 Kathryn D. Held1*
Kathryn D. Held1* Hidemasa Kawamura2,3
Hidemasa Kawamura2,3 Takuya Kaminuma1,2,3
Takuya Kaminuma1,2,3 Athena Evalour S. Paz2
Athena Evalour S. Paz2 Yukari Yoshida2
Yukari Yoshida2 Qi Liu1
Qi Liu1 Henning Willers1
Henning Willers1 Akihisa Takahashi2
Akihisa Takahashi2
- 1Department of Radiation Oncology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA
- 2Gunma University Heavy Ion Medical Center, Gunma, Japan
- 3Department of Radiation Oncology, Gunma University Graduate School of Medicine, Gunma, Japan
The use of charged particle therapy in cancer treatment is growing rapidly, in large part because the exquisite dose localization of charged particles allows for higher radiation doses to be given to tumor tissue while normal tissues are exposed to lower doses and decreased volumes of normal tissues are irradiated. In addition, charged particles heavier than protons have substantial potential clinical advantages because of their additional biological effects, including greater cell killing effectiveness, decreased radiation resistance of hypoxic cells in tumors, and reduced cell cycle dependence of radiation response. These biological advantages depend on many factors, such as endpoint, cell or tissue type, dose, dose rate or fractionation, charged particle type and energy, and oxygen concentration. This review summarizes the unique biological advantages of charged particle therapy and highlights recent research and areas of particular research needs, such as quantification of relative biological effectiveness (RBE) for various tumor types and radiation qualities, role of genetic background of tumor cells in determining response to charged particles, sensitivity of cancer stem-like cells to charged particles, role of charged particles in tumors with hypoxic fractions, and importance of fractionation, including use of hypofractionation, with charged particles.
Introduction
Radiation therapy is a mainstay of cancer treatment, being a common and effective therapy for both curative and palliative treatment of cancer patients. In the last few decades, there has been increasing use of charged particles in radiation therapy. Protons were first proposed for use in cancer therapy by Robert R. Wilson (1), and the number of patients treated with protons has increased dramatically in recent years to a total of over 100,000 patients now treated worldwide (http://www.ptcog.ch/index.php/facilities-in-operation). Radiation treatment of cancer with helium ions began at Berkeley in the late 1950s and was expanded to heavier ions in the 1970s [see a review of the history of charged particles by Skarsgard (2)]. Much of the emphasis has been on carbon ions, with most patients treated in Japan and now totaling over 10,000 patients treated worldwide. The major clinical advantage of protons and heavier charged particles, such as carbon, comes from physics: the Bragg curve provides excellent radiation dose distributions [see reviews in Ref. (3, 4)]. In addition, heavier ions, e.g., carbon, offer the potential of additional biological gains such as increased relative biological effectiveness (RBE) and decreased oxygen enhancement ratio (OER) due to their higher linear energy transfer (LET) in the Bragg peak region, where the tumor is located [reviewed, e.g., in Ref. (3, 5, 6)].
Despite the often-made assumption that the RBE for tumor cells is higher than that for normal cells irradiated under identical conditions, there is only a limited amount of experimental in vitro data that support that assertion (3). However, there have been interesting recent research findings on the differential DNA repair pathways of cancer cells after particle versus photon irradiation, new studies on the effects of charged particles on cancer stem cells, and increasing questions about different responses of tumor and normal cells to hypofractionation, especially with charged particle irradiations, suggest that there may be novel ways to take advantage of differences in characteristics of tumor cells from normal cells to improve or better tailor the use of charged particles in cancer therapy. This review will discuss these issues, with emphasis on data on responses of human tumor cells, largely based on in vitro findings. As discussed in more detail below, RBE is a complex quantity, depending on physical parameters, such as particle type and energy, dose and LET, and biological parameters, including cell/tissue type, cell cycle phase, oxygen level, and endpoint. In vitro assays have limitations compared to in vivo studies and the clinical situation due to lack of 3D architecture and microenvironmental context, including interactions among various cell types, vasculature, and immune system influences. Nevertheless, for studies of RBE, in vitro assays are critical for systematic testing and characterization of effects of various ions, elucidation of DNA damage pathways, and the importance of DNA repair processes and other genetic factors. Furthermore, in vitro studies provide experimental tests for validation of biophysical models, e.g., the local effects model (LEM), prior to clinical application (7), and yield insight on systematic variations in RBE relevant to clinical use (8, 9).
In this review, we start with brief overview sections on the unique biological advantages of charged particle therapy and DNA damage responses that may be important for particle therapy. That introduction is followed by consideration of recent findings on RBEs in human tumor cells, including discussion of the possible roles of genetic factors on RBE, then discussions of new findings on cancer stem cells, hypoxia, and fractionation. In particular, we stress approaches to use the increasing knowledge of the properties of tumors and tumor cells to better advantage when using charged particles in cancer therapy.
An Overview of the Unique Biological Advantages of Charged Particle Therapy
A number of reviews [e.g., in Ref. (3–5)] have discussed the substantial dose distribution advantages of charged particles where, as a result of the Bragg peak, normal tissues can be spared by limiting dose to them, while maximum dose is deposited in the tumor. Heavier ions, such as carbon, have an additional dose distribution advantage over protons because of their reduced lateral scattering compared to protons. However, the major potential advantage of heavier ions in tumor irradiations is their enhanced biological effects, which include increased cell killing, decreased protection by hypoxia, decreased effect of fractionation, and decreased cell cycle dependence. The biological effectiveness of cell killing by higher LET radiations is usually quantified by use of RBE, the ratio of the dose of low-LET radiation (usually X-rays or gamma-rays) to dose of high-LET radiation (e.g., charged particle) for the same biological effect. Many in vitro studies over the years have shown the bell-shaped dependence of RBE for cell killing on LET (6, 10–12) wherein RBE increases with LET to a maximum at about 30–150 keV/μm, then decreases at higher LET. The LET value at which the RBE is maximal depends on the individual ion species, with the peak at higher LET with increasing atomic number of the ions (2). Furthermore, it has also long been recognized that there is great variation in the absolute values of RBE because RBE depends on numerous factors, including particle type and energy, cell type, experimental endpoint, cell cycle phase, dose and dose rate, oxygenation status, culture conditions, etc. (6, 7, 11).
The increased biological effectiveness of radiations with increasing LET lies in the physical dose distribution of the energy of the particles on the micro, and even nano, scale as they traverse matter, the clustering of DNA damages that results from the particle tracks and the increased difficulty cells have in accurately repairing the clustered damage (13–16). As energetic charged particles traverse matter, e.g., cells and tissues of organisms, their electronic interactions with atoms and molecules, mostly through inelastic collisions with atomic electrons, create a path, or track of ionizations before they run out of energy at a finite range, the Bragg peak. The tracks of heavy charged particles are fairly straight, but the electrons ejected from atoms along the track, being much lighter, follow paths that are quite tortuous, with their ranges depend on the energy they acquired when ejected. LET is a measure of the energy imparted to matter by the passage of an ionizing particle. Along the path of a charged particle, the three-dimensional distribution of energy depositions, which cause ionizations and excitations, is called the track structure. For low-LET sparsely ionizing radiations, there are relatively long distances between the energy depositions except at track ends, but with increasing LET, the ionizations along the track become denser and there is lateral spread of the track due to delta-ray electrons, the spectrum of which is determined by the velocity of the heavy charged particle.
If the ionizations from radiation were randomly distributed in cells, the consequences of those energy depositions would likely be minimal, but the non-randomness of the energy depositions accounts for the increased effectiveness of ionizing radiation (14, 17, 18). The clustering of ionizations along radiation tracks occurs on the same scale as the diameter of a DNA molecule and nucleosomes such that if a track traverses DNA it can effectively create clustered DNA damages, such as double-strand breaks (DSBs), clusters of two or more base damages, or clusters of single-strand breaks with base damages. As LET of radiation increases, the clustering becomes more complex, creating, for example, a complex DSB where the break is associated with additional damages, such as base changes or single-strand breaks. Both the proportion and degree of complexity increase with high-LET radiations (19). A number of studies have shown that the complex DNA damages produced by high-LET radiations are repaired less rapidly, less accurately, and less completely than damages from low-LET photons [reviewed recently in Ref. (20, 21)]. Additionally, it is important to bear in mind that track structure has biological relevance not only at the level of DNA damage but also at higher levels of chromatin organization (17): a single high-LET particle track passing through a cell nucleus may cause correlated damages through chromatin structures, such as chromatin fibers, or in adjacent chromosome territories via a string of DSBs along its path, and these correlated damages may result in complex chromosome aberrations. Altogether, the net effect is that complex DNA damages resulting from the greater clustering of ionizations with increasing LET of radiation increases the production of all chromosome aberrations, simple as well as complex.
The increased DNA damage complexity and decreased repair accuracy with radiations of increasing LET not only cause increased cell killing but also result in decreased cell cycle dependence of that killing and play a factor in the decrease in OER. Cells exposed to low-LET radiation show increased resistance when irradiated in late S-phase and increased sensitivity when irradiated in M-phase (22). This fluctuation through the cell cycle decreases with higher LET radiations. However, since in many tumors, the majority of cells are not in the radiation-resistant phases, this effect on treatment outcome in irradiated tumors is likely to be modest (3). The importance of the decreased OER with high LET is discussed below.
Although there has been increasing interest in recent years in the so-called “non-targeted” effects of radiation, including bystander effects and genomic instability in progeny of irradiated cells [for recent reviews, see Ref. (23, 24)], it remains far from clear whether non-targeted effects are similar or different after irradiation with photons versus charged particles (25–27). Furthermore, the role of non-targeted effects or intercellular signaling in response of tumors to radiation remains under investigation (28, 29), with very little work having been done with charged particles. This review is limited to discussion of targeted effects of charge particles.
Overview of DNA Damage Responses Relevant to Charged Particle Biology
Central to any consideration of the effects of charged particles on cells and tissues must be DNA damage response processes. Cells have two main pathways for the repair of radiation-induced DSBs: non-homologous end-joining (NHEJ) and homologous recombination (HR) (30–32). NHEJ is active throughout the cell cycle and is responsible for the repair of most DSBs in cells. NHEJ involves the initial binding of the Ku70/Ku80 heterodimer, recruitment of DNA–PKcs and eventual ligation of the DNA ends by XRCC4–DNA Ligase IV. However, NHEJ is an error-prone repair, and the quality of its repair processes can decrease with increasing levels of DNA damage. HR is active primarily during the S/G2 phases of the cell cycle, when a homologous DNA region is available, and generally results in the preservation of the original DNA sequence. HR involves DSB recognition by the MRN complex (Mre11, Rad50, Nbs1), 3′–5′ DNA resection, DNA stabilization by replication protein A (RPA), Rad51-mediated formation of Holliday junctions, and ultimately resolution of the Holliday junction (31, 33). HR is also involved in the repair and restart of collapsed DNA replication forks (34). At the forks, the BRCA1/2-dependent HR pathway converges with the Fanconi anemia (FA) pathway to resolve the damage (35). It has been suggested that unrepaired clustered DNA damages that collide with replication forks in cells in S-phase require HR for DNA repair and replication restart (36, 37).
It also has been reported that the end-resection activity in cells in the G1 phase may promote micro-homology-mediated end joining (MMEJ) to repair DSBs that cannot be repaired efficiently by NHEJ (38). However, it is unknown how much the activation of HR and MMEJ pathways contribute to escaping cell death in high-LET-irradiated cells. Recently, we showed that targeting and suppressing NHEJ repair yields a high radiosensitivity in cells exposed to carbon-ion beams when compared to the suppression of HR repair (39).
RBEs of Charged Particles in Human Tumor Cells
Experimental studies to determine RBEs have been conducted for many years, with the majority using clonogenic cell survival as the endpoint. It has been felt that lack of clonogenicity is a highly relevant indicator of the efficacy of radiation and its modification because eradication of tumor cells is needed to cure tumors (22). In fact, the shape of curves of tumor control probability, as detected in a clinical context, can be explained from the random nature of tumor cell killing by radiation and the need to kill every cell, as a single cell may give rise to tumor regrowth (22). Furthermore, RBE values, measured or predicted by computer models, are used in clinical treatment planning approaches, which are continually being updated [e.g., Ref. (40, 41)].
It has been argued recently that further studies measuring RBE values may be of limited usefulness because they will have little impact on reducing the uncertainties in ion beam therapy (4, 6). However, determinations of RBEs can help guide understanding of mechanistic underpinnings to the increased effectiveness of higher LET radiations and, thus, may lead to better identification, based on genetic profiles or biomarker evaluation, of patients’ tumors that may benefit most from charged particle therapy.
Shifting the Paradigm of a Generic RBE for Clinical Proton Beam Therapy
Clinical proton beam therapy has been based on the use of a generic RBE of ~1.1 at the center of the spread-out Bragg peak (SOBP) for cancer as well as for normal tissues (8). This RBE value represents an average of a wide range of experimental data in vitro and in vivo and has been intended to be a conservative estimate (8, 42). However, there is now a growing appreciation that the use of a generic value ignores RBE variations that may result, for example, from the heterogeneity of human cancers, LET variations along the SOBP, or the particular clinical endpoint under consideration (42–46). In this section, we will focus primarily on recent data that indicate a dependence of RBE on certain DNA repair defects, with the implication being that proton therapy may have a biological advantage in human tumors that harbor such defects.
There exists very little experimental data on RBE variations in human cancers. In a 2002 review by Paganetti and colleagues (8), the average RBE at the mid-SOBP was estimated as ~1.2 in vitro and ~1.1 in vivo. However, most of the 20 cell lines considered in that analysis were of rodent origin resulting in a somewhat higher in vitro RBE. Only seven human cancer cell lines were included. There is growing evidence for considerable genomic heterogeneity across cancers even of the same type and histology, and it is increasingly appreciated that much of the variations in treatment sensitivity observed clinically are due to genomic heterogeneity, which may include alterations of DNA repair pathways (47–49). Therefore, it is highly doubtful that small numbers of non-representative cell lines are adequate pre-clinical models for assessing clinically relevant variations in RBE values in human cancers. In a recent screen of 17 lung cancer cell lines, RBE estimates at the mid-SOBP of a clinical beam relative to Co60 photons [Co60 equivalent (Eq)] ranged from 0.93 to 1.77 and 1.09 to 1.48 for clonogenic survival fractions of 0.5 and 0.1, respectively (44). In five cell lines (29%), the RBE increase was statistically different from 1.1. Furthermore, in at least three of these cell lines, the RBE increase correlated with defects in the so-called FA/BRCA pathway of DNA repair, and this observation was confirmed in several isogenic cell line models. The FA/BRCA pathway is critical for the maintenance and repair of DNA replication forks [reviewed in Ref. (34, 50)]. Inactivation of any of the FA/BRCA genes has been known to result in hypersensitivity to a variety of anti-cancer agents. However, apart from an involvement of the RAD51 recombinase (FANCR) in the cellular response to proton radiation (43, 51), the importance of the FA/BRCA genes for the repair of proton damage to DNA had been unknown. These observations are clinically significant because genetic or epigenetic defects in the FA/BRCA pathway have been found in large subsets of human cancers (34).
