into osteoblasts. BMC Cancer (2010) 10:298. doi:10.1186/1471-2407-10-298
130. Song H, Kim H, Lee K, Lee DH, Kim TS, Song JY, et al. Ablation of Rassf2 induces bone defects and subsequent haematopoietic anomalies in mice. EMBO J (2012) 31(5):1147–59. doi:10.1038/emboj.2011.480
131. Jung H, Lee MS, Jang EJ, Ahn JH, Kang NS, Yoo SE, et al. Augmentation of PPARgamma-TAZ interaction contributes to the anti-adipogenic activity of KR62980. Biochem Pharmacol (2009) 78(10):1323–9. doi:10.1016/j.bcp.2009.07.001
132. Park BH, Kim DS, Won GW, Jeon HJ, Oh BC, Lee Y, et al. Mammalian ste20-like kinase and SAV1 promote 3T3-L1 adipocyte differentiation by activation of PPARgamma. PLoS One (2012) 7(1):e30983. doi:10.1371/journal.pone.0030983
133. Ishibashi J, Perry RL, Asakura A, Rudnicki MA. MyoD induces myogenic differentiation through cooperation of its NH2- and COOH-terminal regions. J Cell Biol (2005) 171(3):471–82. doi:10.1083/jcb.200502101
134. Rudnicki MA, Jaenisch R. The MyoD family of transcription factors and skeletal myogenesis. Bioessays (1995) 17(3):203–9. doi:10.1002/bies.950170306
135. Bergstrom DA, Tapscott SJ. Molecular distinction between specification and differentiation in the myogenic basic helix-loop-helix transcription factor family. Mol Cell Biol (2001) 21(7):2404–12. doi:10.1128/MCB.21.7.2404-2412.2001
136. Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol (1998) 14:167–96. doi:10.1146/annurev.cellbio.14.1.167
137. Estrella NL, Desjardins CA, Nocco SE, Clark AL, Maksimenko Y, Naya FJ. MEF2 transcription factors regulate distinct gene programs in mammalian skeletal muscle differentiation. J Biol Chem (2015) 290(2):1256–68. doi:10.1074/jbc.M114.589838
138. Tremblay AM, Missiaglia E, Galli GG, Hettmer S, Urcia R, Carrara M, et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell (2014) 26(2):273–87. doi:10.1016/j.ccr.2014.05.029
139. Watt KI, Judson R, Medlow P, Reid K, Kurth TB, Burniston JG, et al. Yap is a novel regulator of C2C12 myogenesis. Biochem Biophys Res Commun (2010) 393(4):619–24. doi:10.1016/j.bbrc.2010.02.034
140. Judson RN, Tremblay AM, Knopp P, White RB, Urcia R, De Bari C, et al. The Hippo pathway member Yap plays a key role in influencing fate decisions in muscle satellite cells. J Cell Sci (2012) 125(Pt 24):6009–19. doi:10.1242/jcs.109546
141. Yoshida T. MCAT elements and the TEF-1 family of transcription factors in muscle development and disease. Arterioscler Thromb Vasc Biol (2008) 28(1):8–17. doi:10.1161/ATVBAHA.107.155788
142. Jeong H, Bae S, An SY, Byun MR, Hwang JH, Yaffe MB, et al. TAZ as a novel enhancer of MyoD-mediated myogenic differentiation. FASEB J (2010) 24(9):3310–20. doi:10.1096/fj.09-151324
143. Park GH, Jeong H, Jeong MG, Jang EJ, Bae MA, Lee YL, et al. Novel TAZ modulators enhance myogenic differentiation and muscle regeneration. Br J Pharmacol (2014) 171(17):4051–61. doi:10.1111/bph.12755
144. Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A (2002) 99(17):11025–30. doi:10.1073/pnas.162172899
145. HaDuong JH, Martin AA, Skapek SX, Mascarenhas L. Sarcomas. Pediatr Clin North Am (2015) 62(1):179–200. doi:10.1016/j.pcl.2014.09.012
146. Bao Y, Hata Y, Ikeda M, Withanage K. Mammalian Hippo pathway: from development to cancer and beyond. J Biochem (2011) 149(4):361–79. doi:10.1093/jb/mvr021
147. McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, et al. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Gene Dev (1998) 12(8):1121–33. doi:10.1101/gad.12.8.1121
148. Nishio M, Hamada K, Kawahara K, Sasaki M, Noguchi F, Chiba S, et al. Cancer susceptibility and embryonic lethality in Mob1a/1b double-mutant mice. J Clin Investig (2012) 122(12):4505–18. doi:10.1172/JCI63735
