 Michael R. Horsman
Michael R. Horsman Peter Vaupel
Peter Vaupel- 1Department of Experimental Clinical Oncology, Aarhus University Hospital, Aarhus, Denmark
- 2Department of Radiooncology and Radiotherapy, Klinikum rechts der Isar, Technische Universität München (TUM), Munich, Germany
Poor microenvironmental conditions are a characteristic feature of solid tumors. Such conditions occur because the tumor vascular supply, which develops from the normal host vasculature by the process of angiogenesis, is generally inadequate in meeting the oxygen and nutrient demands of the growing tumor mass. Regions of low oxygenation (hypoxia) is believed to be the most critical deficiency, since it has been well documented to play a significant role in influencing the response to conventional radiation and chemotherapy treatments, as well as influencing malignant progression in terms of aggressive growth and recurrence of the primary tumor and its metastatic spread. As a result, significant emphasis has been placed on finding clinically applicable approaches to identify those tumors that contain hypoxia and realistic methods to target this hypoxia. However, most studies consider hypoxia as a single entity, yet we now know that it is multifactorial. Furthermore, hypoxia is often associated with other microenvironmental parameters, such as elevated interstitial fluid pressure, glycolysis, low pH, and reduced bioenergetic status, and these can also influence the effects of hypoxia. Here, we review the various aspects of hypoxia, but also discuss the role of the other microenvironmental parameters associated with hypoxia.
Introduction
Most solid tumors are just like normal tissues in that they need a regular supply of oxygen and nutrients to be able to exist, as well as processes for the elimination of the waste products of cellular metabolism. When tumors first appear, this function is provided by the normal blood supply of the host organ in which the tumor arises. However, unlike normal tissues, tumors continually expand in size and a point is reached where the host vascular supply becomes inadequate in supplying these needs. To compensate, tumors will actually develop their own functional vascular supply. This they do from the normal host vessels by the process of angiogenesis. Unfortunately, the tumor neo-vasculature that is formed is not only primitive and chaotic when compared to the normal tissue vascular supply from which it develops, but it also suffers from numerous structural and functional abnormalities. As a result, it is still unable to meet all the demands of the growing tumor mass (with sizes larger than 2–3 mm). Consequently, a hostile microenvironment develops within the tumor and this can be summarized by the so-called “crucial Ps.” These are listed in Table 1 and basically reflect conditions of poor perfusion, oxygen deprivation, nutrient deficiency, severe acidity, and elevated interstitial fluid pressure (IFP).

Table 1. The crucial Ps characterizing the hostile tumor microenvironment.
The tumor cells that exist in this hostile microenvironment are actually still viable. But, as a result of being in these adverse microenvironmental conditions, those same tumor cells can exhibit resistance to conventional cancer therapies, including radiation and certain types of chemotherapy. The poor tumor microenvironment also causes these cells to upregulate the expression of various genes and biosynthesis of proteins, an effect that not only increases their survival potential but can also increase tumor aggressiveness and metastatic spread. Numerous attempts have, and are being made, to identify the microenvironmental conditions within tumors so as to select appropriate therapies to target those cancer cells that thrive in the hostile microenvironment.
Of these poor microenvironmental conditions within tumors, low oxygenation (hypoxia) is the one that has been the focus of most studies and is often considered as the only factor of importance. While hypoxia is clearly critical for outcome of cancer patients, it is generally associated with the other crucial Ps and as such any discussion of the role of hypoxia in tumors must be made in connection with the other hostile microenvironmental parameters. Thus, we will review the general pathophysiological characteristics of the tumor microenvironment, how that microenvironment develops, and what significance that has for cancer.
Factors Influencing the Tumor Microenvironment
Importance of the Tumor Vascular Supply
Angiogenesis is clearly an essential requirement for the growth and development of solid tumors (2–4). This process begins with the release of angiogenic factors, primarily vascular endothelial growth factor (VEGF), by the tumor cells (5). The actual triggers that initiate this process are not fully established. Loss of suppressor gene function and oncogene activation certainly play a role (5, 6), but the development of hypoxia as a result of tumor growth is also a major factor (6). Additional studies with tumor cells grown in culture have shown that the secretion rate of the VEGF protein increases as soon as the oxygen concentration is lowered from 21 to only 5% and that this secretion rate increases as the oxygen concentration decreases reaching maximal levels at around 0.5% and below (7). Release of VEGF and other growth factors set in motion a number of biochemical and physical steps that include enzymatic destruction of the basal membrane of the endothelial cells of the host vasculature, migration of endothelial cells into the extracellular matrix to form sprouts, and endothelial cell division away from the sprout tip (8). Solid strands of endothelial cells are then formed in the extracellular matrix, a lumen develops within those strands, neighboring sprouts fuse to form loops, and from the primary loops new buds and sprouts emerge (8). Finally, functional vessels are established.
Although a functional tumor vascular supply is necessary, the neo-vasculature that develops is actually inadequate to meet all the demands of the growing tumor mass. Endothelial cells divide at a slower rate than tumor cells (9) and as a consequence, the developing tumor vasculature is unable to keep pace with the expanding tumor population. The tumor vasculature that is formed is also very different from that of normal tissues (see Figure 1). Structurally, it is very chaotic. Vascular density is abnormal with increased intervessel distances and the existence of avascular areas. There are contour irregularities reflected by vessels that are elongated, tortuous, and large, and have aberrant branching and blind ends. The pattern of vessel interconnection is also haphazard. Unlike normal vessels, there is a loss of hierarchy. The vessels are also very primitive in nature, having incomplete or missing basement membranes and endothelial lining, and lacking pericytes, smooth muscle, pharmacological receptors, and even innervation. Tumor vessels are often highly permeable allowing significant plasma leakage. Due to an absence of vasomotion and flow regulation, blood flow velocities through tumor vessels can be unstable as can the direction of flow [for review, see Ref. (10)]. It has been estimated that 1–8% of vessels can experience flow stasis (11, 12) and that around 8% of all microvessels show plasma flow only (12). Some of these changes will be mediated through effects of the various blood-borne cells. These include erythrocyte sludging, leukocyte sticking, and blockage of vessels by circulating white blood cells or tumor cells. High IFP can also play a role here. In normal tissues, there exists a perfusion pressure difference of about 20 mmHg between the arterial and venous ends of microvessels and this drives blood flow through capillaries. However, in tumors, the increased leakiness of vessels and the lacking of functional lymphatics result in an increased IFP. Transmural coupling between this high IFP and microvascular pressures can result in abolished perfusion pressure differences between the arterial and venous ends, thus, causing flow stasis and hypoxia (13). However, this is more likely to result in chronic rather than acute hypoxia (13). Finally, the hematocrit within tumor microvessels can be increased by 5–14% and this will also influence flow.
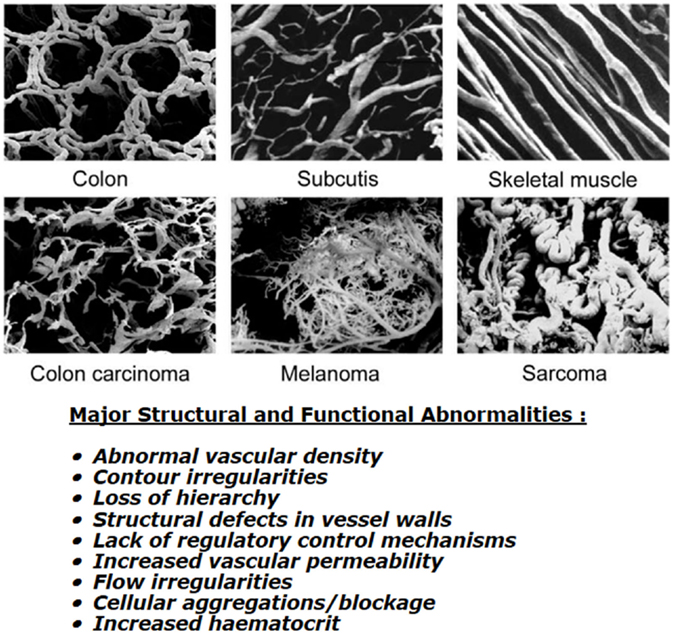
Figure 1. Vascular casting images showing differences in microcirculation between normal tissues (top three panels) and malignant tumors (lower three panels). Specific details of the corrosion casting technique used to produce these images can be found in Konerding et al. (14). Images shown were obtained courtesy of Prof. Konerding, Dept. Functional and Clinical Anatomy, University Medical Center, Mainz, Germany and are from Vaupel (10). Bottom text lists the major structural and functional abnormalities of tumor vessels when compared to normal tissues; composite information based on work by Kimura et al. (12), Reinhold and van der Berg-Blok (15), Vaupel et al. (10, 16), and Baronzio et al. (17).
