 Maria Ferraiuolo
Maria Ferraiuolo Silvia Di Agostino
Silvia Di Agostino Giovanni Blandino
Giovanni Blandino Sabrina Strano
Sabrina Strano- 1Translational Oncogenomics Unit, Department of Molecular Medicine, Regina Elena National Cancer Institute, Rome, Italy
- 2Molecular Chemoprevention Unit, Department of Molecular Medicine, Regina Elena National Cancer Institute, Rome, Italy
The p53 gene family members p53, p73, and p63 display several isoforms derived from the presence of internal promoters and alternative splicing events. They are structural homologs but hold peculiar functional properties. p53, p73, and p63 are tumor suppressor genes that promote differentiation, senescence, and apoptosis. p53, unlike p73 and p63, is frequently mutated in cancer often displaying oncogenic “gain of function” activities correlated with the induction of proliferation, invasion, chemoresistance, and genomic instability in cancer cells. These oncogenic functions are promoted either by the aberrant transcriptional cooperation of mutant p53 (mutp53) with transcription cofactors (e.g., NF-Y, E2F1, Vitamin D Receptor, Ets-1, NF-kB and YAP) or by the interaction with the p53 family members, p73 and p63, determining their functional inactivation. The instauration of these aberrant transcriptional networks leads to increased cell growth, low activation of DNA damage response pathways (DNA damage response and DNA double-strand breaks response), enhanced invasion, and high chemoresistance to different conventional chemotherapeutic treatments. Several studies have clearly shown that different cancers harboring mutant p53 proteins exhibit a poor prognosis when compared to those carrying wild-type p53 (wt-p53) protein. The interference of mutantp53/p73 and/or mutantp53/p63 interactions, thereby restoring p53, p73, and p63 tumor suppression functions, could be among the potential therapeutic strategies for the treatment of mutant p53 human cancers.
Introduction
p53, p73, and p63 proteins belong to a family evolutionarily conserved in animals. They derive from an ancestral gene by duplication and consequent divergence of the original sequence. Functional and phylogenetic analyses reveal that the founding member was p63, followed by p73 and lastly p53 (1–4). In fact, at the sequence level, p63 and p73 display elevated homology to each other, more than to p53 (5–9). Generally, the protein structure consists of a central DNA-binding domain (DBD) (core domain) that binds to response elements of target genes (10–14). The N-terminal transcription-activation domain (TAD) is the binding-site for positive (e.g., p300/CBP and TAFII40/60) or negative regulators (e.g., MDM2 and MDMX) of gene transcription (15). The C-terminal oligomerization domain (OD) is subject to splicing and post-translational modifications, and it has been shown to influence DNA binding and transcriptional activity of the p53 family members (16) (Figure 1).
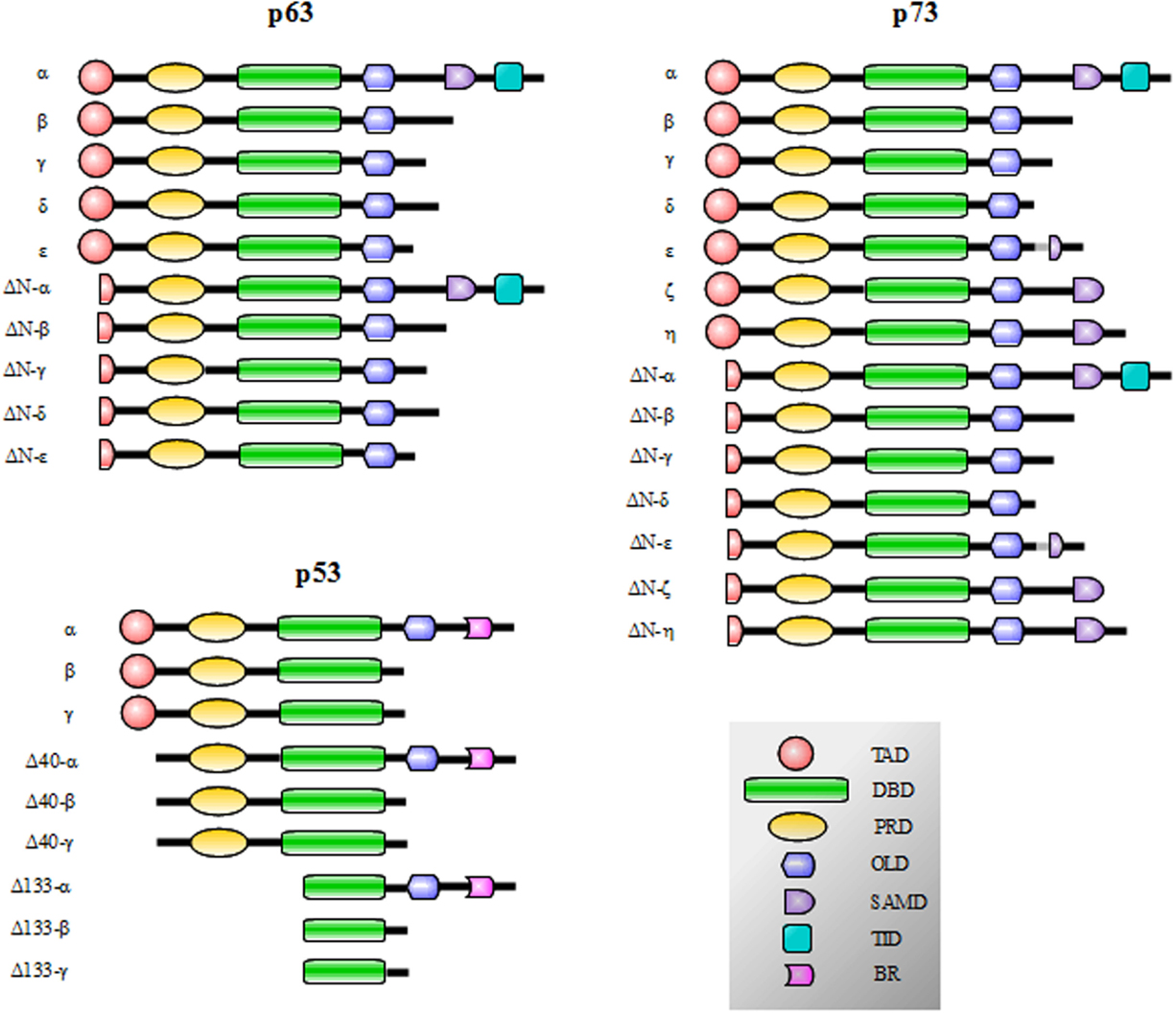
Figure 1. p53, p73, and p63 isoforms obtained by the presence of internal promoters in the sequence (ΔN isoforms) and by alternative splicing. The N-terminal transcription-activation domain (TAD) contains two subdomains (AD1 and AD2) and is the binding-site for regulators of gene transcription; in p63 and p73 proteins, the C-terminal transactivation inhibitory domain (TID) binds to the TAD preventing constitutive transcriptional activity. The Proline-rich sequence recognition domains (PRD) can recognize proline-rich motifs of interacting proteins and has been reported to be essential for the induction of apoptosis driven by p53, p73 and p63. The DNA-binding domain (DBD) is responsible for the binding to DNA consensus of target genes and the oligomerization domain (OD) enables monomers assembly in active oligomers. The sterile alpha motif domain (SAMD) of p63 and p73 is arranged in a small five-helix bundle and is involved in protein–protein interactions. The basic region (BR) in the C-terminal of p53 is involved in the control of the DNA binding affinity.