What are the mechanisms through which the FA/BRCA pathway acts on proton damage? For low-LET radiation, which includes X-rays and protons, it has been estimated that 20–40% of the initial damage is clustered, and the majority of clustered damage is present as non-DSB damage (52, 53). Proton radiation causes slightly more complex clustered DNA damages than photons, which is a reflection of the different LET values, i.e., ~2.5 keV/μm for protons at mid-SOBP versus ~0.3–2.0 keV/μm for different photon radiations. DNA repair-proficient tumor cells and normal cells remove these damages almost equally well, consistent with a proton RBE of 1.1 (Co60Eq). Because the FA genes are specifically involved in replication fork maintenance and repair, it can be inferred that the RBE increase that is seen with defects in this pathway results from impaired repair of forks that collide with clustered proton damages. The requirement for the FA/BRCA pathway is greater for proton damage compared to damage caused by, for example, X-rays, even though the RBE (Co60Eq) and LET of these two radiation modalities are almost identical [RBE(Co60) ~1.1 and LET = 2.0–2.5 keV/μm]. This is illustrated in Figure 1A. Proton-irradiated FA/BRCA-defective cells will accumulate greater numbers of DNA DSB in S-phase and subsequently G2-phase than X-irradiated cells, as has been shown experimentally (44) (Willers et al., unpublished). Interestingly, an increase in the size of DSB-associated foci persisting after proton irradiation has been observed (44), likely signifying unrepaired clustered damages (Figure 1A). It has been proposed that these DSB foci could serve as predictive biomarkers to identify cancers that may be more susceptible to proton beam therapy (44). Alternatively, genetic or epigenetic defects in the FA/BRCA pathway could be detected through genomics techniques in order to identify patients for proton therapy. This approach will require a more detailed knowledge of the genes involved in the cellular response to clustered proton damages. The available data indicate that functional loss of any of several key genes in the FA/BRCA pathway will increase the RBE, with the best current estimate being an average RBE of 1.33 (95% confidence limits, 1.25–1.41) at mid-SOBP as shown in Figure 1B. This is a conservative estimate derived at a surviving fraction of 0.1. For 0.5 survival fraction, which is more applicable to fraction sizes of 2 Gy as used in the clinic and which overlaps with the shoulder of the survival curves, the RBE values of the most proton-sensitive cell lines tended to be even higher than for 0.1 survival fraction. For example, the five most sensitive lung cancer cell lines in the report by Liu et al. (44) had an average RBE of 1.30 (range, 1.22–1.48) and 1.46 (range, 1.31–1.77) at survival fractions of 0.1 and 0.5, respectively.
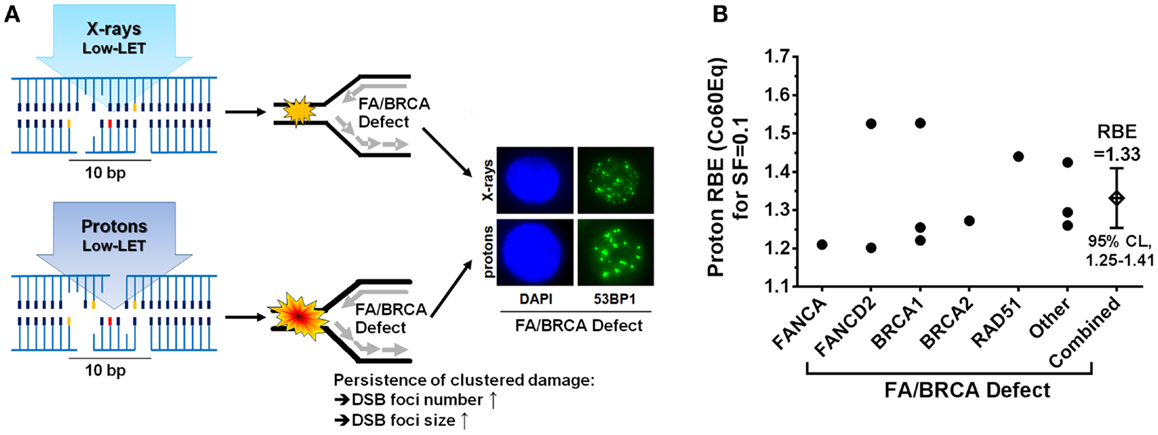
Figure 1. A “New Biology” of proton beam therapy. (A) Illustration of how FA/BRCA defects may sensitize cells to proton irradiation. Left, clustered DNA damages after equal physical doses of X-rays and mid-SOBP protons are slightly different despite similar LET (2–2.5 keV/μm) and identical RBE in repair-proficient cells (~1.1). In the presence of a FA/BRCA defect that affects the repair of replication forks encountering clustered damages, there will be greater unrepaired damage after proton-irradiation, as marked by an increased number and relative size of repair-related protein accumulations of DNA double-strand break markers. Representative immunofluorescence microscopy images showing nucleus (DAPI) and 53BP1 foci (green) in FANCD2-mutant cells are shown on the right. (B) Summary of RBE estimates relative to Co60 photons as a function of defects in the FA/BRCA pathway (44, 45). Other, taken from unpublished data (Willers et al.); CoEq, Co60 equivalent; SF, surviving fraction; CL, confidence limits.
In conclusion, these recent pre-clinical data strongly suggest inter-tumoral heterogeneity of proton RBE that may yield opportunities to identify proton susceptible tumors in the clinic within the next few years. This “New Biology” of protons in cancer coupled with the increasing knowledge of RBE variations as a function of physical proton beam parameters in both cancers and normal tissues is expected to shift the paradigm of a generic proton RBE to a variable RBE.
RBE Determinations with Heavy Charged Particles
The proton studies just described provide a possible DNA repair capacity-based explanation for some of the variation seen in proton RBE values at a given LET. Could a similar finding apply to human tumors exposed to high-LET charged particles? Unfortunately, no single study with a substantial number of cell lines has yet been done for any heavy ion, although many small studies with a few cell lines each have been performed. Some large compilations of cell survival RBE values for many cell types, endpoints and radiation qualities have been published recently (7, 54, 55), and the composite data clearly show that RBEs depend on LET, endpoint, ion, etc. In this section, we focus on analysis of RBE values for human tumor cells exposed to ions heavier than protons. Published papers that describe the cell survival RBE of human tumor cells have been searched by using PubMed; many of these papers are included in the compilations mentioned. A total of 430 RBE values were collected from 36 published papers (56–91). When authors provided RBE values along with dose–response data, those values were used. In cases where authors showed dose–response curves but did not cite any RBE value, an isoeffect line was drawn in the dose–response curves to read corresponding doses of ions and reference photons. As reference beam, 30 papers used X-rays and 6 papers used gamma-rays. For the analyses here, the biological differences in effect between X-rays and gamma-rays were not considered.
Endpoint
Endpoint is one of the major factors, which affects the values of RBE (7, 54, 55, 92). The RBE data as a function of LET sorted by endpoint are shown in Figures 2 and 3. All papers included in Figure 2 presented RBE values for colony formation after exposure to a range of single doses. Within a total of 363 RBE values, 295 values in 31 papers were calculated using an isoeffect dose of 10% survival (D10). The other values that were calculated included D0, D30, D50, D75, ratio of alpha parameters, or isodose effectiveness. The RBE values for D10 ranged from 1.03 to 4.99, showing the “classic” increase in RBE with LET followed by a decrease at higher LET (22) although the range in RBE values at any given LET is substantial in many cases. The RBE values based on D0, D30, D50, and D75 also showed considerable variation at any given LET, but, as expected, there was a trend for higher RBE values at higher levels of survival (22). Some of the highest RBE values were derived using the alpha ratio; this, too, is consistent with higher RBEs at higher survival, since alpha ratios would tend to be derived based on high survival data.
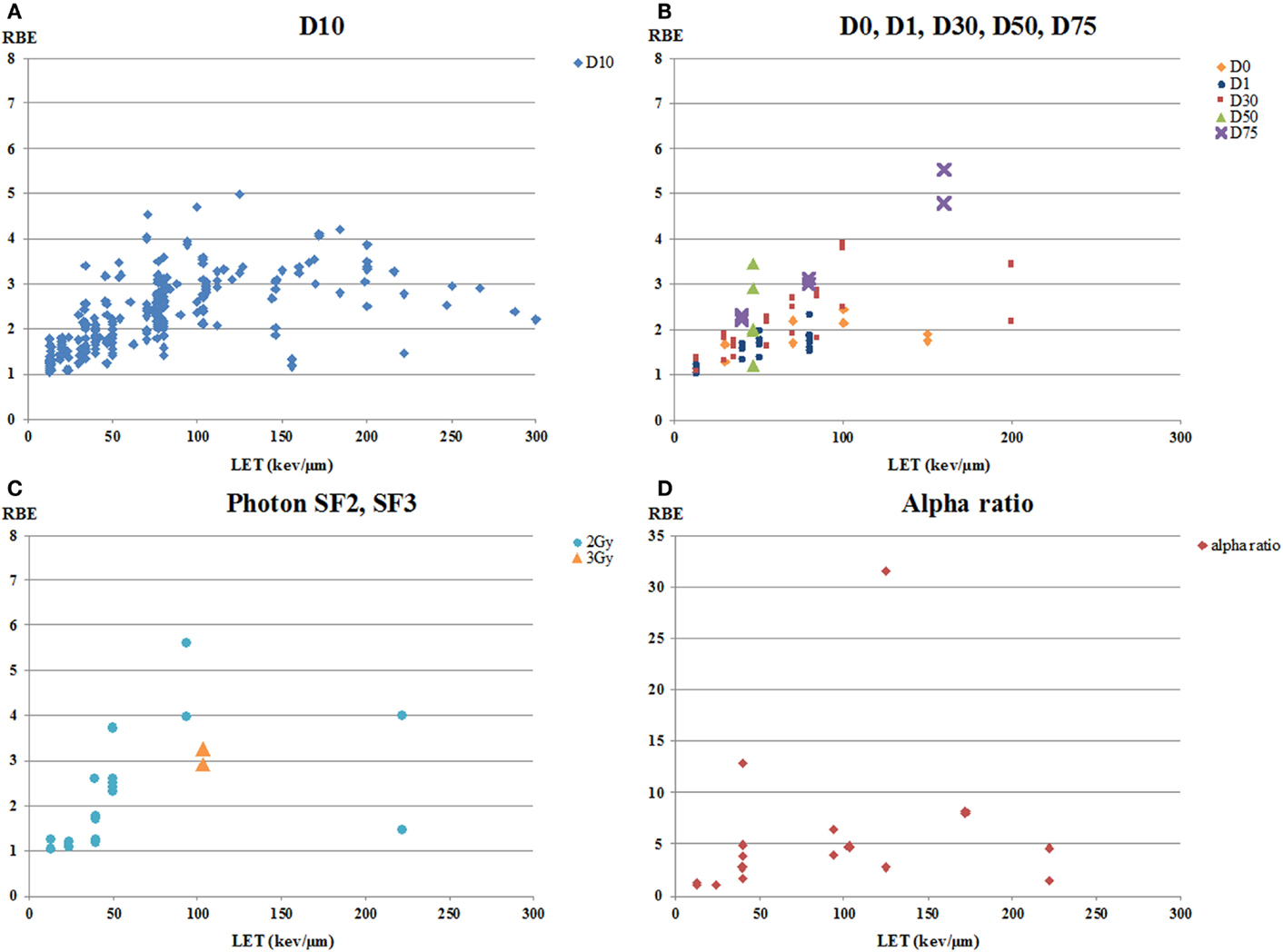
Figure 2. RBE versus LET for human tumor cell lines for various endpoints. RBE values are all based on colony formation assays. (A) RBE values calculated as the ratio of isoeffect doses at 10% survival (D10). (B) RBE values calculated as ratios of doses for D0, D30, D50, and D75. D0 was calculated by fitting the survival curve to the single-hit multi-target (SHMT) model: S/S0 = 1 − (1 − e−D/D0)n. (C) RBE values calculated as the ratio of doses at the level of photon doses of 2 Gy (SF2) or 3 Gy (SF3). (D) RBE values calculated as the ratios of the alpha parameters of survival curves.
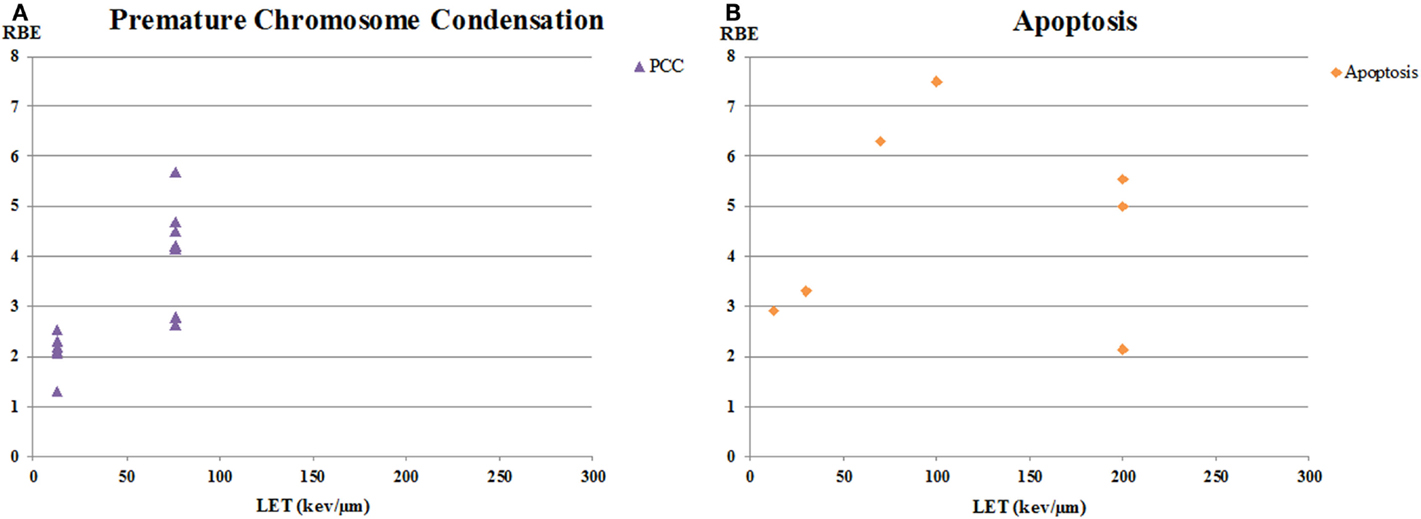
Figure 3. RBE versus LET for human tumor cell lines for chromatin breaks and apoptosis. (A) is from data on unrejoined chromatin breaks in cells after premature chromosome condensation (PCC). Residual unrejoined chromatin breaks were detected using Giemsa staining in cells after chromatin condensation. (B) is RBE values for apoptosis.
The other endpoint that tends to show high RBE values is apoptosis (Figure 3B). This is consistent with the observations that most solid tumor cell lines are resistant to X-ray-induced apoptosis (93) and that apoptosis may be characterized by the alpha-component of the cell survival curve [reviewed, e.g., in Ref. (94)]. In a recent review on proton radiobiology, Tommasino and Durante (95) pointed out that there is a general tendency for an increased apoptotic response with increasing LET and that tumor cells resistant to photon-induced apoptosis may have apoptosis triggered by an alternative pathway by protons, a suggestion that could likely extend to heavier charged particles. However, it should also be pointed out that several groups, including Brown and colleagues (96), have demonstrated that apoptosis induction can be markedly affected by tumor cell genetics and the overall level of cell killing as determined in a clonogenic assay in vitro may not correlate well with apoptosis induction [also reviewed in Ref. (94)].