Other Parameters Affecting the Micromilieu
Apart from the inadequacies of the tumor vascular supply, there are a number of other important factors that can influence the microenvironment within tumors. Chief among these is the oxygen carrying capacity of the blood. This can be substantially reduced under specific conditions, thus making less oxygen available. Such an effect is seen with anemia where the normal hemoglobin levels of 7.5–9.5 mmol/L in females and 8–10 mmol/L in males can be reduced by 50% in anemic patients. Additional studies in which tumor oxygenation status was directly measured with polarographic needle electrodes have reported a correlation between the level of tumor hypoxia and hemoglobin concentration (18). Reduced oxygen availability is also observed in patients who smoke. Smoking impairs the delivery of oxygen to tumors due to the presence of carboxyhemoglobin (HbCO) that is formed by the binding of carbon monoxide (CO) to hemoglobin (19). Heavy smokers can have up to 16–18% HbCO in their blood. This reaction not only decreases the amount of hemoglobin available for oxygen transport but will also shift the oxygen-dissociation curve to the left making it more difficult for the hemoglobin to release oxygen to the cells. Since the affinity of hemoglobin for CO is approximately 250 times the affinity for oxygen, even low concentrations of CO can result in significant levels of HbCO in the blood.
The microenvironment of tumors will also depend on the cellular consumption of oxygen and essential nutrients. As a result of tumor cells close to the vascular supply consuming what they need for growth and survival, less will be available for those cells further away. Consequently, radial oxygen, nutrient, and pH gradients are established (16). The extent of those gradients will depend not only on the rate of consumption but also on what is actually delivered to the cells by the blood supply. Indeed, it has been reported that in the case of oxygen the cells next to the blood vessel can have oxygen concentrations as low as 2% [approximately 15 mmHg; (20)] and this would certainly reduce the oxygen diffusion distance. As it is the case in normal physiology, there is also an intravascular (longitudinal) oxygen partial pressure gradient when the blood moves from the arterial to venous end of the microvessels (21). All these factors, coupled with the structural and functional aberrations described in the previous section, will result in the development of areas within the tumor that can be considered “abnormal and adverse” when compared to those conditions found in normal tissues (10, 16).
Microenvironmental Characteristics of Tumors
Hypoxia
The microenvironmental parameter that has been the most extensively investigated is hypoxia. By definition, hypoxia is a state of reduced oxygenation that influences biological functions (22). The first indirect indication that hypoxia existed in tumors was made by Thomlinson and Gray (23). From histological sections of carcinomas of the bronchus, they typically found viable tumor regions surrounded by vascular stroma, with regions of necrosis evolving in the center of the tumor mass. The thickness of the resulting shell of viable tissue was found to be between 100 and 180 μm. They suggested that as oxygen diffused from the stroma, it was consumed by the cells, and although those beyond the diffusion distance were unable to survive, the cells immediately bordering the necrosis might be viable yet hypoxic; unfortunately, in this concept, the diffusion of glucose and other nutrients is completely ignored. Later, an inverted version of the Thomlinson and Gray model was described, with functional blood vessels surrounded by cords of viable tumor cells outside of which were areas of necrosis [Krogh model of oxygen diffusion; (24)]. This corded structure is the more typical picture found in most solid tumors and is illustrated in Figure 2. As with the Thomlinson and Gray model, an oxygen gradient is created as the oxygen diffuses from the blood vessel, resulting in a region of cells at the edge of the cord that are oxygen deprived and are commonly referred to as diffusion-limited chronic hypoxia. This type of hypoxia has been seen in both animal and human tumors (25). It has been suggested that such hypoxic cells can survive under these adverse conditions for several days (26). Death will also occur as the hypoxic cells move further away from the blood supply as the tumor grows, although this is more likely to result from a glucose deficit rather than just a lack of oxygen.
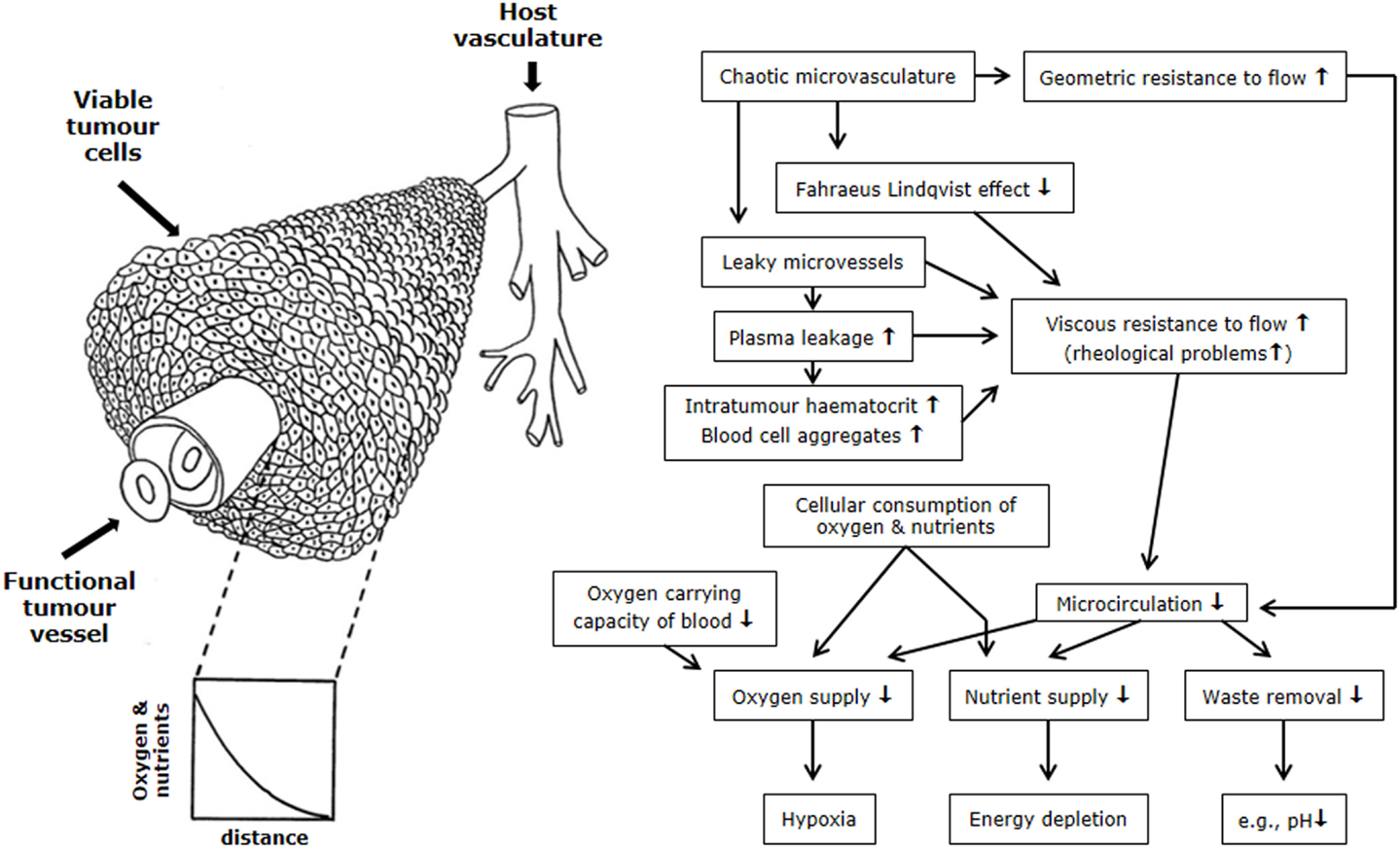
Figure 2. Schematic illustration of the relationship between the tumor vasculature and microenvironment. The left side shows tumor cells growing in a corded structure around a functional vessel from which the cells receive their oxygen and nutrient supply, but as these substances diffuse out from the vessel they are utilized by the cells so that gradients are established. On the right side is a flow chart showing the relationship between the hostile microenvironment of tumors and the factors that give rise to its development. Figure is modified from Ref. (1, 27).