The human TP53 gene (Chr.17p13.1) is about 20 kb, contains 11 exons and encodes for the tumor suppressor p53 protein known as “the guardian of the genome” (17–28). This protein was discovered in 1979 (29–31). p53 is not only able to act as a transcription factor but it is also involved in transcription-independent response, such as apoptosis (32). TP53 is the most frequently mutated gene in human cancers (33–36). It is mostly affected by missense mutations often in the DBD of the protein (37–39). An increasing number of p53-mutated proteins can be distinct in either conformational mutants (when the mutations change the tridimensional structure of the protein) or in DNA-contact defective mutants (when the mutations affect the region designate for the binding to the DNA) (36, 40). These alterations lead to the inability of mutant p53 proteins to fully recognize the DNA consensus sequence for wt-p53 or alter the functional interaction with pro-apoptotic partners, such as WW domain-containing oxidoreductase WOX1 (WWOX). The WWOX/wt-p53 complex is demonstrated to induce apoptosis synergistically and WWOX is essential for p53 activation and apoptosis induction (41). Strikingly, some of the p53-mutated proteins acquire new oncogenic functions [gain of function (GOF)] that strongly contribute to increasing cell proliferation, invasion, angiogenesis, genomic instability and chemoresistance in human cancers. These functions are often promoted by the interaction with sequence-specific transcription factors and the consequent activation/repression of specific target genes diverse from those recruited by wt-p53 (15, 42–58).
The TP73 gene (Chr.1p36.33) is about 65 kb and contains 14 exons. It was discovered in 1997 (59) and similar to its family members, plays an important role at different regulatory checkpoints of the cell cycle (60). However, p73 shows a peculiar function in neuronal differentiation, not shared with p53 (8, 61). p73 is rarely affected by mutation in cancer progression (62, 63) but its expression is deregulated in several human tumors, such as in hepatocellular carcinoma (64), neuroblastoma (65), lung cancer (62), prostate cancer (66) and colorectal carcinoma (67). p73 shares many p53 tumor suppression functions through the activation of the p53-target genes (p21waf1, Bax, PUMA, NOXA, IGF-BP3 and Cyclin G), resulting in the control of cell proliferation, differentiation, development and induction of apoptosis (68–79). Moreover, p73, like p53, can interact with the tumor suppressor WWOX and trigger apoptosis (80, 81). Cells exposure to DNA-damaging agents (e.g., γ-radiation and cisplatin) induces p73 protein activation and accumulation with consequent induction of DNA damage response (DDR), growth arrest and apoptosis (76, 82–88).
The TP63 gene (Chr.3q27–29) is approximately 65 kb and contains 15 exons (89, 90). Like p73, p63 can activate many p53-target genes in response to oncogenic stress or DNA damage (Bax, 14-3-3σ, p53AIP1, IGF-BP3, p21waf1 and cyclin G), it controls cell proliferation, apoptosis, differentiation and development, and shows a tissue-specific localization (79, 91–94). p63 knockout mice exhibit a lethal phenotype soon after birth. They suffer from epithelial abnormalities, concerning skin, glands, teeth and hair follicles (95). Mutations of p63 are extremely rare in malignancies in contrast to p53 mutations. However, alterations in p63 expression patterns play an important role in tumorigenesis (96–98).
The full-length forms of p73 and p63 can also bind to YAP protein in response to DNA-damaging agents and activate pro-apoptotic target genes, such as Bax and p53AIP1 (78, 87, 99–101). Thus, both p73 and p63 can promote p53-independent apoptosis (102).
Protein Structure and Respective Isoforms of the p53 Family Gene Members
p53 family gene members show a high degree of similarity in the exon/intron organization and share a similar modular protein structure previously described (Figure 1). p73 and p63 proteins display 22–29% of homology in the TAD domain, 63% in the DBD, and 42% in the OD of p53. Furthermore, critical residues in the DBD, involved in the folding and binding to target DNA sequences, are strictly conserved (22, 103–106). Moreover, p73 and p63 share a sterile alpha motif domain (SAMD) and a transactivation inhibitory domain (TID); p53, p63, and p73 contain also a proline-rich sequence recognition domain (PRD). p53 also shows an additional basic region (BR) in the C-terminal tail (16, 107, 108) (Figure 1).
p53 protein displays nine isoforms obtained by the presence of cryptic internal promoters and by alternative splicing. The result is the presence of potentially transcript inert isoforms N-terminal deleted (ΔN) and with various C-terminal tails (108) (Figure 1). The same mechanisms occur for p73 and p63: p73 displays 14 isoforms and p63 exhibits 10 isoforms (59, 90, 109–117) (Figure 1). The N-terminal truncated isoforms ΔNp73 and ΔNp63 are highly expressed in the development and display an oncogenic dominant-negative function to p73 and p63 full-length (TAp73 and TAp63, respectively) and wt-p53 (39, 114, 118–124).
mutp53, p73, and p63 Protein Interactions in Cancer
It was observed that human tumor-derived p53 mutants could bind p73α interfering with its transcriptional activity and impeding apoptosis induction (125, 126). Strano et al. (127) demonstrate that p53 mutants (p53R175H and p53D281G) associate with four p73 isoforms in vitro and in vivo (p73α, p73β, p73γ and p73δ). The interactions occur also in physiological conditions in breast cancer cell lines (T47D and SKBR3) and require the DBD of mutp53 and the DBD and OD domains of p73 isoforms. Marin et al. (126) show that the interaction between mutp53 and p73α or p73β is also governed by a polymorphism at the codon 72 of the p53 mutant proteins (e.g., mutp53R175H and mutp53V143A) that encode for Arginine (R) or Proline (P). Particularly, p53 mutants with R72 polymorphism favor binding to p73 more than the P72 polymorphism determining poor response to therapy and poor prognosis in patients (128, 129). Thus, either the type of p53 mutation and 72R/P polymorphism determine mutp53/p73 interaction (126). The mutp53/p63 interaction, in vitro and in tumor cells, is also reported (126). Gaiddon et al. (130) demonstrate that p73α, p73β, p73γ, and p73δ can interact with overexpressed or endogenous p53 mutants (R175H, H179R, Y220C, R248W and R273H) and demonstrate that p53 mutants (R175H, Y220C and R248W) can bind to p63α and p63γ. Low-affinity interactions are observed between mutp53R175H and ΔNp63α or ΔNp63γ. Moreover, they observe that the interaction between p73α and p63α or ΔNp63α is more efficient if p73α is mutated (R292H). Gaiddon et al. (130) confirm that p53 mutants require the DBD domain for the interaction with p73 or p63. Moreover, p53 mutants deleted in several regions, resulting in conformational changes of the DBD, are still able to bind p73 and p63. Thus, also the wild-type DBD of mutp53 can interact with p73 or p63 if it is in a mutant conformation (130, 131). It is also demonstrated that the heat shock protein HSC70 binds those p53 mutants that interact with p73 but not wt-p53 (130, 132), thus, other determinants could affect p53/p73 interaction (130). Strano et al. (46) demonstrate that, under physiological conditions, mutp53 interacts with p63α and p63γ in T47D and HaCat cells and in H1299 cells overexpressing mutp53R273H or mutp53R248W. They observe direct interactions mediated by the DBDs of mutp53 and p63. Mutp53D281G displays a GOF activity, it slightly binds to p73 but does not interact with p63 (46, 126, 127, 133). Moreover, mutp53D281G mutated in the TAD loses its GOF, suggesting that the TAD exerts an important oncogenic role in the GOF of this p53 mutant (15, 134, 135). Santini et al. (136) provide biochemical evidence on the interaction between mutp53R175H and p73. They use atomic force spectroscopy (AFS) (137, 138) and surface plasmons resonance (SPR) (139, 140), identifying a high interaction force and a dissociation equilibrium constant typical for specific bounds. They do not observe any interactions between wt-p53 and p73 (136), confirming the lack of in vivo evidence for the formation of wt-p53/p73 protein complex. Weissmueller et al. (141) confirm that mutant p53 is able to bind p73 and this interaction results in the reduction of p73/NF-Y inhibitory complex in pancreatic ductal adenocarcinoma. This complex displays a tumor suppressor function repressing the oncogenic platelet-derived growth factor receptor b (PDGFRb) that promotes cell invasion and metastasis. Therefore, indirectly, mutant p53 promotes PDGFRb expression disassembling the inhibitory p73/NF-Y complex. Liu et al. (142) show that TopB1 protein promotes mutp53/NF-Y and mutp53/p73/p63 complex formation, inducing chemoresistance and proliferation in cancer cells. Wang and Fersht (143) describe the aggregation kinetics of mutant p53 that co-aggregate in tetramers by trapping also wt-p53, p73, and p63 proteins in the complex. It is worth to mention the role of MDM2 in the mutp53, p63, and p73 interplay: a recent work shows that MDM2, a negative regulator of wt-p53, competes with p63 for binding to mutp53R175H and in this way p63 activity is restored; but, on the other hand, MDM2 forms a trimeric complex with p73 and mutp53R273H strongly inhibiting p73 function (144). All these are clear examples of how different mutations in p53 protein could determine distinct protein–protein interactions and cell responses.
