 Daniela A. Dungl
Daniela A. Dungl Elaina N. Maginn
Elaina N. Maginn Euan A. Stronach
Euan A. Stronach- Molecular Therapy Laboratory, Department of Surgery and Cancer, Ovarian Cancer Action Research Centre, Imperial College London, London, UK
Platinum-based chemotherapy is the cornerstone of ovarian cancer treatment, and its efficacy is dependent on the generation of DNA damage, with subsequent induction of apoptosis. Inappropriate or aberrant activation of the DNA damage response network is associated with resistance to platinum, and defects in DNA repair pathways play critical roles in determining patient response to chemotherapy. In ovarian cancer, tumor cell defects in homologous recombination – a repair pathway activated in response to double-strand DNA breaks (DSB) – are most commonly associated with platinum-sensitive disease. However, despite initial sensitivity, the emergence of resistance is frequent. Here, we review strategies for directly interfering with DNA repair pathways, with particular focus on direct inhibition of non-homologous end joining (NHEJ), another DSB repair pathway. DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is a core component of NHEJ and it has shown considerable promise as a chemosensitization target in numerous cancer types, including ovarian cancer where it functions to promote platinum-induced survival signaling, via AKT activation. The development of pharmacological inhibitors of DNA-PKcs is on-going, and clinic-ready agents offer real hope to patients with chemoresistant disease.
Ovarian Cancer and Chemoresistance
Ovarian cancer is the seventh most common cancer amongst women worldwide with an incidence of 6.1 [age-standardized rate (ASR)] per 100,000, and a mortality rate of 3.7 (ASR) (1). It is the fifth highest cause of cancer-related deaths among women, accounting for more deaths than any other cancer of the female reproductive system. Ovarian cancers are classified into a number of subtypes: serous, mucinous, endometrioid, clear cell, transitional, squamous, mixed, and undifferentiated subtypes (2). Further classification is by histopathological grade, with grade (borderline, grades 1–3) associated with how quickly the tumor is likely to grow. Well differentiated or low-grade (type I) tumors are typically indolent, slow growing tumors that are often detected at early stages, and include low-grade serous, low-grade endometrioid, clear cell, and mucinous carcinomas. Poorly differentiated or high-grade (type II) disease includes high-grade serous, high-grade endometrioid, mixed mesodermal (carcinosarcoma), and undifferentiated carcinomas that tend to grow and spread more quickly (3). The majority of patients (~70%) with ovarian cancer are diagnosed with late stage high-grade serous epithelial ovarian cancer (HGSOC), with dissemination of primary tumor throughout the peritoneal cavity in most cases, and the 5-year survival for these patients is <50% (4–6).
Standard therapy for advanced ovarian cancer usually involves surgical debulking of the tumor mass followed by chemotherapy, including a platinum-containing compound. Optimal tumor debulking is critical, as postoperative residual disease strongly influences patient outcome (7). First-line chemotherapy for ovarian cancer is typically carboplatin, or under certain circumstances cisplatin, given either alone or, more commonly, in combination with paclitaxel (8). Response rates to first-line therapy are favorable; however, the relapse rate is high. The platinum-free interval (PFI: i.e., interval between end of chemotherapy and relapse) is a good indicator of response to subsequent treatment with platinum: >12 month PFI predicts favorable response to retreatment; 6–12 month PFI is regarded as “intermediate”; <6 month PFI is defined as platinum resistant with commensurate poor response rate to retreatment with platinum (9, 10). Other chemotherapeutic options, typically used following platinum-resistant relapse, include topotecan (topoisomerase inhibitor), liposomal doxorubicin (inhibitor of DNA replication), gemcitabine (replaces cytidine during DNA replication leading to tumor growth arrest), and etoposide (forms ternary complexes with DNA and topoisomerase II causing DNA strand breaks), however response rates to such alternatives remain dismal. Accordingly, elucidation of mechanisms underpinning platinum resistance is an urgent priority and may allow the development of precision strategies to reverse resistance.
The biochemical mechanisms of cytotoxicity of cis- and carboplatin involve their binding to DNA and non-DNA targets and induction of cell death through apoptosis, necrosis, or both, within the heterogeneous population of tumor cells (11, 12). Direct binding to genomic DNA (gDNA) can result in a number of lesions: the initial lesion formed is bulky platinum-DNA adducts that can mediate intra- and inter-strand crosslinks. If these are not removed but are encountered by the cells’ transcription or replication machinery, stalling of these processes can lead to the generation of DNA breaks, either single-strand DNA breaks (SSB) or double-strand DNA breaks (DSB). In response to such DNA damage, a cell can either initiate repair, or if the damage is too severe, cell cycle arrest, and/or apoptosis are initiated. This process is required for a successful chemotherapeutic response. Non-DNA targets of cis- and carboplatin include components of the cell membrane lipid bilayer, such as phospholipids and phosphatidylserine, and cytoplasmic targets such as cytoskeletal microfilaments, thiol-containing peptides, proteins, and RNA (11, 13). Furthermore, these compounds can alter the activity of enzymes, receptors, and other proteins through coordination to sulfur atoms of cysteine and/or methionine residues and to nitrogen atoms of histidine residues (14). However, the formation of adducts on gDNA are thought to be the primary lesion underlying the cytotoxic effect of these drugs.
Given the range of targets of cis- and carboplatin, it is not surprising that resistance of ovarian cancer to these compounds is multifactorial and has been reported to involve increased drug inactivation and efflux, decreased drug influx, increased cellular glutathione and metallothionein levels, alterations in cell cycle control, oncogene expression, changes in apoptotic threshold, and increased or inappropriate activation of DNA repair pathways (9, 15, 16). Although only 5–10% of covalently bound cell-associated cisplatin is found in the gDNA, targeting the DNA repair process has shown considerable promise as a therapeutic strategy for platinum-resistant ovarian cancer. Here, we review the DNA repair network as a strategic and rational target for therapeutic intervention in ovarian cancer. In particular, we highlight the non-homologous end joining (NHEJ) pathway as an under-explored target for therapeutic discovery and use DNA-dependent protein kinase catalytic subunit (DNA-PKcs) as an example of how this pathway can be targeted therapeutically (see Figure 1 for graphical summary and Table 1 for a summary of the key messages).
