 Elena Arriazu
Elena Arriazu Raffaella Pippa
Raffaella Pippa María D. Odero
María D. Odero- 1Hematology/Oncology Program, Center for Applied Medical Research (CIMA), University of Navarra, Pamplona, Spain
- 2Centre for Gene Regulation and Expression, University of Dundee, Dundee, UK
- 3Department of Biochemistry and Genetics, University of Navarra, Pamplona, Spain
- 4Instituto de Investigación Sanitaria de Navarra (IdiSNA), Pamplona, Spain
Acute myeloid leukemia (AML) is a heterogeneous malignant disorder of hematopoietic progenitor cells in which several genetic and epigenetic aberrations have been described. Despite progressive advances in our understanding of the molecular biology of this disease, the outcome for most patients is poor. It is, therefore, necessary to develop more effective treatment strategies. Genetic aberrations affecting kinases have been widely studied in AML; however, the role of phosphatases remains underexplored. Inactivation of the tumor-suppressor protein phosphatase 2A (PP2A) is frequent in AML patients, making it a promising target for therapy. There are several PP2A inactivating mechanisms reported in this disease. Deregulation or specific post-translational modifications of PP2A subunits have been identified as a cause of PP2A malfunction, which lead to deregulation of proliferation or apoptosis pathways, depending on the subunit affected. Likewise, overexpression of either SET or cancerous inhibitor of protein phosphatase 2A, endogenous inhibitors of PP2A, is a recurrent event in AML that impairs PP2A activity, contributing to leukemogenesis progression. Interestingly, the anticancer activity of several PP2A-activating drugs (PADs) depends on interaction/sequestration of SET. Preclinical studies show that pharmacological restoration of PP2A activity by PADs effectively antagonizes leukemogenesis, and that these drugs have synergistic cytotoxic effects with conventional chemotherapy and kinase inhibitors, opening new possibilities for personalized treatment in AML patients, especially in cases with SET-dependent inactivation of PP2A. Here, we review the role of PP2A as a druggable tumor suppressor in AML.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous clonal disorder of hematopoietic progenitor cells, which predominantly affects elderly adults. It is characterized by a differentiation blockade of the myeloid hematopoietic progenitor cells accompanied by uncontrolled proliferation. As a consequence, immature cells accumulate in bone marrow and peripheral blood. With the exception of acute promyelocytic leukemia, therapy for AML is not targeted, and the intensity of therapy is driven by the prognostic subgroup. Cytogenetic and molecular genetic aberrations have been postulated as the most powerful markers for survival and therapy response in AML, with patients classified into favorable, intermediate or poor prognosis (1, 2). Although major improvements have been achieved in the overall survival of adult cases ≤60 years, most of the patients are older than 60 years, and in this group only 5–15% are cured (3, 4). Furthermore, the outcome in older patients, who are unable to receive intensive chemotherapy without unacceptable side effects, remains dismal, with a median survival of only 5–10 months (5–7). Therefore, it is necessary to develop more effective treatment strategies for this disease.
The uncontrolled growth of transformed cells is caused by the deregulation of multiple cellular pathways that are involved in normal growth control (8). Reversible phosphorylation is one of the mechanisms that cells use to maintain normal homeostasis, and is involved in several processes, such as proliferation, apoptosis, and differentiation; hence, kinases and phosphatases act as important checkpoint regulators (9). Numerous studies have focused on studying the aberrant kinase behavior in AML; however, although phosphatases are also essential to maintain the correct homeostasis, their role in AML has not been fully considered. In this review, we will focus on the role of protein phosphatase 2A (PP2A), inactivation of which is a recurrent event in AML, as a druggable tumor suppressor.
Protein Phosphatase 2A
Protein phosphatase 2A, one of the main serine/threonine phosphatases in mammalian cells, is a tumor suppressor that regulates several essential functions and counteracts most of the kinase-driven intracellular signaling pathways (8, 10, 11) (Figure 1). PP2A inactivation occurs in several solid and hematological tumors, leading to sustained activation of survival pathways or inhibition of apoptotic pathways (12, 13). Studies with the potent tumor promoter okadaic acid (OA), which inhibits the enzymatic activity of PP2A, have contributed to our understanding of phosphatase functions (14).