Functional Implication of mutp53/p73/p63 Protein Interactions in Cancer
Knockout mice for p53−/−, p73−/− and p63−/− highlight the major physiological roles of these proteins (96), suggesting pivotal functions in the development of nervous and immune systems (mediated by p73), in skin and limb development (mediated by p63) and in tumor suppression (mediated primarily by p53) (111, 145–148).
The role of p73 and p63 in tumorigenesis (p53-dependent or independent) is controversial. In many tumors, these proteins are downregulated, in others they are overexpressed or their genes are amplified. This apparent incongruence is mainly due to the different isoforms, tissue-specific localization and functions exerted by these proteins (121, 130, 149, 150). The decisive effect depends on the ratio TA/ΔN of p73 and p63 isoforms, p73/p63 interactions and p73/p63 binding to the promoters of p53-target genes. (151–155).
The status of p53 in cancer cells is a determining factor in the response to anticancer treatments (156–159). Some of the GOF activities mediated by mutp53 are related to the interaction, and consequent inactivation, of p73 and p63 (Figure 2). For example, the increase in chemoresistance to etoposide or cisplatin might involve the mutp53-dependent inactivation of p73-induced cell death (127, 128, 160–162). Importantly, in the presence of mutant p53 is observed a marked reduction of the transcriptional activity of p73α, p73β, p73γ and p73δ on the p21waf1 promoter (127). Gaiddon et al. (130) show that the interaction between p73 and p53 mutants (R175H, Y220C and R283H) reduces p73 transactivation of the p21waf1 promoter, highlighting the correlation between the capability of p53 mutants to interact with p73 and inhibit its transcriptional activity. Similar results are obtained for those p53 mutants that bind to p63α and p63γ reducing p63 activation of the p21waf1 promoter (130). When mutp53 binds to the OD domain of p73, it causes the functional inhibition of p73, impairing the interactions with other modulators (162). Furthermore, when mutp53 binds to the DBD of p73, it provokes a physical sequester of p73 from the consensus sequences on the target genes (127, 163, 164). Similarly, mutp53/p63 interaction results in p63 impairment of the transcriptional activation of its target genes (Bax, p21waf1, Cyclin G, 14-3-3σ and p53AIP1) (46). The formation of mutp53/p63 complex is also directly related to promoting cell invasion and metastasis in several cancer cell lines through mutp53-dependent inactivation of TAp63 tumor suppression functions (165–167).
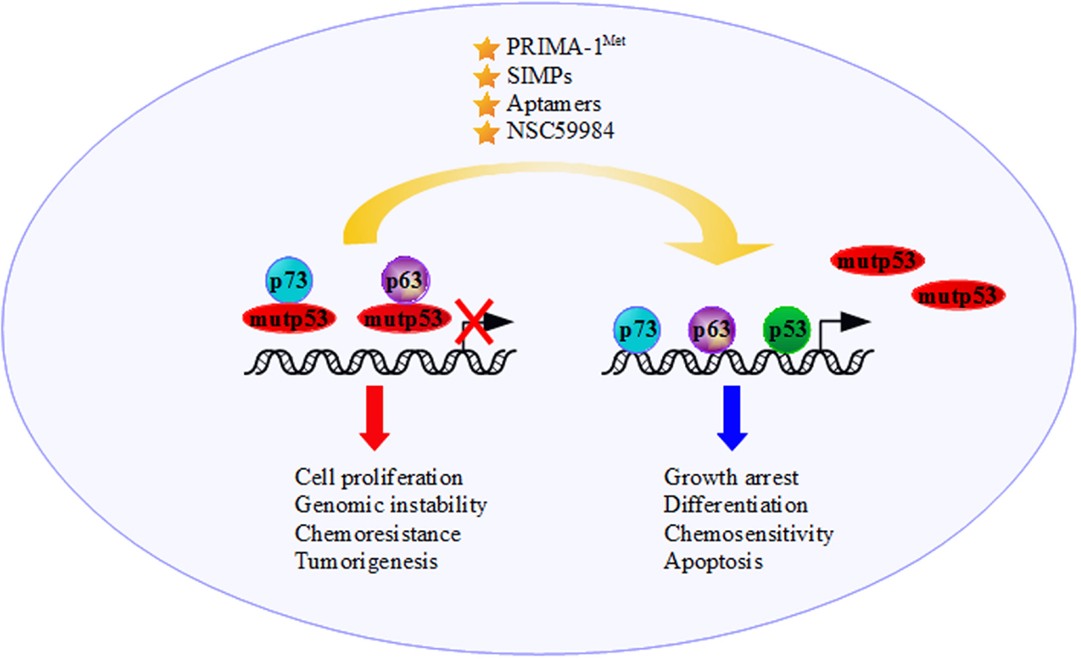
Figure 2. The presence of mutant p53 moves the tumor suppressor functions of p73 and p63 to an oncogenic outcome through binding and inactivating p73 and p63 transcriptional activity. The development of new anticancer strategies, such as increasing p73/p63 activities and employing molecules that interfere with mutantp53/p73 and/or mutantp53/p63 interactions [e.g., the molecule p53 reactivation and induction of massive apoptosis (PRIMA-1Met), short interfering mutp53 peptides (SIMPs), NSC59984 and Aptamers], could restore the p53 family’s tumor suppressor functions.
