 Aizhen Liu1
Aizhen Liu1 Xuan Zong
Xuan Zong- 1Department of Oncology, Yidu Central Hospital, Jinan, China
- 2Department of Oncology Nursing, Yidu Central Hospital, Jinan, China
- 3Department of Oncology, Shandong University School of Medicine, Jinan, China
Glioblastoma is the most fatal brain cancer found in humans. Patients suffering from glioblastoma have a dismal prognosis, with a median survival of 15 months. The tumor may develop rapidly de novo in older patients or through progression from anaplastic astrocytomas in younger patients if glioblastoma is primary or secondary, respectively. During the past decade, significant advances have been made in the understanding of processes leading to glioblastoma, and several important genetic defects that appear to be important for the development and progression of this tumor have been identified. Particularly, the discovery of recurrent mutations in the isocitrate dehydrogenase 1 (IDH1) gene has shed new light on the molecular landscape in glioblastoma. Indeed, emerging research on the consequences of mutant IDH1 protein expression suggests that its neomorphic enzymatic activity catalyzing the production of the oncometabolite 2-hydroxyglutarate influences a range of cellular programs that affect the epigenome and contribute to glioblastoma development. One of the exciting observations is the presence of IDH1 mutation in the vast majority of secondary glioblastoma, while it is almost absent in primary glioblastoma. Growing data indicate that this particular mutation has clinical and prognostic importance and will become a critical early distinction in diagnosis of glioblastoma.
Introduction
Glioblastoma is the most common brain malignancy and one of the most aggressive cancers found in humans. It is divided into primary and secondary types. Both are histologically identical, so clinical features are used to distinguish them. Primary glioblastoma manifests rapidly de novo without recognizable precursor lesions, and is by far the more common, accounting for 80% of cases. It predominates in elderly patients and is characterized by rapid progression and short survival time. Secondary glioblastoma evolves from lower-grade gliomas, such as grade II diffuse astrocytoma or grade II anaplastic astrocytoma, and is typically seen in younger patients. It can be diagnosed with clinical or histological evidence (1). Infiltrating glioblastomas are incurable with current treatment modalities that include surgery, radiation, and chemotherapy. For newly diagnosed patients, the standard treatment is total removal, if possible, followed by the combination of chemotherapy and radiotherapy (2, 3). However, despite this maximum treatment, the prognosis of patients remains dismal with a median survival of 15 months only (2). When compared with other malignancies, there have only been small improvements in the prognosis of glioblastoma patients over recent decades. Nevertheless, understanding of the molecular pathogenesis of glioblastoma has greatly increased and is beginning to match that of other types of cancers. Several important genetic alterations have been known for some time, but new technologies have allowed much deep genetic and epigenetic analysis of large numbers of glioblastoma samples, leading to a number of novel discoveries. One of the most exciting and clinically relevant observations was the discovery that a high percentage of secondary glioblastomas and a very small percentage of primary glioblastoma harbor mutations in the isocitrate dehydrogenase 1 (IDH1) gene. This stunning and unexpected discovery that holds clear clinical implications has led to new insights into glioblastoma biology. Indeed, IDH1 mutation results in gain of function to catalyze the production of hydroxyglutarate (2-HG) (4), a possible oncometabolite that is thought to influence a range of cellular programs involved in epigenetic control and various processes leading to tumor development. Here, we review translational applications of this mutation as well as its incidence in glioblastoma and other cancers. In addition, we highlight importance of epigenetic changes that may need to be considered in future diagnostics and therapy for glioblastoma. Initially, the review presents a brief overview on mutations commonly found in glioblastoma, and prunes the consequences of IDH1 mutation on glioblastoma biology to better understand the potential role of this particular mutation in the development of this tumor.
Common Mutations in Glioblastoma
Although glioblastoma-specific mutations are seen, mutations in common cancer genes, such as TP53 and PTEN, are very frequent in glioblastomas, but are not of prognostic importance (Table 1) (5, 6). EGFR point mutations have also been identified in glioblastoma. The EGFRvIII mutant lacks 267 amino acids in the extracellular part, resulting in a constitutively activated receptor that no longer requires its ligand EGF to signal downstream (5). Although mutations in certain cancer genes, such as BRAF and the RAS genes, have rarely been observed in glioblastoma, inactivating mutations and deletions have been identified in their inhibitory tumor-suppressor gene NF1 (5). Mutations in PIK3CA and PIK3R1 genes, coding, respectively, for the PI3K catalytic subunit p110α and regulatory subunit P85α, have also been described (5, 6).
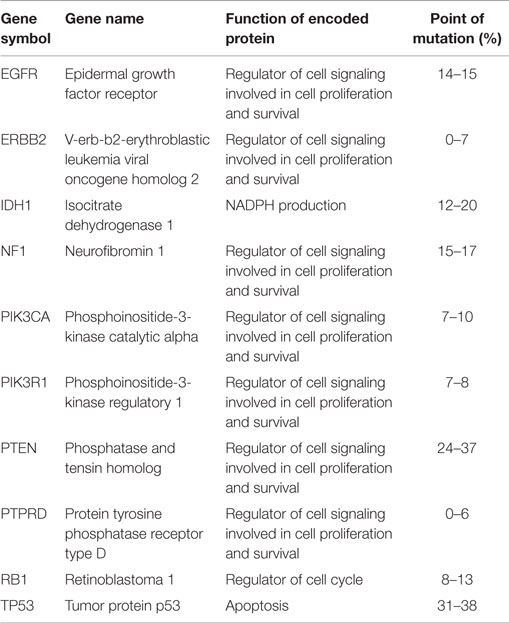
Table 1. Genes commonly mutated in glioblastoma.