Diffusion-limited chronic hypoxia was the working model for hypoxia from the 1950s until around the 1980s when it was then suggested that a second type of hypoxia could exist in tumors and one that was acute in nature (28). This was later confirmed and shown to be the result of the transient stoppages in tumor blood flow described earlier (29). The hypoxia that results was originally called perfusion limited acute hypoxia, although other terms are often used, including cyclic, intermittent, transient, repetitive, or fluctuating, and it is probably the latter term that is the most appropriate description. Evidence for fluctuating hypoxia has been reported in murine tumors (30), human tumor xeongrafts (31), and even in tumors in cancer patients (32–34).
Today, hypoxia is often considered as a single entity even though we know there are at least two types. However, even the concept of chronic and acute/fluctuating hypoxia is an over-simplification of the real picture (13). Acute hypoxia can result from a total or partial shut-down in perfusion (12); a complete shut-down would starve cells of oxygen and nutrients and result in ischemic hypoxia, which would not be the case for a partial shut-down where plasma flow, thus nutrient supply, can occur leading to hypoxemic hypoxia. For chronic hypoxia, the picture is even more complicated. It can result from a diffusion limitation under “normal” conditions (diffusional hypoxia), or be due to reduced oxygen availability as with high HbCO or anemia (anemic hypoxia). But, it will also be dependent on the oxygenation level; cells close to the vessel could be slightly hypoxic, while cells next to necrosis could even be anoxic (a situation where no oxygen can be detected) but still viable if they have sufficient nutrient supply (i.e., glucose). Whatever the description, hypoxia is now known to be a characteristic feature of most solid animal tumor models (35) and numerous human cancers (16, 36).
Interstitial Fluid Pressure
Unlike normal tissues, tumors often contain vessels that are abnormally leaky and also lack a functional lymphatic system (37). These, coupled with a large hydraulic conductivity, results in a significant bulk flow of free fluid in the interstitial space. In normal tissues, water influx into the interstitial compartment has been estimated to be between 0.5 and 1.0% of plasma flow, yet in human tumors values up to 15% can be reached (38). As a result of fluid accumulating in the tumor matrix, there is a build-up of interstitial pressure (39–41). Interstitial fibrosis, contraction of the interstitial space mediated by stromal fibroblasts, and high oncotic pressures within the interstitium may also contribute to the development of interstitial hypertension (42). In most normal tissues, IFP is just above or below atmospheric values (43), but in tumors it can reach 50 or even 100 mmHg (1). IFP is generally uniformally high throughout the center of tumors, but drops steeply in the tumor periphery (44). However, since vascular permeability varies from tumor to tumor and can be heterogeneous within the same tumor, IFP is not constant (1). It can also fluctuate with changes in microvascular pressures (45).
Glycolysis and PH
Warburg’s classic work in the 1920s showed that cancer cells intensively converted glucose to lactate (glycolysis) even in the presence of oxygen [for a review see Ref. (46)]. Today, it is believed that there is no clear evidence that cancer cells are inherently glycolytic, but that some tumors might be glycolytic in vivo as a result of hypoxic response mechanisms (47). Hypoxia will shift the balance of cellular energy production toward glycolysis with the generation and subsequent accumulation of lactate (48). Indeed, several studies have found high median lactate levels of around 7 mM in head and neck cancers (49) and up to 14 mM in uterine cervix (50). Although lactate is generally considered a waste product, there is evidence that the lactate produced by hypoxic cells can be taken up in normoxic cancer cells via the monocarboxylate transporter-1 and can then be utilized for oxidative phosphorylation instead of glucose as a substrate (51). However, cellular lactate production and release will lead to tumor acidosis. What is clear is that like normal cells, tumor cells have efficient mechanisms for exporting protons into the extracellular space (52, 53), thus a pH gradient exists across the tumor cell membrane so that intracellular pH (pHi) remains higher than the extracellular pH (pHe). In normal tissues, this gradient is reversed such that pHi is actually lower than pHe (16, 54–56). The production and release of lactate alone does not fully account for the acidosis found in the extracellular compartment of solid tumors. Other key mechanisms may play an important role, especially ATP hydrolysis, glutaminolysis, carbon dioxide production, and bicarbonate depletion (48).
Bioenergetic Status
Various techniques have been used to monitor the bioenergetic status within tumors. These include ex vivo quantitative bioluminescence (57) and high-performance liquid chromatography [HPLC; (38, 58)], and non-invasive 31P-nuclear magnetic resonance/spectroscopy [NMR/MRS; (59)]. The global concentrations of ATP measured in experimental tumors using HPLC were found to be typically between 0.4 and 2.0 mM (38, 58). These global ATP concentrations and adenylate energy charge only changed marginally provided tumors did not exceed biologically relevant tumor sizes (i.e., 1% of the body weight). With increasing tumor mass, ATP hydrolysis increased. As a result of this increased ATP degradation, an accumulation of purine catabolites, and the final degradation product uric acid, has been observed (38). Using quantitative bioluminescence, the microregional distribution of ATP has been assessed in cryobiopsies of cervix tumors and found to be heterogeneous and comparable to high flow experimental tumors (38). This ATP distribution profile was similar to those seen for both glucose and lactate, but there was no clear-cut correlation between tumor oxygenation and regional ATP levels (38). Bioluminescence measurements of regional ATP distributions in experimental brain tumors reported ATP levels that were similar to normal brain, whereas glucose was slightly lower and lactate substantially higher (38, 60), with these metabolites showing marked tumor heterogeneity (60). Additional studies using NMR have shown that in many human malignancies, high concentrations of phosphomonoesters, phosphodiesters, and inorganic phosphate, as well as low phosphocreatine, are often found. The exception is again in human brain tumors, where no significant differences in 31P-NMR spectra were seen when compared to normal brain tissue (38).
Hypoxia-Driven Adenosine Accumulation
The development of tumor hypoxia is accompanied by a substantial accumulation of the nucleoside adenosine (ADO) in the range of 50–100 μM (61). By contrast, ADO levels in normal tissues have been found to be in the range of 10–100 nM (62, 63). ADO accumulation is preferentially caused by an ATP release from cancer cells into the extracellular space upon hypoxic stress. After transport out of cancer cells, extracellular ATP is converted into ADO by hypoxia/hypoxia-inducible factor (HIF)-sensitive, membrane-bound ectoenzymes CD39 and CD73. Intracellular ADO-formation from AMP by a cytosolic AMP-nucleotidase with subsequent ADO-export into the extracellular space through a nucleoside transporter seems to play a subordinate role. ADO-actions (adenosinergic effects) are mediated upon binding to surface receptors, mainly A2A-receptors on tumor and immune cells. Receptor activation leads to a broad spectrum of strong immune-suppressive properties through modulation of the innate and adaptive immune system, thus facilitating tumor escape from immune control (62, 64–66). Mechanisms include (a) an impaired activity of CD4+ T and CD8+ T, NK cells, and dendritic cells (DCs), a decreased production of immune-stimulatory lymphokines, and (b) an activation of Treg cells, expansion of myeloid-derived suppressor cells (MDSCs), promotion of pro-tumor M2-macrophages, and increased activity of major immune-suppressive cytokines. In addition, ADO can directly stimulate tumor cell proliferation and angiogenesis. Taken together, there is clear evidence that ADO-mechanisms described can thwart anti-tumor immune responses elecited by radiotherapy and fever-range hyperthermia (67).
Significance of the Tumor Microenvironment for Cancer
Radiation
The potential of microenvironmental parameters to influence outcome to radiotherapy was first suggested from experiments in which the radiation response of skin was markedly decreased if the blood flow to the irradiated area was reduced by compression (68). This was followed by a report that tissues in which blood flow was stimulated by diathermia showed a more prominent response to radiation (69). Further experimental observations led Gray and co-workers to finally postulate the role of oxygen deficiency as a major source of radiation resistance (70). This occurs because oxygen is critical for the response of cells to radiation. The mechanism responsible is generally referred to as the “oxygen-fixation hypothesis” (71). When radiation interacts with the cellular target, which is usually DNA, it results in the production of free radicals. These are produced either directly by the radiation itself or indirectly from other molecules that are affected by radiation and then diffuse sufficiently to reach and damage the DNA target. Since water constitutes around 70% of all mammalian cells, most of the indirect radicals are probably produced from water molecules. In the absence of oxygen or in the presence of hydrogen-donating species (i.e., thiols), the free radicals formed in the DNA can react with hydrogen ions and the target is then chemically restored to its original form. However, if oxygen is present, it will react with the free radical to form a product that undergoes further reaction, ultimately producing a chemical change in the target. The damage is now fixed and can only be removed by enzymatic repair processes.