In the past, many studies were dedicated to restoring the wild-type activity of the mutant p53 proteins (168). A great number of small molecules, aiming to restore and stabilize the original DBD conformation of p53, have been developed, such as p53 reactivation and induction of massive apoptosis (PRIMA-1) and maleimide-derived molecule MIRA-1. These compounds showed great promise when tested in cancer cell lines, demonstrating the induction of apoptotic processes (169–172). Unfortunately, the application of these molecules in the clinical practice is far off because the increased activity of p53 subjects non-cancerous cells to apoptosis induction. Further research is needed to minimize the level of cell toxicity (173). Another approach, proposed by Di Agostino et al. (174), refers to the disassembling of the mutp53/p73 complex using short interfering mutp53 peptides (SIMPs) (10–15 residues) that compete specifically with p73 for the binding of mutp53 to the DBD. This results in releasing p73 from the complex, activating apoptosis and rescuing cells from chemosensitivity. Notably, SIMPs have no cytotoxic effects on cells carrying wt-p53 proteins (174).
PRIMA-1MET (APR-246, developed by APREA), a compound very similar to PRIMA-1 but much more active at low dosage, is discovered to restore mutant p53 (R273H and R175H) activity in vitro and in vivo (170, 175). Interestingly, PRIMA-1MET not only restores the pro-apoptotic function of p53 but is also involved in activating downstream target genes of the p53 family (176, 177). PRIMA-1MET alone and in combination with chemotherapeutic drugs are effective to induce apoptosis in vivo (178, 179). PRIMA-1MET has also successfully completed a Phase I clinical trial, showing a promising efficacy (https://www.clinicaltrials.gov/ct2/show/NCT00900614). This molecule seems to lead to the formation of covalent adducts on mutant p53R175H and p53R273H proteins, but the exact mechanism of action has yet to be fully elucidated (180). Moreover, recently it is discovered that a small molecule, NSC59984, can restores wt-p53 signaling via p73 activation and induces p73-dependent cell death in colorectal cancer cells, without evident toxicity toward normal cells (181).
Conclusions
Flores et al. (164) demonstrated that wt-p53, p73 and p63 are recruited onto regulatory regions of the p53-target genes inducing growth arrest, differentiation, senescence and apoptosis (Figure 2). Despite this, the DBD of mutp53, previously regarded as “dead domain” since it could not bind to the wt-p53 binding site of its target genes, acquires a new protein–protein interaction function sequestering and inactivating tumor suppression proteins, including the family members p73 and p63. This mechanism contributes to the GOF activity of mutp53 (9).
Future Perspectives
The precise tackling of GOF activity of mutant p53 might lead to the discovery of drugs with broad anticancer effects. As far as our knowledge on the molecular mechanisms governing mutant p53 oncogenic activities advances, we have learned that mutant p53 proteins are not a single entity but a protein family with high intrinsic complexities. Since mutant p53 is a partner of oncogenic multi-protein complexes, one way to severely defeat its pro-tumorigenic activity may reside in the specific targeting of its key cooperative partners. Along this line of evidence, agents that increase p73 and/or p63 activity promoting chemosensitivity could represent a promising strategy to treat tumors harboring mutant p53 proteins (122, 182, 183) (Figure 2). It could be useful to develop and validate reagents that interfere with mutp53/p73 and mutp53/p63 interactions restoring p53, p73, and p63 tumor suppression functions. NSC59984, PRIMA-1MET, SIMPs and peptide aptamers, which bind specifically to mutant p53, could be a potent strategy in cancer therapy for these tumors (174, 181, 184) (Figure 2). Undoubtedly, greater knowledge must be acquired regarding the determinants of these oncogenic multi-protein complexes in order to design and pave novel therapeutic strategies to successfully treat mutant p53 human tumors.
Author Contributions
The authors, MF and SDA, provided the structure and the writing of the article, which was integrated and supervised by SS and GB.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Tania Merlino for kindly revising the English.
References
1. Yang A, Kaghad M, Wang Y, Gillett E, Fleming MD, Dotsch V, et al. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol Cell (1998) 2:305–16. doi:10.1016/S1097-2765(00)80275-0
2. Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature (1999) 398:714–8. doi:10.1038/19539
3. Yang A, Kaghad M, Caput D, McKeon F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet (2002) 18:90–5. doi:10.1016/S0168-9525(02)02595-7
4. Bergholz J, Xiao ZX. Role of p63 in development, tumorigenesis and cancer progression. Cancer Microenviron (2012) 5:311–22. doi:10.1007/s12307-012-0116-9
6. Kaelin WG Jr. The emerging p53 gene family. J Natl Cancer Inst (1999) 91:594–8. doi:10.1093/jnci/91.7.594
8. Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature (2000) 404:99–103. doi:10.1038/35003607
9. Monti O, Damalas A, Strano S, Blandino G. p73, p63 and mutant p53: members of protein complexes floating in cancer cells. 2007 ed. In: Hainaut P, Wiman KG, editors. 25 Years of p53 Research. Dordrecht: Springer (2007). p. 223–32.
10. Melino G, Lu X, Gasco M, Crook T, Knight RA. Functional regulation of p73 and p63: development and cancer. Trends Biochem Sci (2003) 28:663–70. doi:10.1016/j.tibs.2003.10.004
11. Harms K, Nozell S, Chen X. The common and distinct target genes of the p53 family transcription factors. Cell Mol Life Sci (2004) 61:822–42. doi:10.1007/s00018-003-3304-4
12. Koutsodontis G, Vasilaki E, Chou WC, Papakosta P, Kardassis D. Physical and functional interactions between members of the tumour suppressor p53 and the Sp families of transcription factors: importance for the regulation of genes involved in cell-cycle arrest and apoptosis. Biochem J (2005) 389:443–55. doi:10.1042/BJ20041980
13. Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ (2010) 17:901–11. doi:10.1038/cdd.2010.35
14. Amelio I, Melino G. The p53 family and the hypoxia-inducible factors (HIFs): determinants of cancer progression. Trends Biochem Sci (2015) 40:425–34. doi:10.1016/j.tibs.2015.04.007
15. Lin J, Teresky AK, Levine AJ. Two critical hydrophobic amino acids in the N-terminal domain of the p53 protein are required for the gain of function phenotypes of human p53 mutants. Oncogene (1995) 10:2387–90.
16. Sauer M, Bretz AC, Beinoraviciute-Kellner R, Beitzinger M, Burek C, Rosenwald A, et al. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res (2008) 36:1900–12. doi:10.1093/nar/gkn044
18. Soddu S, Blandino G, Scardigli R, Coen S, Marchetti A, Rizzo MG, et al. Interference with p53 protein inhibits hematopoietic and muscle differentiation. J Cell Biol (1996) 134:193–204. doi:10.1083/jcb.134.1.193
19. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell (1997) 88:323–31. doi:10.1016/S0092-8674(00)81871-1
20. Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev (1998) 12:2973–83. doi:10.1101/gad.12.19.2973
21. Ashcroft M, Kubbutat MH, Vousden KH. Regulation of p53 function and stability by phosphorylation. Mol Cell Biol (1999) 19:1751–8. doi:10.1128/MCB.19.3.1751
22. Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem (1999) 274:36031–4. doi:10.1074/jbc.274.51.36031
23. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature (2000) 408:307–10. doi:10.1038/35042675
25. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer (2006) 6:909–23. doi:10.1038/nrc2012
26. Yang A, Zhu Z, Kapranov P, McKeon F, Church GM, Gingeras TR, et al. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol Cell (2006) 24:593–602. doi:10.1016/j.molcel.2006.10.018
27. Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol (2008) 9:702–12. doi:10.1038/nrm2451
29. Kress M, May E, Cassingena R, May P. Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol (1979) 31:472–83.
30. Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature (1979) 278:261–3. doi:10.1038/278261a0
31. Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell (1979) 17:43–52. doi:10.1016/0092-8674(79)90293-9
32. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell (2009) 137:413–31. doi:10.1016/j.cell.2009.04.037
33. Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res (1994) 54:4855–78.