Two papers reported RBE values calculated for residual unrepaired chromatin breaks using premature chromosome condensation (PCC) (Figure 3A), with the paper by Suzuki et al. using primary cells obtained by biopsy from patients (67, 72). Authors of both studies noted the good correlation between their data on residual chromatin breaks as measured using the PCC technique and colony formation, and concluded that the PCC technique was a potential predictive assay of tumor response to ion therapy. Information on correlation of chromatin breaks using PCC with DNA repair protein foci formation and/or FA/BRCA pathway status, as discussed above for potential use with proton therapy patients, would be helpful for assessment of possible predictive assays.
Ion
The data on RBE values calculated using D10 and sorted by ions are shown in Figure 4. A total of 29 papers reported 247 RBE values for carbon-ion beam, whereas there were 21 RBE values for helium ions in 3 papers, 24 values for neon ion in 2 papers, 6 values for boron ions in 1 paper, 6 values for silicon beam in 2 papers, 5 values for iron beam in 3 papers, 2 values for nitrogen beam in one paper, and 3 values for argon beam in 2 papers. The RBE values showed substantial variation at any given LET, independent of ion species used, but in all cases the RBE increased with LET to a maximum then decreased at high-LET levels. It is well known that the RBE values of carbon ions peak around an LET of 100 keV/μm (7, 54, 55, 92). The other ion beams had peaks between LETs of 100 and 200 keV/μm, with a trend toward a maximum at higher LET with heavier ions.
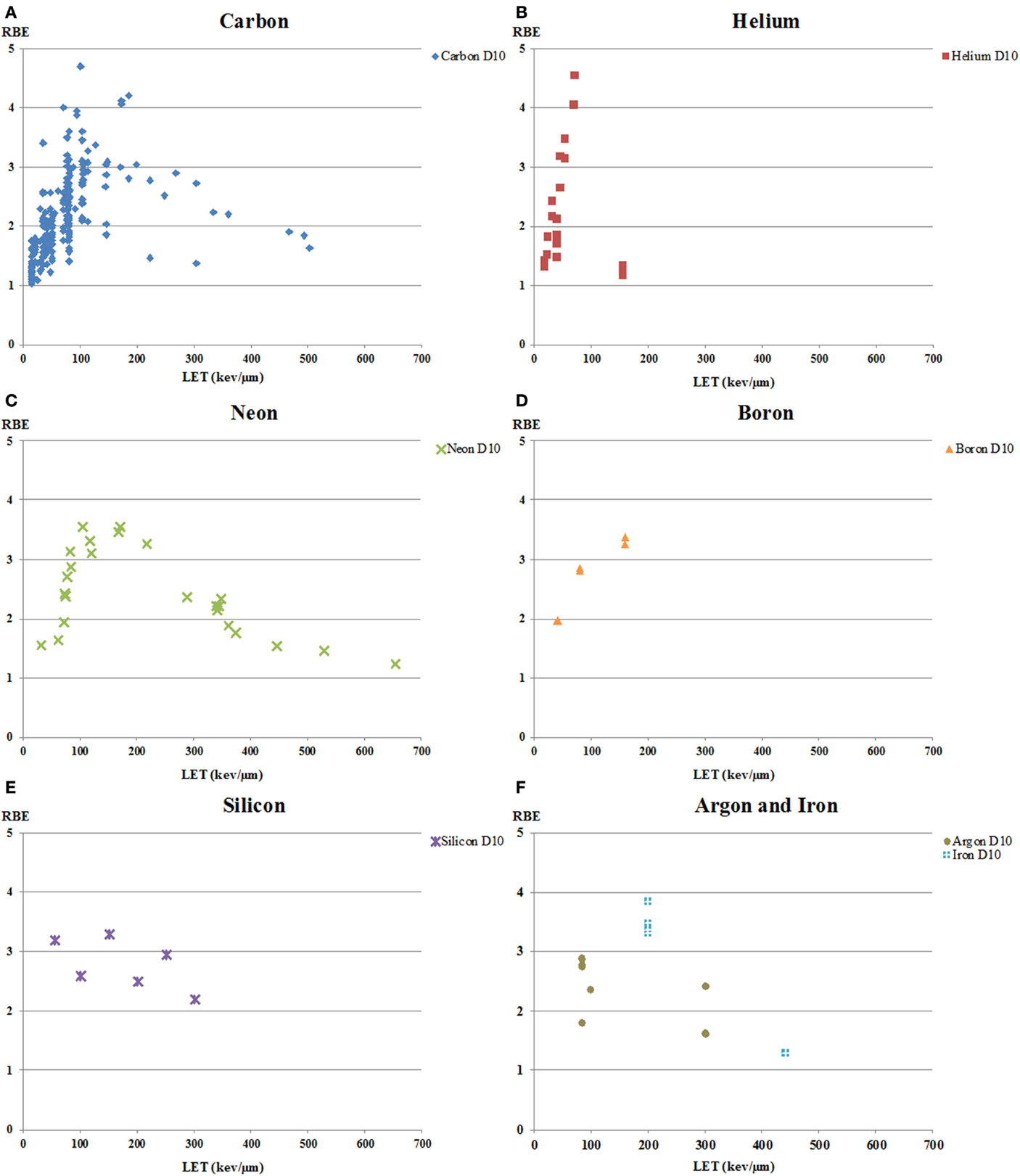
Figure 4. RBE at 10% survival (D10) versus LET for human tumor cell lines exposed to various charged particles heavier than protons. RBE values derived at 10% survival from clonogenic survival curves from all available literature are shown as a function of LET for human tumor cells exposed to (A) carbon ions; (B) helium ions; (C) neon ions; (D) boron ions; (E) silicon ions; and (F) argon and iron ions. The RBE values showed substantial variation at any given LET, independent of ion species, but in all cases the RBE increased with LET to a maximum, then decreased at high-LET levels.
Furusawa et al. (59) exposed human salivary gland tumor cells to carbon, neon, and helium ion beams and calculated the RBE values of each beam. They showed that the RBE values for helium ions were higher than those for the other ions, which seems unexpected. This finding deserves more investigation as there is some interest in development of helium ion beams for cancer therapy since they have less lateral dose than protons (i.e., a better dose distribution) (97), which might make their use particularly relevant in children. Furthermore, in their report, Furusawa et al. show that the peaks of the RBE values shifted to higher LET values with increasing atomic number, an observation that had been made earlier on the basis of work by a number of authors [e.g., Ref. (11, 98, 99)] as reviewed by Skarsgard (2). Such findings deserve emphasis as they highlight the fact that LET is not adequate as the sole descriptor of energy deposition in cells and tissues, but that ion track structure, the nanometer scale distribution of energy, must be considered when evaluating biological effects. In this context, it is interesting to note that NASA’s model for calculating risk of radiation-induced cancer from space radiation takes into account track structure of heavy ions rather than simply LET (100). In a clinical context in heavy ion therapy, the LEM, which is used for RBE prediction, also provides particle species and LET-specific RBE values that are then propagated, using a treatment planning system, to a representative RBE value at each position in the irradiated field (9, 101), a process needed because ion fragmentation produces a mixed radiation field.
With regard to ions, it is worth pointing out that we did not include data with oxygen ions in Figure 4 because we found only one study using oxygen ions, and that work used only a single LET (87). That work reported that for four human hepatocellular carcinoma cell lines irradiated in the SOPB of oxygen ions with a mean energy of 154 MeV/u (LET of 146 keV/μm), the clonogenic RBE10 values ranged from 1.9 to 3.1, with the values not being significantly different from those obtained in the same study using 130 MeV/u carbon ions (LET of 112 keV/μm). However, this study is noteworthy because of the current interest in using oxygen ions, with their lower OER, in treating tumors with large hypoxic fractions (102).
Type of Tumor Cells
The data on RBE values as a function of LET for carbon-ion beam only, calculated using D10 and sorted by tumor type, are shown in Figure 5. Figure 6 shows a subset of the data separated out by adenocarcinoma and squamous cell carcinoma. The graphs show data only for LET < 100 keV/μm. The number of data points, or cell lines, varies greatly with tumor type. Generally, the brain tumors (composite slope of 0.018) and adenocarcinomas (composite slope of 0.018) appear to have lower slopes for the RBE versus LET curves than do squamous cell carcinomas (composite slope of 0.024 or higher). It should be noted, however, that data from Suzuki et al. (71) for cervical cancer included in the squamous cell carcinoma graph were derived from primary cultured cells from biopsies from patients, the only data from primary cultures included in this analysis. These primary culture data appear to have lower slopes than the other squamous cell data, although it should also be noted that the steeper slopes for the established squamous carcinoma cell lines are determined by only four data points at high-LET values. Thus, it is not possible to determine whether there is a systematic difference between primary squamous cell cultures compared to established tumor cell lines or between squamous cell carcinomas and adenocarcinomas. It is not clear from clinical data with carbon ions whether a difference exists between sensitivity of squamous tumors and adenocarcinomas, suggesting an area for further in vitro, in vivo, and/or clinical study. For comparison, it can be pointed out that in a similar analysis approach, Ando (54) found that the RBE versus LET plot for cultured human fibroblasts had a slope of 0.027, which the author noted was steeper than the composite slope for the human tumor data he analyzed.
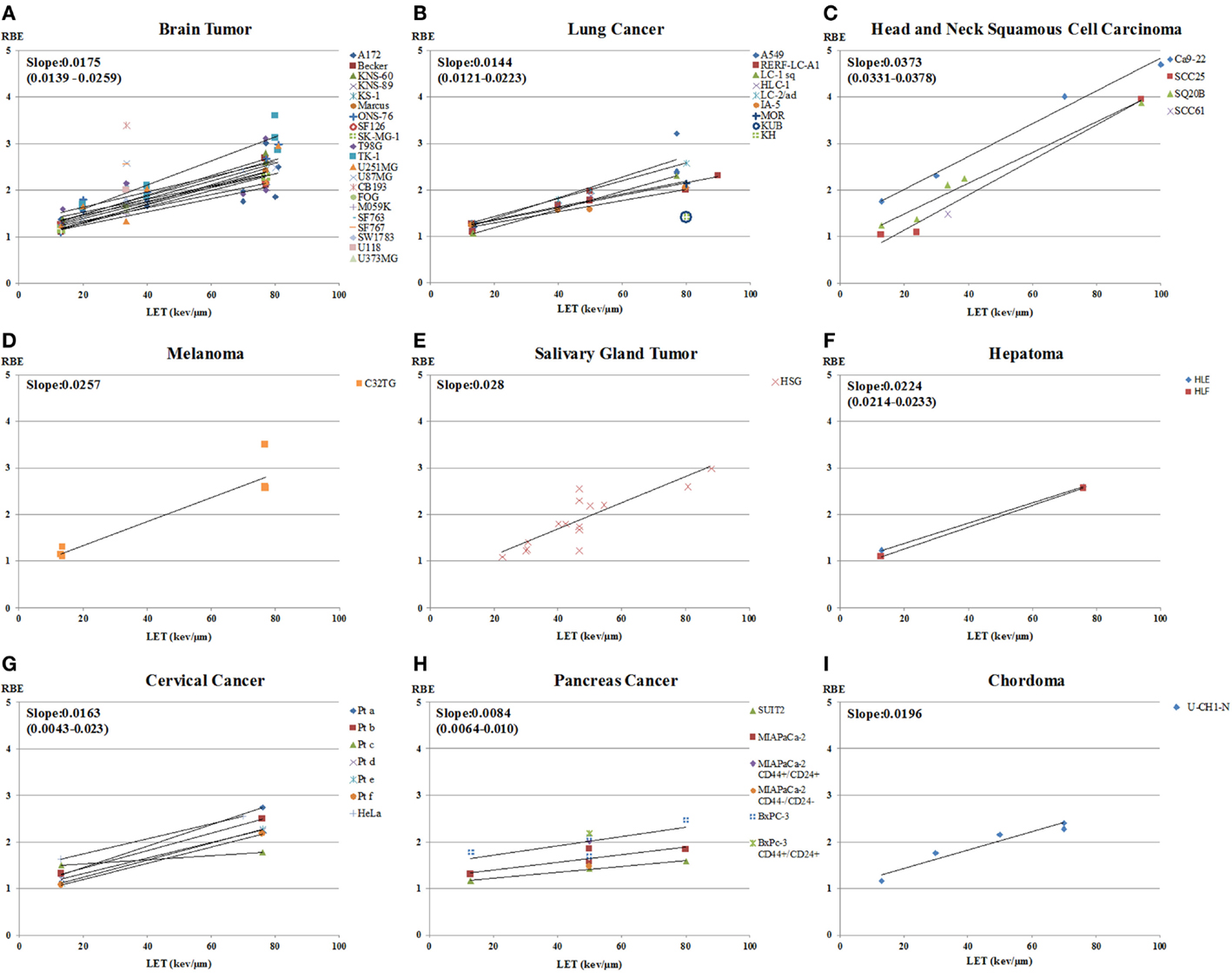
Figure 5. RBE at 10% survival versus LET for cells from various types of human tumors exposed to carbon ions. RBE values as a function of LET for carbon-ion beam only, calculated using D10 and sorted by tumor type, are shown. Tumor types included are: (A) brain tumor; (B) lung cancer; (C) head and neck squamous cell carcinoma; (D) melanoma; (E) salivary gland tumor; (F) hepatoma; (G) cervical cancer; (H) pancreatic cancer and (I) chordoma. The graphs include data only for LET < 100 keV/μm. The number of data points, or cell lines, varies greatly with tumor type. The slopes of the RBE versus LET curves are calculated for each cell line and tumor type.
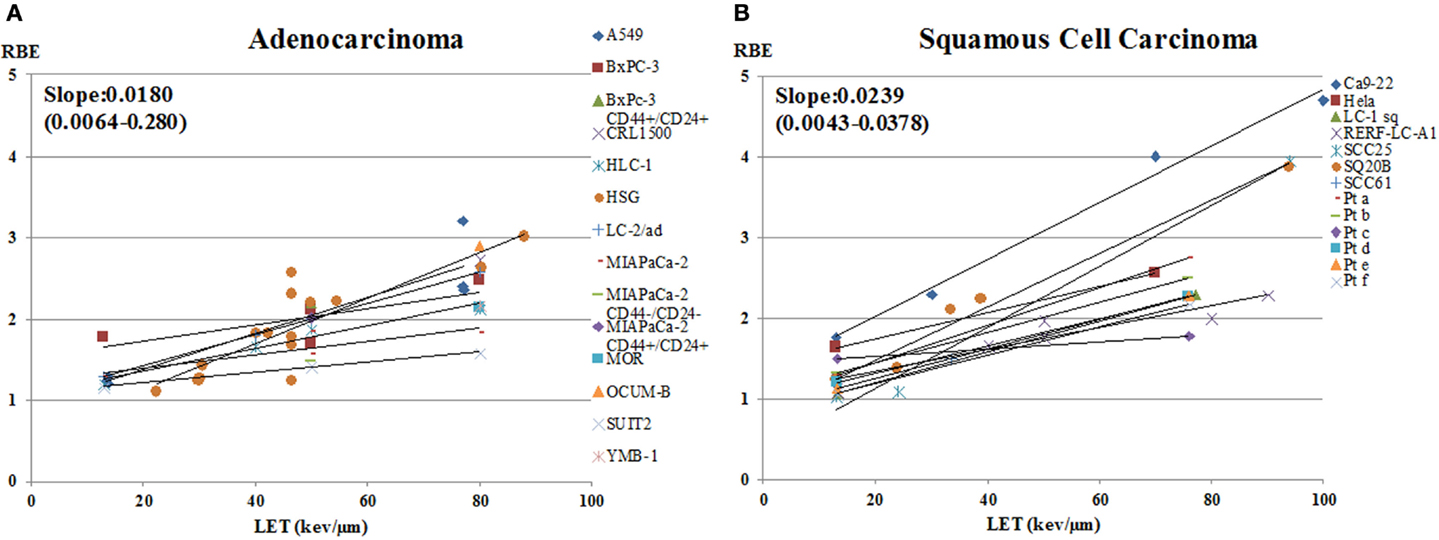
Figure 6. RBE at 10% survival versus LET for cells from human adenocarcinomas and squamous cell carcinomas exposed to carbon ions. RBE values as a function of LET for carbon ion beam only, calculated using D10 for (A) adenocarcinomas and (B) squamous cell carcinomas. The graphs show data only for LET < 100 keV/μm. The slopes of the curves are shown for each cell line.