It has been demonstrated from rapid-mix studies that the oxygen effect occurs only if oxygen is present either during irradiation or within a few milliseconds thereafter (72, 73). The amount of oxygen is also critical. An almost maximum enhancement of radiation is seen with oxygen partial pressures above around 20 mmHg (approximately 3%). Below this partial pressure radiation sensitivity decreases in an oxygen-dependent fashion (71); in the absence of oxygen roughly three times as much radiation is required to kill the same number of cells as seen under normoxic conditions. This effect is generally referred to as the “oxygen enhancement ratio” (OER; ratio of the radiation dose in hypoxia/anoxia to that in air, to give the same biological effect). For radiation of higher energy than X-rays produced by modern radiotherapy units the OER actually decreases (74).
Numerous animal studies have demonstrated that hypoxia in tumors can influence radiation response. Three classical assays have been used (35, 75). They are (a) the clamped clonogenic survival assay, in which tumors are excised after treatment and cell survival measured in culture; (b) the clamped tumor growth delay assay, where measurements are made of the time taken for tumors to reach a specific size after treatment; and (c) the clamped tumor control assay, whereafter the percentage of animals showing local tumor control at a certain time after treatment is recorded. For each technique, it is necessary to produce full radiation dose–response curves under air breathing and fully anoxic (clamped tumor) conditions. The results of such assays not only demonstrate that hypoxia in tumors influences radiation response, but the degree of displacement of the dose–response curves also allows us to calculate the actual percentage of radiobiological cells in the tumor. Using these assays, the hypoxic fractions have been estimated to range from 1% to well over 50% of the viable tumor cells in animal tumors and human tumor xenografts (35).
Demonstrating that hypoxia can influence the radiation response of human tumors is more difficult, since none of the above approaches are applicable to humans. Although numerous other methods have been developed to try and identify hypoxia in human tumors (25, 76, 77), not all have been used to demonstrate the relationship between hypoxia and radiation response. The earliest attempts to do the latter were based on indirect approaches (78), and involved estimates of tumor vascularization, using such endpoints as intercapillary distance, vascular density and the distance from tumor cells to the nearest blood vessel (79–81). All showed that patients with less well vascularized tumors, and presumably more hypoxic, had a poorer outcome to radiation therapy. More direct approaches have used exogenous markers that are injected into the host and bind to regions of tumor hypoxia, or endogenous markers that are genes/proteins upregulated under hypoxia. The former include nitroimidazole or copper-based derivatives, which can be identified immunohistochemically from histological sections or non-invasively using positron emission tomography, SPECT, or magnetic resonance spectroscopy (25). Although such exogenous markers can be used to identify tumor hypoxia and even associated with outcome following radiation therapy in head-and-neck carcinoma patients (82), there has never been a proper radomized trial. The endogenous markers include such factors as carbonic anhydrase IX, GLUT-1, HIF-1, and osteopontin (83–86). These can be measured from biopsy material using protein immunohistochemistry or as gene expression, or proteins identified from blood samples. Although endogenous markers have been correlated with outcome to radiation therapy in some studies, it is not a universal finding (82), which probably reflects the fact that many of these endogenous markers are not hypoxia-specific rather than any indication that hypoxia does not play a role in influencing radiation response.
Probably the most direct method for estimating tumor hypoxia and one that has certainly been used to show the negative influence of such hypoxia on radiation response is the measurement of oxygen partial pressure (pO2) distributions with polarographic electrodes. Early attempts to achieve this used “home-made” glass electrodes which were cumbersome, fragile, and only a few pO2 values 3–4 mm below the surface of the tumor were possible. Nevertheless, clinical data were obtained in cervix (79) and head and neck (87) that clearly demonstrated a relationship between such oxygenation measurements and outcome to radiation therapy, in that those patients with tumors that were better oxygenated had a significantly superior local response to irradiation.
This whole area was revolutionized with the development of the Eppendorf histography system, which had two distinct improvements. The first was having the oxygen microsensor inside a metal needle and the second the attachment of this needle to a stepping motor that allowed for multiple measurements along the needle track through the tumor. Numerous clinical studies were, thus, undertaken in a variety of human tumor types. The results clearly showed that hypoxia was to be found in virtually all human tumors investigated, although the degree of hypoxia could be variable (16, 36, 88, 89). Probably the most significant finding from these studies was the confirmation that hypoxia influenced outcome to therapy, especially where radiation was given. This has been reported for cervix (90–95), head and neck (96–100), soft tissue sarcomas (101, 102), and prostate (103, 104). Results for all four tumor types are summarized in Table 2 and clearly illustrate that the patients with more hypoxic tumors had a poorer outcome response.
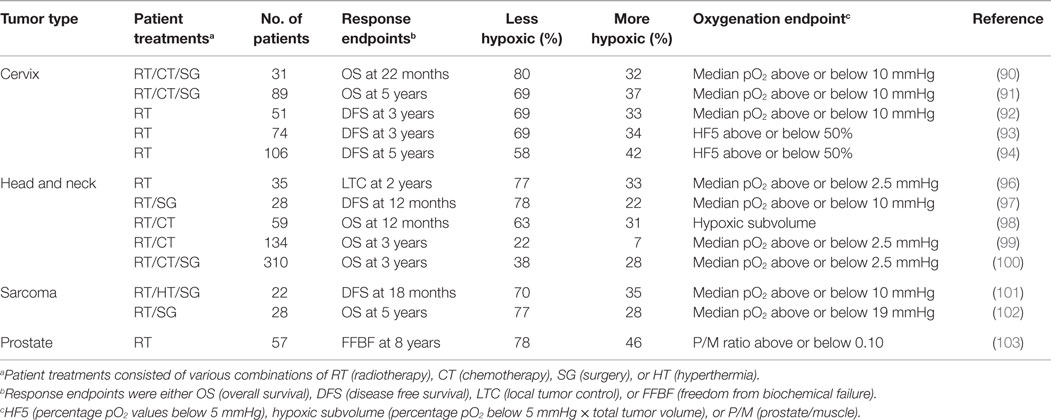
Table 2. Relationship between tumor oxygenation estimated prior to therapy using the Eppendorf histograph and outcome to therapy.
One major focus of current cancer research is the role of cancer stem cells in tumorigenesis and therapy. Such cells amount to around 1–25% of the total viable tumor cell population (105), but they are believed to be the cells that must be completely eliminated to obtain tumor control (106). Significant effort is currently being made to identify these cells and specifically target them. However, recent evidence suggests a possible link between hypoxia and cancer stem cells (106). Hypoxia may affect cancer stem cell generation and maintenance through the upregulation of hypoxia-induced factors (105, 106). Pre-clinical studies have also shown an inverse correlation between hypoxia and local tumor control after irradiation (107, 108), suggesting that hypoxia may also actually protect the cancer stem cells from the lethal effects of radiation. If the link between cancer stem cells and hypoxia is proven, then hypoxia may be an even more important issue for radiation response than we currently believe.
Chemotherapy
The pathophysiological characteristics of tumors play a significant role in influencing the response of the tumor cells to chemotherapy (109). An inadequate vascular supply will naturally be expected to hinder blood-borne drug delivery. A decrease in drug availability will certainly be seen in areas where flow fluctuates, especially where complete cessations in flow occur (110). In addition, the mean vascular density in tumors is lower than that found in normal tissues and, thus, diffusion distances are enlarged (1). Thus, transport of drugs from tumor microvessels to tumor cells that are distant from them will be compromised. The high IFP within solid tumors will also decrease extravasation (111). IFP at the tumor to normal tissue interface is low and as a result interstitial fluid oozes out of the tumor into the surrounding normal tissue. At the same time, it will also carry away anti-cancer drugs (37).
Cells most distant from the vascular supply will also be cycling at a reduced rate and this can act as a protective mechanism against a number of chemotherapeutic agents that work by interacting with cellular DNA and only kill the cell when it divides. Such cells are also exposed to hypoxia and acidic conditions, factors which are known to influence chemotherapeutic agent activity (111). However, these adverse microenvironmental parameters do not always have a negative effect of drug activity; some drugs are actually more effective under such conditions as illustrated in Table 3.
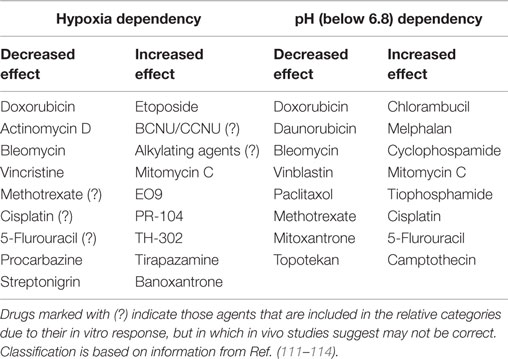
Table 3. Influence of the hostile tumor microenvironment on the activity of chemotherapeutic drugs.