34. Hainaut P, Soussi T, Shomer B, Hollstein M, Greenblatt M, Hovig E, et al. Database of p53 gene somatic mutations in human tumors and cell lines: updated compilation and future prospects. Nucleic Acids Res (1997) 25:151–7. doi:10.1093/nar/25.1.151
35. Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer (2009) 9:701–13. doi:10.1038/nrc2693
36. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev (2012) 26:1268–86. doi:10.1101/gad.190678.112
37. Prives C, Hall PA. The p53 pathway. J Pathol (1999) 187:112–26. doi:10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.3.CO;2-V
38. Wong KB, DeDecker BS, Freund SM, Proctor MR, Bycroft M, Fersht AR. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc Natl Acad Sci U S A (1999) 96:8438–42. doi:10.1073/pnas.96.15.8438
39. Chan WM, Siu WY, Lau A, Poon RY. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol (2004) 24:3536–51. doi:10.1128/MCB.24.8.3536-3551.2004
40. Joerger AC, Fersht AR. Structural biology of the tumor suppressor p53 and cancer-associated mutants. Adv Cancer Res (2007) 97:1–23. doi:10.1016/S0065-230X(06)97001-8
41. Chang NS, Doherty J, Ensign A, Schultz L, Hsu LJ, Hong Q. WOX1 is essential for tumor necrosis factor-, UV light-, staurosporine-, and p53-mediated cell death, and its tyrosine 33-phosphorylated form binds and stabilizes serine 46-phosphorylated p53. J Biol Chem (2005) 280:43100–8. doi:10.1074/jbc.M505590200
42. Blandino G, Levine AJ, Oren M. Mutant p53 gain of function: differential effects of different p53 mutants on resistance of cultured cells to chemotherapy. Oncogene (1999) 18:477–85. doi:10.1038/sj.onc.1202314
43. Murphy KL, Dennis AP, Rosen JM. A gain of function p53 mutant promotes both genomic instability and cell survival in a novel p53-null mammary epithelial cell model. FASEB J (2000) 14:2291–302. doi:10.1096/fj.00-0128com
44. Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res (2000) 60:6788–93.
45. Matas D, Sigal A, Stambolsky P, Milyavsky M, Weisz L, Schwartz D, et al. Integrity of the N-terminal transcription domain of p53 is required for mutant p53 interference with drug-induced apoptosis. EMBO J (2001) 20:4163–72. doi:10.1093/emboj/20.15.4163
46. Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O, Baccarini A, et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J Biol Chem (2002) 277:18817–26. doi:10.1074/jbc.M201405200
47. Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell (2006) 10:191–202. doi:10.1016/j.ccr.2006.08.013
48. Li Y, Prives C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene (2007) 26:2220–5. doi:10.1038/sj.onc.1210311
49. Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell (2007) 12:303–12. doi:10.1016/j.ccr.2007.10.001
50. Strano S, Dell’Orso S, Di Agostino S, Fontemaggi G, Sacchi A, Blandino G. Mutant p53: an oncogenic transcription factor. Oncogene (2007) 26:2212–9. doi:10.1038/sj.onc.1210296
51. Weisz L, Damalas A, Liontos M, Karakaidos P, Fontemaggi G, Maor-Aloni R, et al. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res (2007) 67:2396–401. doi:10.1158/0008-5472.CAN-06-2425
52. Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene (2007) 26:2202–11. doi:10.1038/sj.onc.1210294
53. Fontemaggi G, Dell’Orso S, Trisciuoglio D, Shay T, Melucci E, Fazi F, et al. The execution of the transcriptional axis mutant p53, E2F1 and ID4 promotes tumor neo-angiogenesis. Nat Struct Mol Biol (2009) 16:1086–93. doi:10.1038/nsmb.1669
54. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell (2009) 139:1327–41. doi:10.1016/j.cell.2009.11.026
55. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol (2010) 2:a001107. doi:10.1101/cshperspect.a001107
56. Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell (2010) 17:273–85. doi:10.1016/j.ccr.2009.11.025
57. Masciarelli S, Fontemaggi G, Di Agostino S, Donzelli S, Carcarino E, Strano S, et al. Gain-of-function mutant p53 downregulates miR-223 contributing to chemoresistance of cultured tumor cells. Oncogene (2014) 33:1601–8. doi:10.1038/onc.2013.106
58. Valenti F, Ganci F, Fontemaggi G, Sacconi A, Strano S, Blandino G, et al. Gain of function mutant p53 proteins cooperate with E2F4 to transcriptionally downregulate RAD17 and BRCA1 gene expression. Oncotarget (2015) 6:5547–66. doi:10.18632/oncotarget.2587
59. Schmale H, Bamberger C. A novel protein with strong homology to the tumor suppressor p53. Oncogene (1997) 15:1363–7. doi:10.1038/sj.onc.1201500
60. Melino G, De Laurenzi V, Vousden KH. p73: friend or foe in tumorigenesis. Nat Rev Cancer (2002) 2:605–15. doi:10.1038/nrc861
61. Cancino GI, Miller FD, Kaplan DR. p73 haploinsufficiency causes tau hyperphosphorylation and tau kinase dysregulation in mouse models of aging and Alzheimer’s disease. Neurobiol Aging (2013) 34:387–99. doi:10.1016/j.neurobiolaging.2012.04.010
62. Mai M, Yokomizo A, Qian C, Yang P, Tindall DJ, Smith DI, et al. Activation of p73 silent allele in lung cancer. Cancer Res (1998) 58:2347–9.
63. Irwin MS, Kaelin WG Jr. Role of the newer p53 family proteins in malignancy. Apoptosis (2001) 6:17–29. doi:10.1023/A:1009663809458
64. Bantel H, Simon HU. DeltaNp73beta is oncogenic in hepatocellular carcinoma by blocking apoptosis signaling via death receptors and mitochondria. Cell Cycle (2010) 9:2710–1. doi:10.4161/cc.9.14.12592
65. Casciano I, Ponzoni M, Lo Cunsolo C, Tonini GP, Romani M. Different p73 splicing variants are expressed in distinct tumour areas of a multifocal neuroblastoma. Cell Death Differ (1999) 6:391–3. doi:10.1038/sj.cdd.4400522
66. Carastro LM, Lin HY, Park HY, Kim D, Radlein S, Hampton KK, et al. Role of p73 dinucleotide polymorphism in prostate cancer and p73 protein isoform balance. Prostate Cancer (2014) 2014:129582. doi:10.1155/2014/129582
67. Lee KE, Hong YS, Kim BG, Kim NY, Lee KM, Kwak JY, et al. p73 G4C14 to A4T14 polymorphism is associated with colorectal cancer risk and survival. World J Gastroenterol (2010) 16:4448–54. doi:10.3748/wjg.v16.i35.4448
68. Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, et al. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature (1995) 377:646–9. doi:10.1038/377646a0
69. Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell (1995) 80:293–9. doi:10.1016/0092-8674(95)90412-3
70. Jost CA, Marin MC, Kaelin WG Jr. p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature (1997) 389:191–4. doi:10.1038/38298
71. Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell (1997) 90:809–19. doi:10.1016/S0092-8674(00)80540-1
72. Zhu J, Jiang J, Zhou W, Chen X. The potential tumor suppressor p73 differentially regulates cellular p53 target genes. Cancer Res (1998) 58:5061–5.