149. St John MAR, Tao WF, Fei XL, Fukumoto R, Carcangiu ML, Brownstein DG, et al. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat Genet (1999) 21(2):182–6. doi:10.1038/5965
150. Longhi A, Errani C, De Paolis M, Mercuri M, Bacci G. Primary bone osteosarcoma in the pediatric age: state of the art. Cancer Treat Rev (2006) 32(6):423–36. doi:10.1016/j.ctrv.2006.05.005
151. Martin JW, Squire JA, Zielenska M. The genetics of osteosarcoma. Sarcoma (2012) 2012:627254. doi:10.1155/2012/627254
152. Sadikovic B, Yoshimoto M, Chilton-MacNeill S, Thorner P, Squire JA, Zielenska M. Identification of interactive networks of gene expression associated with osteosarcoma oncogenesis by integrated molecular profiling. Hum Mol Genet (2009) 18(11):1962–75. doi:10.1093/hmg/ddp117
153. Zhang YH, Li B, Shen L, Shen Y, Chen XD. The role and clinical significance of YES-associated protein 1 in human osteosarcoma. Int J Immunopathol Pharmacol (2013) 26(1):157–67.
154. Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia (2007) 9(2):166–80. doi:10.1593/neo.07112
155. Yang Z, Zhang M, Xu K, Liu L, Hou WK, Cai YZ, et al. Knockdown of YAP1 inhibits the proliferation of osteosarcoma cells in vitro and in vivo. Oncol Rep (2014) 32(3):1265–72. doi:10.3892/or.2014.3305
156. Richter AM, Walesch SK, Wurl P, Taubert H, Dammann RH. The tumor suppressor RASSF10 is upregulated upon contact inhibition and frequently epigenetically silenced in cancer. Oncogenesis (2012) 1:e18. doi:10.1038/oncsis.2012.18
157. Zhou XH, Yang CQ, Zhang CL, Gao Y, Yuan HB, Wang C. RASSF5 inhibits growth and invasion and induces apoptosis in osteosarcoma cells through activation of MST1/LATS1 signaling. Oncol Rep (2014) 32(4):1505–12. doi:10.3892/or.2014.3387
158. Park J, Kang SI, Lee SY, Zhang XF, Kim MS, Beers LF, et al. Tumor suppressor Ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J Biol Chem (2010) 285(45):35029–38. doi:10.1074/jbc.M110.165506
159. Zhang Y, Xia H, Ge X, Chen Q, Yuan D, Chen Q, et al. CD44 acts through RhoA to regulate YAP signaling. Cell Signal (2014) 26(11):2504–13. doi:10.1016/j.cellsig.2014.07.031
160. Gvozdenovic A, Arlt MJ, Campanile C, Brennecke P, Husmann K, Born W, et al. Silencing of CD44 gene expression in human 143-B osteosarcoma cells promotes metastasis of intratibial tumors in SCID mice. PLoS One (2013) 8(4):e60329. doi:10.1371/journal.pone.0060329
161. Jiang Y, Ludwig J, Janku F. Targeted therapies for advanced Ewing sarcoma family of tumors. Cancer Treat Rev (2015) 41(5):391–400. doi:10.1016/j.ctrv.2015.03.008
162. Uren A, Toretsky JA. Ewing’s sarcoma oncoprotein EWS-FLI1: the perfect target without a therapeutic agent. Future Oncol (2005) 1(4):521–8. doi:10.2217/14796694.1.4.521
163. Hsu JH, Lawlor ER. BMI-1 suppresses contact inhibition and stabilizes YAP in Ewing sarcoma. Oncogene (2011) 30(17):2077–85. doi:10.1038/onc.2010.571
164. Douglas D, Hsu JH, Hung L, Cooper A, Abdueva D, van Doorninck J, et al. BMI-1 promotes ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res (2008) 68(16):6507–15. doi:10.1158/0008-5472.CAN-07-6152
165. Gharanei S, Brini AT, Vaiyapuri S, Alholle A, Dallol A, Arrigoni E, et al. RASSF2 methylation is a strong prognostic marker in younger age patients with Ewing sarcoma. Epigenetics (2013) 8(9):893–8. doi:10.4161/epi.25617
166. Avigad S, Shukla S, Naumov I, Cohen IJ, Ash S, Meller I, et al. Aberrant methylation and reduced expression of RASSF1A in Ewing sarcoma. Pediatr Blood Cancer (2009) 53(6):1023–8. doi:10.1002/pbc.22115