Other Tumor Therapies
Hyperthermia is a less conventional therapy, but is one example where the more deficient the tumor vasculature and the more deprived the tumors cells, the better the tumor response. Blood flow, being one of the major means by which heat is dissipated from tissues, will affect the ability to heat tumors (115). Thus, the poorer the blood supply, the easier it should be to heat. This has been demonstrated in vivo in which blood flow was compromised using agents that could reduce tumor blood flow (116, 117). In vitro studies have also reported that cells under hypoxic conditions were more sensitive to killing by hyperthermia than the same cells in a well-oxygenated environment (118, 119). However, this is not a consequence of hypoxia per se because under well-defined nutrient conditions, acute hypoxia does not significantly alter cellular response to heat (119). However, cells under prolonged oxygen deprivation will show an increased sensitivity to heat, an effect that is the result of chronically hypoxic cells becoming acidic (118).
Another treatment modality in which the tumor microenvironment influences response is photodynamic therapy (PDT). It involves the administration of a photosensitizing agent and its subsequent activation by light. This reaction is strongly dependent on oxygen concentration (120–123). Cell killing by PDT appears to be complete at normal oxygen levels and above, but decreases as the oxygen concentration drops below 5% (123). This is perhaps not surprising since the mechanism of action of PDT involves the generation of singlet oxygen (124).
Malignant Progression
One of the most striking results found with the Eppendorf electrode measurements made in cervix cancers and soft tissue sarcomas was that hypoxia influenced outcome in patients in which surgery was the primary or only treatment (90, 91, 102): this suggested that hypoxia could also influence malignant progression, especially metastatic spread. In fact, one other study in cervix was able to show that the primary tumors of patients with metastases were indeed less oxygenated than those of patients without metastases (125).
Pre-clinical studies also support the importance of hypoxia in inducing metastatic spread. The earliest studies involved exposing murine fibrosarcoma cells in culture to different oxygen concentrations for up to 24 h and then intravenously injecting these cells and observing the number of lung metastasis that develop (126). That study clearly showed that the more hypoxic the exposure, the greater the number of lung metastases. In vivo attempts to investigate this issue have involved either correlating the constitutive level of hypoxia in primary tumors or exposing the mice to different oxygen environments to change the level of tumor hypoxia, and then determining the number of metastases formed (127–130). The results generally show that the greater the degree of hypoxia, the more metastases observed and that acute hypoxia was better at inducing metastases than chronic. These hypoxia-induced effects on malignant progression can be the result of changes at the transcriptional level in which a range of different genes are over-expressed or at the translational level with various proteins being upregulated. Such effects can be mediated via activation of various oxygen-sensitive signaling pathways (130, 131).
Other microenvironment factors found in tumors have also been shown to influence malignant progression. Tumor cells exposed to glucose deprivation and acidosis prior to intravenous injection into mice resulted in more lung metastases than cells not exposed to these conditions (132, 133). The deleterious effects of glucose deprivation in vivo have made it impossible to investigate its potential effects on malignancy in animals, but there have been attempts to relate pHe in vivo with metastases formation. Those studies involved either experimentally increasing tumor acidity (134) or simply making probe measurements of pHe in untreated tumors (135). But in both situations, no correlations were found between pH and metastases. This is perhaps somewhat surprising because one of the major factors causing acidity is lactate production and clinical studies have shown high tumor lactate concentrations to be associated with an increased risk of nodal and distant metastases (49), and a shorter overall and disease-free survival (50).
The relationship between IFP and metastatic dissemination is also controversial. Using melanoma xenografts, an association was found between high IFP and the development of pulmonary and lymph node metastases (136). Another study from the same research group reported that IFP was significantly higher in intramuscularly implanted TS-415 cervix carcinoma xenografts that metastasized than those that did not (135). However, that same study showed this not to be the case for CK-160 cervical xenografts. Furthermore, a lack of any correlation between IFP and metastatic spread was also reported using two other cervical xenograft models (ME180 and SiHa) regardless of whether they were grown intramuscularly or orthotopically (137). Clinical studies have been less controversial, with IFP measurements in cervix cancer patients being shown to predict for survival (41) and for both nodal and distant metastases (94). A possible relationship between IFP and metastatic disease in other tumor sites is not yet known.
Conclusion
The pathophysiological characteristics of the tumor microenvironment are very different from those conditions found in normal tissues. In many respects, the tumor microenvironment can be considered abnormal and hostile. These adverse pathophysiological conditions, especially hypoxia, are now known to play a significant role in determining the tumor response to therapy and influencing the metastatic potential of tumors. Clearly, the future requirement is the application of methods by which one can accurately and reliably image the various important microenvironmental parameters, especially using techniques that are routinely available in the clinic. With such information, it should be possible to identify those patients, on an individual basis, that would be expected to have a poor prognosis and, thus, select appropriate additional treatments to dramatically improve that prognosis.
Author Contributions
Both authors contributed equally to the concept, development, and the writing of this manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Financial support from the Danish Cancer Society and the Danish Council for Independent Research: Medical Sciences is gratefully acknowledged (MH).
References
1. Vaupel P. Pathophysiology of human tumors. In: Osinsky S, Friese H, Vaupel P, editors. Tumor Hypoxia in the Clinical Setting. Kiev: Natl. Acad. Sci. Ukraine (2011). p. 21–64.
2. Brem S, Brem H, Folkman J, Finkelstein D, Patz A. Prolonged tumor dormancy by prevention of neovascularization in the vitreous. Cancer Res (1976) 36:2807–12.
3. Folkman J. How is blood vessel growth regulated in normal and neoplastic tissue? Cancer Res (1986) 46:467–73.
4. Hahnfeldt P, Panigrahy D, Folkman J, Hlatky L. Tumor development under angiogenic signaling: a dynamic theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res (1999) 59:4770–5.
5. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med (2003) 9:669–76. doi: 10.1038/nm0603-669
6. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med (2003) 9:677–84. doi:10.1038/nm0603-677
7. Leith JT, Michelson S. Secretion rates and levels of vascular endothelial growth factor in clone A or HCT-8 human colon tumour cells as a function of oxygen concentration. Cell Prolif (1995) 28:415–30. doi:10.1111/j.1365-2184.1995.tb00082.x
8. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer (2003) 3:401–10. doi:10.1038/nrc1093
9. Denekamp J, Hobson B. Endothelial-cell proliferation in experimental tumours. Br J Cancer (1982) 46:711–20. doi:10.1038/bjc.1982.263
10. Vaupel P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin Radiat Oncol (2004) 14:198–206. doi:10.1016/j.semradonc.2004.04.008
11. Chaplin DJ, Trotter MJ. Chemical modifiers of tumor blood flow. In: Vaupel P, Jain RK, editors. Tumor Blood Supply and Metabolic Microenvironment. Stuttgart; New York: Gustav Fischer Verlag (1991). p. 65–85.
12. Kimura H, Braun RD, Ong ET, Hsu R, Secomb TW, Papahadjopoulos D, et al. Fluctuations in red cell flux in tumor microvessels can lead to transient hypoxia and reoxygenation in tumor parenchyma. Cancer Res (1996) 56:5522–8.
13. Bayer C, Shi K, Astner ST, Maftei CA, Vaupel P. Acute versus chronic hypoxia: why a simplified classification is simply not enough. Int J Radiat Oncol Biol Phys (2011) 80:965–8. doi:10.1016/j.ijrobp.2011.02.049
14. Konerding MA, Malkusch W, Klapthor B, van Ackern C, Fait E, Hill SA, et al. Evidence for characteristic vascular patterns in solid tumours: quantitative studies using corrosion casts. Br J Cancer (1999) 80:724–32. doi:10.1038/sj.bjc.6690416
15. Reinhold HS, van der Berg-Blok AE. Circulation physiology of tumors. In: Kallman RF, editor. Rodent Tumor Models in Experimental Cancer Therapy. New York: Pergamon Press (1987). p. 39–42.
16. Vaupel P, Kallinowski F, Okunieff P. Blood flow, oxygen and nutrient supply, and metabolic micro-environment of human tumors: a review. Cancer Res (1989) 49:6449–65.