73. Okamoto K, Prives C. A role of cyclin G in the process of apoptosis. Oncogene (1999) 18:4606–15. doi:10.1038/sj.onc.1202821
74. Zaika AI, Kovalev S, Marchenko ND, Moll UM. Overexpression of the wild type p73 gene in breast cancer tissues and cell lines. Cancer Res (1999) 59:3257–63.
75. Fontemaggi G, Gurtner A, Strano S, Higashi Y, Sacchi A, Piaggio G, et al. The transcriptional repressor ZEB regulates p73 expression at the crossroad between proliferation and differentiation. Mol Cell Biol (2001) 21:8461–70. doi:10.1128/MCB.21.24.8461-8470.2001
76. Fontemaggi G, Kela I, Amariglio N, Rechavi G, Krishnamurthy J, Strano S, et al. Identification of direct p73 target genes combining DNA microarray and chromatin immunoprecipitation analyses. J Biol Chem (2002) 277:43359–68. doi:10.1074/jbc.M205573200
77. Strano S, Rossi M, Fontemaggi G, Munarriz E, Soddu S, Sacchi A, et al. From p63 to p53 across p73. FEBS Lett (2001) 490:163–70. doi:10.1016/S0014-5793(01)02119-6
78. Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, et al. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem (2001) 276:15164–73. doi:10.1074/jbc.M010484200
79. Lapi E, Iovino A, Fontemaggi G, Soliera AR, Iacovelli S, Sacchi A, et al. S100A2 gene is a direct transcriptional target of p53 homologues during keratinocyte differentiation. Oncogene (2006) 25:3628–37. doi:10.1038/sj.onc.1209401
80. Aqeilan RI, Pekarsky Y, Herrero JJ, Palamarchuk A, Letofsky J, Druck T, et al. Functional association between Wwox tumor suppressor protein and p73, a p53 homolog. Proc Natl Acad Sci U S A (2004) 101:4401–6. doi:10.1073/pnas.0400805101
81. Lin D, Cui Z, Kong L, Cheng F, Xu J, Lan F. p73 participates in WWOX-mediated apoptosis in leukemia cells. Int J Mol Med (2013) 31:849–54. doi:10.3892/ijmm.2013.1289
82. Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature (1999) 399:809–13. doi:10.1038/21697
83. Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG Jr, Levrero M, et al. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature (1999) 399:806–9. doi:10.1038/21690
85. Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, et al. p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage. Nature (1999) 399:814–7. doi:10.1038/21704
86. Costanzo A, Merlo P, Pediconi N, Fulco M, Sartorelli V, Cole PA, et al. DNA damage-dependent acetylation of p73 dictates the selective activation of apoptotic target genes. Mol Cell (2002) 9:175–86. doi:10.1016/S1097-2765(02)00431-8
87. Strano S, Monti O, Pediconi N, Baccarini A, Fontemaggi G, Lapi E, et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol Cell (2005) 18:447–59. doi:10.1016/j.molcel.2005.04.008
88. Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med (2014) 20:599–606. doi:10.1038/nm.3562
89. Senoo M, Seki N, Ohira M, Sugano S, Watanabe M, Inuzuka S, et al. A second p53-related protein, p73L, with high homology to p73. Biochem Biophys Res Commun (1998) 248:603–7. doi:10.1006/bbrc.1998.9013
90. Trink B, Okami K, Wu L, Sriuranpong V, Jen J, Sidransky D. A new human p53 homologue. Nat Med (1998) 4:747–8. doi:10.1038/nm0798-747
91. Dohn M, Zhang S, Chen X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene (2001) 20:3193–205. doi:10.1038/sj.onc.1204427
92. Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol (2006) 8:551–61. doi:10.1038/ncb1420
93. Nguyen BC, Lefort K, Mandinova A, Antonini D, Devgan V, Della Gatta G, et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev (2006) 20:1028–42. doi:10.1101/gad.1406006
94. Xu-Monette ZY, Zhang S, Li X, Manyam GC, Wang XX, Xia Y, et al. p63 expression confers significantly better survival outcomes in high-risk diffuse large B-cell lymphoma and demonstrates p53-like and p53-independent tumor suppressor function. Aging (Albany NY) (2016) 8(2):345–65.
95. Vanbokhoven H, Melino G, Candi E, Declercq W. p63, a story of mice and men. J Invest Dermatol (2011) 131:1196–207. doi:10.1038/jid.2011.84
96. Flores ER, Sengupta S, Miller JB, Newman JJ, Bronson R, Crowley D, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell (2005) 7:363–73. doi:10.1016/j.ccr.2005.02.019
97. Keyes WM, Vogel H, Koster MI, Guo X, Qi Y, Petherbridge KM, et al. p63 heterozygous mutant mice are not prone to spontaneous or chemically induced tumors. Proc Natl Acad Sci U S A (2006) 103:8435–40. doi:10.1073/pnas.0602477103
98. Su X, Chakravarti D, Flores ER. p63 steps into the limelight: crucial roles in the suppression of tumorigenesis and metastasis. Nat Rev Cancer (2013) 13:136–43. doi:10.1038/nrc3446
99. Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell (2003) 11:11–23. doi:10.1016/S1097-2765(02)00776-1
100. Lapi E, Di Agostino S, Donzelli S, Gal H, Domany E, Rechavi G, et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol Cell (2008) 32:803–14. doi:10.1016/j.molcel.2008.11.019
101. Xiao Q, Qian Z, Zhang W, Liu J, Hu E, Zhang J, et al. Depletion of CABYR-a/b sensitizes lung cancer cells to TRAIL-induced apoptosis through YAP/p73-mediated DR5 upregulation. Oncotarget (2016). doi:10.18632/oncotarget.7069
102. Blandino G, Dobbelstein M. p73 and p63: why do we still need them? Cell Cycle (2004) 3:886–94. doi:10.4161/cc.3.7.996
103. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science (1994) 265:346–55. doi:10.1126/science.8023157
104. De Laurenzi V, Melino G. Evolution of functions within the p53/p63/p73 family. Ann N Y Acad Sci (2000) 926:90–100. doi:10.1111/j.1749-6632.2000.tb05602.x
105. Marin MC, Kaelin WG Jr. p63 and p73: old members of a new family. Biochim Biophys Acta (2000) 1470:M93–100.
106. Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ (2006) 13:962–72. doi:10.1038/sj.cdd.4401914
107. Serber Z, Lai HC, Yang A, Ou HD, Sigal MS, Kelly AE, et al. A C-terminal inhibitory domain controls the activity of p63 by an intramolecular mechanism. Mol Cell Biol (2002) 22:8601–11. doi:10.1128/MCB.22.24.8601-8611.2002
108. Khoury MP, Bourdon JC. The isoforms of the p53 protein. Cold Spring Harb Perspect Biol (2010) 2:a000927. doi:10.1101/cshperspect.a000927
109. De Laurenzi V, Costanzo A, Barcaroli D, Terrinoni A, Falco M, Annicchiarico-Petruzzelli M, et al. Two new p73 splice variants, gamma and delta, with different transcriptional activity. J Exp Med (1998) 188:1763–8. doi:10.1084/jem.188.9.1763
110. De Laurenzi VD, Catani MV, Terrinoni A, Corazzari M, Melino G, Costanzo A, et al. Additional complexity in p73: induction by mitogens in lymphoid cells and identification of two new splicing variants epsilon and zeta. Cell Death Differ (1999) 6:389–90. doi:10.1038/sj.cdd.4400521
111. Pozniak CD, Radinovic S, Yang A, McKeon F, Kaplan DR, Miller FD. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science (2000) 289:304–6. doi:10.1126/science.289.5477.304
112. Fillippovich I, Sorokina N, Gatei M, Haupt Y, Hobson K, Moallem E, et al. Transactivation-deficient p73alpha (p73Deltaexon2) inhibits apoptosis and competes with p53. Oncogene (2001) 20:514–22. doi:10.1038/sj.onc.1204118
113. Ishimoto O, Kawahara C, Enjo K, Obinata M, Nukiwa T, Ikawa S. Possible oncogenic potential of DeltaNp73: a newly identified isoform of human p73. Cancer Res (2002) 62:636–41.