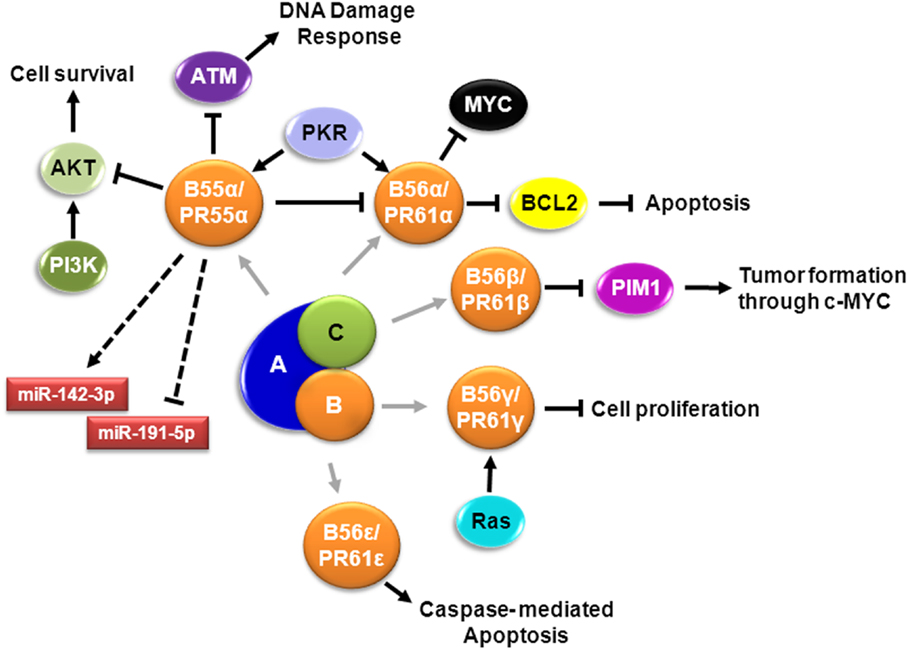
Figure 1. Signaling pathways involving PP2A in AML. Schematic representation of known PP2A complexes involving different B regulatory subunits in AML cells. Cell survival is regulated by PR55α/B55α – mediated dephosphorylation of AKT (15, 16). PR55α/B55α also supports expression of miR-142-3p and suppresses expression of miR-191-5p, relevant miRNAs in AML (17). DNA damage response is impaired by dephosphorylation of ATM by PP2A-PR55α/B55α, which translocate to the nucleus by PKR (18). Extracellular survival signals activate SRC that suppresses the B subunit; when SRC is suppressed, PR55α/B55α is expressed, resulting in dephosphorylation of PKCα and suppression of PR61α/B56α protein expression, with concomitant induction of MYC (19). Apoptosis is regulated by activation of PR61α/B56α by PKR leading to dephosphorylation of BCL2 (20). PR61β/B56β regulates PIM1 contributing to tumor formation (21). PP2A-B56γ function in G2 is crucial to sustain normal G0/G1 control and this G2 PP2A function involves modulation of endogenous RAS signaling (22). PR61ϵ/B56ϵ, which is downregulated in AML, controls caspase-mediated apoptosis (23).
Protein phosphatase 2A exists in two different forms: as a dimer and in a trimeric form (24). The dimeric form is known as the core enzyme and consists of a structural A subunit and a catalytic C subunit. These subunits are coded by two different genes, which may further generate two distinct isoforms: PP2A-A (PPP2R1A/Aα and PPP2R1B/Aβ) and PP2A-C (PPP2CA/Cα and PPP2CB/Cβ) (Table 1). The trimeric form is an active holoenzyme complex that consists of three subunits: the above-mentioned scaffold (PP2A-A) and catalytic (PP2A-C) subunits, and a regulatory B subunit (PP2A-B) (Figure 1; Table 1). In its heterotrimeric form, the structural subunit mediates the interaction between the catalytic subunit with a variety of regulatory PP2A-B subunits; whereas in the dimeric form, PP2A-A acts as a regulator by changing the catalytic specificity (25). Four unrelated families of regulatory PP2A-B subunits (B/PR55/B55, B′/PR61/B56, B′′/PR72, and B′′′/the striatins, STRN) have been identified, including at least 23 different alternative transcripts and spliced forms, which determine the substrate specificity and the intracellular location of the PP2A complex (Figure 1; Table 1) (12, 26, 27). The actual challenge is not only to explore the potential therapeutic value of PP2A activators (28–30), but also to identify the particular PP2A complexes affected in each disease, in order to develop more efficient therapeutic strategies (31).