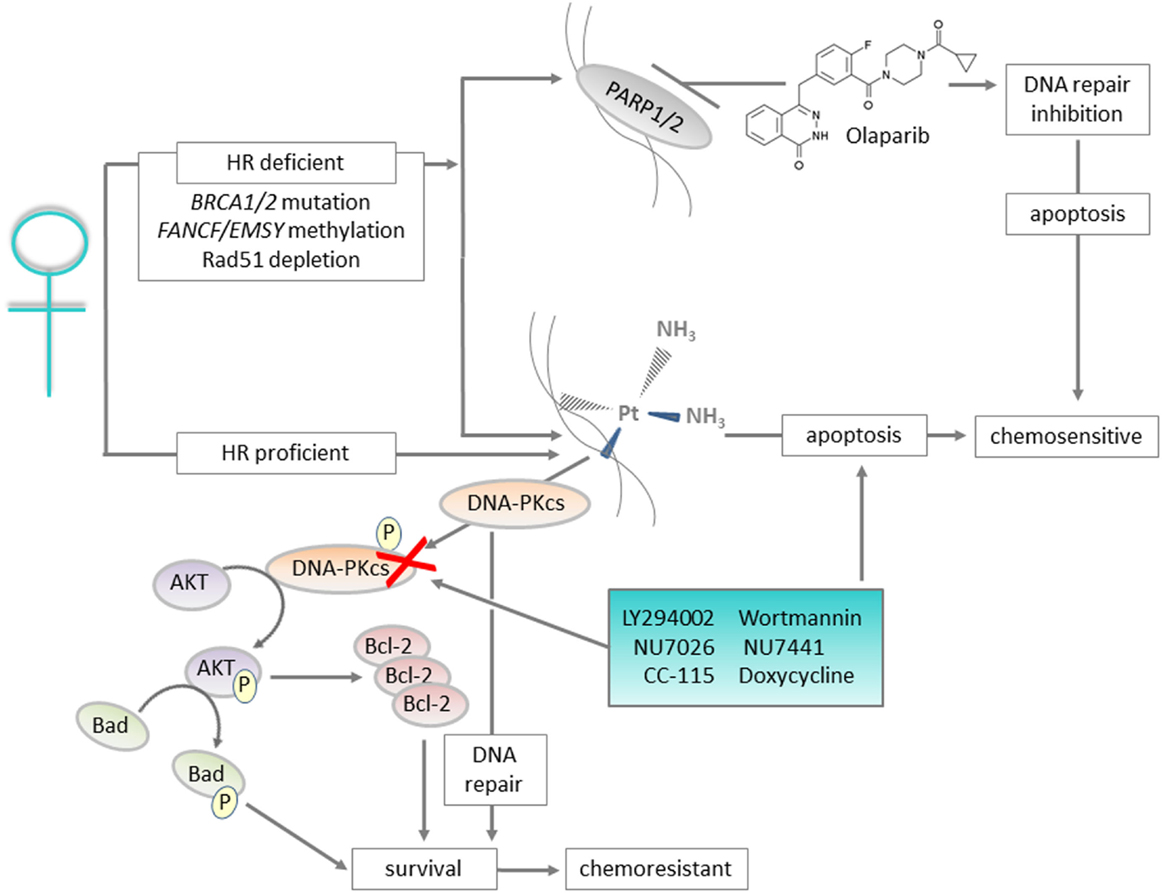
Figure 1. Graphical summary: DNA repair proteins such as DNA-PKcs can be targeted to improve outcomes for patients with ovarian cancer. Ovarian cancers with defects in the homologous recombination (HR) pathway are initially sensitive to platinum treatment, and also respond to Olaparib that targets the base excision repair pathway protein PARP. For the majority of cases, which are HR proficient, platinum-based chemotherapy is still utilized but resistance is likely. For patients with platinum-resistant disease, inhibition of DNA-PKcs, a key component of the non-homologous end joining pathway, represents a targeted approach to prevent the pro-survival AKT and anti-apoptotic signaling associated with resistance.
Table 1. Section summary and key “take home” messages.
DNA Repair Proteins as Therapeutic Targets
The generation of DNA damage is not an exclusive effect of chemotherapeutic agents, and occurs in response to numerous “natural” events such as replication errors or the production of reactive oxygen species after exposure to UV light. In order to respond to the various types of damage that can occur, cells possess an arsenal of DNA repair mechanisms, listed in Table 2. A more detailed description of these processes and their key components is reviewed elsewhere (17). Although DNA repair pathways are crucial for normal cell survival, defects in the execution and control of these mechanisms have been linked both with the development of cancers and response to chemotherapy. In particular, defects in nucleotide excision repair (NER), mismatch repair (MMR), and DSB repair pathways have been linked to platinum sensitivity and resistance (18, 19). Ovarian cancer together with pancreatic and breast cancers have been defined as the top three cancers in which DNA repair pathways are defective (20). As such, disruption of these pathways has been identified as a strategic approach to increase therapeutic responses to DNA-damaging agents (21–23).
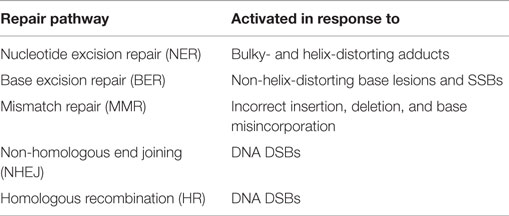
Table 2. DNA repair pathways and associated lesions.
Homologous Recombination Repair Deficiency is Associated with Chemoresponse
Homologous recombination (HR) is one of the major pathways for repair of DSBs, and requires an intact sister chromatid to use as a template to repair the damage. As such, it is restricted to the S and G2 phases of the cell cycle. HR deficiency (HRD) most commonly occurs in HGSOC, and is detected in 44% of these patients (24). In addition, HR defects have been suggested to drive HGSOC development (25). The majority of HRD is linked to germline or somatic alterations in the HR-associated genes BRCA1 and BRCA2 (~20% mutation; 10% methylation) (6, 24). Germline mutations in these genes represent significant risk factors for developing HGSOC: for a woman with a BRCA1 mutation, the risk of developing epithelial ovarian cancer is 39–46%, and with a BRCA2 mutation, 12–20% (26). HRD phenotype is also associated with sensitivity to platinum-based chemotherapy. Additionally, these patients are responsive to poly(ADP-ribose) polymerase (PARP) inhibitors, which are the most successful drugs targeting DNA repair proteins developed to date. PARP functions in the base excision repair (BER) pathway to repair SSBs, and inhibitors have been found to stabilize or regress ovarian cancer with BRCA1/BRCA2 mutations (27–29). The biological basis for this is synthetic lethality due to loss of both BER and HR, resulting in simultaneous inhibition of SSB and DSB repair. The PARP inhibitor Olaparib was approved by the European Union for maintenance treatment of BRCA mutant, platinum-sensitive ovarian cancer in December 2014; the first-in-class approval for a PARP inhibitor. However, some patients without BRCA1/2 mutations also respond to PARP inhibition, implying the presence of other HR defects. For example, BRCA pathway inactivation may also occur through methylation of FANCF and/or EMSY amplification (30, 31). Additionally, RAD51 depletion has been found to sensitize ovarian cancer cells to PARP inhibitor-based combination chemotherapy (32). For a thorough review of strategies to target HR processes, or to exploit inherent deficiencies in associated genes, with the aim of improving ovarian cancer response to platinum-based chemotherapy, see Wiedemeyer et al. (33).