An interesting gene found to contain mutations in glioblastoma is IDH1, which encodes IDH1 and is involved in energy metabolism. This gene shows differential expression between primary and secondary glioblastoma, while PTEN loss, EGFR amplification, and loss of heterozygosity (LOH) of chromosome 10 are associated with primary glioblastoma, and ATRX mutation, loss of p53, and LOH of chromosome 19 are common in secondary glioblastoma. The IDH1 mutation predicts secondary glioblastoma better than these other mutations predict their respective glioblastoma subtype. Indeed, IDH1 mutations have been predominantly identified in secondary glioblastoma and low-grade gliomas, with mutations in more than 70% of cases (5, 6). They are found only sporadically in primary glioblastoma (5, 6). Because patients with IDH1-mutated primary glioblastoma are generally younger and have longer median survival and wild-type EGFR, which are characteristics of secondary glioblastoma, it is hypothesized that these are in fact secondary glioblastomas for which no histological evidence of evolution from a less malignant glioma is found. Therefore, IDH1 mutation could be used to differentiate primary from secondary glioblastoma.
Biological Consequences of IDH1 Mutation
Isocitrate dehydrogenase 1 is a metabolic enzyme that converts isocitrate into α-ketoglutarate (α-KG) via oxidative decarboxylation using NADP+ as an electron acceptor, and producing NADPH (7). While NADPH is thought to be important for limiting cellular oxidative damage and for lipid biosynthesis, α-KG is considered as an essential intermediate in the Kreb’s cycle. Under hypoxia conditions, IDH1 catalyzes the inverse reaction, converting α-KG to isocitrate which can in turn be converted to acetyl-CoA for lipid metabolism and many biochemical reactions.
Since 2008, with the discovery of recurrent mutations in the IDH1 coding gene by the Vogelstein group analyzing the DNA sequence of the glioblastoma genome (6), substantial progress has been made to understand how such genetic modification leads IDH1 to play a role in the tumorigenesis. The major finding was the discovery that the IDH1 mutation is a gain-of-function mutation, conferring neomorphic activity to IDH1. Initially, a pivotal study profiling IDH1 wild-type and mutant glioblastoma cells with liquid chromatography–mass spectrometry has reported high levels of the metabolite 2-HG in mutant cells (4). Then, it has been demonstrated that mutant IDH1 catalyzes the reduction of α-KG to the R-enantiomer of the metabolite 2-HG (R-2-HG) (8). Thus, rather than catalyze the NADP+-dependent production of α-KG, mutant IDH1 catalyzes the NADPH-dependent reduction of α-KG to produce only the R-enantiomer of 2-HG, indicating a gain of neomorphic function. Specifically, authors have demonstrated that the mutation reduces the affinity of the IDH1 active site for isocitrate while concomitantly increasing it for NADPH and α-KG (8). Reduced affinity for isocitrate occurs as a result of alterations to a binding site residue that forms hydrogen bonds between the alpha and beta groups of isocitrate (8). Consequently, the conversion of α-KG is favorized, but rather than isocitrate, the mutant enzyme converts α-KG into R-2-HG, an oncometabolite that promotes tumorigenesis through inhibition of α-KG-dependent (Figure 1).
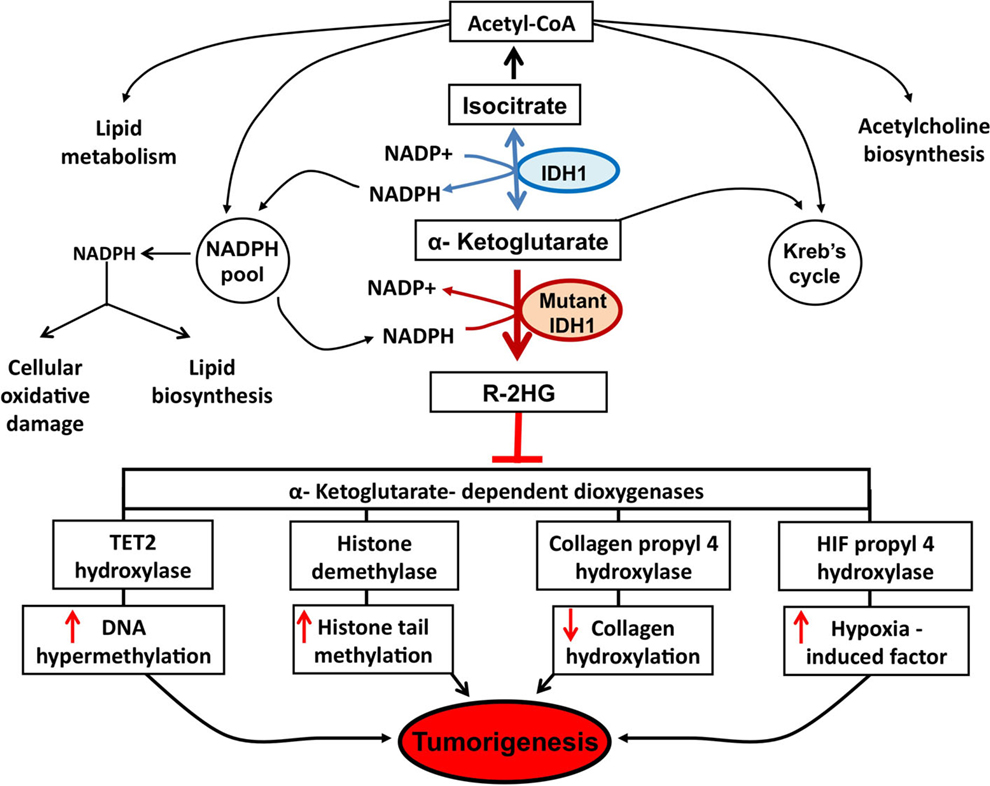
Figure 1. Potential mechanisms implicated in tumor formation induced by IDH1 mutation. A number of potential mechanisms have been proposed to explain how R-2-HG produced by mutant IDH1 enzyme promotes tumor formation. Epigenetic modification, via inhibition of α-KG-dependent dioxygenases leading to DNA and histone hypermethylation, has been at the forefront of research efforts. Specifically, the inhibition of tet methylcytosine dioxygenase 2 (TET2) leads to increased DNA hypermethylation and the inhibition of histone demethylases leads to increased histone tail methylation. Additional mechanisms include inhibition of several groups of prolyl hydroxylases, leading to hypoxia-induced factor (HIF) activation and alterations in collagen formation.