114. Stiewe T, Theseling CC, Putzer BM. Transactivation-deficient Delta TA-p73 inhibits p53 by direct competition for DNA binding: implications for tumorigenesis. J Biol Chem (2002) 277:14177–85. doi:10.1074/jbc.M200480200
115. Stiewe T, Zimmermann S, Frilling A, Esche H, Putzer BM. Transactivation-deficient DeltaTA-p73 acts as an oncogene. Cancer Res (2002) 62:3598–602.
116. Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, et al. p53 isoforms can regulate p53 transcriptional activity. Genes Dev (2005) 19:2122–37. doi:10.1101/gad.1339905
117. Mangiulli M, Valletti A, Caratozzolo MF, Tullo A, Sbisa E, Pesole G, et al. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res (2009) 37:6092–104. doi:10.1093/nar/gkp674
118. Yang A, McKeon F. P63 and P73: P53 mimics, menaces and more. Nat Rev Mol Cell Biol (2000) 1:199–207. doi:10.1038/35044563
119. Senoo M, Matsumura Y, Habu S. TAp63gamma (p51A) and dNp63alpha (p73L), two major isoforms of the p63 gene, exert opposite effects on the vascular endothelial growth factor (VEGF) gene expression. Oncogene (2002) 21:2455–65. doi:10.1038/sj.onc.1205330
120. Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell (2006) 9:45–56. doi:10.1016/j.ccr.2005.12.013
121. Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene (2007) 26:5169–83. doi:10.1038/sj.onc.1210337
122. Bisso A, Collavin L, Del Sal G. p73 as a pharmaceutical target for cancer therapy. Curr Pharm Des (2011) 17:578–90. doi:10.2174/138161211795222667
123. Memmi EM, Sanarico AG, Giacobbe A, Peschiaroli A, Frezza V, Cicalese A, et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci U S A (2015) 112:3499–504. doi:10.1073/pnas.1500762112
124. Orzol P, Nekulova M, Holcakova J, Muller P, Votesek B, Coates PJ. DeltaNp63 regulates cell proliferation, differentiation, adhesion, and migration in the BL2 subtype of basal-like breast cancer. Tumour Biol (2016). doi:10.1007/s13277-016-4880-x
125. Di Como CJ, Gaiddon C, Prives C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol Cell Biol (1999) 19:1438–49. doi:10.1128/MCB.19.2.1438
126. Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, Tidy JA, et al. A common polymorphism acts as an intragenic modifier of mutant p53 behaviour. Nat Genet (2000) 25:47–54. doi:10.1038/75586
127. Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y, Castagnoli L, et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J Biol Chem (2000) 275:29503–12. doi:10.1074/jbc.M003360200
128. Bergamaschi D, Gasco M, Hiller L, Sullivan A, Syed N, Trigiante G, et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell (2003) 3:387–402. doi:10.1016/S1535-6108(03)00079-5
129. Dumont P, Leu JI, Della Pietra AC III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet (2003) 33:357–65. doi:10.1038/ng1093
130. Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol (2001) 21:1874–87. doi:10.1128/MCB.21.5.1874-1887.2001
131. Bensaad K, Le Bras M, Unsal K, Strano S, Blandino G, Tominaga O, et al. Change of conformation of the DNA-binding domain of p53 is the only key element for binding of and interference with p73. J Biol Chem (2003) 278:10546–55. doi:10.1074/jbc.M208233200
132. Hinds PW, Finlay CA, Frey AB, Levine AJ. Immunological evidence for the association of p53 with a heat shock protein, hsc70, in p53-plus-ras-transformed cell lines. Mol Cell Biol (1987) 7:2863–9. doi:10.1128/MCB.7.8.2863
133. Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet (1993) 4:42–6. doi:10.1038/ng0593-42
134. Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol (1998) 18:3735–43. doi:10.1128/MCB.18.7.3735
135. Gualberto A, Aldape K, Kozakiewicz K, Tlsty TD. An oncogenic form of p53 confers a dominant, gain-of-function phenotype that disrupts spindle checkpoint control. Proc Natl Acad Sci U S A (1998) 95:5166–71. doi:10.1073/pnas.95.9.5166
136. Santini S, Di Agostino S, Coppari E, Bizzarri AR, Blandino G, Cannistraro S. Interaction of mutant p53 with p73: a surface plasmon resonance and atomic force spectroscopy study. Biochim Biophys Acta (2014) 1840:1958–64. doi:10.1016/j.bbagen.2014.02.014
137. Bizzarri AR, Cannistraro S. Atomic force spectroscopy in biological complex formation: strategies and perspectives. J Phys Chem B (2009) 113:16449–64. doi:10.1021/jp902421r
138. Bizzarri AR, Cannistraro S. The application of atomic force spectroscopy to the study of biological complexes undergoing a biorecognition process. Chem Soc Rev (2010) 39:734–49. doi:10.1039/b811426a
139. Cooper MA. Label-free screening of bio-molecular interactions. Anal Bioanal Chem (2003) 377:834–42. doi:10.1007/s00216-003-2111-y
140. Homola J. Surface plasmon resonance sensors for detection of chemical and biological species. Chem Rev (2008) 108:462–93. doi:10.1021/cr068107d
141. Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell (2014) 157:382–94. doi:10.1016/j.cell.2014.01.066
142. Liu K, Ling S, Lin WC. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol Cell Biol (2011) 31:4464–81. doi:10.1128/MCB.05574-11
143. Wang G, Fersht AR. Propagation of aggregated p53: cross-reaction and coaggregation vs. seeding. Proc Natl Acad Sci U S A (2015) 112:2443–8. doi:10.1073/pnas.1500262112
144. Stindt MH, Muller PA, Ludwig RL, Kehrloesser S, Dotsch V, Vousden KH. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene (2015) 34:4300–10. doi:10.1038/onc.2014.359
145. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature (1992) 356:215–21. doi:10.1038/356215a0
146. Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell (1999) 99:143–53. doi:10.1016/S0092-8674(00)81646-3
147. Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature (1999) 398:708–13. doi:10.1038/19531
149. Fomenkov A, Zangen R, Huang YP, Osada M, Guo Z, Fomenkov T, et al. RACK1 and stratifin target DeltaNp63alpha for a proteasome degradation in head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle (2004) 3:1285–95. doi:10.4161/cc.3.10.1155
150. Zangen R, Ratovitski E, Sidransky D. DeltaNp63alpha levels correlate with clinical tumor response to cisplatin. Cell Cycle (2005) 4:1313–5. doi:10.4161/cc.4.10.2066
151. Davison TS, Vagner C, Kaghad M, Ayed A, Caput D, Arrowsmith CH. p73 and p63 are homotetramers capable of weak heterotypic interactions with each other but not with p53. J Biol Chem (1999) 274:18709–14. doi:10.1074/jbc.274.26.18709