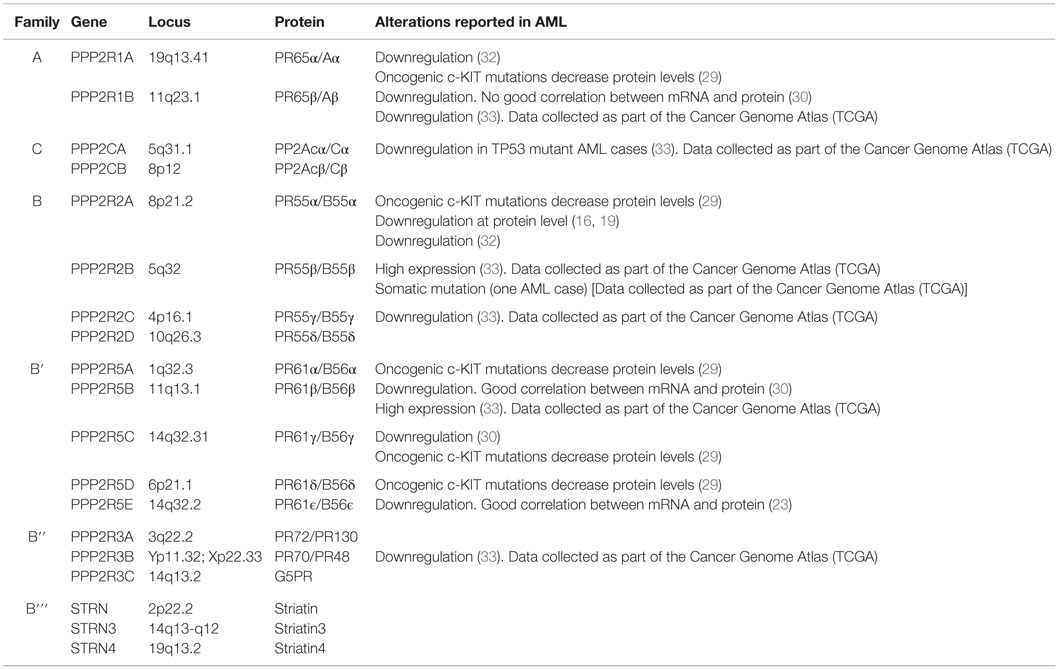
Table 1. PP2A subunits and reported alterations in AML.
The precise mechanism of assembly of active PP2A holoenzyme is still incompletely understood [reviewed in Ref. (34)]. The activity of PP2A can be regulated by post-translational modifications. Methylation and phosphorylation of residues from PP2A-C subunits modulate the formation of the complex. For instance, methylation of leucine 309 (L309) in the catalytic PP2A-C subunit, by the leucine carboxyl methyltransferase 1 (LCMT1), is indispensable for binding the PR55/B55 subunit, although it is not an essential requisite for other B families (34–36). Phosphorylation of tyrosine 307 (Y307) dramatically impairs PP2A phosphatase activity by inhibiting the interaction of PP2A-C with the PR55/B55 and PR61/B56αβγϵ subunits; whereas threonine 304 (T304) phosphorylation prevents the assembly of PR55/B55 to the core enzyme (34). Interestingly, increased phosphorylation of Y307 of PP2A-C is a common event in both cell lines and AML patient samples (30). Additionally, post-translational modification of B subunits can affect the sub-cellular localization of PP2A, influencing which proteins are targeted (9).
We and others have reported that PP2A inhibition is a recurrent event in AML, and that restoration of PP2A phosphatase activity by treatment with PP2A-activating drugs (PADs) has antileukemic effects in both c-KIT wild-type (c-KIT−) and c-KIT mutated (c-KIT+) AML cells, inducing cell growth arrest and caspase-dependent apoptosis (12, 13, 29, 30, 33, 37). Furthermore, we have shown that PADs can be used alone or in association with either kinase inhibitors or traditional chemotherapy in AML, suggesting that PP2A rescue could represent an innovative therapeutic target in this disease (29, 30, 37–41).
Mechanisms of PP2A Inactivation in AML
Transformed cells display a wide variety of mechanisms to inactivate PP2A. Several somatic mutations in PP2A subunits have been described in different types of tumors, such as melanoma, lung, colon, and breast cancers (31, 42–47). Mutations of structural PP2A-Aα and/or PP2A-Aβ subunits cause a defective binding of B and C subunits, inhibiting PP2A activity and contributing to cell transformation (44, 45). However, this seems to be an uncommon mechanism in AML. Our analysis of data from the Cancer Genome Atlas Research Network (https://tcga-data.nci.nih.gov/tcga), which analyzed the genomes of 200 patients with AML (50 with the use of whole-genome sequencing and 150 with the use of whole-exome sequencing), show that only one case has somatic mutations in PPP2R2B, the gene encoding the PP2A subunit PR55β (48).
We and other groups have shown that deregulation of some PP2A subunits, deregulated expression of the endogenous PP2A inhibitors SET or cancerous inhibitor of protein phosphatase 2A (CIP2A), or overexpression of SETBP1, contribute to PP2A inhibition in AML (28, 30, 40, 49, 50).