Incidence of IDH1 Mutation in Glioblastoma and Other Cancers
Extensive genomic profiling has revealed that about 90% of IDH1 mutations involve exon 4 at codon 132, replacing arginine with histidine (R132H). Of the remaining 10% of IDH1 mutations, 4.7% are due to arginine being replaced with cysteine (R132C), 2.1% with glycine (R132G), 1.7% with serine (R132S), 0.8% with leucine (R132L), and 0.3% with glutamine (R132Q) (9).
Following the first observation of recurrent IDH1 mutation in glioblastoma, several groups have begun to clarify the frequency and distribution of IDH1 mutation across all brain tumors, including gliomas and other subtypes. Data summarized from many studies show that only approximately 5.6% of primary glioblastoma are IDH1 mutant, while more than 76% of secondary glioblastomas carry the IDH1 mutation. The reported rates of IDH1 mutation in lower-grade gliomas are comparable with those of secondary glioblastoma (Table 2) (10).
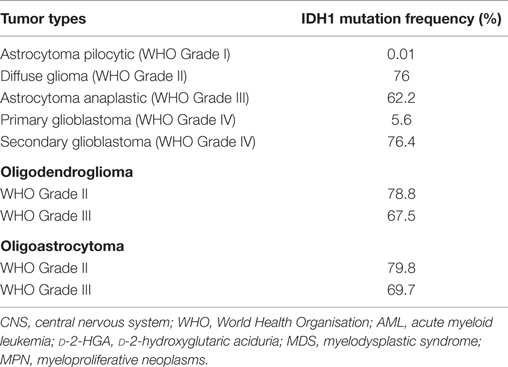
Table 2. Frequency of IDH1 mutation in various tumors.
Other brain tumors harboring IDH1 mutations with moderate frequency include gangliogliomas, giant cell glioblastomas, and primitive neuroectodermal tumors, although only small numbers of these tumors have been studied (11).
Isocitrate dehydrogenase mutations are also present in some tumors originating in cells outside of the central nervous system. Indeed, about 7.7% of acute myeloid leukemia (AML) possessed the IDH1 mutation (12), but prevalence rates vary between 15 and 33% if IDH2 are also considered (13). Approximately 50% of d-2-hydroxyglutaric aciduria (d-2-HGA), a rare inherited neurometabolic disorder, has also been found to display IDH1 mutations (14), as well as 10% of intrahepatic cholangiocarcinomas (15), 5% of myelodysplastic syndrome (MDS), 8.8% of myeloproliferative neoplasms (MPN), and fewer than 10% of secondary AML (14). In their investigation, Amary and colleagues found that nearly 60% of central and periosteal cartilaginous tumors displayed DH1mutations (16). The same group has also identified IDH1 mutation to occur in patients with Ollier disease and Maffucci syndrome (17). The majority of patients exhibited the R132C IDH1 mutation, in contrast to most secondary glioblastoma, which harbor the R132H mutation.
Translational Applications of Mutant IDH1
In a relatively short time after the discovery of IDH1 mutation in glioblastoma, a tremendous amount of work has been performed on the clinical relevance of this mutation regarding particularly its applications in the diagnosis, prognosis, and treatment of patients suffering from glioblastoma.
Diagnostic Applications
The determination of IDH1 mutation status is highly relevant for the diagnosis of primary brain tumors, and strongly supports the differential diagnosis between an anaplastic glioma and a glioblastoma.
Traditionally, IDH1 mutation status was detected through classic Sanger sequencing and polymerase chain reaction (PCR). Although this has the clear advantage of being able to detect non-R132H mutation, it is time consuming and requires there to be at least 20% mutant allele frequency within the tissue specimen for reliable detection (18). Pyrosequencing is an alternative that allows for rapid high-throughput analysis of IDH1 mutation, and has demonstrated an advantage over classic Sanger sequencing in that it can detect mutated allele frequencies down to 5% (19). Derived cleaved amplified polymorphic sequence analysis is another alternative to DNA sequencing that uses mismatched primers for specific mutations, which, following PCR amplification will create differing restriction endonuclease sites dependent on the presence of the mutation (20). The advantage of this technique is that it uses supplies commonly found in most laboratories, obviating expensive sequencing equipment. However, unlike sequencing, the method is limited in that it can only detect mutations being queried. Other PCR-based techniques include coamplification at lower temperature (COLD) PCR with high-resolution melting (HRM) and real-time PCR, and post-PCR fluorescent melting curve analysis (FMCA). Through COLD PCR combined with HRM, Boisselier and colleagues were able to detect mutant allele concentrations of 0.25% in a span of only 3 h (21). However, because the technique requires the new mutation to have a melting temperature (Tm) that is lower than IDH1 wild type, it theoretically may not be able to detect R132G mutation. Regarding real-time PCR with post-PCR FMCA, this technique was shown to be more sensitive than Sanger sequencing with detection rate of as little as 10% mutant DNA and a processing time of 80 min.
Other technologies currently in use to detect mutations in the IDH1 gene include SNaPshot (22) and Oncomap (23), both of which can be used with paraffin-embedded tissue, using base pair extension that results in an allele-specific probe that is read out by either fluorescence detection (SNaPshot) or mass spectrometry (Oncomap). In addition to these approaches, Boisselier and colleagues demonstrated evidence of principle in detection of IDH1 mutations from the plasma of patients with mutant glioma (24). Although the potential application for monitoring disease non-invasively is compelling, the sensitivity attained using this assay was 60%. Further work on the nature of circulating tumor material will be necessary to determine whether it will be possible to monitor the IDH1 mutation status in the peripheral blood of all patients with mutant gliomas.