DNA repair defects other than BRCA mutations are also probable influencers of platinum sensitivity in HGSOC; we and others have previously described a BRCA2 reversion mutation, which does not equate to full cisplatin resistance in a HGSOC cell line series (34). The PEO1/4 cell line set were initially derived from a HGSOC patient during the platinum-sensitive and -resistant phases of the disease, respectively (35). This patient presented with a germline BRCA2 truncating mutation, which was absent in the platinum-resistant PEO4 cell line. However, reversion of this mutation has been reported in PEO1 cells, by us and others (34, 36), and although this restores BRCA2 functionality, the platinum-resistant phenotype is not fully recapitulated; indeed, we have reported a 10-fold difference in cisplatin IC50 values between BRCA revertant PEO1 cells and PEO4 cells (34). Based on this, a continuum of platinum sensitivity can be proposed: from extreme (HR defective/BRCA mutant) to intermediate (HR competent i.e., BRCA revertant) to resistant (i.e., active resistance mechanisms). These latter mechanisms may be driven by components of other DNA repair pathway components, for example, DNA-PKcs that drives AKT survival signaling in HGSOC (37), and will be discussed now.
DNA-PKcs as a Therapeutic Target for Ovarian Cancer
DNA-PKcs Structure and Regulation
In comparison to HR that is restricted to post-DNA replication phases of the cell cycle, the NHEJ pathway can respond to DSBs throughout the cell cycle. This is due to its lack of requirement for a template DNA strand to use in repair. A key mediator of NHEJ is DNA-PKcs, a DNA-activated serine/threonine protein kinase that is abundantly expressed in almost all mammalian cells. DNA-PKcs is a member of the phosphatidylinositol-3-OH kinase (PI(3)K)-related protein (PIKK) superfamily, and is encoded on chromosome 8q11.21 by the PRKDC gene with a size of 187.07 kb and 86 exons. The DNA-PKcs protein consists of 4129 amino acids (~469 kDa), and contains a number of regions, including a catalytic domain and DNA-binding and Ku-binding domains (38).
DNA-PKcs functions as the catalytic subunit of the DNA-PK holoenzyme, which although best known for its role in NHEJ has other reported roles, including regulating apoptosis, maintaining telomere length, cell cycle control, and regulation of mitochondrial protein function (39–41). The holoenzyme is composed of DNA-PKcs and the Ku70/80 proteins. These are not constitutively associated; instead the holoenzyme is assembled in response to contact with DNA. The ring-like structure of the Ku70/80 dimer first binds to the DNA break, encircling the end and allowing the dimer to translocate along the duplex. DNA-PKcs is then recruited, and the complex acts as a scaffold, providing binding sites for downstream components of NHEJ. The kinase activity of DNA-PKcs is activated following its association with both the Ku70/80 dimer and a DNA terminus and autophosphorylation triggers a conformational change leading to the release of the DNA ends, making them available to other factors participating in DNA end-processing or ligation. As such, DNA-PKcs functions as a “gatekeeper” to protect DNA ends from premature processing, ligation, and degradation until the two broken ends are properly positioned (42–44). Although DNA-PKcs is most potently activated in response to DSBs, DNA-independent activation of DNA-PKcs due to its interaction with other proteins has also been reported, e.g., C1D, HSF1, and Lyn (45–47). The physiological significance of these DNA-independent activities of DNA-PKcs is less well understood.
DNA-PKcs contains multiple phosphorylation sites, and modifications to these alter the activity, conformation, and stability of the protein. Autophosphorylation events are initiated following binding of DNA-PKcs to DNA, and function reciprocally to coordinate the control of NHEJ. For example, phosphorylation at the ABCDE cluster, located between Thr2609 and Thr2647, promotes DNA end-processing, whereas phosphorylation at the PQR cluster between Ser2023 and Ser2056 prevents this by decreasing accessibility. End-processing of complex DSBs (e.g., hairpins) also requires trans-phosphorylation of DNA-PKcs by ATM, and this serves to recruit the endonuclease Artemis to the site of damage. Subsequent to this, autophosphorylation is required to remove DNA-PKcs in order for end-ligation to proceed (48). Autophosphorylation of DNA-PKcs at its extreme N terminus, on Ser56 and Ser72, may be involved in holoenzyme complex stability, while phosphorylation in the T-loop of the kinase domain at Thr3950 results in kinase inactivation without affecting complex stability. DNA-PKcs phosphorylation may also affect DSB repair pathway choice: cells deficient in DNA-PKcs show increased HR-mediated repair of DSBs, and autophosphorylation of DNA-PKcs at the Thr946 and Ser1004 cluster seems to serve as a switch between NHEJ and HR (49–51).
The choice of pathway may additionally be affected by the interaction of DNA-PKcs with other proteins. For example, the kinase and proto-oncogene c-Abl is phosphorylated by ATM in response to ionizing radiation (IR) and certain other DNA-damaging agents, and has been shown to interact with DNA-PKcs. Phosphorylation of DNA-PKcs by c-Abl inhibits its ability to form a complex with DNA, whereas phosphorylation of c-Abl by DNA-PKcs potentially activates c-Abl kinase activity in response to IR exposure. This suggests the existence of an autoregulatory negative feed-back loop that might lead to repression of DNA-PKcs activity after the appropriate DNA damage signaling and/or repair pathways have been initiated (52). The protein kinase activity of DNA-PKcs can also be stimulated by PARP independently of the Ku70/80 complex (53) suggesting that PARP, in addition to its key role in BER, may additionally facilitate DNA DSB repair via regulation of DNA-PKcs.
A number of mechanisms by which DNA-PKcs activity is attenuated have been reported. DNA-PKcs inactivation via cleavage by the ICE family cysteine proteases, for example, caspase-3, may occur in apoptotic cells to prevent repair of the fragmented gDNA that is produced during the final steps of the apoptotic pathway (54, 55). Degradation of DNA-PKcs via an ubiquitin-mediated proteasome pathway has also been reported following Herpes simplex virus type 1 infection, possibly as a mechanism for aiding virus replication (56).
DNA-PKcs and Chemoresistance
Numerous studies have shown a correlation between DNA-PKcs expression and activity with response to radio- and chemotherapy. This is unsurprising given that DNA-PKcs plays a crucial role in the repair of the DSBs generated by these treatments. Additionally, chemoresponse has been linked to other activities of DNA-PKcs, including cell cycle control and regulation of mitochondrial heat-shock proteins (39, 41). However, predictive association of DNA-PKcs expression/activity in human cancer is controversial and differs between stage and pathological type. For example, cells with defective DNA-PKcs activity show increased radiosensitivity, and in lung carcinoma cell lines after γ-irradiation, the lowest DNA-PKcs protein content and kinase activity was found in the most radiosensitive cells, U-1285 and H-69, while the highest was found in the most radioresistant cells U-1810 (57, 58). DNA-PKcs activity in peripheral blood lymphocytes from untreated patients with advanced breast and uterine cervix cancers have been found to be significantly lower than in those with early stage disease. However, other studies have shown correlation between increased DNA-PKcs expression and advanced tumor stage. A genome-wide copy number and expression microarray analysis of gastric cancer revealed diverse chromosomal region alterations for different stages or histological subtypes of this disease: copy number gains at chromosome 8q11-q24 were very frequent (63%) and candidate genes such as PRKDC showed co-regulation with increased expression levels (59).