17. Baronzio G, Freitas I, Kwaan HC. Tumor microenvironment and hemo-rheological abnormalities. Semin Thromb Hemost (2003) 29:489–97. doi:10.1055/s-2003-44557
18. Vaupel P, Mayer A, Höckel M. Impact of hemoglobin levels on tumor oxygenation: the higher, the better? Strahlenther Onkol (2006) 182:63–71. doi:10.1007/s00066-006-1543-7
19. Overgaard J, Nielsen JE, Grau C. Effect of carboxyhemoglobin on tumor oxygenation unloading capacity in patients with squamous cell carcinoma of the head and neck. Int J Radiat Oncol Biol Phys (1992) 22:407–10. doi:10.1016/0360-3016(92)90842-6
20. Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med (1997) 3:177–82. doi:10.1038/nm0297-177
21. Dewhirst MW, Cao Y, Moeller B. Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat Rev Cancer (2008) 8:425–37. doi:10.1038/nrc2397
22. Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic and molecular aspects. J Natl Cancer Inst (2001) 93:266–76. doi:10.1093/jnci/93.4.266
23. Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer (1955) 9:539–549.
24. Tannock IF. The relationship between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br J Cancer (1968) 22:258–273.
25. Horsman MR, Mortensen LS, Petersen JB, Busk M, Overgaard J. Imaging hypoxia to improve radiotherapy outcome. Nat Rev Clin Oncol (2012) 9:674–87. doi:10.1038/nrclinonc.2012.171
26. Durand RE, Sham E. The lifetime of hypoxic tumor cells. Int J Radiat Oncol Biol Phys (1998) 42:711–5. doi:10.1016/S0360-3016(98)00305-8
27. Hokland SL, Nielsen T, Busk M, Horsman MR. Imaging tumour physiology and vasculature to predict and assess response to heat. Int J Hyperthermia (2010) 26:264–72. doi:10.3109/02656730903585982
28. Brown JM. Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br J Radiol (1979) 52:650–656.
29. Chaplin DJ, Olive PL, Durand RE. Intermittent blood flow in a murine tumor: radiobiological effects. Cancer Res (1987) 47:597–601.
30. Horsman MR. Nicotinamide and other benzamide analogs as agents for overcoming hypoxic cell radiation resistance in tumours. Acta Oncol (1995) 34:571–587.
31. Maftei CA, Bayer C, Shi K, Astner ST, Vaupel P. Quantitative assessment of hypoxia subtypes in microcirculatory supply units of malignant tumors using (immune-)fluorescence techniques. Strahlenther Onkol (2011) 187:260–6. doi:10.1007/s00066-010-2216-0
32. Powell ME, Hill SA, Saunders MI, Hoskin PJ, Chaplin DJ. Human tumor blood flow is enhanced by nicotinamide and carbogen breathing. Cancer Res (1997) 57:5261–4.
33. Begg AC, Janssen H, Sprong D, Hofland I, Blommestijn G, Raleigh JA, et al. Hypoxia and perfusion measurements in human tumours – initial experience with pimonidazole and IUdR. Acta Oncol (2001) 40:924–8. doi:10.1080/02841860152708198
34. Janssen HL, Haustermans KM, Sprong D, Blommestijn G, Hofland I, Hoebers FJ, et al. HIF-1A, pimonidazole, and iododeoxyuridine to estimate hypoxia and perfusion in human head-and-neck tumors. Int J Radiat Oncol Biol Phys (2002) 54:1537–49. doi:10.1016/S0360-3016(02)03935-4
35. Moulder JE, Rockwell S. Hypoxic fractions of solid tumour. Int J Radiat Oncol Biol Phys (1984) 10:695–712.
36. Vaupel P, Höckel M, Mayer A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal (2007) 9:1221–35. doi:10.1089/ars.2007.1628
37. Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem (2007) 101:937–49. doi:10.1002/jcb.21187
38. Vaupel P. Blood Flow, Oxygenation, Tissue pH Distribution and Bioenergetic Status of Tumors. Berlin: Ernst Schering Research Foundation (1994). Lecture 23.
39. Gutmann R, Leunig M, Feyh J, Goetz AE, Messmer K, Kastenbauer E, et al. Interstital hypertension in head and neck tumors in patients: correlation with tumor size. Cancer Res (1992) 52:1993–5.
40. Less JR, Posner MC, Boucher Y, Borochovitz D, Wolmark N, Jain RK. Interstitial hypertension in human breast and colorectal tumors. Cancer Res (1992) 52:6371–4.
41. Milosevic M, Fyles A, Hedley D, Pintilie M, Levin W, Manchul L, et al. Interstitial fluid pressure predicts survival in patients with cervix cancer independent of clinical prognostic factors and tumor oxygen measurements. Cancer Res (2001) 61:6400–5.
42. Heldin C-H, Rubin K, Pietras K, Östman A. High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer (2004) 4:806–13.
43. Guyton AC, Hall JE. Textbook of Medical Physiology. 11th edit ed. Philadelphia: Elsevier (2006).
44. Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res (1990) 50:4478–84.
45. Netti PA, Baxter LT, Boucher Y, Skalak R, Jain RK. Time-dependent behaviour of interstitial fluid pressure in solid tumors: implications for drug delivery. Cancer Res (1995) 55:5451–8.
46. vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirement of cell proliferation. Science (2009) 324:1029–33. doi:10.1126/science.1160809
47. Zu XL, Guppy M. Cancer metabolism: facts, fantasy and fiction. Biochem Biophys Res Commun (2004) 313:459–65. doi:10.1016/j.bbrc.2003.11.136
48. Vaupel P. Pathophysiology of solid tumors. In: Molls M, Vaupel P, Nieder C, Anscher MS, editors. The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies. Berlin; New York: Springer (2009). p. 51–92.
49. Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, et al. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys (2001) 51:349–53. doi:10.1016/S0360-3016(01)01630-3
50. Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol (2004) 14:267–74. doi:10.1016/j.semradonc.2004.04.004
51. Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, et al. Targeting lactate-fueled respiration selectively kills hypoxic cells in mice. J Clin Invest (2008) 118:3930–42. doi:10.1172/JCI36843
52. Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res (1989) 49:4373–84.
53. Stubbs M, McSheehy PM, Griffiths JR, Bashford CL. Causes and consequences of tumour acidity and implications for treatment. Mol Med Today (2000) 6:15–9. doi:10.1016/S1357-4310(99)01615-9
54. Vaupel P. Physiological properties of malignant tumours. NMR Biomed (1992) 5:220–5. doi:10.1002/nbm.1940050505
56. Gerweck LE. Tumor pH: implications for treatment and novel drug design. Semin Radiat Oncol (1998) 8:176–82. doi:10.1016/S1053-4296(98)80043-X
57. Mueller-Klieser W, Walenta S. Geographical mapping of metabolites in biological tissue with quantitative bioluminescence and single photon imaging. Histochem J (1993) 25:407–20. doi:10.1007/BF00157805
58. Vaupel P, Kelleher DK, Engel T. Stable bioenergetic status despite sunstantial changes in blood flow and tissue oxygenation. Br J Cancer (1994) 69:46–9. doi:10.1038/bjc.1994.7
59. Griffiths JR, Cady E, Edwards RH, McCready VR, Wilkie DR, Wiltshaw E. 31P-NMR studies of a human tumour in situ. Lancet (1983) 1:1435–6. doi:10.1016/S0140-6736(83)92375-9
60. Hossmann KA, Niebuhr I, Tamura M. Local cerebral blood flow and glucose consumption of rats with experimental gliomas. J Cereb Blood Flow Metab (1982) 2:25–32. doi:10.1038/jcbfm.1982.3
61. Busse M, Vaupel P. Accumulation of purine catabolites in solid tumors exposed to therapeutic hyperthermia. Experientia (1996) 52:469–73. doi:10.1007/BF01919318
62. Vaupel P, Mayer A. Can respiratory hyperoxia mitigate adenosine-driven suppression of antitumor immunity? Ann Transl Med (2015) 3:292. doi:10.3978/j.issn.2305-5839.2015.09.23
63. Vaupel P, Mayer A. Hypoxia-driven adenosine accumulation: a crucial microenvironmental factor promoting tumor progression. Adv Exp Med Biol (2016) 876:177–83. doi:10.1007/978-1-4939-3023-4-22
64. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res (1997) 57:2602–5.