Recently, a new technique called amplification-refractory mutation system (ARMS) has been developed (25) to identify R140Q mutations in IDH2 and such novel methods can be of great help in detecting IDH1 mutations. Recently, a novel strategy of PCR clamping was employed for qualitative detection of seven different mutations in IDH1 and five mutations in IDH2 in a single PCR assay (26).
A monoclonal antibody specific for the IDH-R132H mutation (mIDH1R132H), developed by Capper’s group, recognizes the mutant protein with a high degree of sensitivity and specificity (27). Using this antibody, 90% of IDH1 mutation can be detected on paraffin sections, and it is recommended to test the remaining 10% by sequencing. Nevertheless, proponents of immunohistochemistry-based antibody staining argue that the use of mIDH1R132H antibody to identify IDH1 mutation may even be superior to direct sequencing because there are reported cases in which this antibody detects mutations missed by direct sequencing (28).
Beyond determination of IDH1 mutation status, the histopathological utility of the mIDH1R132H antibody has extended to additional clinical scenarios such as the discrimination between diffuse astrocytoma and reactive astrocytosis when combined with a panel of key molecular feature (29). Indeed, seeing that IDH1 mutations are specific to gliomas, this antibody can be used to help differentiate between diffuse gliomas and areas of reactive gliosis. Accordingly, the detection of positive cells by IDH1-R132H immounohistochemistry allows a clear and safe separation between low-grade glioma and reactive gliosis, and clearly supports the diagnosis of a diffusely infiltrating glioma as well as the differential diagnosis between an anaplastic glioma and a glioblastoma. This approach allows narrowing down the possible diagnosis to the group of diffusely infiltrating of WHO grade II and III and secondary glioblastoma and to a certain extent to primary glioblastoma. IDH1-R132H immounohistochemistry has also been shown to be effective in separating out oligodendrogliomas from several other similar entities, such as central neurocytomas, tanycytic ependymomas, and pilocytic astrocytomas (27).
Another area of investigation has focused on the detection of the 2-HG metabolite rather than on the specific sequence of the IDH1 gene or protein product. In theory, sensitive and specific detection of 2-HG is sequence independent in that 2-HG should be present regardless of the type of mutation in IDH1 or IDH2. High levels of 2-HG have been detected in ex vivo tissue samples by using two approaches. In the first approach, combination gas or liquid chromatography/mass spectrometry was used to identify 2-HG in frozen or paraffin-embedded glioma tissue. However, these extraction-based approaches do not preserve the integrity of the sampled tissue. In the second approach, proton high-resolution magic angle spinning (HRMAS) MRS is used to determine the metabolic profiles in ex vivo tissue. This technique does not require alteration of tissue samples, and identified 2-HG in ex vivo specimens with high sensitivity and specificity. Unlike the case with AML, wherein 2-HG can be detected in the blood of patients with IDH-mutant AML (30), its presence in peripheral blood is similar between patients with IDH1-mutant and wild-type tumors (31).
Detection of 2-HG by MRS represents a completed non-invasive method with which to determine the presence of IDH mutations in gliomas, irrespective of the sequence of the mutation or the mutation maps to IDH1 or IDH2. Importantly, this approach represents the only example in human cancer in which a genomic feature can be identified specifically by using imaging-based metabolic profiling. Prior work completed by MRS-based approach has demonstrated that IDH-mutant tumors display characteristic imaging findings. The IDH1-mutant tumors display reduced contrast enhancement, less surrounding edema, cystic components, and often found in the frontal lobe compared with IDH-wild-type tumors (32). Because it is recognized that the spectrum of 2-HG has some overlap with other commonly found metabolites, such as glutamate and glutamine, the methodology for obtaining and analyzing in vivo MRS is critical. One study to demonstrate 2-HG detection in glioma by MRS used the acquisition of a point-resolved spectroscopy (PRESS) sequence (33). There was good concordance between sequence-based mutation status and 2-HG detection, although several false positives and false negatives were reported. Additional studies in which spectral-editing analysis (34) or 2D-correlation MRS with spectral editing (35) were used demonstrated that IDH mutation status can be identified non-invasively by MRS techniques with a high levels of sensitivity and specificity.
Prognostic Implications
Extensive research has been performed to determine the prognostic value of IDH1 mutations, and a better prognosis has been generally reported in glioblastoma patients carrying an IDH1 mutation. Studies have shown that IDH1 mutation convey an improved prognosis with respect to both overall survival and progression-free survival for the rare glioblastoma patients who express this mutation. Indeed, since the publication of the first report on improved survival in glioblastoma patients with mutation by Parsons and colleagues, indicating 45.6 months overall survival in IDH1-mutant versus 13.2 months overall survival in IDH1 wild type (6), numerous groups have been able to replicate similar findings (36–41). Besides improved overall survival, Sanson et al. (37) were able to demonstrate improved progressive-free survival (PFS) as well in their set of patients with glioblastoma, with 55 months PFS in patients with IDH1 mutation versus 8.8 months PFS in those without this mutation. The analysis was extended to anaplastic (WHO grade III) tumors because many groups were readily able to show an improved overall survival in grade III tumors that harbored the IDH1 mutation compared with those that did not in both univariate (36) and multivariate analyses (37, 40). With anaplastic astrocytomas, patients harboring IDH1 mutation had an overall survival of 65 months compared to 20 months for patients without IDH1 mutation (38). The survival benefit also extended to grade II gliomas, showing a median overall survival of 12.6 years in patients with IDH1 mutation versus 5.5 years in patients devoid of this mutation. In a prospective analysis, Wick and colleagues found that grade III astrocytomas that possessed the IDH1 mutation were associated with greater PFS regardless of the treatment (42).