The RBE values for the pancreas cancer cells are the lowest in all the data (64). The slope of the graph of pancreas is 0.0084, which is gentler than the others. This might suggest that pancreatic cancer would not be a good candidate for carbon-ion therapy, yet clinical trials of carbon ions for pancreas cancer in Japan have shown promising results (4, 103). The clinical results may reflect properties of the human tumors in situ, such as high hypoxia, radioresistance (high cancer stem cell component?), and anatomic location, that might not be evident in studies of isolated tumor cells.
It is noteworthy that there are few tumor cell data on RBE values with charged particles for prostate cancer or bone and soft tissue cancers, which are the two cancer types with the most patients treated to-date with carbon ions at NIRS in Japan (103). Furthermore, we found no experimental RBE data for human cell lines of mucosal malignant melanoma, adenoid cystic carcinoma, or rectal carcinoma, which are all being treated with carbon ions at NIRS with favorable outcomes (103).
D10 has been used as the parameter for calculating RBE values in this analysis by tumor type (Figures 5 and 6) because that is the parameter most frequently reported in the literature. However, the use of D10 may have minimized the ability to see differences between tumor cell types, resulting in the relatively similar values of the slopes of the RBE versus LET curves for the various tumor cells. Generally, inherent photon radiosensitivity differences between cell types become most evident at high and low cell survival levels, and it has long been recognized that RBE values are larger at high survival levels than at low ones because of the “shoulder” on photon survival curves (22). For example, this is consistent with the data shown in Figure 2 where RBEs based on alpha ratio (generally reflecting high survival, low dose results) tend to be higher than those based on D10. Since it has been shown that photon dose–response curves for different tumor cell types have significant differences [e.g., Ref. (104, 105)], one might expect that the RBE versus LET curves would also differ in a manner consistent with the photon sensitivity. The finding here (Figures 5 and 6) that the differences seem small may reflect the use of the less discriminating parameter, D10. If sufficient data existed to do this analysis with a parameter more weighted toward lower or, especially, higher survival levels, e.g., alpha ratio, greater differences in dependence of RBE on LET for various cell types might be seen.
Role of Genetic Background of Tumor Cells in Response to Charged Particles
In light of the proton data, discussed above, indicating a correlation between cell lines with higher proton RBE values and defects in DNA repair, specifically in HR repair, we wondered whether the same finding would extend to heavier ions, notably carbon ions. Although the literature data on RBEs for human tumor cell lines shows substantial variations at any given LET, even just for carbon ions (Figures 2–6), we could not find any information in the literature on possible DNA repair deficiencies, particularly in HR repair, for the cell lines with the highest RBEs after carbon-ion irradiation, e.g., TK-1 brain tumor, Ca9–22 gingival squamous cell carcinoma, SQ20B head-and-neck cancer. Therefore, experiments to ascertain carbon RBE values for human tumor cell lines known to be defective in the FA/BRCA DNA repair pathway are warranted. Furthermore, both the proton data of Liu et al. (44) and the carbon-ion data of Suzuki et al. (70) on residual unrepaired DNA damage (assays of 53BP1 foci and DNA damage revealed by PCC, respectively) suggest that such assays may be useful biodosimeters to select patients for charged particle therapy.
What would be the clinical application of increased tumor RBE values in subsets of patients? Identifying patients with proton- and/or heavy ion-sensitive tumors may allow us to: (a) de-escalate the physical dose of charged particles if normal tissue damage is a particular concern; (b) select patients for proton or heavy ion treatment slots who would have not otherwise had the opportunity to be treated with such radiations, thereby increasing the odds of local tumor control; or (c) biologically optimize tumor-directed therapy, for example, by employing intensity-modulated ion therapy algorithms to superimpose an LET increase on the already pre-existing RBE advantage, thereby further improving local tumor control. Because RBE values tend to increase with increasing fractionation sensitivity of tumors (i.e., decreasing alpha/beta values) (42), there exists additional opportunity to improve the outcome of ion beam therapy in tumors with low alpha/beta values, such as prostate or breast cancer. However, this approach will require better knowledge of the inter-tumoral variation of alpha/beta values and the development of predictive biomarkers to identify appropriate tumors.
Sensitivity of Cancer Stem-Like Cells to Charged Particles
In recent years, considerable interest has developed in the possibility that cancer stem-like cells (CSCs) in human tumors could be major contributors to resistance of tumors to conventional photon radiotherapy (RT) (106–108). However, intriguing data also suggest that the presence of CSCs might be overcome by carbon-ion therapy (89, 109). In this section, we discuss such a potential from a radiobiological perspective.
Cancer stem-like cells, also called cancer-initiating cells (CICs), are tumorigenic and have the potential to give rise to all cell types identified in hematological cancers and in several types of solid tumors (110). CSCs are regarded as “roots of cancer,” analogous to normal stem cells in hierarchical tissues, although the origin of CSCs is still not clear and various theories have been proposed to explain their origin (111). It is believed that tumor growth is driven by a discrete subpopulation of CSCs that are defined by their capacity for self-renewal and their ability to generate heterogeneous lineages of cancer cells (110). The CSCs can survive and usually persist in tumors for a substantial length of time as a distinct population and can eventually cause cancer recurrence after treatment and tumor metastasis. It seems reasonable to suggest that cancer cure can be achieved only if this population is eliminated.
There is growing evidence that CSCs are inherently resistant to conventional fractionated RT. This radioresistant phenomenon of CSCs has been described within the framework of the four Rs of radiobiology: (i) repair, (ii) redistribution, (iii) reoxygenation, and (iv) repopulation (112).
(i) Regarding DNA repair, CSCs exhibit fewer DNA DSBs after exposure to ionizing radiation than non-tumorigenic cancer cells, which has been correlated with efficient DNA repair machinery due to constitutive hyperphosphorylation of the DNA checkpoint kinases Chk1 and Chk2 (106).
(ii) Regarding redistribution, quiescent or slowly cycling cells, normal or cancer stem cells, generally are radioresistant, although dose fractionation can cause redistribution of radioresistant S-phase cells into a more sensitive phase of the cell cycle. If this happens only in tumor cells, it could result in a therapeutic benefit for slowly cycling normal cells, sparing late responding normal tissues during fractionation. However, if tumors also have a significant proportion of CSCs that are slowly cycling, any benefit from redistribution may not apply (112).
(iii) Regarding reoxygenation, if the niche in which CSCs reside is hypoxic, during radiation fractionation the quiescent CSCs may be exposed to increasing oxygen levels causing increasing radiosensitivity due to transition of cells into an activated, proliferative state. It appears that in some cases, the CSC niche may be in perivascular regions (113, 114) where they may be exposed to rapidly changing cycles of hypoxia-reoxygenation (112). During reoxygenation, the cells would become more radiosensitive, and reoxygenation triggers metabolic processes that generate damaging reactive oxygen species (ROS). However, CSCs manifest enhanced protection against ROS (107, 108). It was reported that expression of the CSC marker CD44, in particular that of a variant isoform (CD44v), contributes to ROS defense by promoting the synthesis of glutathione (GSH), a primary intracellular antioxidant radical scavenger (115). Hence, the roles of hypoxia, reoxygenation, and ROS defenses in CSCs appear quite complex, and more research is required to elucidate their roles in radiation response.
(iv) Regarding repopulation, it was reported that developmental signaling pathways, such as TGF-β, Notch, Wnt/B-catenin, and Sonic hedgehog pathways greatly contribute to maintenance of CSCs, as they do with normal tissue stem cells (112). Intrinsic inter-conversion and dynamic equilibrium between CSCs and non-stem cancer cells (NSCCs) exist under normal and irradiation conditions, and TGF-β might have important roles in the equilibrium (116).
In addition to the four Rs of radiobiology, it has been shown that CSCs can acquire radioresistance through activation of anti-apoptotic Bcl-2 (117) and serine/threonine protein kinase B (PKB, also known as AKT) survival signaling (118, 119). Hence, there is substantial reason to believe that CSCs are a radiation-resistant cell population in at least some tumors exposed to photon irradiation.
On the other hand, intriguing studies have reported that CSCs may be more effectively killed by carbon ions compared to photons in colon and pancreas cancers both in vitro and in vivo (89, 109, 120), and CSCs from colon and breast cancers may be more efficiently eliminated by proton irradiation than photon treatment, at least in vitro (121, 122). One or more of several processes may explain the observations that ion beams have biological advantages for killing CSCs compared to photons. These include the diminished capacity for NHEJ repair, which may play an important role in the quiescent G0 cell cycle phase, after heavy ion exposure (39); a decreased OER with heavy ions (59, 123), and an efficacy in dealing with radioresistant tumor cells (TP53-mutated and BCL2-overexpressing cells) (124) compared with results produced by photon beams. We demonstrated that heavy ion beams depress AKT-related survival signaling (125). Therefore, we speculate that heavy ion beams may target CSCs via depression of AKT survival signaling. Indeed, we demonstrated that the population of CSCs is only slightly increased or unchanged after carbon-ion irradiation because carbon ions may simultaneously kill CSCs and non-CSCs, while X-rays have less effect on CSCs than on the bulk cancer cells (126). These results suggest that carbon ions may enhance apoptosis and autophagy through activation of death signaling and may target CSCs via the depression of AKT survival signaling (Figure 7). However, it should be noted that the observations of CSCs being preferentially more sensitive to charged particles is not universal as it has been reported that head-and-neck cancer CSCs are resistant to both photon and carbon-ion irradiation (127). Clearly, more detailed studies are necessary, for example, using tumor samples from carbon- and photon-irradiated patients, to understand the potential significant therapeutic benefit of heavier charged particles on CSCs. It is also worth investigating whether, or how, the enhanced DNA repair advantages in CSCs might relate to the potential for development of biomarkers based on residual DNA damage for identifying patients whose cancers might be treated more efficaciously using charged particles, as discussed in the section above.
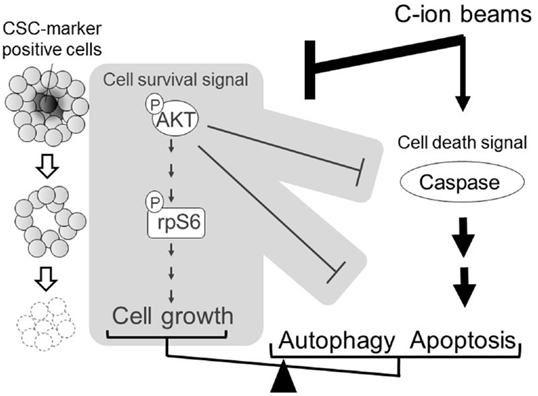
Figure 7. A model for carbon ion-induced apoptosis and autophagy through the enhancement of death signals and the depression of survival signals. The model is based on AKT survival signaling as shown in our work (126). An arrow “→” indicates enhancement; a sidewise “⊣” indicates depression.
Sensitivity of Hypoxic Tumor Cells to Charged Particles
A long-recognized property of tumors is their development of hypoxic regions. It has also been documented, for many years, that hypoxic cells are resistant to photons, but that resistance is reduced when hypoxic cells are irradiated with higher LET particles (22). Suit et al. (3) postulated that the potential gain from high-LET radiations in the clinic may be due principally to the lower OER (ratio of doses for a given endpoint in hypoxic to well-aerated cells). This section discusses the reduced hypoxic protection with carbon-ion therapy and how that might be exploited in cancer therapy.
Cellular sensitivity to low-LET radiations (photons, clinical energy protons) depends on the degree of hypoxia at the time of irradiation, increasing in a sigmoid fashion from an OER = 1.0 (no difference between anoxic and well-aerated cells) at very low oxygen levels to a maximum (OER ~ 3.0) usually obtained by about 2–3% oxygen (22, 128). Sensitivity also depends on the duration of exposure to the hypoxic conditions. There are two distinct mechanisms that promote oxygen deficiency in tumor cells; each exposes cells to different periods of hypoxia. Acute or perfusion-limited hypoxia is caused by poorly formed or dysfunctional vasculature that can cause transient closing of blood vessels that deprives the surrounding cells of an appropriate oxygen supply (129). On the other hand, in chronic or diffusion-limited hypoxia, the imbalance of oxygen supply and consumption in actively proliferating tumor cells causes cells far from blood vessels to experience a deficiency in oxygenation for long periods of time (130). Historically, most studies of hypoxic radioresistance have dealt with chronic hypoxia, but experiments investigating the influence of acute and chronic oxygenation conditions on cell response have shown increased radioresistance for the acute case (131–133). Ma and colleagues demonstrated that for both X-ray and carbon-ion irradiation, cells under acute anoxia were more radioresistant than those under chronic anoxia, whereas cells subjected to acute and chronic hypoxia (0.5% O2) exhibited no significant difference in sensitivity (131, 132). They argued that prolonged exposure to anoxia induced a breakdown in cellular energy metabolism, which led to delays in cell cycle progression. They found that cells were arrested in the G1 phase of the cell cycle with a significant decrease in the number of active S-phase cells after 24 h of hypoxia. However, abrupt changes in the oxygenation status did not result in changes in the cell cycle distribution. The energy deficiency of cells also has been associated with the reduction of DNA damage repair (133). Therefore, chronically hypoxic cells were found to be more vulnerable to radiation damage.