Some relationship may exist between the NHEJ pathway and cellular sensitivity or resistance to DNA damage; however, this relationship does not appear to be universal and may be cell or tumor-type specific. No strong correlation between radiosensitivity and DNA-PKcs was found in a study of sporadic human ovarian cancer cell lines. The data suggested that DNA-PKcs copy number, expression level, or kinase activities are not reliable predictors of radiosensitivity in ovarian cancer (60). However, inhibition of DNA-PKcs has been shown by us to reverse cisplatin resistance in a panel of ovarian cancer cell lines (37). This study used paired cell lines derived from patients before and after the emergence of platinum resistance. Interestingly, we found that DNA-PKcs inhibition did not increase the apoptotic effect of platinum in already sensitive cells. The mechanism underpinning the response to DNA-PKcs inhibition was shown to result from direct modulation of AKT survival signaling by DNA-PKcs. In platinum-resistant cells only, AKT was activated in response to cisplatin-mediated DNA damage by phosphorylation on serine 473 by DNA-PKcs. This was prevented by DNA-PKcs inhibition and doing so reversed the platinum-resistant phenotype, identifying a specific role for DNA-PKcs in mediating platinum resistance in HGSOC. DNA-PKcs-driven AKT signaling has also been identified to underlie doxorubicin resistance in glioblastoma cell lines (61).
Pharmacological DNA-PKcs Inhibitors
Despite the current lack of a robust predictive biomarker of response to DNA-PKcs inhibition, many small-molecule inhibitors for this protein have been developed. The chemistry of these compounds varies, and some of these have selectivity for DNA-PKcs while others are broad-range PI3K family member inhibitors. The most common PIKK proteins targeted by these compounds are mTOR, PI3K, ATM, and ATR. The first identified inhibitors of DNA-PKcs were the PI3K inhibitors wortmannin and the quercetin derivative LY294002. Wortmannin is a fungal metabolite, and was found to inhibit DNA-PKcs in a non-ATP-competitive manner, to potentiate IR-induced cytotoxicity and to inhibit DSB repair at concentrations that inhibit DNA-PKcs activity. However, the IC50 required for this is ~2-fold higher than that required for PI3K inhibition, indicating the effects observed may be due to multiple protein targeting (62). The cell-permeable compound LY294002, on the other hand, acts on the ATP-binding site of PI3K proteins and inhibits DNA-PKcs in a competitive manner. The apoptotic rate of X-ray irradiated HeLa cells after pre-treatment with LY294002 was found to be significantly higher than that of untreated cells, with a prolonged G2/M delay also observed in these cells (63). IC87361 is a morpholino-flavonoid that has been derived from LY294002, and which is 50-fold more selective for DNA-PKcs than for PI3K and other kinases. It enhances radiation sensitivity in wild-type C57BL6 mouse pulmonary endothelial cells but not in SCID mouse cells, which lack DNA-PKcs. Furthermore, it was found to increase irradiation-induced apoptosis in lung cancer and melanoma cells, and caused significant growth delay in Lewis lung carcinoma xenografts treated with radiation and IC87361 in comparison to tumors treated with radiation alone (64). Other compounds developed and that have been studied as radiosensitizers are PI-103 (65), IC86621 (66), and AMA37 (67).
Two of the most widely studied DNA-PKcs inhibitors are NU7026 and NU7441. NU7026 is an ATP-competitive inhibitor of DNA-PKcs which displays selectivity over other PIKK family enzymes. It was developed using LY294002 as a template, shows a 60-fold greater potency against DNA-PKcs than PI3K, and is inactive against both ATM and ATR (IC50 0.23 μM for DNA-PK, 13 μM for PI3K, >100 μM for ATM and ATR). It has been shown to sensitize mouse embryonic fibroblasts and Chinese hamster ovary cells to radiation in vitro, and is not cytotoxic itself (68, 69). Additionally, NU7026 has shown chemosensitization activity, and has been shown to have synergistic cytotoxic activity when combined with chlorambucil in chronic lymphocytic leukemia cells, and this chemosensitization was found to correlate with inhibition of DNA-PKcs phosphorylation (70). In pancreatic ductal adenocarcinoma cells, inhibition of NHEJ with NU7026 has been shown to result in accumulation of DNA damage, inhibition of growth, and ultimately apoptosis even in the absence of exogenous DNA-damaging agents (71). Our previous data have used NU7026 to demonstrate the potential of DNA-PKcs inhibition as a chemosensitization strategy in HGSOC cell lines derived from patients with clinically platinum-resistant disease (37).
NU7441 is more potent than NU7026, and is a specific inhibitor of DNA-PKcs, with at least 100-fold selectivity for this enzyme compared to other PIKK family kinases (72, 73). NU7441 has been found to increase the response of many cancer cell types to both chemo- and radiotherapy (73–88). The specificity of this compound was initially illustrated by the finding that it increases chemo- and radiosensitivity in MO59 glioblastoma cells, but not in their DNA-PKcs-deficient MO59-J counterparts. However, the mechanism by which NU7441 increases drug sensitivity has recently been shown to also result from inhibition of multidrug-resistance protein 1 (MDR1), resulting in increased nuclear accumulation of cytotoxic agents that are substrates for this drug efflux pump and thus adding complexity to the mechanistic interpretation of response data obtained using this compound (85). Interestingly, MDR1 upregulation itself has been associated with DNA-PKcs activity in glioblastoma cell lines with acquired doxorubicin resistance (61).
To date, only one pharmacological inhibitor of DNA-PKcs has been assessed clinically. CC-115 is a dual DNA-PKcs/mTOR inhibitor which has been used in a phase 1 trial for patients with advanced malignancies (NCT01353625; http://clinicaltrials.gov). The results of this study are expected in 2016. DNA-PKcs has also been targeted in another recently completed phase 1 trial, combining DT-01, a DSB mimetic, with radiation in metastatic melanoma (NCT01469455). DT-01 is comprised of small DNA molecules, known as Dbait, which sequester DNA-PKcs and PARP, thereby impairing the DNA repair response to radiotherapy (89). Intriguingly, DNA-PKcs has recently been identified as key protein downregulated in breast cancer stem cells in response to treatment with the broad spectrum antibiotic doxycycline (90). Moreover, this was associated with increased radiosensitivity and decreased mammosphere formation in vitro, and suggests a potentially immediate way to incorporate DNA-PKcs inhibition into clinical practice.