In studies pooling low-grade astrocytomas and oligodendrogliomas, the IDH1 mutation status was prognostic for overall and PFS. In primary glioblastoma, IDH1 mutational status has been reported to be the only factor that showed significant association with patient survival times. However, the evidence for low-grade gliomas and the prognostic value of IDH1 mutation is slightly more controversial. Two independent groups found that IDH1 mutations in low-grade gliomas were associated with significant improved overall survival (43, 44), whereas others could not find any significant association (36, 45). Nevertheless, the consistent finding of a more favorable outcome of diffuse gliomas patients with IDH1 mutation implies that IDH1 testing might be useful for prognostic considerations in the clinical setting.
Predictive Implications
Isocitrate dehydrogenase 1 mutation was correlated with a higher rate of response to up-front temozolomide in low-grade glioma patients. Furthermore, evidence for differential responsiveness to genotoxic therapy of IDH1 mutant versus IDH1 wild-type low-grade gliomas has been provided. Indeed, the presence of IDH1 mutation was associated with favorable progression-free and overall survival in WHO grade II gliomas who received radio- or chemotherapy. Current studies aim to validate and clarify the predictive value of IDH1 mutation in the different glioma types.
It is still unclear if IDH1 mutational status is a prognostic indicator or predictive measure of response to treatment. Houillier and colleagues stratified a cohort of low-grade gliomas into three groups based on prognostic factors according to the presence of 1p19q deletion, IDH1 mutation, or both together (46). They found that each of these factors was an independent predictor of improved clinical outcome in response to treatment with the chemotherapeutic agent temozolomide and that the group of patients with both mutations had the best treatment response. Objective response was in 80% with both mutations, 61% of IDH1-mutants without 1p19q deletion, and 17% without either mutation. In a similar fashion, Hartman and colleagues found that, in their cohort of patients receiving adjuvant therapy, IDH1 mutation status was the single most important predictor of PFS and overall survival. This was not seen in their cohort of patients that did not receive adjuvant therapy (47). These findings support the notion that IDH1 mutation may be an important predictor to treatment response.
Treatment Implications
The identification of IDH1 mutation and the rapid characterization of its protein products present a therapeutic opportunity to treat the IDH1-mutant tumors. From a medical standpoint, it will be important to determine whether these mutant tumors would be sensitive to molecules that inhibit the mutant enzyme. Although there are no published studies to date addressing this possibility, one study performed by Jin and colleagues suggested that mutant tumor cells may depend on the continued expression of the mutant enzyme and/or its resulting 2-HG metabolite. In their investigation, the authors showed that a cell line expressing endogenous mutant IDH1 required its expression for survival and anchorage-independent growth (48), suggesting that pharmacological inhibition of mutant IDH1 may recapitulate this result. With respect to mutant IDH1-mediated biology, this result would also suggest that 2-HG-induced changes, such as global DNA hypermethylation, are either potentially reversible or, if not, are insufficient for tumor maintenance. It may be anticipated that inhibition of this pathway would increase patient survival. Although there has been some discussion of whether it is prudent to inhibit mutant IDH1 because patients with mutant tumors have a better survival than patients with wild-type tumors, it is not expected that inhibiting the mutant enzyme would make mutant tumors behave like their more aggressive wild-type counterparts. The differences in survival most likely stem from the fact that IDH1-mutant and wild-type tumors arise from distinct lineages and ontogenies and, thus, represent entirely different neoplastic disease processes.
Epigenetic Changes in Glioblastoma
Epigenetic is the mitotically heritable changes in gene expression that are not due to changes in the DNA sequence, and has emerged as hallmark of human cancers. Indeed, aberrant epigenetic mechanisms, such as DNA methylation, histone modifications, chromatin remodeling, or altered non-coding RNA expression, are currently recognized as relevant events in tumor formation (49). Until now, most studies about the epigenetic changes in glioblastoma have focused on DNA methylation, including hypermethylation, gene-specific hypomethylation, and genome-wide hypomethylation (50). The leading mechanism attributed to the observed hypermethylation phenotype in IDH1 mutant involves silencing of the α-KG-dependent DNA modifying enzyme, Tet methylcytosine dioxygenase 2 (TET2) (Figure 1).
Since the first report demonstrating that a subset of glioblastoma exhibits a global decrease in 5-methylcytosine, subsequent follow-up studies have revealed not only that genome-wide or global hypomethylation occurs at a frequency of 80% in primary glioblastoma, but also that the level of hypomethylation varies between glioblastomas, ranging from near normal levels to approximately 50% of normal in about 20% of cases (51). The most severe globally hypomethylated primary glioblastomas are also the most proliferative and display dramatic hypomethylation (22–50% of normal brain) of the tandem repeat satellite 2 (Sat2) located at the juxtacentromeric region of chromosomes 1, 9, and 16, and moderate hypomethylation (71–80%) of the D4Z4 located at the subtelomeric regions of chromosomes 4q35 and 10q26 (51). Moreover, glioblastoma with hypomethylated Sat2 also harbored copy number alterations of adjacent euchromatic sequences, specifically near the pericentromeric region of chromosome 1. These data suggest that one consequence of hypomethylated repetitive sequences in glioblastoma is predisposition to chromosomal breakage and copy number alteration. Although the full consequences of genomic hypomethylation are unknown, murine models of defective imprinting provide evidence for a causal role of DNA methylation alteration in tumorigenesis.