The poor performance of photons in curing hypoxic tumor cells has prompted researchers to turn to high-LET radiation, such as ion beams that have lower OERs [reviewed, e.g., in Ref. (22, 128, 134, 135)]. Radiation damage from low-LET beams is mostly mediated by free radicals (indirect effects), i.e., secondary electrons generated from the ionizations interact with molecules, such as water, to produce free radicals which in turn damage the DNA. In contrast to their low-LET counterparts, the contribution of radiation damage by direct ionizations in DNA is higher for high-LET beams. Here, the secondary electrons directly interact with the critical target, thus producing, at least in part, different damage. Hence, the oxygen effect can be explained, at least in part, by differences in induction and repair of DNA damage. Hirayama et al. reported that the rejoining kinetics of DNA DSBs incurred from carbon-ion irradiation were the same for cells in oxic and hypoxic conditions (136). This led them to postulate that DNA DSBs produced by carbon ions are the same for the two oxygenation conditions. However, their results for X-ray irradiation showed a dependence of the repair dynamics on the oxygen level, with DSBs generated under oxic conditions rejoined more efficiently than those produced under hypoxia. They postulated that this resulted from different mechanisms for DNA damage depending on oxygenation, namely, that in the presence of oxygen, oxygen-reacting radicals could cause additional DNA DSBs but in hypoxia more damage is produced by direct ionizations or by radicals irrelevant to oxygen. Furthermore, the repair times were longer after carbon-ion irradiation and more unrepaired DNA DSBs remained after 5 h while for X-rays almost all DSBs were efficiently rejoined. This can be explained by the high ionization density generated along the track of heavy charged particles that produces complex DNA damages, making repair more difficult. Therefore, the OER decreases with increasing LET values, with the OER of carbon ions about half that with X-rays. Typical survival curves obtained using carbon-ion and X-ray irradiation under oxic and hypoxic conditions are illustrated in Figure 8. The difference with oxygenation status is diminished with the high-LET carbon ions and the survival curves tend to converge. By contrast, the larger variation in the cell response seen for X-ray irradiation is reflected by the higher OER value. A consequence of the enhanced radioresistance observed in X-ray survival curves under hypoxia is that RBEhypoxic generally exceeds RBEoxic. In vitro studies have also shown that OER approaches unity at dose-averaged LETs of ~300 keV/μm (59, 134). Oxygen ions, with their high-LET values within therapeutic fields, might be advantageous for tumors with significant hypoxic fractions. Scifoni et al. (135) compared computed OER values in a tumor irradiated with oxygen or carbon ions, and showed that, assuming the same dose in the entrance region, there was a dramatic decrease in OER for the oxygen ions.
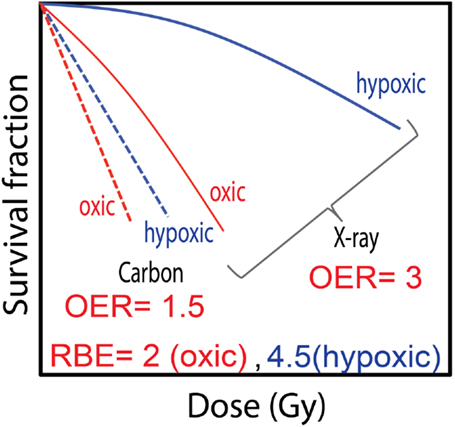
Figure 8. The dependence of survival curves on oxygen concentration typically observed after exposure to X-rays and carbon ions. The importance of the oxygen effect is reduced with high-LET carbon-ion irradiation as is apparent in the small separation of the survival curves compared to that seen with X-rays. The large difference between the cell response in air and hypoxia for X-rays results in a RBEhypoxic that is greater than RBEoxic.
The advantage of high-LET carbon ions over photons in treating tumor hypoxia has been confirmed in the clinical setting by Nakano et al. (137). They measured the intratumor oxygen partial pressure of uterine cervical cancer patients prior to and at the fifth day of treatment with either photons or carbon ions using a polarographic electrode. The 4-year local control rates were found to be independent of the oxygenation condition for carbon-ion treatment, whereas the control rate for photon therapy of patients with high pO2 status was more than twice that with low pO2.
It has been suggested that further improvements in treatment outcome with carbon therapy can be achieved by considering the time course of reoxygenation of hypoxic areas in the tumor. According to Antonovic et al. (138), the number of fractions and the dose per fraction for carbon therapy can be optimized by taking into account the effect of local oxygenation changes on tumor control probability. In the future, more detailed studies are necessary to take into account the OER and rates of reoxygenation in treatment planning for carbon-ion RT, as are underway (134, 135).
Dose Fractionation with Charged Particles
Fractionated irradiation is a valuable tool in conventional RT to reduce early and late effects in normal tissue by allowing repair of sublethal damage or increase tumor response due to reoxygenation of a hypoxic tumor. The linear quadratic (LQ) model describes cell killing using single-hit and double-hit components (22). The shape of the curve is determined by:
The α parameter describes the linear component of the curve, while the β component describes the quadratic portion of the curve. The α/β ratio, the point at which linear cell killing is equivalent to quadratic cell killing, is an important parameter used to model cell killing by radiation. Presently, this ratio is used as a staple for predicting the clinical effects in response to RT despite various limitations. A high α/β ratio, seen in many human tumors, suggests a predominance of the α component, implying a decreased response to fractionation and, therefore, clinical benefit from hypofractionation (decreased number of fractions of larger dose per fraction). A lower α/β ratio is usually associated with late responding normal tissue and is the basis for the therapeutic gain achieved using hyperfractionation (increased number of fractions of small dose per fraction), which allows for greater repair/recovery of normal tissues (139). However, some human tumors, e.g., prostate cancer, melanoma, and some sarcomas, may have α/β values similar to late responding tissues (140–142).
In image-guided RT, intensity-modulated RT, and X-ray stereotactic body RT (SBRT), there are tendencies to reduce the number of fractions and increase the dose per fraction (i.e., hypofractionation) (143, 144). With carbon-ion RT, superiority of the physical dose distribution can lead to a reduction in the number of fractions (145), allowing hypofractionation. There are few relevant experimental data using human tumor cells on hypofractionation effects with high-LET charged particles. Experiments involving high-LET fast neutron beams demonstrated that increasing the dose per fraction tended to decrease the RBE for both tumor and normal tissues (146). However, the dose-dependent decrease in the RBE for the tumor was less pronounced than that for normal tissues, such as skin and lung (147). These experiments led to the assumption that the therapeutic gain of carbon-ion RT would increase when the dose per fraction increased. This assumption was confirmed in animal experiments that compared RBE for carbon ions between tumor and skin (148). In additional studies with high-LET radiation, RBE depends on dose and dose per fraction: dose-dependent decrease of RBE was reported after fast neutrons to normal skin, intestine, growing cartilage, and hematopoietic tissues (149), and after Ne-ions to the skin of mice and hamsters (150). The change in dose dependence is caused by the higher α/β ratio of target cells after high-LET radiation than after photons (151).
The value of the α term increases with increasing LET in both tumor and normal tissue, while the issue of whether the value of the β term changes with LET remains controversial (148, 152). In our study (153) by evaluating the therapeutic gain of carbon-ion fractionation using intestinal crypt survival and tumor growth delay (TGD) assays, the values of the α and β terms for the mouse fibrosarcoma (NFSa) tumor are close to those reported by Ando et al. (148), while those for normal tissues are different (Figure 9). In addition, the LET-dependent increase (e.g., slope of the regression line) of the α term for NFSa is similar to that for human salivary gland tumor cells (148, 154). LET-dependent increase of α terms for crypt is greater than that for the early skin reaction after daily fractionated doses to leg skin (148), whereas it is similar to that for the late skin reaction after 4-h interval fractionations to foot skin (154). These results indicate that therapeutic gain for carbon-ion RT depends on the normal tissue and fractionation schedule. Further studies with mouse skin and rat spinal cord where the normal tissues were exposed to varying numbers of fractions and doses per fraction of γ-rays and carbon ions have shown that the magnitude of damage repair depends on both the number of fractions and the size of dose per fraction for high-LET radiation (155, 156). It was concluded that repair of radiation injury is much reduced with dose per fraction, especially with 125 keV/μm carbon ions. Unfortunately, few studies of fractionation effects with carbon ions have been performed with tumors, especially human tumors.
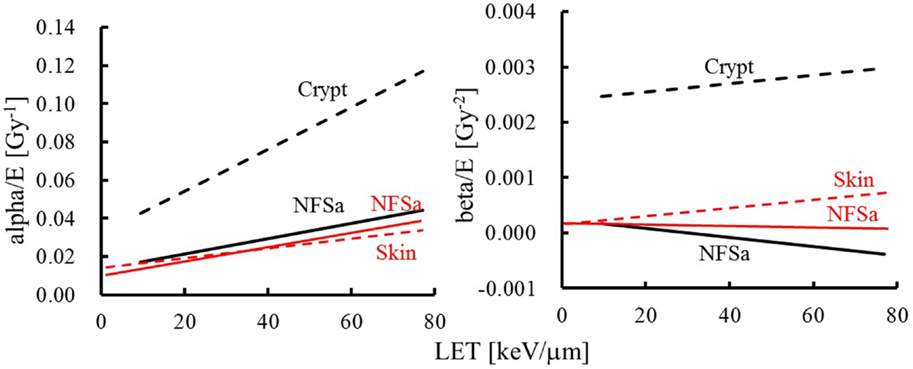
Figure 9. α and β terms for mouse normal tissues and tumors plotted against LET. The comparison of survival parameters of Ando et al. (red) (148) and our study (153) using crypt survival and tumor growth delay assays (black). The solid lines show NFSa tumor and broken lines show the normal tissues (crypt or skin).
As discussed above, hypoxia is one of the main factors reducing local control in some solid tumors, and fractionation in RT may have an advantage because of reoxygenation of the hypoxic areas. It has been reported that reoxygenation in several tumors irradiated with carbon ions occurs earlier than that in those irradiated with photons (157, 158). Reoxygenation in the NFSa fibrosarcoma was observed at 4 days, 1 day, and within 0.5 days after irradiation with photons, low-LET carbon ions (14 and 18 keV/μm) and high-LET carbon ions (43, 58, and 74 keV/μm), respectively (157). Thus, short-term fractionated irradiation with carbon ions may be effective in the treatment of tumors, at least in part, because of altered reoxygenation.
The clinical RBE is replaced by an LET-dependent RBE for in vitro cell killing data determined in single-dose experiments and is employed to design the SOBP and in the Japanese treatment planning system for carbon-ion RT (159, 160). A question remains as to whether the biological effects with fractionated doses are also uniform within the SOBP. Uzawa et al. evaluated uniformity of a new ridge filter that was designed based on α and β values for various LETs to cause mouse foot skin reaction by carbon-ion fractionated irradiation (154). The physical dose distribution of the new ridge filter was almost identical to the ridge filter designed based on in vitro cell kill. While the LQ model is useful for conversion between relatively low radiation doses as used in conventional RT, it has been suggested that it is not applicable to higher fractional doses or smaller fraction numbers (6, 161). It has been questioned whether the LQ model is applicable to hypofractionated carbon-ion RT. For establishment of the optimal fractionation strategy in carbon-ion RT, applicability of the LQ model should be investigated in future studies.
With photon RT, the rapidly expanding use of hypofractionation even to the extreme of single fractions as used in stereotactic radiosurgery (SRS) and SBRT has lead to recent discussion about whether “new” biology should be advanced to explain the greater than expected anti-tumor efficacy of some hypofractionation regimens. Some have proposed that consideration of only the clonogenic survival of only the tumor cells is not sufficient to account for the observed responses [e.g., Ref. (162, 163)], although not all agree [e.g., Ref. (164–166)]. Brown et al. reviewed the clinical data for early-stage NSCLC and suggested that radiobiological modeling with the LQ model is adequate to explain the efficacy of SRS and SBRT (166). Fowler showed the potential advantages of hypofractionation for prostate cancer by using the LQ model and concluded that use of the LQ model can yield consistent results, for example, the remarkable agreement for tumor effects of some of the best schedules in regular use (167). It is likely that the same considerations apply to carbon-ion therapy, although few data exist, especially for human tumors in experimental situations. Here, we briefly review some aspects that may pertain.
In some situations, vascular damage may be a dominant pathway for tumor suppression. Irradiation of human tumor xenografts or rodent tumors with 5–10 Gy in a single dose causes relatively mild vascular damages. On the other hand, numerous studies with experimental tumors indicate that irradiation with doses higher than 10 Gy in a single fraction or 20–60 Gy in limited numbers of fractions causes severe vascular damage, including endothelial cell apoptosis, leading to the deterioration of the intratumor microenvironment and indirect death of tumor cells (163, 168, 169). Little is known about the vascular changes in human tumors treated with high-dose hypofractionation, particularly with heavy ions, but experimentation is indicated to address whether radiation-induced vascular damage and the resulting indirect death of tumor cells may play important roles in the response of tumors to high-dose hypofractionation with charged particles.
In addition to potential vascular effects, it has been suggested that high-dose irradiation evokes immune reactions and thereby eradicates tumor cells that escaped radiation-induced death (170, 171). In support of such notion, a recent report showed that ablative RT dramatically increased T-cell priming in lymphoid tissues, leading to reduction/eradication of the primary tumor or distant metastasis in a CD8+ T-cell dependent fashion (170). Several studies have shown that carbon ions induce anti-tumor immunity (172–176), although the effects of high-LET radiation on immune function have not been studied in detail. Hence, enhanced immune reactions might be involved in the response of tumors to high-dose hypofractionation, especially with charged particles (177).
It is also noteworthy that, unlike photon irradiation, particle irradiation may suppress the metastatic potential of cancer cells (172, 178), and a recent paper has shown that there is a decrease in metastasis with decreasing fraction number of carbon ions (179). Clearly, further studies are warranted to gain better insights into the effects of high-dose hypofractionation with heavy ions on tumor vasculature, immune system, and metastasis, and how such biology might impact human tumor RBE values and therapeutic gain.
Conclusion
In a recent review of charged particle therapy, Loeffler and Durante (4) stated that “Considering the current uncertainties in clinical results [with charged particles] and the difficulties in performing clinical trials, research in physics and radiobiology should reduce the cost/benefit ratio.” In this review, we have focused on discussion of selected aspects of radiobiological data with human tumors exposed to protons and heavier charged particles, raising specific instances where further laboratory research may contribute to improving particle therapy. With increasing understanding of the genetic heterogeneity in human tumors, particularly with regard to alterations in DNA repair pathways, a fruitful research area appears to be elucidation of DNA repair pathways selectively involved in repair of the unique clustered DNA damages caused by charged particles. With increases in such knowledge, the differences can be exploited to identify patients who may be better treated with particles because of characteristics of their tumors and to develop novel pharmacologic approaches that capitalize on the differences in DNA damage and repair. Another area ripe for charged particle biology study with implications to clinical advances is in cancer stem cells. The intriguing observations that cancer stem cells from human tumors may be more effectively killed by carbon ions than by photons begs for further study on mechanisms involved – altered DNA repair? location in a hypoxic niche? – and consideration of how to exploit such a difference to the advantage of ion therapy. Finally, the biology underlying the notable clinical effectiveness of high dose, hypofractionated charged particles, which may be explained by radiosensitivity of tumor cells themselves at high doses or may involve vasculature and/or immune system responses, requires further elucidation.
This article has focused on data from in vitro studies of human tumor cells, for reasons described in the Section “Introduction” and recognizing that there are limitations when applying in vitro findings to the in vivo and clinical situations. However, it is also clear that because of the stochastic natures of radiation-induced cell killing and tumor cure and the, albeit simplistic, relationship of the two endpoints via TCP = e−(SF × M) (where TCP is tumor control probability, SF is surviving fraction, and M is number of clonogens), understanding effects of radiations of varying qualities on the tumor cells themselves can be informative. The questions and issues raised herein require follow-up in vivo studies leading to transfer of knowledge to the clinic, but guidance from the in vitro work, e.g., on use of DNA damage assays and exploiting DNA repair as biomarkers for patient selection or using in vitro survival α/β information to help guide design of hypofractionation protocols in vivo and in the clinic, is critical.
Author Contributions
KH, HK, TK, AP, YY, QL, HW, and AT contributed to the conception, drafting, and revising of the review article.