DNA-PKcs Inhibition as a Platinum Sensitization Strategy
Although the majority of HGSOC patients initially respond to platinum-based chemotherapy, the emergence of resistance is a major barrier to its long-term effectiveness. Therefore, identifying a targeted strategy to restore response may offer real promise in the treatment of this disease. As discussed here, DNA-PKcs inhibition has been shown to be effective in restoring platinum cytotoxicity in HGSOC (37). 4,5-Dimethoxy-2-nitrobenzaldehyde (DMNB), a cell-permeable vanillin derivative has also been reported to sensitize A2780 human ovarian carcinoma cells to cisplatin via inhibition of DNA-PKcs (91). Loss of DNA-PKcs has also been associated with increased cisplatin response in cervical cancer and gliomas (92, 93). The therapeutic activity of DNA-PKcs inhibition has also been linked to mechanisms independent of its role in DNA-damage repair, and with respect to restoring platinum sensitivity. This may partly be related to increased drug accumulation due to MDR1 protein inhibition; however, additionally, we have shown that DNA-PKcs is involved in DNA-damage-mediated activation of AKT cell survival signaling (37). In this study, we found that platinum exposure induces an AKT-dependent pro-survival DNA damage response in clinically platinum-resistant but not platinum-sensitive HGSOC. In this system, AKT is phosphorylated specifically on Ser473 by DNA-PKcs in the nucleus of platinum resistant but not sensitive cells. Inhibition of DNA-PKcs or AKT, but not mTORC2, was found to restore platinum sensitivity. Together, these data indicate that DNA-PKcs inhibition might be a clinical useful strategy for the prevention of platinum-induced AKT activation without interfering with normal glucose homeostasis. Although DNA-PKcs inhibitors are not yet ready for full clinical implementation, we have been able to test our laboratory hypothesis that preventing DNA-PKcs-mediated AKT signaling will reverse platinum resistance clinically by using an AKT inhibitor. A clinical trial led by our center reported a 37% overall response rate to daily oral AKT inhibition using GSK2110183 in combination with six cycles of 3-weekly carboplatin (AUC5) and paclitaxel (175 mg/m2) in clinically platinum-resistant ovarian cancer: a patient group with an expected response rate to carboplatin/taxol of only 10–14% (94).
In vitro studies have also shown the potential of DNA-PKcs inhibition as a sensitizing strategy for platinum compounds in other cancers, including osteosarcoma, breast, and pancreatic (37, 95, 96), and therefore development of such inhibitors may present broad clinical applicability. In particular, they may be particularly beneficial in lung cancer, where platinum resistance is also associated with aberrant DDR mechanisms (97). A role for DNA-PKcs in driving metastatic processes, i.e., angiogenesis, migration, and invasion, has been recently identified, suggesting an additional benefit of DNA-PKcs inhibitor-based chemotherapy in treating metastatic cancers (98).
The promise of PARP inhibition strategies and DNA-PKcs as a therapeutic target highlights the importance of understanding the clinical significance of the functioning and defective DNA repair mechanisms in cancer. There remains the need to identify reliable biomarkers of tumor cell response and resistance to therapies targeting DNA repair proteins and indeed to identify and validate new therapeutic targets from this critical but insufficiently mined resource. The identification of patient subgroups who will benefit most from such strategies is also required – DDR proteins such as DNA-PKcs have been suggested to have a tumor-suppressive role in the early stages of carcinogenesis where ineffective DDR may contribute to the generation of genomic instability that drives tumor progression (99). As such, the development of DNA-PKcs inhibitions, and indeed other DDR targeted therapies, should be mindful of DNA damage thresholds that can be either oncogenic or tumor-suppressive, depending on the tumor stage. Together, such knowledge and understanding will translate into the development of new directed strategies that will help overcome clinical platinum resistance in ovarian cancer, and by that reduce patient mortality.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Ovarian Cancer Action (ES) and Plum’s Fund (EM) for funding.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer (2014) 136(5):E359–86. doi: 10.1002/ijc.29210
2. Soslow RA. Histologic subtypes of ovarian carcinoma: an overview. Int J Gynecol Pathol (2008) 27(2):161–74. doi:10.1097/PGP.0b013e31815ea812
3. Shih Ie M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol (2004) 64(5):1511–8. doi:10.1016/S0002-9440(10)63708-X
4. Herzog TJ. Recurrent ovarian cancer: how important is it to treat to disease progression? Clin Cancer Res (2004) 10(22):7439–49. doi:10.1158/1078-0432.CCR-04-0683
5. Berns EM, Bowtell DD. The changing view of high-grade serous ovarian cancer. Cancer Res (2012) 72(11):2701–4. doi:10.1158/0008-5472.CAN-11-3911
6. The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature (2012) 474:609–15. doi:10.1038/nature10166
7. Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol (2002) 20(5):1248–59. doi:10.1200/JCO.20.5.1248
8. Ledermann JA, Raja FA, Fotopoulou C, Gonzalez-Martin A, Colombo N, Sessa C, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol (2013) 24(Suppl 6):vi24–32. doi:10.1093/annonc/mdt333
9. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer (2003) 3(7):502–16. doi:10.1038/nrc1123
10. Cooke SL, Brenton JD. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol (2011) 12(12):1169–74. doi:10.1016/S1470-2045(11)70123-1
11. Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci (2000) 57(8–9):1229–35. doi:10.1007/PL00000762
12. Cepeda V, Fuertes MA, Castilla J, Alonso C, Quevedo C, Pérez JM. Biochemical mechanisms of cisplatin cytotoxicity. Anticancer Agents Med Chem (2007) 7(1):3–18. doi:10.2174/187152007779314044
13. Speelmans G, Staffhorst RW, Versluis K, Reedijk J, de Kruijff B. Cisplatin complexes with phosphatidylserine in membranes. Biochemistry (1997) 36(34):10545–50. doi:10.1021/bi9703047
14. Peleg-Shulman T, Gibson D. Cisplatin-protein adducts are efficiently removed by glutathione but not by 5’-guanosine monophosphate. J Am Chem Soc (2001) 123(13):3171–2. doi:10.1021/ja005854y
15. Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev (2007) 33(1):9–23. doi:10.1016/j.ctrv.2006.09.006
16. Sherman-Baust CA, Becker KG, Wood Iii WH, Zhang Y, Morin PJ. Gene expression and pathway analysis of ovarian cancer cells selected for resistance to cisplatin, paclitaxel, or doxorubicin. J Ovarian Res (2011) 4(1):21. doi:10.1186/1757-2215-4-21
17. Maginn EN, de Sousa CH, Wasan HS, Stronach EA. Opportunities for translation: targeting DNA repair pathways in pancreatic cancer. Biochim Biophys Acta (2014) 1846(1):45–54. doi:10.1016/j.bbcan.2014.04.002
18. Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol (2006) 24(23):3799–808. doi:10.1200/JCO.2005.05.4171
19. Martin LP, Hamilton TC, Schilder RJ. Platinum resistance: the role of DNA repair pathways. Clin Cancer Res (2008) 14(5):1291–5. doi:10.1158/1078-0432.CCR-07-2238
20. Cowley MJ, Chang DK, Pajic M, Johns AL, Waddell N, Grimmond SM, et al. Understanding pancreatic cancer genomes. J Hepatobiliary Pancreat Sci (2013) 20(6):549–56. doi:10.1007/s00534-013-0610-6
21. Al-Ejeh F, Kumar R, Wiegmans A, Lakhani SR, Brown MP, Khanna KK. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene (2010) 29(46):6085–98. doi:10.1038/onc.2010.407
22. Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer (2012) 12(9):587–98. doi:10.1038/nrc3342
23. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature (2012) 481(7381):287–94. doi:10.1038/nature10760
24. Cunningham JM, Cicek MS, Larson NB, Davila J, Wang C, Larson MC, et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci Rep (2014) 4:4026. doi:10.1038/srep04026
25. Bowtell DD. The genesis and evolution of high-grade serous ovarian cancer. Nat Rev Cancer (2010) 10(11):803–8. doi:10.1038/nrc2946
26. George SH, Shaw P. BRCA and early events in the development of serous ovarian cancer. Front Oncol (2014) 4:5. doi:10.3389/fonc.2014.00005
27. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361(2):123–34. doi:10.1056/NEJMoa0900212
28. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med (2012) 366(15):1382–92. doi:10.1056/NEJMoa1105535
29. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol (2014) 15(8):852–61. doi:10.1016/S1470-2045(14)70228-1
30. Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin SF, et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell (2003) 115(5):523–35. doi:10.1016/S0092-8674(03)00930-9
31. Wang Z, Li M, Lu S, Zhang Y, Wang H. Promoter hypermethylation of FANCF plays an important role in the occurrence of ovarian cancer through disrupting Fanconi anemia-BRCA pathway. Cancer Biol Ther (2006) 5(3):256–60. doi:10.4161/cbt.5.3.2380
32. Huehls AM, Wagner JM, Huntoon CJ, Karnitz LM. Identification of DNA repair pathways that affect the survival of ovarian cancer cells treated with a poly(ADP-ribose) polymerase inhibitor in a novel drug combination. Mol Pharmacol (2012) 82(4):767–76. doi:10.1124/mol.112.080614
33. Wiedemeyer WR, Beach JA, Karlan BY. Reversing platinum resistance in high-grade serous ovarian carcinoma: targeting BRCA and the homologous recombination system. Front Oncol (2014) 4:34. doi:10.3389/fonc.2014.00034
34. Stronach EA, Alfraidi A, Rama N, Datler C, Studd JB, Agarwal R, et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res (2011) 71(13):4412–22. doi:10.1158/0008-5472.CAN-10-4111
35. Langdon SP, Lawrie SS, Hay FG, Hawkes MM, McDonald A, Hayward IP, et al. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res (1988) 48(21):6166–72.
36. Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res (2009) 69(16):6381–6. doi:10.1158/0008-5472.CAN-09-1178
37. Stronach EA, Chen M, Maginn EN, Agarwal R, Mills GB, Wasan H, et al. DNA-PK mediates AKT activation and apoptosis inhibition in clinically acquired platinum resistance. Neoplasia (2011) 13(11):1069–80. doi:10.1593/neo.111032
38. Jette N, Lees-Miller SP. The DNA-dependent protein kinase: a multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog Biophys Mol Biol (2014) 117(2–3):194–205. doi:10.1016/j.pbiomolbio.2014.12.003
39. Um JH, Kang CD, Hwang BW, Ha MY, Hur JG, Kim DW, et al. Involvement of DNA-dependent protein kinase in regulation of the mitochondrial heat shock proteins. Leuk Res (2003) 27(6):509–16. doi:10.1016/S0145-2126(02)00264-3
40. Dip R, Naegeli H. More than just strand breaks: the recognition of structural DNA discontinuities by DNA-dependent protein kinase catalytic subunit. FASEB J (2005) 19(7):704–15. doi:10.1096/fj.04-3041rev
41. Gustafsson AS, Abramenkovs A, Stenerlöw B. Suppression of DNA-dependent protein kinase sensitise cells to radiation without affecting DSB repair. Mutat Res (2014) 769:1–10. doi:10.1016/j.mrfmmm.2014.06.004
42. Weterings E, van Gent DC. The mechanism of non-homologous end-joining: a synopsis of synapsis. DNA Repair (Amst) (2004) 3:1425–35. doi:10.1016/j.dnarep.2004.06.003
43. Costantini S, Woodbine L, Andreoli L, Jeggo PA, Vindigni A. Interaction of the Ku heterodimer with the DNA ligase IV/Xrcc4 complex and its regulation by DNA-PK. DNA Repair (Amst) (2007) 6(6):712–22. doi:10.1016/j.dnarep.2006.12.007
44. Pawelczak KS, Turchi JJ. A mechanism for DNA-PK activation requiring unique contributions from each strand of a DNA terminus and implications for microhomology-mediated nonhomologous DNA end joining. Nucleic Acids Res (2008) 36(12):4022–31. doi:10.1093/nar/gkn344
45. Kumar S, Pandey P, Bharti A, Jin S, Weichselbaum R, Weaver D, et al. Regulation of DNA-dependent protein kinase by the Lyn tyrosine kinase. J Biol Chem (1998) 273(40):25654–8. doi:10.1074/jbc.273.40.25654
46. Yavuzer U, Smith GC, Bliss T, Werner D, Jackson SP. DNA end-independent activation of DNA-PK mediated via association with the DNA-binding protein C1D. Genes Dev (1998) 12(14):2188–99. doi:10.1101/gad.12.14.2188
47. Mi J, Dziegielewski J. Activation of DNA-PK by ionizing radiation is mediated by protein phosphatase 6. PLoS One (2009) 4(2):e4395. doi:10.1371/journal.pone.0004395
48. Jiang W, Crowe JL, Liu X, Nakajima S, Wang Y, Li C, et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol Cell (2015) 58(1):172–85. doi:10.1016/j.molcel.2015.02.024
49. Allen C, Kurimasa A, Brenneman MA, Chen DJ, Nickoloff JA. DNA-dependent protein kinase suppresses double-strand break-induced and spontaneous homologous recombination. Proc Natl Acad Sci U S A (2002) 99:3758–63. doi:10.1073/pnas.052545899
50. Cui X, Yu Y, Gupta S, Cho YM, Lees-Miller SP, Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol Cell Biol (2005) 25(24):10842–52. doi:10.1128/MCB.25.24.10842-10852.2005
51. Neal JA, Meek K. Choosing the right path: does DNA-PK help make the decision? Mutat Res (2011) 711(1–2):73–86. doi:10.1016/j.mrfmmm.2011.02.010
52. Kharbanda S, Pandey P, Jin S, Inoue S, Bharti A, Yuan ZM, et al. Functional interaction between DNA-PK and c-Abl in response to DNA damage. Nature (1997) 386(6626):732–5. doi:10.1038/386732a0
53. Ruscetti T, Lehnert BE, Halbrook J, Le Trong H, Hoekstra MF, Chen DJ, et al. Stimulation of the DNA-dependent protein kinase by poly(ADP-ribose) polymerase. J Biol Chem (1998) 273(23):14461–7. doi:10.1074/jbc.273.23.14461
54. Han Z, Malik N, Carter T, Reeves WH, Wyche JH, Hendrickson EA. DNA-dependent protein kinase is a target for a CPP32-like apoptotic protease. J Biol Chem (1996) 271(40):25035–40. doi:10.1074/jbc.271.40.25035
55. Song Q, Lees-Miller SP, Kumar S, Zhang Z, Chan DW, Smith GC, et al. DNA-dependent protein kinase catalytic subunit: a target for an ICE-like protease in apoptosis. EMBO J (1996) 15(13):3238–46.