152. DeYoung MP, Johannessen CM, Leong CO, Faquin W, Rocco JW, Ellisen LW. Tumor-specific p73 up-regulation mediates p63 dependence in squamous cell carcinoma. Cancer Res (2006) 66:9362–8. doi:10.1158/0008-5472.CAN-06-1619
153. Di C, Sun C, Li H, Si J, Zhang H, Han L, et al. Diallyl disulfide enhances carbon ion beams-induced apoptotic cell death in cervical cancer cells through regulating Tap73/DeltaNp73. Cell Cycle (2015) 14:3725–33. doi:10.1080/15384101.2015.1104438
154. Gonfloni S, Caputo V, Iannizzotto V. P63 in health and cancer. Int J Dev Biol (2015) 59:87–93. doi:10.1387/ijdb.150045sg
155. Lucena-Araujo AR, Kim HT, Thome C, Jacomo RH, Melo RA, Bittencourt R, et al. High DeltaNp73/TAp73 ratio is associated with poor prognosis in acute promyelocytic leukemia. Blood (2015) 126:2302–6. doi:10.1182/blood-2015-01-623330
156. Strano S, Dell’Orso S, Mongiovi AM, Monti O, Lapi E, Di Agostino S, et al. Mutant p53 proteins: between loss and gain of function. Head Neck (2007) 29:488–96. doi:10.1002/hed.20531
157. Suzuki K, Matsubara H. Recent advances in p53 research and cancer treatment. J Biomed Biotechnol (2011) 2011:978312. doi:10.1155/2011/978312
158. Mirzayans R, Andrais B, Scott A, Murray D. New insights into p53 signaling and cancer cell response to DNA damage: implications for cancer therapy. J Biomed Biotechnol (2012) 2012:170325. doi:10.1155/2012/170325
159. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell (2014) 25:304–17. doi:10.1016/j.ccr.2014.01.021
160. Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG Jr. Chemosensitivity linked to p73 function. Cancer Cell (2003) 3:403–10. doi:10.1016/S1535-6108(03)00078-3
161. Strano S, Blandino G. p73-mediated chemosensitivity: a preferential target of oncogenic mutant p53. Cell Cycle (2003) 2:348–9. doi:10.4161/cc.2.4.426
162. Irwin MS. Family feud in chemosensitvity: p73 and mutant p53. Cell Cycle (2004) 3:319–23. doi:10.4161/cc.3.3.768
163. Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell (2001) 7:683–94. doi:10.1016/S1097-2765(01)00214-3
164. Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature (2002) 416:560–4. doi:10.1038/416560a
165. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell (2009) 137:87–98. doi:10.1016/j.cell.2009.01.039
166. Marine JC, Berx G. Transforming growth factor-beta and mutant p53 conspire to induce metastasis by antagonizing p63: a (ternary) complex affair. Breast Cancer Res (2009) 11:304. doi:10.1186/bcr2337
167. Neilsen PM, Noll JE, Suetani RJ, Schulz RB, Al-Ejeh F, Evdokiou A, et al. Mutant p53 uses p63 as a molecular chaperone to alter gene expression and induce a pro-invasive secretome. Oncotarget (2011) 2:1203–17. doi:10.18632/oncotarget.382
168. Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer (2001) 1:68–76. doi:10.1038/35094077
169. Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med (2002) 8:282–8. doi:10.1038/nm0302-282
170. Bykov VJ, Zache N, Stridh H, Westman J, Bergman J, Selivanova G, et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene (2005) 24:3484–91. doi:10.1038/sj.onc.1208419
171. Lambert JM, Gorzov P, Veprintsev DB, Soderqvist M, Segerback D, Bergman J, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell (2009) 15:376–88. doi:10.1016/j.ccr.2009.03.003
172. Saha MN, Chen Y, Chen MH, Chen G, Chang H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br J Cancer (2014) 110:2224–31. doi:10.1038/bjc.2014.164
173. Mandinova A, Lee SW. The p53 pathway as a target in cancer therapeutics: obstacles and promise. Sci Transl Med (2011) 3:64rv61. doi:10.1126/scitranslmed.3001366
174. Di Agostino S, Cortese G, Monti O, Dell’Orso S, Sacchi A, Eisenstein M, et al. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle (2008) 7:3440–7. doi:10.4161/cc.7.21.6995
175. Zache N, Lambert JM, Wiman KG, Bykov VJ. PRIMA-1MET inhibits growth of mouse tumors carrying mutant p53. Cell Oncol (2008) 30:411–8.
176. Saha MN, Jiang H, Yang Y, Reece D, Chang H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol Cancer Ther (2013) 12:2331–41. doi:10.1158/1535-7163.MCT-12-1166
177. Sobhani M, Abdi J, Manujendra SN, Chen C, Chang H. PRIMA-1Met induces apoptosis in Waldenstrom’s Macroglobulinemia cells independent of p53. Cancer Biol Ther (2015) 16:799–806. doi:10.1080/15384047.2015.1026482
178. Piantino CB, Reis ST, Viana NI, Silva IA, Morais DR, Antunes AA, et al. Prima-1 induces apoptosis in bladder cancer cell lines by activating p53. Clinics (2013) 68:297–303. doi:10.6061/clinics/2013(03)OA03
179. Shalom-Feuerstein R, Serror L, Aberdam E, Muller FJ, van Bokhoven H, Wiman KG, et al. Impaired epithelial differentiation of induced pluripotent stem cells from ectodermal dysplasia-related patients is rescued by the small compound APR-246/PRIMA-1MET. Proc Natl Acad Sci U S A (2013) 110:2152–6. doi:10.1073/pnas.1201753109
180. Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol (2012) 30:3633–9. doi:10.1200/JCO.2011.40.7783
181. Zhang S, Zhou L, Hong B, van den Heuvel AP, Prabhu VV, Warfel NA, et al. Small-molecule NSC59984 restores p53 pathway signaling and antitumor effects against colorectal cancer via p73 activation and degradation of mutant p53. Cancer Res (2015) 75:3842–52. doi:10.1158/0008-5472.CAN-13-1079
182. Leong CO, Vidnovic N, DeYoung MP, Sgroi D, Ellisen LW. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J Clin Invest (2007) 117:1370–80. doi:10.1172/JCI30866
183. Maslon MM, Hupp TR. Drug discovery and mutant p53. Trends Cell Biol (2010) 20:542–55. doi:10.1016/j.tcb.2010.06.005
Keywords: p53 gene family members, gain of function, homology, isoforms, protein–protein interaction, target genes, apoptosis, differentiation
Citation: Ferraiuolo M, Di Agostino S, Blandino G and Strano S (2016) Oncogenic Intra-p53 Family Member Interactions in Human Cancers. Front. Oncol. 6:77. doi: 10.3389/fonc.2016.00077
Received: 12 February 2016; Accepted: 21 March 2016;
Published: 31 March 2016
Edited by:
Ala-Eddin Al Moustafa, Qatar University, Qatar; McGill University, Canada; Concordia University, CanadaReviewed by:
Janice Anne Royds, University of Otago, New ZealandNan-Shan Chang, National Cheng Kung University, Taiwan
Copyright: © 2016 Ferraiuolo, Di Agostino, Blandino and Strano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabrina Strano, c3RyYW5vQGlmby5pdA==