Conflict of Interest Statement
The authors declare that preparation of this article was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health under Award Number R21CA182259 (KH), the Gunma University Initiative for Advanced Research (KH), Federal Share of program income earned by Massachusetts General Hospital on C06 CA059267, Proton Therapy Research and Treatment Center (HW and KH), Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) Number 15H05945 (AT), Gunma University Program for Cultivating Global Leaders in Heavy Ion Therapeutics and Engineering of the MEXT (AP), Research Project with heavy ions at the Gunma University Heavy Ion Medical Center (HK, TK, AP, YY, and AT). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
2. Skarsgard LD. Radiobiology with heavy charged particles: a historical review. Phys Med (1998) 14(Suppl. 1):1–19.
3. Suit H, DeLaney T, Goldberg S, Paganetti H, Clasie B, Gerweck L, et al. Proton vs carbon ion beams in the definitive radiation treatment of cancer patients. Radiother Oncol (2010) 95:3–22. doi:10.1016/j.radonc.2010.01.015
4. Loeffler JS, Durante M. Charged particle therapy – optimization, challenges and future directions. Nat Rev Clin Oncol (2013) 10:411–24. doi:10.1038/nrclinonc.2013.79
5. Durante M, Loeffler JS. Charged particles in radiation oncology. Nat Rev Clin Oncol (2010) 7:37–43. doi:10.1038/nrclinonc.2009.183
6. Durante M. New challenges in high-energy particle radiobiology. Br J Radiol (2014) 87(1035):20130626. doi:10.1259/bjr.20130626
7. Friedrich T, Scholz U, Elsasser T, Durante M, Scholz M. Systematic analysis of RBE and related quantities using a database of cell survival experiments with ion beam irradiation. J Radiat Res (2013) 54(4):494–514. doi:10.1093/jrr/rrs114
8. Paganetti H, Niemierko A, Ancukiewicz M, Gerweck LE, Goitein M, Loeffler JS, et al. Relative biological effectiveness (RBE) values for proton beam therapy. Int J Radiat Oncol Biol Phys (2002) 53:407–21. doi:10.1016/S0360-3016(02)02754-2
9. Friedrich T, Grun R, Scholz U, Elsasser T, Durante M, Scholz M. Sensitivity analysis of the relative biological effectiveness predicted by the local effect model. Phys Med Biol (2013) 58(19):6827–49. doi:10.1088/0031-9155/58/19/6827
10. Barendsen GW. Responses of cultured cells, tumors, and normal tissues to radiation of different linear energy transfer. Curr Top Radiat Res Q (1968) 4:293–356.
11. Blakely EA, Ngo FQH, Curtis SB, Tobias CA. Heavy-ion radiobiology: cellular studies. Adv Radiat Biol (1984) 11:295–389. doi:10.1016/B978-0-12-035411-5.50013-7
12. Sorensen BS, Overgaard J, Bassler N. In vitro RBE-LET dependence for multiple particle types. Acta Oncol (2011) 50:757–62. doi:10.3109/0284186X.2011.582518
13. Kraft G, Kramer M, Scholz M. LET, track structure and models. A review. Radiat Environ Biophys (1992) 31:161–80. doi:10.1007/BF01214825
14. Goodhead DT. Initial events in the cellular effects of ionizing radiations: clustered damage in DNA. Int J Radiat Biol (1994) 65:7–17. doi:10.1080/09553009414550021
15. Ward JF. The complexity of DNA damage: relevance to biological consequences. Int J Radiat Biol (1994) 66:427–32. doi:10.1080/09553009414551401
16. Hada M, Sutherland BM. Spectrum of complex DNA damages depends on the incident radiation. Radiat Res (2006) 165:223–30. doi:10.1667/RR3498.1
17. Goodhead DT. Mechanisms for the biological effectiveness of high-LET radiations. J Radiat Res (1999) 40(Suppl):1–13. doi:10.1269/jrr.40.S1
18. Goodhead DT. Fifth Warren K. Sinclair keynote address: issues in quantifying the effects of low-level radiation. Health Phys (2009) 97(5):394–406. doi:10.1097/HP.0b013e3181ae8acf
19. Nikjoo H, O’Neill P, Wilson WE, Goodhead DT. Computational approach for determining the spectrum of DNA damage induced by ionizing radiation. Radiat Res (2001) 156:577–83. doi:10.1667/0033-7587(2001)156[0577:CAFDTS]2.0.CO;2
20. Georgakilas AG, O’Neill P, Stewart RD. Induction and repair of clustered DNA lesions: what do we know so far? Radiat Res (2013) 180(1):100–9. doi:10.1667/RR3041.1
21. Sridharan DM, Asaithamby A, Bailey SM, Costes SV, Doetsch PW, Dynan WS, et al. Understanding cancer development processes after HZE-particle exposure: roles of ROS, DNA damage repair and inflammation. Radiat Res (2015) 183(1):1–26. doi:10.1667/RR13804.1
22. Hall EJ, Giaccia A. Radiobiology for the Radiologist. Philadelphia, PA: Lippincott Williams & Wilkins Publishers (2012).
23. Morgan WF, Sowa MB. Non-targeted effects induced by ionizing radiation: mechanisms and potential impact on radiation induced health effects. Cancer Lett (2015) 356(1):17–21. doi:10.1016/j.canlet.2013.09.009
24. Kadhim MA, Hill MA. Non-targeted effects of radiation exposure: recent advances and implications. Radiat Prot Dosimetry (2015) 166(1–4):118–24. doi:10.1093/rpd/ncv167
25. Sowa MB, Goetz W, Baulch JE, Pyles DN, Dziegielewski J, Yovino S, et al. Lack of evidence for low-LET radiation induced bystander response in normal human fibroblasts and colon carcinoma cells. Int J Radiat Biol (2010) 86(2):102–13. doi:10.3109/09553000903419957
26. Yang H, Asaad N, Held KD. Medium-mediated intercellular communication is involved in bystander responses of X-irradiated normal human fibroblasts. Oncogene (2005) 24:2096–103. doi:10.1038/sj.onc.1208439
27. de Toledo SM, Buonanno M, Li M, Asaad N, Qin Y, Gonon G, et al. The impact of adaptive and non-targeted effects in the biological responses to low dose/low fluence ionizing radiation: the modulating effect of linear energy transfer. Health Phys (2011) 100(3):290–2. doi:10.1097/HP.0b013e31820832d8
28. McMahon SJ, McGarry CK, Butterworth KT, Jain S, O’Sullivan JM, Hounsell AR, et al. Cellular signalling effects in high precision radiotherapy. Phys Med Biol (2015) 60(11):4551–64. doi:10.1088/0031-9155/60/11/4551
29. Asur R, Butterworth KT, Penagaricano JA, Prise KM, Griffin RJ. High dose bystander effects in spatially fractionated radiation therapy. Cancer Lett (2015) 356(1):52–7. doi:10.1016/j.canlet.2013.10.032
30. Willers H, Dahm-Daphi J, Powell SN. Repair of radiation damage to DNA. Br J Cancer (2004) 90:1297–301. doi:10.1038/sj.bjc.6601729
31. Hiom K. Coping with DNA double strand breaks. DNA Repair (Amst) (2010) 9(12):1256–63. doi:10.1016/j.dnarep.2010.09.018
32. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem (2010) 79:181–211. doi:10.1146/annurev.biochem.052308.093131
33. Jeggo PA, Geuting V, Lobrich M. The role of homologous recombination in radiation-induced double-strand break repair. Radiother Oncol (2011) 101(1):7–12. doi:10.1016/j.radonc.2011.06.019
34. Willers H, Pfaffle HN, Zou L. Targeting homologous recombination repair in cancer. In: Kelley MR, editor. DNA Repair in Cancer Therapy: Molecular Targets and Clinical Applications. Waltham, MA: Academic Press (2012). p. 119–60.
35. Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, et al. XPF-ERCC1 acts in unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell (2014) 54(3):460–71. doi:10.1016/j.molcel.2014.03.015
36. Frankenberg-Schwager M, Gebauer A, Koppe C, Wolf H, Pralle E, Frankenberg D. Single-strand annealing, conservative homologous recombination, nonhomologous DNA end joining, and the cell cycle-dependent repair of DNA double-strand breaks induced by sparsely or densely ionizing radiation. Radiat Res (2009) 171(3):265–73. doi:10.1667/RR0784.1
37. Zafar F, Seidler SB, Kronenberg A, Schild D, Wiese C. Homologous recombination contributes to the repair of DNA double-strand breaks induced by high-energy iron ions. Radiat Res (2010) 173(1):27–39. doi:10.1667/RR1910.1
38. Yajima H, Fujisawa H, Nakajima NI, Hirakawa H, Jeggo PA, Okayasu R, et al. The complexity of DNA double strand breaks is a critical factor enhancing end-resection. DNA Repair (Amst) (2013) 12(11):936–46. doi:10.1016/j.dnarep.2013.08.009
39. Takahashi A, Kubo M, Ma H, Nakagawa A, Yoshida Y, Isono M, et al. Nonhomologous end-joining repair plays a more important role than homologous recombination repair in defining radiosensitivity after exposure to high-LET radiation. Radiat Res (2014) 182(3):338–44. doi:10.1667/RR13782.1
40. Steinstrater O, Scholz U, Friedrich T, Kramer M, Grun R, Durante M, et al. Integration of a model-independent interface for RBE predictions in a treatment planning system for active particle beam scanning. Phys Med Biol (2015) 60(17):6811–31. doi:10.1088/0031-9155/60/17/6811
41. Inaniwa T, Kanematsu N, Matsufuji N, Kanai T, Shirai T, Noda K, et al. Reformulation of a clinical-dose system for carbon-ion radiotherapy treatment planning at the National Institute of Radiological Sciences, Japan. Phys Med Biol (2015) 60(8):3271–86. doi:10.1088/0031-9155/60/8/3271
42. Paganetti H. Relative biological effectiveness (RBE) values for proton beam therapy. Variations as a function of biological endpoint, dose, and linear energy transfer. Phys Med Biol (2014) 59(22):R419–72. doi:10.1088/0031-9155/59/22/R419
43. Grosse N, Fontana A, Hug EB, Lomax A, Coray A, Augsburger M, et al. Deficiency in homologous recombination renders mammalian cells more sensitive to proton versus photon irradiation. Int J Radiat Oncol Biol Phys (2014) 88(1):175–81. doi:10.1016/j.ijrobp.2013.09.041
44. Liu Q, Ghosh P, Magpayo N, Testa M, Tang S, Gheorghiu L, et al. Lung cancer cell line screen links Fanconi anemia/BRCA pathway defects to increased relative biological effectiveness of proton radiation. Int J Radiat Oncol Biol Phys (2015) 91(5):1081–9. doi:10.1016/j.ijrobp.2014.12.046
45. Fontana AO, Augsburger MA, Grosse N, Guckenberger M, Lomax AJ, Sartori AA, et al. Differential DNA repair pathway choice in cancer cells after proton- and photon-irradiation. Radiother Oncol (2015) 116(3):374–80. doi:10.1016/j.radonc.2015.08.014
46. Guan F, Bronk L, Titt U, Lin SH, Mirkovic D, Kerr MD, et al. Spatial mapping of the biologic effectiveness of scanned particle beams: towards biologically optimized particle therapy. Sci Rep (2015) 5:9850. doi:10.1038/srep09850
47. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature (2012) 483(7391):570–5. doi:10.1038/nature11005
48. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell (2012) 150(6):1107–20. doi:10.1016/j.cell.2012.08.029
49. Network CGAR. Comprehensive genomic characterization of squamous cell lung cancers. Nature (2012) 489(7417):519–25. doi:10.1038/nature11404
50. Wang AT, Smogorzewska A. SnapShot: Fanconi anemia and associated proteins. Cell (2015) 160(1–2):354–e1. doi:10.1016/j.cell.2014.12.031
51. Rostek C, Turner EL, Robbins M, Rightnar S, Xiao W, Obenaus A, et al. Involvement of homologous recombination repair after proton-induced DNA damage. Mutagenesis (2008) 23(2):119–29. doi:10.1093/mutage/gem055
52. Gulston M, Fulford J, Jenner T, de Lara C, O’Neill P. Clustered DNA damage induced by gamma radiation in human fibroblasts (HF19), hamster (V79-4) cells and plasmid DNA is revealed as Fpg and Nth sensitive sites. Nucleic Acids Res (2002) 30(15):3464–72. doi:10.1093/nar/gkf467
53. Sutherland BM, Bennett PV, Sutherland JC, Laval J. Clustered DNA damages induced by X-rays in human cells. Radiat Res (2002) 157:611–6. doi:10.1667/0033-7587(2002)157[0611:CDDIBX]2.0.CO;2
54. Ando K. Review of RBE data on ion beams from Chiba: influence of LET and biological system. Dose Reporting in Ion Beam Therapy. Vienna: International Atomic Energy Agency (2007). p. 89–98.