56. Parkinson J, Lees-Miller SP, Everett RD. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol (1999) 73(1):650–7.
57. Kirchgessner CU, Patil CK, Evans JW, Cuomo CA, Fried LM, Carter T, et al. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science (1995) 267(5201):1178–83. doi:10.1126/science.7855601
58. Sirzén F, Nilsson A, Zhivotovsky B, Lewensohn R. DNA-dependent protein kinase content and activity in lung carcinoma cell lines: correlation with intrinsic radiosensitivity. Eur J Cancer (1999) 35(1):111–6. doi:10.1016/S0959-8049(98)00289-5
59. Cheng L, Wang P, Yang S, Yang Y, Zhang Q, Zhang W, et al. Identification of genes with a correlation between copy number and expression in gastric cancer. BMC Med Genomics (2012) 5:14. doi:10.1186/1755-8794-5-14
60. Langland GT, Yannone SM, Langland RA, Nakao A, Guan Y, Long SB, et al. Radiosensitivity profiles from a panel of ovarian cancer cell lines exhibiting genetic alterations in p53 and disparate DNA-dependent protein kinase activities. Oncol Rep (2010) 23(4):1021–6. doi:10.3892/or_00000728
61. Xi G, Hayes E, Lewis R, Ichi S, Mania-Farnell B, Shim K, et al. CD133 and DNA-PK regulate MDR1 via the PI3K- or Akt-NF-κB pathway in multidrug-resistant glioblastoma cells in vitro. Oncogene (2015) 1–10. doi:10.1038/onc.2015.78
62. Izzard RA, Jackson SP, Smith GCM. Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res (1999) 59(11):2581–6.
63. Zhuang L, Cao Y, Xiong H, Gao Q, Cao Z, Liu F, et al. Suppression of DNA-PKcs and Ku80 individually and in combination: different effects of radiobiology in HeLa cells. Int J Oncol (2011) 39(2):443–51. doi:10.3892/ijo.2011.1041
64. Shinohara ET, Geng L, Tan J, Chen H, Chen H, Shir Y, et al. DNA-dependent protein kinase is a molecular target for the development of noncytotoxic radiation-sensitizing drugs. Cancer Res (2005) 65(12):4987–92. doi:10.1158/0008-5472.CAN-04-4250
65. Westhoff MA, Kandenwein JA, Karl S, Vellanki SH, Braun V, Eramo A, et al. The pyridinylfuranopyrimidine inhibitor, PI-103, chemosensitizes glioblastoma cells for apoptosis by inhibiting DNA repair. Oncogene (2009) 28(40):3586–96. doi:10.1038/onc.2009.215
66. Peddi P, Loftin CW, Dickey JS, Hair JM, Burns KJ, Aziz K, et al. DNA-PKcs deficiency leads to persistence of oxidatively induced clustered DNA lesions in human tumor cells. Free Radic Biol Med (2010) 48(10):1435–43. doi:10.1016/j.freeradbiomed.2010.02.033
67. Sturgeon CM, Knight ZA, Shokat KM, Roberge M. Effect of combined DNA repair inhibition and G2 checkpoint inhibition on cell cycle progression after DNA damage. Mol Cancer Ther (2006) 5(4):885–92. doi:10.1158/1535-7163.MCT-05-0358
68. Veuger SJ, Curtin NJ, Richardson CJ, Smith GC, Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res (2003) 63(18):6008–15.
69. Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, et al. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood (2004) 103(12):4659–65. doi:10.1182/blood-2003-07-2527
70. Amrein L, Loignon M, Goulet AC, Dunn M, Jean-Claude B, Aloyz R, et al. Chlorambucil cytotoxicity in malignant B lymphocytes is synergistically increased by 2-(morpholin-4-yl)-benzo[h]chomen-4-one (NU7026)-mediated inhibition of DNA double-strand break repair via inhibition of DNA-dependent protein kinase. J Pharmacol Exp Ther (2007) 321(3):848–55. doi:10.1124/jpet.106.118356
71. Li YH, Wang X, Pan Y, Lee DH, Chowdhury D, Kimmelman AC. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS One (2012) 7(6):e39588. doi:10.1371/journal.pone.0039588
72. Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett (2004) 14(24):6083–7. doi:10.1016/j.bmcl.2004.09.060
73. Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res (2006) 66(10):5354–62. doi:10.1158/0008-5472.CAN-05-4275
74. Willmore E, Elliott SL, Mainou-Fowler T, Summerfield GP, Jackson GH, O’Neill F, et al. DNA-dependent protein kinase is a therapeutic target and an indicator of poor prognosis in B-cell chronic lymphocytic leukemia. Clin Cancer Res (2008) 14(12):3984–92. doi:10.1158/1078-0432.CCR-07-5158
75. Cano C, Barbeau OR, Bailey C, Cockcroft XL, Curtin NJ, Duggan H, et al. DNA-dependent protein kinase (DNA-PK) inhibitors. Synthesis and biological activity of quinolin-4-one and pyridopyrimidin-4-one surrogates for the chromen-4-one chemotype. J Med Chem (2010) 53(24):8498–507. doi:10.1021/jm100608j
76. Elliott SL, Crawford C, Mulligan E, Summerfield G, Newton P, Wallis J, et al. Mitoxantrone in combination with an inhibitor of DNA-dependent protein kinase: a potential therapy for high risk B-cell chronic lymphocytic leukaemia. Br J Haematol (2011) 152(1):61–71. doi:10.1111/j.1365-2141.2010.08425.x
77. Guo L, Liu X, Jiang Y, Nishikawa K, Plunkett W. DNA-dependent protein kinase and ataxia telangiectasia mutated (ATM) promote cell survival in response to NK314, a topoisomerase IIα inhibitor. Mol Pharmacol (2011) 80(2):321–7. doi:10.1124/mol.109.057125
78. Shaheen FS, Znojek P, Fisher A, Webster M, Plummer R, Gaughan L, et al. Targeting the DNA double strand break repair machinery in prostate cancer. PLoS One (2011) 6(5):e20311. doi:10.1371/journal.pone.0020311
79. Tavecchio M, Munck JM, Cano C, Newell DR, Curtin NJ. Further characterisation of the cellular activity of the DNA-PK inhibitor, NU7441, reveals potential cross-talk with homologous recombination. Cancer Chemother Pharmacol (2012) 69(1):155–64. doi:10.1007/s00280-011-1662-4