65. Ohta A, Sitkovsky M. Role of G- protein-coupled adenosine-receptors in downregulation of inflammation and protection from tissue damage. Nature (2001) 414:916–20. doi:10.1038/414916a
66. Antonioli L, Blandizzi C, Pacher P, Haskó G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer (2013) 13:842–57. doi:10.1038/nrc3613
67. Vaupel P, Multhoff G. Adenosine can thwart antitumor immune responses elicited by ionizing radiation? Strahlenther Onkol (2016) doi:10.1007/s00066-016-0948-1
68. Schwarz G. Über Desensibiliserung gegen Röntgen– und Radiumstrahlen. Münchener Med Wschr (1909) 24:1–2.
69. Müller C. Eine neue Behandlungsmethode bösartiger Geschwülste. Münchener Med Wschr (1910) 28:1490–1493.
70. Gray LH, Conger AD, Ebert M, Hornsey S, Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol (1953) 26:638–648.
71. Horsman MR, Wouters BG, Joiner MC, Overgaard J. The oxygen effect and fractionated radiotherapy. 4th ed. In: van der Kogel AJ, Joiner M, editors. Basic Clinical Radiobiology for Radiation Oncologists. London: Hodder Arnold (2009). p. 207–16.
72. Howard-Flanders P, Moore D. The time interval after pulsed irradiation within which injury to bacteria can be modified by dissolved oxygen. I. A search for an effect of oxygen 0.02 second after pulsed irradiation. Radiat Res (1958) 9:422–37. doi:10.2307/3570768
73. Michael BD, Adams GE, Hewitt HB, Jones WB, Watts ME. A posteffect of oxygen in irradiated bacteria: a submillisecond fast mixing study. Radiat Res (1973) 54:239–51. doi:10.2307/3573702
74. Joiner MC. Linear energy transfer and relative biological effectiveness. 4th ed. In: van der Kogel AJ, Joiner M, editors. Basic Clinical Radiobiology for Radiation Oncologists. London: Hodder Arnold (2009). p. 68–77.
75. Horsman MR. Hypoxia in tumours: its relevance, identification and modification. In: Beck-Bornholdt HP, editor. Current Topics in Clinical Radiobiology of Tumours. Berlin: Springer-Verlag (1993). p. 99–112.
76. Raleigh JA, Dewhirst MW, Thrall DE. Measuring tumor hypoxia. Semin Radiat Oncol (1996) 6:37–45. doi:10.1016/S1053-4296(96)80034-8
77. Krohn KA, Link JM, Mason RP. Molecular imaging of hypoxia. J Nucl Med (2008) 49(Suppl 2):129S–48S. doi:10.2967/jnumed.107.045914
78. Horsman MR. Measurement of tumor oxygenation. Int J Radiat Oncol Biol Phys (1998) 42:701–4. doi:10.1016/S0360-3016(98)00332-0
79. Kolstad P. Intercapillary distance, oxygen tension and local recurrence in cervix cancer. Scand J Clin Lab Invest (1968) 106:145–57. doi:10.3109/00365516809168210
80. Lauk S, Skates S, Goodman M, Suit HD. Morphometric study of the vascularity of oral squamous cell carcinomas and its relation to outcome of radiation therapy. Eur J Cancer Clin Oncol (1989) 25:1431–40. doi:10.1016/0277-5379(89)90101-6
81. Révész L, Siracka E, Siracky J, Delides G, Pavlaki K. Variation of vascular density within and between tumors of the uterine cervix and its predictive value for radiotherapy. Int J Radiat Oncol Biol Phys (1989) 16:1161–3. doi:10.1016/0360-3016(89)90274-5
82. Bussink J, Kaanders JH, van der Kogel AJ. Tumor hypoxia at the micro-regional level: clinical relevance and predictive value of exogenous and endogenous hypoxic cell markers. Radiother Oncol (2003) 67:3–15. doi:10.1016/S0167-8140(03)00011-2
83. Hui EP, Chan AT, Pezzella F, Turley H, To KF, Poon TC, et al. Coexpression of hypoxia-inducible factors 1α and 2α, carbonic anhydrase IX, and vascular endothelial growth factor in nasopharyngeal carcinoma and relationship to survival. Clin Cancer Res (2002) 8:2595–604.
84. Airley RE, Loncaster J, Raleigh JA, Harris AL, Davidson SE, Hunter RD, et al. GLUT-1 and CAIX as intrinsic markers of hypoxia in carcinoma of the cervix: relationship to pimonidazole binding. Int J Cancer (2003) 104:85–91. doi:10.1002/ijc.10904
85. Le QT, Sutphin PD, Raychaudhuri S, Yu SC, Terris DJ, Lin HS, et al. Identification of osteopontin as a prognostic plasma marker for head and neck squamous cell carcinomas. Clin Cancer Res (2003) 9:59–67.
86. Overgaard J, Eriksen JG, Nordsmark M, Alsner J, Horsman MR. Plasma osteopontin predicts hypoxia and response to the hypoxic sensitizer nimoprazole in radiotherapy of head and neck cancer. Results from the randomized DAHANCA 5 trial. Lancet Oncol (2005) 6:757–64. doi:10.1016/S1470-2045(05)70292-8
87. Gatenby RA, Kessler HB, Rosenblum JS, Coia LR, Moldofsky PJ, Hartz WH, et al. Oxygen distribution in squamous cell carcinoma metastases and its relationship to outcome of radiation therapy. Int J Radiat Oncol Biol Phys (1988) 14:831–838.
88. Vaupel P, Schlenger K, Knoop M, Hoeckel M. Oxygenation of human tumors: evaluation of tissue oxygen distribution in breast cancers by computerized O2 tension measurements. Cancer Res (1991) 51:3316–22.
89. Höckel M, Schlenger K, Knoop C, Vaupel P. Oxygenation of carcinomas of the uterine cervix: evaluation by computerized O2 tension measurements. Cancer Res (1991) 51:6096–102.
90. Höckel M, Knoop C, Schlenger K, Vorndran B, Baussmann E, Mitze M, et al. Intratumoral pO2 predicts survival in advanced cancer of the uterine cervix. Radiother Oncol (1993) 26:45–50.
91. Höckel M, Schlenger K, Aral B, Mitze M, Schäffer U, Vaupel P. Association between tumor hypoxia and malignant progression in advanced cancer of the uterine cervix. Cancer Res (1996) 56:4509–15.
92. Knocke TH, Weitmann HD, Feldmann HJ, Selzer E, Pötter R. Intratumoral pO2-measurements as predictive assay in the treatment of carcinoma of the uterine cervix. Radiother Oncol (1999) 53:99–104. doi:10.1016/S0167-8140(99)00139-5
93. Fyles AW, Milosevic M, Wong R, Kavanagh MC, Pintilie M, Sun A, et al. Oxygenation predicts radiation response and survival in patients with cervix cancer. Radiother Oncol (1998) 48:149–56. doi:10.1016/S0167-8140(98)00044-9
94. Fyles A, Milosevic M, Pintilie M, Syed A, Levin W, Manchul L, et al. Long-term performance of interstitial fluid pressure and hypoxia as prognostic factors in cervix cancer. Radiother Oncol (2006) 80:132–7. doi:10.1016/j.radonc.2006.07.014
95. Lyng H, Sundfør K, Rofstad EK. Changes in tumor oxygen tension during radiotherapy of uterine cervical cancer: relationships to changes in vascular density, cell density, and frequency of mitosis and apoptosis. Int J Radiat Oncol Biol Phys (2000) 46:935–46. doi:10.1016/S0360-3016(99)00497-6
96. Nordsmark M, Overgaard M, Overgaard J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother Oncol (1996) 41:31–9. doi:10.1016/S0167-8140(96)01811-7
97. Brizel DM, Sibley GS, Prosnitz LR, Scher RL, Dewhirst MW. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int J Radiat Oncol Biol Phys (1997) 38:285–9. doi:10.1016/S0360-3016(97)00101-6
98. Stadler P, Becker A, Feldmann HJ, Hänsgen G, Dunst J, Würschmidt F, et al. Influence of the hypoxic subvolume on the survival of patients with head and neck cancer. Int J Radiat Oncol Biol Phys (1999) 44:749–54. doi:10.1016/S0360-3016(99)00115-7
99. Rudat V, Stadler P, Becker A, Vanselow B, Dietz A, Wannenmacher M, et al. Predictive value of the tumor oxygenation by means of pO2 histography in patients with advanced head and neck cancer. Strahlenther Onkol (2001) 177:462–8. doi:10.1007/PL00002427
100. Nordsmark M, Bentzen SM, Rudat V, Brizel D, Lartigau E, Stadler P, et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother Oncol (2005) 77:18–24. doi:10.1016/j.radonc.2005.06.038
101. Brizel DM, Scully SP, Harrelson JM, Layfield LJ, Bean JM, Prosnitz LR, et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res (1996) 56:941–3.