DNA methylation has been shown to play critical roles in the control of gene activity and the architecture of the nucleus of the cell. In humans, DNA methylation occurs in cytosines that precede the guanines to create 5-methylcytosine and these are commonly called dinucleotide CpGs (52). These dinucleotides are not randomly distributed in the genome, but instead are present as CpG-rich regions referred to as CpG islands. Hypermethylation of CpG islands in the promoter regions of tumor-suppressor genes is a major event in the origin of many cancers. In glioblastoma, CpG promoter hypermethylation occurs at genes with diverse functions related to tumorigenesis and tumor progression, including cell cycle regulation (CDK2A-p16INK4a and CDK2B-p15INK4b) tumor suppression (RB1, VHL, EMP3, RASSF1A, and BLU), DNA repair [methylguanine DNA methyltransferase (MGMT) and MLH1], inhibition of apoptosis (DAPK1, TIMP3, CDH1), and genes associated with angiogenesis, regulation of tumor invasion, and drug resistance (53). Many new tumor-suppressor candidates have been identified, including the cell motility regulator testis-derived transcript (TES) as well as many polycomb repressor complex 2 (PRC2) target genes (54), and epithelial membrane protein 3 (EMP3), a myelin-related gene involved in cell proliferation and cell–cell interaction that is silenced by hypermethylation in glioblastoma (55).
Promoter hypermethylation has been demonstrated to regulate the oncogenic and proliferation-promoting transforming growth factor (TGF)-beta signaling pathway in aggressive, highly proliferative glioblastomas. High levels of TGF-beta signaling are normally associated with poor prognosis. TGF-beta signaling promotes proliferation through the induction of platelet-derived growth factor-beta (PDGF-B). However, epigenetic silencing of PDGF-B can override the increased proliferative effects of TGF-beta signaling. Specifically, PDGF-B promoter hypermethylation prevents PDGF-B transcriptional activation by TGF-beta-induced Smad proteins (56). The oncogenic effect of the TGF-beta pathway is, therefore, blocked by epigenetic alteration of one of its targets.
Genes involved in invasion and metastasis can also be affected by promoter hypermethylation in glioblastoma. Approximately 87% of glioblastoma exhibit CpG hypermethylation of the protocadherin-gamma subfamily A11 (PCDH-gamma-A11) gene, which is thought to be important in invasion of cancer cells into normal brain parenchyma (57).
The hypermethylation of promoter can also modulate sensitivity to drugs and radiotherapy in glioblastoma. The best known example is the O6-MGMT promoter methylation and response to alkylating agents, but there is also the gene suppressor of cytokine signaling 1 (SOCS1), which in some glioblastomas enhanced resistance to ionizing radiation and decreased activation of MAPKs associated with the ERK pathway after transcriptional silencing by hypermethylation (58). This suggests that epigenetic profiling might be one way to categorize glioblastomas and to rationally apply patient-specific therapy.
For certain sets of genes, promoter hypermethylation might not be causal or may not be required for gene silencing in glioblastoma. There is some evidence for deregulation of gene controlling histone modifications in this tumor. The gene encoding BMI-1, a member of the polycomb group complex that regulates histone H3K27 methylation, is subject to frequent copy number alterations in both low- and high-grade gliomas (59). Expression of some histone deacetylase (HDAC) proteins is reported to be altered in glioblastoma. Class II and IV HDACs displayed decreased mRNA expression in glioblastoma compared to low-grade astrocytomas and normal brain, and overall histone H3 was more acetylated in glioblastoma (60). Large-scale sequencing of protein-coding genes in glioblastoma revealed mutations in many genes involved in epigenetic regulation, including HDACs, HDAC2 and HDAC9, histone demethylases, JMD1A and JMD1B, histone methyltransferases, SET7, SETD7, MLL, MLL4, and methyl-CpG binding domain protein1 (MBD1) (6). These intriguing initial studies suggest that alteration in epigenetic mechanisms could be a major defect in glioblastoma.
Conclusion
Following the discovery of IDH1 mutation, our understanding of the biochemistry, genetics, and epigenetics as well as the prevalence and pathogenic role of this mutation has grown at a rapid rate, and a tremendous amount of work has been performed on its translational relevance in a relative short time. It is, henceforth, clear that IDH1 status is a major determinant of survival, and many ways have been developed to identify the IDH mutation as well as the oncometabolite 2-HG from clinical samples, using non-invasive procedures. The determination of IDH1 status in glioblastoma will likely be an early step in treatment algorithms for patients suffering from this tumor.