167. Harada K, Toyooka S, Maitra A, Maruyama R, Toyooka KO, Timmons CF, et al. Aberrant promoter methylation and silencing of the RASSF1A gene in pediatric tumors and cell lines. Oncogene (2002) 21(27):4345–9. doi:10.1038/sj.onc.1205446
168. Patel N, Black J, Chen X, Marcondes AM, Grady WM, Lawlor ER, et al. DNA methylation and gene expression profiling of ewing sarcoma primary tumors reveal genes that are potential targets of epigenetic inactivation. Sarcoma (2012) 2012:498472. doi:10.1155/2012/498472
169. Tomazou EM, Sheffield NC, Schmidl C, Schuster M, Schonegger A, Datlinger P, et al. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep (2015) 10(7):1082–95. doi:10.1016/j.celrep.2015.01.042
170. Loeb DM, Thornton K, Shokek O. Pediatric soft tissue sarcomas. Surg Clin North Am (2008) 88(3):615–627. doi:10.1016/j.suc.2008.03.008
171. Meza JL, Anderson J, Pappo AS, Meyer WH. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children’s Oncology Group. J Clin Oncol (2006) 24(24):3844–51. doi:10.1200/JCO.2005.05.3801
172. Oberlin O, Rey A, Lyden E, Bisogno G, Stevens MC, Meyer WH, et al. Prognostic factors in metastatic rhabdomyosarcomas: results of a pooled analysis from United States and European cooperative groups. J Clin Oncol (2008) 26(14):2384–9. doi:10.1200/JCO.2007.14.7207
173. Davicioni E, Anderson JR, Buckley JD, Meyer WH, Triche TJ. Gene expression profiling for survival prediction in pediatric rhabdomyosarcomas: a report from the children’s oncology group. J Clin Oncol (2010) 28(7):1240–6. doi:10.1200/JCO.2008.21.1268
174. Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov (2014) 4(2):216–31. doi:10.1158/2159-8290.CD-13-0639
175. Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene (2001) 20(40):5736–46. doi:10.1038/sj.onc.1204599
176. Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol (2012) 30(14):1670–7. doi:10.1200/JCO.2011.38.5591
177. Williamson D, Missiaglia E, de Reynies A, Pierron G, Thuille B, Palenzuela G, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol (2010) 28(13):2151–8. doi:10.1200/JCO.2009.26.3814
178. Crose LE, Galindo KA, Kephart JG, Chen C, Fitamant J, Bardeesy N, et al. Alveolar rhabdomyosarcoma-associated PAX3-FOXO1 promotes tumorigenesis via Hippo pathway suppression. J Clin Invest (2014) 124(1):285–96. doi:10.1172/JCI67087
179. Judson RN, Gray SR, Walker C, Carroll AM, Itzstein C, Lionikas A, et al. Constitutive expression of Yes-associated protein (Yap) in adult skeletal muscle fibres induces muscle atrophy and myopathy. PLoS One (2013) 8(3):e59622. doi:10.1371/journal.pone.0059622
180. Chen L, Shern JF, Wei JS, Yohe ME, Song YK, Hurd L, et al. Clonality and evolutionary history of rhabdomyosarcoma. PLoS Genet (2015) 11(3):e1005075. doi:10.1371/journal.pgen.1005075
181. Mosquera JM, Sboner A, Zhang L, Kitabayashi N, Chen CL, Sung YS, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes Chromosomes Cancer (2013) 52(6):538–50. doi:10.1002/gcc.22050
182. Carroll SJ, Nodit L. Spindle cell rhabdomyosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med (2013) 137(8):1155–8. doi:10.5858/arpa.2012-0465-RS
183. Panagopoulos I, Gorunova L, Bjerkehagen B, Boye K, Heim S. Chromosome aberrations and HEY1-NCOA2 fusion gene in a mesenchymal chondrosarcoma. Oncol Rep (2014) 32(1):40–4. doi:10.3892/or.2014.3180
184. Pappo AS, Devidas M, Jenkins J, Rao B, Marcus R, Thomas P, et al. Phase II trial of neoadjuvant vincristine, ifosfamide, and doxorubicin with granulocyte colony-stimulating factor support in children and adolescents with advanced-stage nonrhabdomyosarcomatous soft tissue sarcomas: a Pediatric Oncology Group Study. J Clin Oncol (2005) 23(18):4031–8. doi:10.1200/JCO.2005.03.209
185. Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer (2003) 3(9):685–94. doi:10.1038/nrc1168
186. Eisinger-Mathason TS, Mucaj V, Biju KM, Nakazawa MS, Gohil M, Cash TP, et al. Deregulation of the Hippo pathway in soft-tissue sarcoma promotes FOXM1 expression and tumorigenesis. Proc Natl Acad Sci U S A (2015) 112(26):E3402–11. doi:10.1073/pnas.1420005112
187. Seidel C, Schagdarsurengin U, Bluemke K, Wuerl P, Pfeifer GP, Hauptmann S, et al. Frequent hypermethylation of MST1 and MST2 in soft tissue sarcoma. Mol Carcinog (2007) 46(10):865–71. doi:10.1002/mc.20317
188. Hisaoka M, Tanaka A, Hashimoto H. Molecular alterations of h-warts/LATS1 tumor suppressor in human soft tissue sarcoma. Lab Invest (2002) 82(10):1427–35. doi:10.1097/01.LAB.0000032381.68634.CA
189. Seidel C, Bartel F, Rastetter M, Bluemke K, Wurl P, Taubert H, et al. Alterations of cancer-related genes in soft tissue sarcomas: hypermethylation of RASSF1A is frequently detected in leiomyosarcoma and associated with poor prognosis in sarcoma. Int J Cancer (2005) 114(3):442–7. doi:10.1002/ijc.20707
190. Halperin DS, Pan C, Lusis AJ, Tontonoz P. Vestigial-like 3 is an inhibitor of adipocyte differentiation. J Lipid Res (2013) 54(2):473–81. doi:10.1194/jlr.M032755
191. Helias-Rodzewicz Z, Perot G, Chibon F, Ferreira C, Lagarde P, Terrier P, et al. YAP1 and VGLL3, encoding two cofactors of TEAD transcription factors, are amplified and overexpressed in a subset of soft tissue sarcomas. Genes Chromosomes Cancer (2010) 49(12):1161–71. doi:10.1002/gcc.20825
192. Hallor KH, Sciot R, Staaf J, Heidenblad M, Rydholm A, Bauer HC, et al. Two genetic pathways, t(1;10) and amplification of 3p11-12, in myxoinflammatory fibroblastic sarcoma, haemosiderotic fibrolipomatous tumour, and morphologically similar lesions. J Pathol (2009) 217(5):716–27. doi:10.1002/path.2513
193. Tanas MR, Sboner A, Oliveira AM, Erickson-Johnson MR, Hespelt J, Hanwright PJ, et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci Transl Med (2011) 3(98):98ra82. doi:10.1126/scitranslmed.3002409
194. Errani C, Zhang L, Sung YS, Hajdu M, Singer S, Maki RG, et al. A novel WWTR1-CAMTA1 gene fusion is a consistent abnormality in epithelioid hemangioendothelioma of different anatomic sites. Genes Chromosomes Cancer (2011) 50(8):644–53. doi:10.1002/gcc.20886
195. Antonescu CR, Le Loarer F, Mosquera JM, Sboner A, Zhang L, Chen CL, et al. Novel YAP1-TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer (2013) 52(8):775–84. doi:10.1002/gcc.22073
196. Flucke U, Vogels RJ, de Saint Aubain Somerhausen N, Creytens DH, Riedl RG, van Gorp JM, et al. Epithelioid Hemangioendothelioma: clinicopathologic, immunhistochemical, and molecular genetic analysis of 39 cases. Diagn Pathol (2014) 9:131. doi:10.1186/1746-1596-9-131
197. Patel NR, Salim AA, Sayeed H, Sarabia SF, Hollingsworth F, Warren M, et al. Molecular characterization of epithelioid haemangioendotheliomas identifies novel WWTR1-CAMTA1 fusion variants. Histopathology (2015). doi:10.1111/his.12697
198. Robanus-Maandag E, Giovannini M, van der Valk M, Niwa-Kawakita M, Abramowski V, Antonescu C, et al. Synergy of Nf2 and p53 mutations in development of malignant tumours of neural crest origin. Oncogene (2004) 23(39):6541–7. doi:10.1038/sj.onc.1207858
199. Giovannini M, Robanus-Maandag E, van der Valk M, Niwa-Kawakita M, Abramowski V, Goutebroze L, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Gene Dev (2000) 14(13):1617–30. doi:10.1101/gad.14.13.1617