80. Cano C, Saravanan K, Bailey C, Bardos J, Curtin NJ, Frigerio M, et al. 1-substituted (Dibenzo[b,d]thiophen-4-yl)-2-morpholino-4H-chromen-4-ones endowed with dual DNA-PK/PI3-K inhibitory activity. J Med Chem (2013) 56(16):6386–401. doi:10.1021/jm400915j
81. Song H, Hedayati M, Hobbs RF, Shao C, Bruchertseifer F, Morgenstern A, et al. Targeting aberrant DNA double-strand break repair in triple-negative breast cancer with alpha-particle emitter radiolabeled anti-EGFR antibody. Mol Cancer Ther (2013) 12(10):2043–54. doi:10.1158/1535-7163.MCT-13-0108
82. Tichy A, Durisova K, Salovska B, Pejchal J, Zarybnicka L, Vavrova J, et al. Radio-sensitization of human leukaemic MOLT-4 cells by DNA-dependent protein kinase inhibitor, NU7441. Radiat Environ Biophys (2014) 53(1):83–92. doi:10.1007/s00411-013-0494-5
83. Ciszewski WM, Tavecchio M, Dastych J, Curtin NJ. DNA-PK inhibition by NU7441 sensitizes breast cancer cells to ionizing radiation and doxorubicin. Breast Cancer Res Treat (2014) 143(1):47–55. doi:10.1007/s10549-013-2785-6
84. Azad A, Bukczynska P, Jackson S, Haupt Y, Cullinane C, McArthur GA, et al. Co-targeting deoxyribonucleic acid-dependent protein kinase and poly(adenosine diphosphate-ribose) polymerase-1 promotes accelerated senescence of irradiated cancer cells. Int J Radiat Oncol Biol Phys (2014) 88(2):385–94. doi:10.1016/j.ijrobp.2013.10.043
85. Mould E, Berry P, Jamieson D, Hill C, Cano C, Tan N, et al. Identification of dual DNA-PK MDR1 inhibitors for the potentiation of cytotoxic drug activity. Biochem Pharmacol (2014) 88(1):58–65. doi:10.1016/j.bcp.2014.01.001
86. Chua ML, Horn S, Somaiah N, Davies S, Gothard L, A’Hern R, et al. DNA double-strand break repair and induction of apoptosis in ex vivo irradiated blood lymphocytes in relation to late normal tissue reactions following breast radiotherapy. Radiat Environ Biophys (2014) 53(2):355–64. doi:10.1007/s00411-014-0531-z
87. Cornell L, Munck JM, Alsinet C, Villanueva A, Ogle L, Willoughby CE, et al. DNA-PK-A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin Cancer Res (2015) 21(4):925–33. doi:10.1158/1078-0432.CCR-14-0842
88. Yu L, Shang ZF, Hsu FM, Zhang Z, Tumati V, Lin YF, et al. NSCLC cells demonstrate differential mode of cell death in response to the combined treatment of radiation and a DNA-PKcs inhibitor. Oncotarget (2015) 6(6):3848–60. doi:10.18632/oncotarget.2975
89. Biau J, Devun F, Jdey W, Kotula E, Quanz M, Chautard E, et al. A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia (2014) 16(10):835–44. doi:10.1016/j.neo.2014.08.008
90. Lamb R, Fiorillo M, Chadwick A, Ozsvari B, Reeves KJ, Smith DL, et al. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: implications for more effective radiation therapy. Oncotarget (2015) 6(16):14005–25. doi:10.18632/oncotarget.4159
91. Durant S, Karran P. Vanillins – a novel family of DNA-PK inhibitors. Nucleic Acids Res (2003) 31(19):5501–12. doi:10.1093/nar/gkg753
92. Dhandapani KM, Mahesh VB, Brann DW. Curcumin suppresses growth and chemoresistance of human glioblastoma cells via AP-1 and NFκB transcription factors. J Neurochem (2007) 102(2):522–38. doi:10.1111/j.1471-4159.2007.04633.x
93. Tian X, Chen G, Xing H, Weng D, Guo Y, Ma D. The relationship between the down-regulation of DNA-PKcs or Ku70 and the chemosensitization in human cervical carcinoma cell line HeLa. Oncol Rep (2007) 18(4):927–32. doi:10.3892/or.18.4.927
94. Blagden S, Hamilton A, Mileshkin L, Hall M, Meniawy T, Wong S, et al. Afuresertib (GSK2110183), an oral AKT kinase inhibitor, in combination with carboplatin and paclitaxel in recurrent ovarian cancer. Eur J Cancer (2014) 50(Suppl 6):7. doi:10.1016/S0959-8049(14)70129-7
95. Li X, Tian J, Bo Q, Li K, Wang H, Liu T, et al. Targeting DNA-PKcs increased anticancer drug sensitivity by suppressing DNA damage repair in osteosarcoma cell line MG63. Tumour Biol (2015). doi:10.1007/s13277-015-3642-5
96. Albarakati N, Abdel-Fatah TM, Doherty R, Russell R, Agarwal D, Moseley P, et al. Targeting BRCA1-BER deficient breast cancer by ATM or DNA-PKcs blockade either alone or in combination with cisplatin for personalized therapy. Mol Oncol (2015) 9(1):204–17. doi:10.1016/j.molonc.2014.08.001
97. O’Grady S, Finn SP, Cuffe S, Richard DJ, O’Byrne KJ, Barr MP. The role of DNA repair pathways in cisplatin resistant lung cancer. Cancer Treat Rev (2014) 40(10):1161–70. doi:10.1016/j.ctrv.2014.10.003
98. Kotula E, Berthault N, Agrario C, Lienafa MC, Simon A, Dingli F, et al. DNA-PKcs plays role in cancer metastasis through regulation of secreted proteins involved in migration and invasion. Cell Cycle (2015) 14(12):1961–72. doi:10.1080/15384101.2015.1026522
Keywords: DNA-PKcs, platinum resistance, ovarian cancer, DNA repair, chemosensitization
Citation: Dungl DA, Maginn EN and Stronach EA (2015) Preventing damage limitation: targeting DNA-PKcs and DNA double-strand break repair pathways for ovarian cancer therapy. Front. Oncol. 5:240. doi: 10.3389/fonc.2015.00240
Received: 24 June 2015; Accepted: 10 October 2015;
Published: 26 October 2015
Edited by:
Ashani Weeraratna, The Wistar Institute, USAReviewed by:
Parvin Mehdipour, Tehran University of Medical Sciences, IranMassimo Broggini, Istituto di Ricerche Farmacologiche Mario Negri, Italy
Copyright: © 2015 Dungl, Maginn and Stronach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elaina N. Maginn, ZS5tYWdpbm5AaW1wZXJpYWwuYWMudWs=
†Daniela A. Dungl and Elaina N. Maginn have contributed equally to this work.