55. Ando K, Kase Y. Biological characteristics of carbon-ion therapy. Int J Radiat Biol (2009) 85(9):715–28. doi:10.1080/09553000903072470
56. Ando S, Nojima K, Ishihara H, Suzuki M, Ando M, Majima H, et al. Induction by carbon-ion irradiation of the expression of vascular endothelial growth factor in lung carcinoma cells. Int J Radiat Biol (2000) 76(8):1121–7. doi:10.1080/09553000050111596
57. Belli M, Bettega D, Calzolari P, Cherubini R, Cuttone G, Durante M, et al. Effectiveness of monoenergetic and spread-out Bragg peak carbon-ions for inactivation of various normal and tumour human cell lines. J Radiat Res (2008) 49(6):597–607. doi:10.1269/jrr.08052
58. Combs SE, Bohl J, Elsasser T, Weber KJ, Schulz-Ertner D, Debus J, et al. Radiobiological evaluation and correlation with the local effect model (LEM) of carbon ion radiation therapy and temozolomide in glioblastoma cell lines. Int J Radiat Biol (2009) 85(2):126–37. doi:10.1080/09553000802641151
59. Furusawa Y, Fukutsu K, Aoki M, Itsukaichi H, Eguchi-Kasai K, Ohara H, et al. Inactivation of aerobic and hypoxic cells from three different cell lines by accelerated (3)He-, (12)C- and (20)Ne-ion beams. Radiat Res (2000) 154(5):485–96. doi:10.1667/0033-7587(2000)154[0485:IOAAHC]2.0.CO;2
60. Ito A, Nakano H, Kusano Y, Hirayama R, Furusawa Y, Murayama C, et al. Contribution of indirect action to radiation-induced mammalian cell inactivation: dependence on photon energy and heavy-ion LET. Radiat Res (2006) 165(6):703–12. doi:10.1667/RR3557.1
61. Iwadate Y, Mizoe J, Osaka Y, Yamaura A, Tsujii H. High linear energy transfer carbon radiation effectively kills cultured glioma cells with either mutant or wild-type p53. Int J Radiat Oncol Biol Phys (2001) 50(3):803–8. doi:10.1016/S0360-3016(01)01514-0
62. Kagawa K, Murakami M, Hishikawa Y, Abe M, Akagi T, Yanou T, et al. Preclinical biological assessment of proton and carbon ion beams at Hyogo Ion Beam Medical Center. Int J Radiat Oncol Biol Phys (2002) 54(3):928–38. doi:10.1016/S0360-3016(02)02949-8
63. Kamlah F, Hanze J, Arenz A, Seay U, Hasan D, Juricko J, et al. Comparison of the effects of carbon ion and photon irradiation on the angiogenic response in human lung adenocarcinoma cells. Int J Radiat Oncol Biol Phys (2011) 80(5):1541–9. doi:10.1016/j.ijrobp.2011.03.033
64. Matsui Y, Asano T, Kenmochi T, Iwakawa M, Imai T, Ochiai T. Effects of carbon-ion beams on human pancreatic cancer cell lines that differ in genetic status. Am J Clin Oncol (2004) 27(1):24–8. doi:10.1097/01.coc.0000046037.75545.AD
65. Matsumoto Y, Iwakawa M, Furusawa Y, Ishikawa K, Aoki M, Imadome K, et al. Gene expression analysis in human malignant melanoma cell lines exposed to carbon ions. Int J Radiat Biol (2008) 84:299–314. doi:10.1080/09553000801953334
66. Matsuzaki H, Miyamoto T, Miyazawa Y, Okazumi S, Koide Y, Isono K. Biological effects of heavy ion beam on human breast cancers. Breast Cancer (1998) 5(3):261–8. doi:10.1007/BF02966706
67. Ofuchi T, Suzuki M, Kase Y, Ando K, Isono K, Ochiai T. Chromosome breakage and cell lethality in human hepatoma cells irradiated with X rays and carbon-ion beams. J Radiat Res (1999) 40(2):125–33. doi:10.1269/jrr.40.125
68. Persson LM, Edgren MR, Stenerlow B, Lind BK, Hedlof I, Jernberg AR, et al. Relative biological effectiveness of boron ions on human melanoma cells. Int J Radiat Biol (2002) 78(8):743–8. doi:10.1080/09553000210140091
69. Stenerlow B, Pettersson OA, Essand M, Blomquist E, Carlsson J. Irregular variations in radiation sensitivity when the linear energy transfer is increased. Radiother Oncol (1995) 36(2):133–42. doi:10.1016/0167-8140(95)01591-4
70. Suzuki M, Kase Y, Yamaguchi H, Kanai T, Ando K. Relative biological effectiveness for cell-killing effect on various human cell lines irradiated with heavy-ion medical accelerator in Chiba (HIMAC) carbon-ion beams. Int J Radiat Oncol Biol Phys (2000) 48(1):241–50. doi:10.1016/S0360-3016(00)00568-X
71. Suzuki M, Kase Y, Nakano T, Kanai T, Ando K. Residual chromatin breaks as biodosimetry for cell killing by carbon ions. Adv Space Res (1998) 22(12):1663–71. doi:10.1016/S0273-1177(99)00031-9
72. Suzuki M, Kase Y, Kanai T, Ando K. Correlation between cell killing and residual chromatin breaks measured by PCC in six human cell lines irradiated with different radiation types. Int J Radiat Biol (2000) 76(9):1189–96. doi:10.1080/09553000050134429
73. Suzuki M, Kase Y, Kanai T, Ando K. Change in radiosensitivity with fractionated-dose irradiation of carbon-ion beams in five different human cell lines. Int J Radiat Oncol Biol Phys (2000) 48(1):251–8. doi:10.1016/S0360-3016(00)00606-4
74. Takahashi A, Ohnishi K, Tsuji K, Matsumoto H, Aoki H, Wang X, et al. WAF1 accumulation by carbon-ion beam and alpha-particle irradiation in human glioblastoma cultured cells. Int J Radiat Biol (2000) 76(3):335–41. doi:10.1080/095530000138673
75. Takahashi A, Ohnishi K, Ota I, Asakawa I, Tamamoto T, Furusawa Y, et al. p53-dependent thermal enhancement of cellular sensitivity in human squamous cell carcinomas in relation to LET. Int J Radiat Biol (2001) 77(10):1043–51. doi:10.1080/09553000110066095
76. Takahashi A, Matsumoto H, Yuki K, Yasumoto J, Kajiwara A, Aoki M, et al. High-LET radiation enhanced apoptosis but not necrosis regardless of p53 status. Int J Radiat Oncol Biol Phys (2004) 60(2):591–7. doi:10.1016/j.ijrobp.2004.05.062
77. Takahashi A, Matsumoto H, Furusawa Y, Ohnishi K, Ishioka N, Ohnishi T. Apoptosis induced by high-LET radiations is not affected by cellular p53 gene status. Int J Radiat Biol (2005) 81:581–6. doi:10.1080/09553000500280484
78. Tsuboi K, Tsuchida Y, Nose T, Ando K. Cytotoxic effect of accelerated carbon beams on glioblastoma cell lines with p53 mutation: clonogenic survival and cell-cycle analysis. Int J Radiat Biol (1998) 74(1):71–9. doi:10.1080/095530098141744
79. Tsuboi K, Moritake T, Tsuchida Y, Tokuuye K, Matsumura A, Ando K. Cell cycle checkpoint and apoptosis induction in glioblastoma cells and fibroblasts irradiated with carbon beam. J Radiat Res (2007) 48(4):317–25. doi:10.1269/jrr.06081
80. Tsuchida Y, Tsuboi K, Ohyama H, Ohno T, Nose T, Ando K. Cell death induced by high-linear-energy transfer carbon beams in human glioblastoma cell lines. Brain Tumor Pathol (1998) 15(2):71–6. doi:10.1007/BF02478886
81. Yamakawa N, Takahashi A, Mori E, Imai Y, Furusawa Y, Ohnishi K, et al. High LET radiation enhances apoptosis in mutated p53 cancer cells through caspase-9 activation. Cancer Sci (2008) 99(7):1455–60. doi:10.1111/j.1349-7006.2008.00818.x
82. Ferrandon S, Magne N, Battiston-Montagne P, Hau-Desbat NH, Diaz O, Beuve M, et al. Cellular and molecular portrait of eleven human glioblastoma cell lines under photon and carbon ion irradiation. Cancer Lett (2015) 360(1):10–6. doi:10.1016/j.canlet.2015.01.025
83. Schlaich F, Brons S, Haberer T, Debus J, Combs SE, Weber KJ. Comparison of the effects of photon versus carbon ion irradiation when combined with chemotherapy in vitro. Radiat Oncol (2013) 8:260. doi:10.1186/1748-717X-8-260
84. Beuve M, Alphonse G, Maalouf M, Colliaux A, Battiston-Montagne P, Jalade P, et al. Radiobiologic parameters and local effect model predictions for head-and-neck squamous cell carcinomas exposed to high linear energy transfer ions. Int J Radiat Oncol Biol Phys (2008) 71(2):635–42. doi:10.1016/j.ijrobp.2007.10.050
85. Kato TA, Tsuda A, Uesaka M, Fujimori A, Kamada T, Tsujii H, et al. In vitro characterization of cells derived from chordoma cell line U-CH1 following treatment with X-rays, heavy ions and chemotherapeutic drugs. Radiat Oncol (2011) 6:116. doi:10.1186/1748-717X-6-116
86. Takiguchi Y, Miyamoto T, Nagao K, Kuriyama T. Assessment of the homogeneous efficacy of carbon ions in the spread-out Bragg peak for human lung cancer cell lines. Radiat Med (2007) 25(6):272–7. doi:10.1007/s11604-007-0134-6
87. Habermehl D, Ilicic K, Dehne S, Rieken S, Orschiedt L, Brons S, et al. The relative biological effectiveness for carbon and oxygen ion beams using the raster-scanning technique in hepatocellular carcinoma cell lines. PLoS One (2014) 9(12):e113591. doi:10.1371/journal.pone.0113591
88. Fujisawa H, Genik PC, Kitamura H, Fujimori A, Uesaka M, Kato TA. Comparison of human chordoma cell-kill for 290 MeV/n carbon ions versus 70 MeV protons in vitro. Radiat Oncol (2013) 8:91. doi:10.1186/1748-717X-8-91
89. Oonishi K, Cui X, Hirakawa H, Fujimori A, Kamijo T, Yamada S, et al. Different effects of carbon ion beams and X-rays on clonogenic survival and DNA repair in human pancreatic cancer stem-like cells. Radiother Oncol (2012) 105(2):258–65. doi:10.1016/j.radonc.2012.08.009
90. El Shafie RA, Habermehl D, Rieken S, Mairani A, Orschiedt L, Brons S, et al. In vitro evaluation of photon and raster-scanned carbon ion radiotherapy in combination with gemcitabine in pancreatic cancer cell lines. J Radiat Res (2013) 54(Suppl 1):i113–9. doi:10.1093/jrr/rrt052
91. Combs SE, Zipp L, Rieken S, Habermehl D, Brons S, Winter M, et al. In vitro evaluation of photon and carbon ion radiotherapy in combination with chemotherapy in glioblastoma cells. Radiat Oncol (2012) 7:9. doi:10.1186/1748-717X-7-9
92. Ando K, Kase Y, Matsufuji N. Light ion radiation biology. In: Brahme A, editor. Comprehensive Biomedical Physics. Amsterdam: Elsevier (2014). p. 195–210.
93. Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer (2005) 5(3):231–7. doi:10.1038/nrc1560
94. Held KD. Radiation-induced apoptosis and its relationship to loss of clonogenic survival. Apoptosis (1997) 2:265–82. doi:10.1023/A:1026485003280
95. Tommasino F, Durante M. Proton radiobiology. Cancers (2015) 7(1):353–81. doi:10.3390/cancers7010353
96. Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res (1999) 59:1391–9.
97. Grun R, Friedrich T, Kramer M, Zink K, Durante M, Engenhart-Cabillic R, et al. Assessment of potential advantages of relevant ions for particle therapy: a model based study. Med Phys (2015) 42(2):1037–47. doi:10.1118/1.4905374
98. Wulf H, Kraft-Weyrather W, Miltenburger HG, Blakely EA, Tobias CA, Kraft G. Heavy-ion effects on mammalian cells: inactivation measurements with different cell lines. Radiat Res Suppl (1985) 8:S122–34. doi:10.2307/3576639
99. Belli M, Cera F, Cherubini R, Haque AMI, Ianzini F, Moschini G, et al. The RBE of protons for cell inactivation: the experience with V79 cells. In: Amaldi U, Larsson B, editors. Hadrontherapy in Oncology. Amsterdam: Elsevier Science (1994). p. 702–5.
100. Cucinotta FA, Kim MH, Chappell LJ. Space Radiation Cancer Risk Projections and Uncertainties – 2012. NASA/TP-2013-217375 (2013). Available from: http://ston.jsc.nasa.gov/collections/TRS
101. Friedrich T, Scholz U, Elsasser T, Durante M, Scholz M. Calculation of the biological effects of ion beams based on the microscopic spatial damage distribution pattern. Int J Radiat Biol (2012) 88(1–2):103–7. doi:10.3109/09553002.2011.611213
102. Bassler N, Toftegaard J, Luhr A, Sorensen BS, Scifoni E, Kramer M, et al. LET-painting increases tumour control probability in hypoxic tumours. Acta Oncol (2014) 53(1):25–32. doi:10.3109/0284186X.2013.832835
103. Kamada T, Tsujii H, Blakely EA, Debus J, De Neve W, Durante M, et al. Carbon ion radiotherapy in Japan: an assessment of 20 years of clinical experience. Lancet Oncol (2015) 16(2):e93–100. doi:10.1016/S1470-2045(14)70412-7
104. Steel GG, McMillan TJ, Peacock JH. The radiobiology of human cells and tissues. In vitro radiosensitivity. The picture has changed in the 1980s. Int J Radiat Biol (1989) 56(5):525–37. doi:10.1080/09553008914551691
105. Steel GG, Peacock JH. Why are some human tumours more radiosensitive than others? Radiother Oncol (1989) 15(1):63–72. doi:10.1016/0167-8140(89)90119-9
106. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature (2006) 444:756–60. doi:10.1038/nature05236
107. Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst (2006) 98:1777–85. doi:10.1093/jnci/djj495
108. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature (2009) 458(7239):780–3. doi:10.1038/nature07733
109. Cui X, Oonishi K, Tsujii H, Yasuda T, Matsumoto Y, Furusawa Y, et al. Effects of carbon ion beam on putative colon cancer stem cells and its comparison with X-rays. Cancer Res (2011) 71(10):3676–87. doi:10.1158/0008-5472.CAN-10-2926
110. Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer (2003) 3:895–902. doi:10.1038/nrc1232
111. Khan IN, Al-Karim S, Bora RS, Chaudhary AG, Saini KS. Cancer stem cells: a challenging paradigm for designing targeted drug therapies. Drug Discov Today (2015) 20(10):1205–16. doi:10.1016/j.drudis.2015.06.013
112. Pajonk F, Vlashi E, McBride WH. Radiation resistance of cancer stem cells: the 4 R’s of radiobiology revisited. Stem Cells (2010) 28:639–48. doi:10.1002/stem.318
113. Vlashi E, Kim K, Lagadec C, Donna LD, McDonald JT, Eghbali M, et al. In vivo imaging, tracking, and targeting of cancer stem cells. J Natl Cancer Inst (2009) 101(5):350–9. doi:10.1093/jnci/djn509
114. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell (2007) 11(1):69–82. doi:10.1016/j.ccr.2006.11.020
115. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell (2011) 19(3):387–400. doi:10.1016/j.ccr.2011.01.038
116. Yang G, Quan Y, Wang W, Fu Q, Wu J, Mei T, et al. Dynamic equilibrium between cancer stem cells and non-stem cancer cells in human SW620 and MCF-7 cancer cell populations. Br J Cancer (2012) 106(9):1512–9. doi:10.1038/bjc.2012.126
117. Piao LS, Hur W, Kim TK, Hong SW, Kim SW, Choi JE, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett (2012) 315(2):129–37. doi:10.1016/j.canlet.2011.10.012
118. Shimura T, Noma N, Oikawa T, Ochiai Y, Kakuda S, Kuwahara Y, et al. Activation of the AKT/cyclin D1/Cdk4 survival signaling pathway in radioresistant cancer stem cells. Oncogenesis (2012) 1:e12. doi:10.1038/oncsis.2012.12
119. Xia P, Xu XY. PI3K/Akt/mTOR signaling pathway in cancer stem cells: from basic research to clinical application. Am J Cancer Res (2015) 5(5):1602–9.