200. Renner M, Wolf T, Meyer H, Hartmann W, Penzel R, Ulrich A, et al. Integrative DNA methylation and gene expression analysis in high-grade soft tissue sarcomas. Genome Biol (2013) 14(12):r137. doi:10.1186/gb-2013-14-12-r137
201. Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov (2014) 13(1):63–79. doi:10.1038/nrd4161
202. Park HW, Guan KL. Regulation of the Hippo pathway and implications for anticancer drug development. Trends Pharmacol Sci (2013) 34(10):581–9. doi:10.1016/j.tips.2013.08.006
203. Verteporfin in Photodynamic Therapy Study Group. Verteporfin therapy of subfoveal choroidal neovascularization in age-related macular degeneration: two-year results of a randomized clinical trial including lesions with occult with no classic choroidal neovascularization – verteporfin in photodynamic therapy report 2. Am J Ophthalmol (2001) 131(5):541–60. doi:10.1016/S0002-9394(01)00967-9
204. Brodowska K, Al-Moujahed A, Marmalidou A, Meyer Zu Horste M, Cichy J, Miller JW, et al. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res (2014) 124:67–73. doi:10.1016/j.exer.2014.04.011
205. Zhou Z, Hu T, Xu Z, Lin Z, Zhang Z, Feng T, et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J (2015) 29(2):724–32. doi:10.1096/fj.14-262980
206. Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Gene Dev (2010) 24(21):2383–8. doi:10.1101/gad.1978810
207. Errico A. Targeted therapies: Hippo effector YAP1 inhibition-towards a new therapeutic option to overcome drug resistance. Nat Rev Clin Oncol (2015) 12(4):190. doi:10.1038/nrclinonc.2015.31
208. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet (2015) 47(3):250–6. doi:10.1038/ng.3218
209. Song S, Honjo S, Jin J, Chang SS, Scott AW, Chen Q, et al. The Hippo coactivator YAP1 mediates EGFR overexpression and confers chemo-resistance in esophageal cancer. Clin Cancer Res (2015) 21(11):2580–90. doi:10.1158/1078-0432.CCR-14-2191
210. Swingle MR, Amable L, Lawhorn BG, Buck SB, Burke CP, Ratti P, et al. Structure-activity relationship studies of fostriecin, cytostatin, and key analogs, with PP1, PP2A, PP5, and(beta12-beta13)-chimeras (PP1/PP2A and PP5/PP2A), provide further insight into the inhibitory actions of fostriecin family inhibitors. J Pharmacol Exp Ther (2009) 331(1):45–53. doi:10.1124/jpet.109.155630
211. McConnell JL, Wadzinski BE. Targeting protein serine/threonine phosphatases for drug development. Mol Pharmacol (2009) 75(6):1249–61. doi:10.1124/mol.108.053140
212. Anand R, Maksimoska J, Pagano N, Wong EY, Gimotty PA, Diamond SL, et al. Toward the development of a potent and selective organoruthenium mammalian sterile 20 kinase inhibitor. J Med Chem (2009) 52(6):1602–11. doi:10.1021/jm8005806
213. Basu D, Lettan R, Damodaran K, Strellec S, Reyes-Mugica M, Rebbaa A. Identification, mechanism of action, and antitumor activity of a small molecule inhibitor of Hippo, TGF-beta, and Wnt signaling pathways. Mol Cancer Ther (2014) 13(6):1457–67. doi:10.1158/1535-7163.MCT-13-0918
214. Jang EJ, Jeong H, Kang JO, Kim NJ, Kim MS, Choi SH, et al. TM-25659 enhances osteogenic differentiation and suppresses adipogenic differentiation by modulating the transcriptional co-activator TAZ. Br J Pharmacol (2012) 165(5):1584–94. doi:10.1111/j.1476-5381.2011.01664.x
215. Zhao J, Meyerkord CL, Du Y, Khuri FR, Fu H. 14-3-3 proteins as potential therapeutic targets. Semin Cell Dev Biol (2011) 22(7):705–12. doi:10.1016/j.semcdb.2011.09.012
216. Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell (2014) 25(2):166–80. doi:10.1016/j.ccr.2014.01.010
217. Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell (2013) 23(1):121–8. doi:10.1016/j.ccr.2012.11.007