Author Contributions
The authors collected and analyzed the information and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol (2007) 170:1445–53. doi:10.2353/ajpath.2007.070011
2. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352:987–96. doi:10.1056/NEJMoa043330
3. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10:459–66. doi:10.1016/S1470-2045(09)70025-7
4. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature (2009) 462:739–44. doi:10.1038/nature08617
5. Cancer Genome Atlas Research. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature (2008) 455:1061–8. doi:10.1038/nature07385
6. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321:1807–12. doi:10.1126/science.1164382
7. Geisbrecht BV, Liang X, Morrell JC, Schulz H, Gould SJ. The mouse gene PDCR encodes a peroxisomal delta(2), delta(4)-dienoyl-CoA reductase. J Biol Chem (1999) 274:25814–20. doi:10.1074/jbc.274.36.25814
8. Pietrak B, Zhao H, Qi H, Quinn C, Gao E, Boyer JG, et al. A tale of two subunits: how the neomorphic R132H IDH1 mutation enhances production of alphaHG. Biochemistry (2011) 50:4804–12. doi:10.1021/bi200499m
9. Pusch S, Schweizer L, Beck AC, Lehmler JM, Weissert S, Balss J, et al. D-2-Hydroxyglutarate producing neo-enzymatic activity inversely correlates with frequency of the type of isocitrate dehydrogenase 1 mutations found in glioma. Acta Neuropathol Commun (2014) 2:19. doi:10.1186/2051-5960-2-19
10. Dunn GP, Andronesi OC, Cahill DP. From genomics to the clinic: biological and translational insights of mutant IDH1/2 in glioma. Neurosurg Focus (2013) 34:E2. doi:10.3171/2012.12.FOCUS12355
11. Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol (2008) 116:597–602. doi:10.1007/s00401-008-0455-2
12. Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med (2009) 361:1058–66. doi:10.1056/NEJMoa0903840
13. Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders JE, Zeilemaker A, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood (2010) 116:2122–6. doi:10.1182/blood-2009-11-250878
14. Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B, et al. Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia (2010) 24:1094–6. doi:10.1038/leu.2010.52
15. Wang P, Dong Q, Zhang C, Kuan PF, Liu Y, Jeck WR, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene (2013) 32:3091–100. doi:10.1038/onc.2012.315
16. Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol (2011) 224:334–43. doi:10.1002/path.2913
17. Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, et al. Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet (2011) 43:1262–5. doi:10.1038/ng.994
18. von Deimling A, Korshunov A, Hartmann C. The next generation of glioma biomarkers: MGMT methylation, BRAF fusions and IDH1 mutations. Brain Pathol (2011) 21:74–87. doi:10.1111/j.1750-3639.2010.00454.x
19. Setty P, Hammes J, Rothamel T, Vladimirova V, Kramm CM, Pietsch T, et al. A pyrosequencing-based assay for the rapid detection of IDH1 mutations in clinical samples. J Mol Diagn (2010) 12:750–6. doi:10.2353/jmoldx.2010.090237
20. Meyer J, Pusch S, Balss J, Capper D, Mueller W, Christians A, et al. PCR- and restriction endonuclease-based detection of IDH1 mutations. Brain Pathol (2010) 20:298–300. doi:10.1111/j.1750-3639.2009.00327.x
21. Boisselier B, Marie Y, Labussiere M, Ciccarino P, Desestret V, Wang X, et al. COLD PCR HRM: a highly sensitive detection method for IDH1 mutations. Hum Mutat (2010) 31:1360–5. doi:10.1002/humu.21365
22. Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med (2010) 2:146–58. doi:10.1002/emmm.201000070
23. MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L, et al. Profiling critical cancer gene mutations in clinical tumor samples. PLoS One (2009) 4:e7887. doi:10.1371/journal.pone.0007887
24. Boisselier B, Gallego Perez-Larraya J, Rossetto M, Labussiere M, Ciccarino P, Marie Y, et al. Detection of IDH1 mutation in the plasma of patients with glioma. Neurology (2012) 79:1693–8. doi:10.1212/WNL.0b013e31826e9b0a
25. Berenstein R, Blau IW, Kar A, Cay R, Sindram A, Seide C, et al. Comparative examination of various PCR-based methods for DNMT3A and IDH1/2 mutations identification in acute myeloid leukemia. J Exp Clin Cancer Res (2014) 33:44. doi:10.1186/1756-9966-33-44
26. Catteau A, Girardi H, Monville F, Poggionovo C, Carpentier S, Frayssinet V, et al. A new sensitive PCR assay for one-step detection of 12 IDH1/2 mutations in glioma. Acta Neuropathol Commun (2014) 2:58. doi:10.1186/2051-5960-2-58
27. Capper D, Reuss D, Schittenhelm J, Hartmann C, Bremer J, Sahm F, et al. Mutation-specific IDH1 antibody differentiates oligodendrogliomas and oligoastrocytomas from other brain tumors with oligodendroglioma-like morphology. Acta Neuropathol (2011) 121:241–52. doi:10.1007/s00401-010-0770-2
28. Takano S, Tian W, Matsuda M, Yamamoto T, Ishikawa E, Kaneko MK, et al. Detection of IDH1 mutation in human gliomas: comparison of immunohistochemistry and sequencing. Brain Tumor Pathol (2011) 28:115–23. doi:10.1007/s10014-011-0023-7
29. Camelo-Piragua S, Jansen M, Ganguly A, Kim JC, Cosper AK, Dias-Santagata D, et al. A sensitive and specific diagnostic panel to distinguish diffuse astrocytoma from astrocytosis: chromosome 7 gain with mutant isocitrate dehydrogenase 1 and p53. J Neuropathol Exp Neurol (2011) 70:110–5. doi:10.1097/NEN.0b013e31820565f9
30. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell (2010) 17:225–34. doi:10.1016/j.ccr.2010.01.020
31. Capper D, Simon M, Langhans CD, Okun JG, Tonn JC, Weller M, et al. 2-Hydroxyglutarate concentration in serum from patients with gliomas does not correlate with IDH1/2 mutation status or tumor size. Int J Cancer (2012) 131:766–8. doi:10.1002/ijc.26425
32. Carrillo JA, Lai A, Nghiemphu PL, Kim HJ, Phillips HS, Kharbanda S, et al. Relationship between tumor enhancement, edema, IDH1 mutational status, MGMT promoter methylation, and survival in glioblastoma. AJNR Am J Neuroradiol (2012) 33:1349–55. doi:10.3174/ajnr.A2950