102. Nordsmark M, Alsner J, Keller J, Nielsen OS, Jensen OM, Horsman MR, et al. Hypoxia in soft tissue sarcomes: adverse impact on survival and no association with p53 mutations. Br J Cancer (2001) 84:1070–5. doi:10.1054/bjoc.2001.1728
103. Turaka A, Buyyounouski MK, Hanlon AL, Horwitz EM, Greenberg RE, Movsas B. Hypoxic prostate/muscle PO2 ratio predicts for outcome in patients with localized prostate cancer: long-term results. Int J Radiat Oncol Biol Phys (2012) 82:433–9. doi:10.1016/j.ijrobp.2011.05.037
104. Milosevic M, Warde P, Ménard C, Chung P, Toi A, Ishkanian A, et al. Tumor hypoxia predicts biochemical failure following radiotherapy for clinically localized prostate cancer. Clin Cancer Res (2012) 18:2108–14. doi:10.1158/1078-0432.CCR-11-2711
105. Hill RP, Marie-Egyptienne DT, Hedley DW. Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol (2009) 19:106–11. doi:10.1016/j.semradonc.2008.12.002
106. Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer (2008) 8:545–54. doi:10.1038/nrc2419
107. Horsman MR, Khalil AA, Nordsmark M, Grau C, Overgaard J. Relationship between radiobiological hypoxia and direct estimates of tumour oxygenation in a mouse tumour model. Radiother Oncol (1993) 28:69–71. doi:10.1016/0167-8140(93)90188-E
108. Yaromina A, Zips D, Thames HD, Eicheler W, Krause M, Rosner A, et al. Pimonidazole labeling and response to fractionated irradiation of five human squamous cell carcinoma (hSCC) lines in nude mice: the need for a multivariate approach in biomarker studies. Radiother Oncol (2006) 81:122–9. doi:10.1016/j.radonc.2006.08.010
109. Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer (2006) 6:583–92. doi:10.1038/nrc1893
110. Durand RE. Keynote address: the influence of microenvironmental factors on the activity of radiation and drugs. Int J Radiat Oncol Biol Phys (1991) 20:253–8. doi:10.1016/0360-3016(91)90100-I
111. Vaupel P, Osinsky S. Tumor hypoxia and acidosis, and the response to chemotherapy. In: Osinsky S, Friese H, Vaupel P, editors. Tumor Hypoxia in the Clinical Setting. Kiev: Natl. Acad. Sci. Ukraine (2011). p. 93–110.
112. Teicher BA, Lazo JS, Sartorelli AC. Classification of antineoplastic agents by their selective toxicities toward oxygenated and hypoxic tumor cells. Cancer Res (1981) 41:73–81.
113. Grau C, Overgaard J. Effect of cancer chemotherapy on the hypoxic fraction of a solid tumor measured using a local tumor control assay. Radiother Oncol (1988) 13:301–9. doi:10.1016/0167-8140(88)90225-3
114. Tannock I. Response of aerobic and hypoxic cells in a solid tumor to adriamycin and cyclophosphamide and interaction of the drugs with radiation. Cancer Res (1982) 42:4921–6.
115. Horsman MR, Siemann DW. Pathophysiological effects of vascular targeting agents and the implications for combination therapies. Cancer Res (2006) 66:11520–39. doi:10.1158/0008-5472.CAN-06-2848
116. Voorhees WD, Babbs CF. Hydralazine-enhanced selective heating of transmissible venereal tumor implants in dogs. Eur J Cancer Clin Oncol (1982) 19:1027–33. doi:10.1016/0277-5379(82)90252-8
117. Horsman MR, Christensen KL, Overgaard J. Hydralazine-induced enhancement of hyperthermic damage in a C3H mammary carcinoma in vivo. Int J Hyperthermia (1989) 5:123–36. doi:10.3109/02656738909140442
118. Overgaard J, Bichel P. The influence of hypoxia and acidity on the hyperthermic response of malignant cells in vitro. Radiology (1977) 123:511–4. doi:10.1148/123.2.511
119. Gerweck LE, Nygaard TG, Burlett M. Response of cells to hyperthermia under acute and chronic hypoxic conditions. Cancer Res (1979) 39:966–72.
120. Mitchell JB, McPherson S, DeGraff W, Gamson J, Zabell A, Russo A. Oxygen dependence of hematoporphyrin derivative-induced photoinactivation of Chinese hamster cells. Cancer Res (1985) 45:2008–11.
121. Moan J, Sommer S. Oxygen dependence of the photosensitizing effect of hematoporphyrin derivative in NHIK 3025 cells. Cancer Res (1985) 45:1608–10.
122. Henderson BW, Miller AC. Effects of scavengers of reactive oxygen and radical species on cell survival following photodynamic treatment in vitro: comparison to ionizing radiation. Radiat Res (1986) 108:196–205. doi:10.2307/3576825
123. Henderson BW, Fingar VH. Relationship of tumor hypoxia and response to photodynamic treatment in an experimental mouse tumor. Cancer Res (1987) 47:3110–4.
124. Dougherty TJ. Photosensitizers: therapy and detection of malignant tumors. Photochem Photobiol (1987) 45:879–89. doi:10.1111/j.1751-1097.1987.tb07898.x
125. Sundfør K, Lyng H, Rofstad EK. Tumour hypoxia and vascular density as predictors of metastasis in squamous cell carcinoma of the uterine cervix. Br J Cancer (1998) 78:822–7. doi:10.1038/bjc.1998.586
126. Young SD, Marshall RS, Hill RP. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc Natl Acad Sci U S A (1988) 85:9533–7. doi:10.1073/pnas.85.24.9533
127. Rofstad EK, Rasmussen H, Galappathi K, Mathiesen B, Nilsen K, Graff BA. Hypoxia promotes lymph node metastasis in human melanoma xenografts by up-regulating the urokinase-type plasminogen activator receptor. Cancer Res (2002) 62:1847–53.
128. Rofstad EK, Galappathi K, Mathiesen B, Ruud E-BM. Fluctuating and diffusion-limited hypoxia in hypoxia-induced metastasis. Clin Cancer Res (2007) 13:19711978. doi:10.1158/1078-0432.CCR-06-1967
129. Rofstad EK, Gaustad J-V, Egeland TA, Mathiesen B, Galappathi K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int J Cancer (2010) 127:1535–46. doi:10.1002/ijc.25176
130. Lunt SJ, Chaudary N, Hill RP. The tumor microenvironment and metastatic disease. Clin Exp Metastasis (2009) 26:19–34. doi:10.1007/s10585-008-9182-2
131. Vaupel P. Metabolic microenvironment of tumor cells: a key factor in malignant progression. Exp Oncol (2010) 32:125–7.
132. Schlappack OK, Zimmermann A, Hill RP. Glucose starvation and acidosis: effect on experimental metastatic potential, DNA content and MTX resistance of murine tumour cells. Br J Cancer (1991) 64:663–70. doi:10.1038/bjc.1991.378
133. Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res (2006) 66:6699–707. doi:10.1158/0008-5472.CAN-06-0983
134. Kalliomaki T, Hill RP. Effects of tumour acidification with glucose+MIBG on the spontaneous metastatic potential of two murine cell lines. Br J Cancer (2004) 90:1842–9.
135. Ellingsen C, Hompland T, Mathiesen B, Rofstad EK. Microenvironment-associated lymph node metastasis of human cervical carcinoma xenografts. Acta Oncol (2012) 51:465–72. doi:10.3109/0284186X.2011.636751
136. Rofstad EK, Tunheim SH, Mathiesen B, Graff BA, Halsør EF, Nilsen K, et al. Pulmonary and lymph node metastasis is associated with primary tumor interstitial fluid pressure in human melanoma xenografts. Cancer Res (2002) 62:661–4.
Keywords: hypoxia, tumor microenvironment, radiotherapy, chemotherapy, malignant progression
Citation: Horsman MR and Vaupel P (2016) Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front. Oncol. 6:66. doi: 10.3389/fonc.2016.00066
Received: 17 December 2015; Accepted: 07 March 2016;
Published: 12 April 2016
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Alexandre Arcaro, University of Bern, SwitzerlandDaniele Tibullo, University of Catania, Italy
Copyright: © 2016 Horsman and Vaupel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael R. Horsman, bWlrZUBvbmNvbG9neS5hdS5kaw==