218. Yu FX, Zhang Y, Park HW, Jewell JL, Chen Q, Deng Y, et al. Protein kinase A activates the Hippo pathway to modulate cell proliferation and differentiation. Genes Dev (2013) 27(11):1223–32. doi:10.1101/gad.219402.113
219. Bao Y, Nakagawa K, Yang Z, Ikeda M, Withanage K, Ishigami-Yuasa M, et al. A cell-based assay to screen stimulators of the Hippo pathway reveals the inhibitory effect of dobutamine on the YAP-dependent gene transcription. J Biochem (2011) 150(2):199–208. doi:10.1093/jb/mvr063
220. Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM, et al. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol (2012) 19(8):955–62. doi:10.1016/j.chembiol.2012.07.005
221. Gamble JR, Xia P, Hahn CN, Drew JJ, Drogemuller CJ, Brown D, et al. Phenoxodiol, an experimental anticancer drug, shows potent antiangiogenic properties in addition to its antitumour effects. Int J Cancer (2006) 118(10):2412–20. doi:10.1002/ijc.21682
222. Jiang G, Xu Y, Fujiwara Y, Tsukahara T, Tsukahara R, Gajewiak J, et al. Alpha-substituted phosphonate analogues of lysophosphatidic acid (LPA) selectively inhibit production and action of LPA. ChemMedChem (2007) 2(5):679–90. doi:10.1002/cmdc.200600280
223. Mo JS, Yu FX, Gong R, Brown JH, Guan KL. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev (2012) 26(19):2138–43. doi:10.1101/gad.197582.112
224. Fleming JK, Wojciak JM, Campbell MA, Huxford T. Biochemical and structural characterization of lysophosphatidic Acid binding by a humanized monoclonal antibody. J Mol Biol (2011) 408(3):462–76. doi:10.1016/j.jmb.2011.02.061
225. Ponnusamy S, Selvam SP, Mehrotra S, Kawamori T, Snider AJ, Obeid LM, et al. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol Med (2012) 4(8):761–75. doi:10.1002/emmm.201200244
226. Wojciak JM, Zhu N, Schuerenberg KT, Moreno K, Shestowsky WS, Hiraiwa M, et al. The crystal structure of sphingosine-1-phosphate in complex with a Fab fragment reveals metal bridging of an antibody and its antigen. Proc Natl Acad Sci U S A (2009) 106(42):17717–22. doi:10.1073/pnas.0906153106
227. Kishi Y, Ohta S, Kasuya N, Sakita S, Ashikaga T, Isobe M. Ibudilast: a non-selective PDE inhibitor with multiple actions on blood cells and the vascular wall. Cardiovasc Drug Rev (2001) 19(3):215–25. doi:10.1111/j.1527-3466.2001.tb00066.x
228. Martinez A, Gil C. cAMP-specific phosphodiesterase inhibitors: promising drugs for inflammatory and neurological diseases. Expert Opin Ther Pat (2014) 24(12):1311–21. doi:10.1517/13543776.2014.968127
229. Wilde C, Barth H, Sehr P, Han L, Schmidt M, Just I, et al. Interaction of the Rho-ADP-ribosylating C3 exoenzyme with RalA. J Biol Chem (2002) 277(17):14771–6. doi:10.1074/jbc.M201072200
Keywords: Hippo, sarcoma, osteosarcoma, Ewing sarcoma, rhabdomyosarcoma, mesenchymal, targeted therapy, pediatric cancers
Citation: Deel MD, Li JJ, Crose LES and Linardic CM (2015) A review: molecular aberrations within Hippo signaling in bone and soft-tissue sarcomas. Front. Oncol. 5:190. doi: 10.3389/fonc.2015.00190
Received: 22 May 2015; Accepted: 10 August 2015;
Published: 02 September 2015
Edited by:
Thomas Grunewald, Ludwig Maximilian University of Munich, GermanyReviewed by:
Rimas J. Orentas, Lenten Technology Inc., USASimone Hettmer, Zentrum für Kinderheilkunde und Jugendmedizin, Germany
Copyright: © 2015 Deel, Li, Crose and Linardic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Corinne M. Linardic, Division of Hematology-Oncology, Department of Pediatrics, Duke University School of Medicine, Box 102382 DUMC, Durham, NC 27710, USA,Y29yaW5uZS5saW5hcmRpY0BkbS5kdWtlLmVkdQ==