33. Pope WB, Prins RM, Albert Thomas M, Nagarajan R, Yen KE, Bittinger MA, et al. Non-invasive detection of 2-hydroxyglutarate and other metabolites in IDH1 mutant glioma patients using magnetic resonance spectroscopy. J Neurooncol (2012) 107:197–205. doi:10.1007/s11060-011-0737-8
34. Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, et al. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med (2012) 18:624–9. doi:10.1038/nm.2682
35. Andronesi OC, Kim GS, Gerstner E, Batchelor T, Tzika AA, Fantin VR, et al. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med (2012) 4:116ra114. doi:10.1126/scitranslmed.3002693
36. Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol (2009) 11:341–7. doi:10.1215/15228517-2009-025
37. Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol (2009) 27:4150–4. doi:10.1200/JCO.2009.21.9832
38. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med (2009) 360:765–73. doi:10.1056/NEJMoa0808710
39. Gravendeel LA, Kloosterhof NK, Bralten LB, van Marion R, Dubbink HJ, Dinjens W, et al. Segregation of non-p.R132H mutations in IDH1 in distinct molecular subtypes of glioma. Hum Mutat (2010) 31:E1186–99. doi:10.1002/humu.21201
40. Labussiere M, Idbaih A, Wang XW, Marie Y, Boisselier B, Falet C, et al. All the 1p19q codeleted gliomas are mutated on IDH1 or IDH2. Neurology (2010) 74:1886–90. doi:10.1212/WNL.0b013e3181e1cf3a
41. van den Bent MJ, Dubbink HJ, Marie Y, Brandes AA, Taphoorn MJ, Wesseling P, et al. IDH1 and IDH2 mutations are prognostic but not predictive for outcome in anaplastic oligodendroglial tumors: a report of the European Organization for Research and Treatment of Cancer Brain Tumor Group. Clin Cancer Res (2010) 16:1597–604. doi:10.1158/1078-0432.CCR-09-2902
42. Wick W, Hartmann C, Engel C, Stoffels M, Felsberg J, Stockhammer F, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol (2009) 27:5874–80. doi:10.1200/JCO.2009.23.6497
43. Dubbink HJ, Taal W, van Marion R, Kros JM, van Heuvel I, Bromberg JE, et al. IDH1 mutations in low-grade astrocytomas predict survival but not response to temozolomide. Neurology (2009) 73:1792–5. doi:10.1212/WNL.0b013e3181c34ace
44. Metellus P, Coulibaly B, Colin C, de Paula AM, Vasiljevic A, Taieb D, et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol (2010) 120:719–29. doi:10.1007/s00401-010-0777-8
45. Kim YH, Nobusawa S, Mittelbronn M, Paulus W, Brokinkel B, Keyvani K, et al. Molecular classification of low-grade diffuse gliomas. Am J Pathol (2010) 177:2708–14. doi:10.2353/ajpath.2010.100680
46. Houillier C, Wang X, Kaloshi G, Mokhtari K, Guillevin R, Laffaire J, et al. IDH1 or IDH2 mutations predict longer survival and response to temozolomide in low-grade gliomas. Neurology (2010) 75:1560–6. doi:10.1212/WNL.0b013e3181f96282
47. Hartmann C, Hentschel B, Tatagiba M, Schramm J, Schnell O, Seidel C, et al. Molecular markers in low-grade gliomas: predictive or prognostic? Clin Cancer Res (2011) 17:4588–99. doi:10.1158/1078-0432.CCR-10-3194
48. Jin G, Pirozzi CJ, Chen LH, Lopez GY, Duncan CG, Feng J, et al. Mutant IDH1 is required for IDH1 mutated tumor cell growth. Oncotarget (2012) 3:774–82. doi:10.18632/oncotarget.577
49. Jones PA, Baylin SB. The epigenomics of cancer. Cell (2007) 128:683–92. doi:10.1016/j.cell.2007.01.029
50. Kloosterhof NK, de Rooi JJ, Kros M, Eilers PH, Sillevis Smitt PA, van den Bent MJ, et al. Molecular subtypes of glioma identified by genome-wide methylation profiling. Genes Chromosomes Cancer (2013) 52:665–74. doi:10.1002/gcc.22062
51. Cadieux B, Ching TT, VandenBerg SR, Costello JF. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res (2006) 66:8469–76. doi:10.1158/0008-5472.CAN-06-1547
52. Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet (2007) 39:457–66. doi:10.1038/ng1990
53. Caren H, Pollard SM, Beck S. The good, the bad and the ugly: epigenetic mechanisms in glioblastoma. Mol Aspects Med (2013) 34:849–62. doi:10.1016/j.mam.2012.06.007
54. Martinez R, Martin-Subero JI, Rohde V, Kirsch M, Alaminos M, Fernandez AF, et al. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics (2009) 4:255–64. doi:10.4161/epi.9130
55. Gotze S, Wolter M, Reifenberger G, Muller O, Sievers S. Frequent promoter hypermethylation of Wnt pathway inhibitor genes in malignant astrocytic gliomas. Int J Cancer (2010) 126:2584–93. doi:10.1002/ijc.24981
56. Bruna A, Darken RS, Rojo F, Ocana A, Penuelas S, Arias A, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell (2007) 11:147–60. doi:10.1016/j.ccr.2006.11.023
57. Waha A, Guntner S, Huang TH, Yan PS, Arslan B, Pietsch T, et al. Epigenetic silencing of the protocadherin family member PCDH-gamma-A11 in astrocytomas. Neoplasia (2005) 7:193–9. doi:10.1593/neo.04490
58. Zhou H, Miki R, Eeva M, Fike FM, Seligson D, Yang L, et al. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin Cancer Res (2007) 13:2344–53. doi:10.1158/1078-0432.CCR-06-2303
59. Hayry V, Tanner M, Blom T, Tynninen O, Roselli A, Ollikainen M, et al. Copy number alterations of the polycomb gene BMI1 in gliomas. Acta Neuropathol (2008) 116:97–102. doi:10.1007/s00401-008-0376-0
Keywords: glioblastoma, genetics, epigenetics, IDH1 mutation
Citation: Liu A, Hou C, Chen H, Zong X and Zong P (2016) Genetics and Epigenetics of Glioblastoma: Applications and Overall Incidence of IDH1 Mutation. Front. Oncol. 6:16. doi: 10.3389/fonc.2016.00016
Received: 14 November 2015; Accepted: 16 January 2016;
Published: 29 January 2016
Edited by:
Kerrie Leanne McDonald, UNSW Australia, AustraliaReviewed by:
Keith Giles, New York University School of Medicine, USAKamalakannan Palanichamy, The Ohio State University Medical Center, USA
Copyright: © 2016 Liu, Hou, Chen, Zong and Zong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuan Zong, bHVsaWFud2VpNjY2NjY2QDE2My5jb20=