120. Sai S, Wakai T, Vares G, Yamada S, Kamijo T, Kamada T, et al. Combination of carbon ion beam and gemcitabine causes irreparable DNA damage and death of radioresistant pancreatic cancer stem-like cells in vitro and in vivo. Oncotarget (2015) 6(8):5517–35. doi:10.18632/oncotarget.3584
121. Quan Y, Wang W, Fu Q, Mei T, Wu J, Li J, et al. Accumulation efficiency of cancer stem-like cells post γ-ray and proton irradiation. Nucl Instr Meth Phys Res B (2012) 286:341–5. doi:10.1016/j.nimb.2011.11.019
122. Fu Q, Quan Y, Wang W, Mei T, Wu J, Li J, et al. Response of cancer stem-like cells and non-stem cancer cells to proton and γ-ray irradiation. Nucl Instr Meth Phys Res B (2012) 286:346–50. doi:10.1016/j.nimb.2012.01.032
123. Blakely EA, Tobias CA, Yang TCH, Smith KC, Lyman JT. Inactivation of human kidney cells by high-energy monoenergetic heavy-ion beams. Radiat Res (1979) 80:122–60. doi:10.2307/3575121
124. Nakagawa Y, Takahashi A, Kajihara A, Yamakawa N, Imai Y, Ota I, et al. Depression of p53-independent Akt survival signals in human oral cancer cells bearing mutated p53 gene after exposure to high-LET radiation. Biochem Biophys Res Commun (2012) 423(4):654–60. doi:10.1016/j.bbrc.2012.06.004
125. Hamada N, Imaoka T, Masunaga S, Ogata T, Okayasu R, Takahashi A, et al. Recent advances in the biology of heavy-ion cancer therapy. J Radiat Res (2010) 51(4):365–83. doi:10.1269/jrr.09137
126. Takahashi A, Ma H, Nakagawa A, Yoshida Y, Kanai T, Ohno T, et al. Carbon-ion beams efficiently induce cell killing in X-ray resistant human squamous tongue cancer cells. Int J Med Phys Clin Eng Radiat Oncol (2014) 3:133–42. doi:10.4236/ijmpcero.2014.33019
127. Bertrand G, Maalouf M, Boivin A, Battiston-Montagne P, Beuve M, Levy A, et al. Targeting head and neck cancer stem cells to overcome resistance to photon and carbon ion radiation. Stem Cell Rev (2014) 10(1):114–26. doi:10.1007/s12015-013-9467-y
129. Fleming IN, Manavaki R, Blower PJ, West C, Williams KJ, Harris AL, et al. Imaging tumour hypoxia with positron emission tomography. Br J Cancer (2015) 112(2):238–50. doi:10.1038/bjc.2014.610
130. Vaupel P, Harrison L. Tumor hypoxia: causative factors, compensatory mechanisms, and cellular response. Oncologist (2004) 9(Suppl 5):4–9. doi:10.1634/theoncologist.9-90005-4
131. Tinganelli W, Ma NY, Von Neubeck C, Maier A, Schicker C, Kraft-Weyrather W, et al. Influence of acute hypoxia and radiation quality on cell survival. J Radiat Res (2013) 54(Suppl 1):i23–30. doi:10.1093/jrr/rrt065
132. Ma NY, Tinganelli W, Maier A, Durante M, Kraft-Weyrather W. Influence of chronic hypoxia and radiation quality on cell survival. J Radiat Res (2013) 54(Suppl 1):i13–22. doi:10.1093/jrr/rrs135
133. Zolzer F, Streffer C. Increased radiosensitivity with chronic hypoxia in four human tumor cell lines. Int J Radiat Oncol Biol Phys (2002) 54(3):910–20. doi:10.1016/S0360-3016(02)02963-2
134. Wenzl T, Wilkens JJ. Modelling of the oxygen enhancement ratio for ion beam radiation therapy. Phys Med Biol (2011) 56(11):3251–68. doi:10.1088/0031-9155/56/11/006
135. Scifoni E, Tinganelli W, Weyrather WK, Durante M, Maier A, Kramer M. Including oxygen enhancement ratio in ion beam treatment planning: model implementation and experimental verification. Phys Med Biol (2013) 58(11):3871–95. doi:10.1088/0031-9155/58/11/3871
136. Hirayama R, Furusawa Y, Fukawa T, Ando K. Repair kinetics of DNA-DSB induced by X-rays or carbon ions under oxic and hypoxic conditions. J Radiat Res (2005) 46(3):325–32. doi:10.1269/jrr.46.325
137. Nakano T, Suzuki Y, Ohno T, Kato S, Suzuki M, Morita S, et al. Carbon beam therapy overcomes the radiation resistance of uterine cervical cancer originating from hypoxia. Clin Cancer Res (2006) 12(7 Pt 1):2185–90. doi:10.1158/1078-0432.CCR-05-1907
138. Antonovic L, Lindblom E, Dasu A, Bassler N, Furusawa Y, Toma-Dasu I. Clinical oxygen enhancement ratio of tumors in carbon ion radiotherapy: the influence of local oxygenation changes. J Radiat Res (2014) 55(5):902–11. doi:10.1093/jrr/rru020
139. Schlaff CD, Krauze A, Belard A, O’Connell JJ, Camphausen KA. Bringing the heavy: carbon ion therapy in the radiobiological and clinical context. Radiat Oncol (2014) 9(1):88. doi:10.1186/1748-717X-9-88
140. Bentzen SM, Ritter MA. The alpha/beta ratio for prostate cancer: what is it, really? Radiother Oncol (2005) 76(1):1–3. doi:10.1016/j.radonc.2005.06.009
141. Bentzen SM, Overgaard J, Thames HD, Overgaard M, Vejby Hansen P, von der Maase H, et al. Clinical radiobiology of malignant melanoma. Radiother Oncol (1989) 16(3):169–82.
142. Williams MV, Denekamp J, Fowler JF. A review of alpha/beta ratios for experimental tumors: implications for clinical studies of altered fractionation. Int J Radiat Oncol Biol Phys (1985) 11(1):87–96. doi:10.1016/0360-3016(85)90366-9
143. Lo SS, Fakiris AJ, Chang EL, Mayr NA, Wang JZ, Papiez L, et al. Stereotactic body radiation therapy: a novel treatment modality. Nat Rev Clin Oncol (2010) 7(1):44–54. doi:10.1038/nrclinonc.2009.188
144. Kupelian PA, Thakkar VV, Khuntia D, Reddy CA, Klein EA, Mahadevan A. Hypofractionated intensity-modulated radiotherapy (70 gy at 2.5 Gy per fraction) for localized prostate cancer: long-term outcomes. Int J Radiat Oncol Biol Phys (2005) 63(5):1463–8. doi:10.1016/j.ijrobp.2005.05.054
145. Schulz-Ertner D, Nikoghosyan A, Hof H, Didinger B, Combs SE, Jakel O, et al. Carbon ion radiotherapy of skull base chondrosarcomas. Int J Radiat Oncol Biol Phys (2007) 67(1):171–7. doi:10.1016/j.ijrobp.2006.08.027
146. Denekamp J, Waites T, Fowler JF. Predicting realistic RBE values for clinically relevant radiotherapy schedules. Int J Radiat Biol (1997) 71(6):681–94. doi:10.1080/095530097143699
147. Denekamp J, Harris SR, Morris C, Field SB. The response of a transplantable tumor to fractionated irradiation. II. Fast neutrons. Radiat Res (1976) 68(1):93–103. doi:10.2307/3574537
148. Ando K, Koike S, Uzawa A, Takai N, Fukawa T, Furusawa Y, et al. Biological gain of carbon-ion radiotherapy for the early response of tumor growth delay and against early response of skin reaction in mice. J Radiat Res (2005) 46(1):51–7. doi:10.1269/jrr.46.51
149. Field SB, Hornsey S. RBE values for cyclotron neutrons for effects on normal tissues and tumours as a function of dose and dose fractionation. Eur J Cancer (1971) 7(2):161–9. doi:10.1016/0014-2964(71)90011-9
150. Leith JT, Woodruff KH, Lyman JT. Early effects of single doses of 375 MeV/nucleon 20Neon ions on the skin of mice and hamsters. Radiat Res (1976) 65(3):440–50. doi:10.2307/3574375
151. Joiner MC. Linear energy transfer and relative biological effectiveness. 4th ed. In: Joiner MC, van der Kogel AJ, editors. Basic Clinical Radiobiology. London: Hodder Arnold (2009). p. 68–77.
152. Jones B. The apparent increase in the {beta}-parameter of the linear quadratic model with increased linear energy transfer during fast neutron irradiation. Br J Radiol (2010) 83(989):433–6. doi:10.1259/bjr/68792966
153. Yoshida Y, Ando K, Ando K, Murata K, Yoshimoto Y, Musha A, et al. Evaluation of therapeutic gain for fractionated carbon-ion radiotherapy using the tumor growth delay and crypt survival assays. Radiother Oncol (2015) 117(2):351–7. doi:10.1016/j.radonc.2015.09.027
154. Uzawa A, Ando K, Kase Y, Hirayama R, Matsumoto Y, Matsufuji N, et al. Designing a ridge filter based on a mouse foot skin reaction to spread out Bragg-peaks for carbon-ion radiotherapy. Radiother Oncol (2015) 115(2):279–83. doi:10.1016/j.radonc.2015.04.007
155. Ando K, Koike S, Uzawa A, Takai N, Fukawa T, Furusawa Y, et al. Repair of skin damage during fractionated irradiation with gamma rays and low-LET carbon ions. J Radiat Res (2006) 47(2):167–74. doi:10.1269/jrr.47.167
156. Karger CP, Peschke P, Sanchez-Brandelik R, Scholz M, Debus J. Radiation tolerance of the rat spinal cord after 6 and 18 fractions of photons and carbon ions: experimental results and clinical implications. Int J Radiat Oncol Biol Phys (2006) 66(5):1488–97. doi:10.1016/j.ijrobp.2006.08.045
157. Ando K, Koike S, Ohira C, Chen YJ, Nojima K, Ando S, et al. Accelerated reoxygenation of a murine fibrosarcoma after carbon-ion radiation. Int J Radiat Biol (1999) 75(4):505–12. doi:10.1080/095530099140438
158. Oya N, Sasai K, Shibata T, Takagi T, Shibuya K, Koike S, et al. Time course of reoxygenation in experimental murine tumors after carbon-beam and X-ray irradiation. J Radiat Res (2001) 42(2):131–41. doi:10.1269/jrr.42.131
159. Kanai T, Matsufuji N, Miyamoto T, Mizoe J, Kamada T, Tsuji H, et al. Examination of GyE system for HIMAC carbon therapy. Int J Radiat Oncol Biol Phys (2006) 64(2):650–6. doi:10.1016/j.ijrobp.2005.09.043
160. Kanai T, Furusawa Y, Fukutsu K, Itsukaichi H, Eguchi-Kasai K, Ohara H. Irradiation of mixed beam and design of spread-out Bragg peak for heavy-ion radiotherapy. Radiat Res (1997) 147(1):78–85. doi:10.2307/3579446
161. Shibamoto Y, Otsuka S, Iwata H, Sugie C, Ogino H, Tomita N. Radiobiological evaluation of the radiation dose as used in high-precision radiotherapy: effect of prolonged delivery time and applicability of the linear-quadratic model. J Radiat Res (2012) 53(1):1–9. doi:10.1269/jrr.11095
162. Song CW, Kim MS, Cho LC, Dusenbery K, Sperduto PW. Radiobiological basis of SBRT and SRS. Int J Clin Oncol (2014) 19(4):570–8. doi:10.1007/s10147-014-0717-z
163. Park HJ, Griffin RJ, Hui S, Levitt SH, Song CW. Radiation-induced vascular damage in tumors: implications of vascular damage in ablative hypofractionated radiotherapy (SBRT and SRS). Radiat Res (2012) 177(3):311–27. doi:10.1667/RR2773.1
164. Williams JP, Brown SL, Georges GE, Hauer-Jensen M, Hill RP, Huser AK, et al. Animal models for medical countermeasures to radiation exposure. Radiat Res (2010) 173:557–78. doi:10.1667/RR1880.1
165. Brown JM, Carlson DJ, Brenner DJ. Dose escalation, not “new biology,” can account for the efficacy of stereotactic body radiation therapy with non-small cell lung cancer. In reply to Rao et al. Int J Radiat Oncol Biol Phys (2014) 89(3):693–4. doi:10.1016/j.ijrobp.2014.03.014
166. Brown JM, Carlson DJ, Brenner DJ. The tumor radiobiology of SRS and SBRT: are more than the 5 Rs involved? Int J Radiat Oncol Biol Phys (2014) 88(2):254–62. doi:10.1016/j.ijrobp.2013.07.022
167. Fowler JF. The radiobiology of prostate cancer including new aspects of fractionated radiotherapy. Acta Oncol (2005) 44(3):265–76. doi:10.1080/02841860410002824
168. Garcia-Barros M, Paris F, Cordon-Cardo C, Lyden D, Rafii S, Haimovitz-Friedman A, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science (2003) 300(5622):1155–9. doi:10.1126/science.1082504
169. Fuks Z, Kolesnick R. Engaging the vascular component of the tumor response. Cancer Cell (2005) 8(2):89–91. doi:10.1016/j.ccr.2005.07.014
170. Lee Y, Auh SL, Wang Y, Burnette B, Meng Y, Beckett M, et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood (2009) 114(3):589–95. doi:10.1182/blood-2009-02-206870
171. Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol (2008) 181(5):3099–107. doi:10.4049/jimmunol.181.5.3099
172. Ogata T, Teshima T, Kagawa K, Hishikawa Y, Takahashi Y, Kawaguchi A, et al. Particle irradiation suppresses metastatic potential of cancer cells. Cancer Res (2005) 65(1):113–20.
173. Tamaki T, Iwakawa M, Ohno T, Imadome K, Nakawatari M, Sakai M, et al. Application of carbon-ion beams or gamma-rays on primary tumors does not change the expression profiles of metastatic tumors in an in vivo murine model. Int J Radiat Oncol Biol Phys (2009) 74(1):210–8. doi:10.1016/j.ijrobp.2008.12.078
174. Matsunaga A, Ueda Y, Yamada S, Harada Y, Shimada H, Hasegawa M, et al. Carbon-ion beam treatment induces systemic antitumor immunity against murine squamous cell carcinoma. Cancer (2010) 116(15):3740–8. doi:10.1002/cncr.25134
175. Ohkubo Y, Iwakawa M, Seino K, Nakawatari M, Wada H, Kamijuku H, et al. Combining carbon ion radiotherapy and local injection of alpha-galactosylceramide-pulsed dendritic cells inhibits lung metastases in an in vivo murine model. Int J Radiat Oncol Biol Phys (2010) 78(5):1524–31. doi:10.1016/j.ijrobp.2010.06.048
176. Yoshimoto Y, Oike T, Okonogi N, Suzuki Y, Ando K, Sato H, et al. Carbon-ion beams induce production of an immune mediator protein, high mobility group box 1, at levels comparable with X-ray irradiation. J Radiat Res (2015) 56(3):509–14. doi:10.1093/jrr/rrv007
177. Durante M, Reppingen N, Held KD. Immunologically augmented cancer treatment using modern radiotherapy. Trends Mol Med (2013) 19(9):565–82. doi:10.1016/j.molmed.2013.05.007
178. Ogata T, Teshima T, Inaoka M, Minami K, Tsuchiya T, Isono M, et al. Carbon ion irradiation suppresses metastatic potential of human non-small cell lung cancer A549 cells through the phosphatidylinositol-3-kinase/Akt signaling pathway. J Radiat Res (2011) 52(3):374–9. doi:10.1269/jrr.10102
Keywords: charged particles, proton therapy, carbon-ion therapy, relative biological effectiveness, clustered DNA damage, cancer stem cells, hypoxic radioresistance, altered fractionation
Citation: Held KD, Kawamura H, Kaminuma T, Paz AES, Yoshida Y, Liu Q, Willers H and Takahashi A (2016) Effects of Charged Particles on Human Tumor Cells. Front. Oncol. 6:23. doi: 10.3389/fonc.2016.00023
Received: 23 October 2015; Accepted: 21 January 2016;
Published: 12 February 2016
Edited by:
Marco Durante, GSI Helmholtz Centre for Heavy Ion Research, GermanyReviewed by:
Kevin Prise, Queen’s University Belfast, UKThomas Friedrich, GSI Helmholtz Centre for Heavy Ion Research Darmstadt, Germany
Copyright: © 2016 Held, Kawamura, Kaminuma, Paz, Yoshida, Liu, Willers and Takahashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathryn D. Held, a2hlbGRAbWdoLmhhcnZhcmQuZWR1