Deregulation of PP2A Subunits
Alterations of the PP2A subunits have been found in AML with different genetic backgrounds, contributing to the malignant process (Table 1). Downregulation of the Aβ subunit is a common event in AML (30). Most cellular PP2A holoenzymes contain the Aα isoform of the scaffold subunit, but a small fraction (10%) contain a second isoform termed Aβ. As indicated above, mutations that disrupt the ability of Aβ to form holoenzymes in vitro were identified in several types of cancer, but Sablina et al. provide the first hard evidence that loss of functional Aβ due to these cancer-associated mutations contributes to transformation (8, 51). Suppression of PP2A Aβ permits immortalized human cells to achieve a tumorigenic state through the deregulation of RaIA GTPase activity. Cancer-associated Aβ mutants fail to reverse this tumorigenic phenotype, indicating that these mutants function as null alleles (51). In addition, both Aα mutants and Aα downregulation lead to a functional haploinsufficiency that seems to induce human cell transformation by activating the AKT/PI3K signaling pathway (51, 52). However, it is likely that different sets of genetic aberrations during tumor formation require the loss of different PP2A holoenzyme complexes for the tumor progression, and this would involve the regulatory subunits, which are playing a key role directing PP2A to dephosphorylate and regulate key tumor suppressors or oncogenes (26).
Altered expression of the scaffold as well as the regulatory subunits has been reported in AML patients with c-KIT mutations, which is associated with poor outcome in AML (Table 1) (32, 53). c-KIT is a type 3 receptor tyrosine kinase, the activation of which induces proliferation, differentiation, and survival. Oncogenic c-KIT mutations reduce PP2A activity by decreasing protein levels of PR65α, PR55α, PR61α, PR61δ, and PR61γ (Table 1) (29). c-KIT-mediated growth and survival may be prevented by overexpressing PP2A-Aα in myeloid c-KIT+ cells, suggesting that restoration of PP2A activity in c-KIT+ AML patients may represent a good therapeutic strategy to overcome drug resistance (29).
The PP2A-PR55/B55 family consists of four different isoforms (α, β, γ, and δ) associated with several core-signaling pathways, including ARF/MDM2/p53, PI3K/AKT, Raf/MEK/ERK, TGFBR1/TGF-β, and Wnt/β-catenin (54), and the regulation of the cell cycle and mitosis (55, 56). In AML cells, PP2A-PR55α dephosphorylates AKT on threonine 308 (T308) (15, 16). Interestingly, increased phosphorylation of AKT correlates with poor outcome in AML (57). Ruvolo et al. quantified the expression of several transcripts in 30 newly diagnosed patients with AML, and found that the expression of PPP2CA, PPP2CB (catalytic C subunits), and PPP2R2A (regulatory PR55α subunit) was elevated in blast cells. However, when they looked at protein expression, the levels of PR55α were low in the blast cells from the AML patients, suggesting different rates of translation, degradation, cleavage, or post-translational inactivation (Table 1) (16). As expected, they saw a link between expression of PR55α and AKT dephosphorylation. This suggests that strategies to promote PR55α inactivation of AKT may be useful for the therapy of AML (16). Apart from its function regulating cell survival, PR55α has been discovered as a modulator of the expression of microRNA relevant for AML, such as miR-142-3p and miR-142-5p. These miRs are found to be mutated in 2% of AML patients, although the mechanisms and the implications are still unknown (17, 19). In addition, a recent study demonstrated that PR55α is also implicated in DNA damage response in AML. Cheng et al. found that the double-stranded RNA-activated protein kinase PKR activates PP2A by promoting the nuclear localization of PR55α/B55α. Activated PP2A in turn antagonizes autophosphorylation and activation of ATM and its association with downstream targets, preventing DNA damage response and contributing to transformed phenotype (18). Significantly, PKR is involved in AML progression (58, 59), and high PKR expression is associated with poor overall survival and shortened remission duration for AML patients (18).
Several members of the PR61/B56 family of regulatory PP2A subunits appear to have a main role in directing PP2A potential tumor-suppressive activity (21, 60–63). The PP2A-PR61/B56 regulatory family has five different isoforms (α, β, γ, δ, and ϵ) (49), which can bind directly to the core enzyme and be regulated by phosphorylation from kinases (20, 64). In acute lymphoblastic leukemia cells, it has been described that PKR promotes the mitochondrial localization of PP2A–PR61α, leading to BCL2 dephosphorylation and inactivation and, therefore, contributing to apoptosis (20). Downregulation of PP2A–PR61β and PP2A–PR61γ seems to be a common event in AML cases, leading to the inactivation of PP2A, and consequently contributing to malignant cell proliferation (Table 1) (30). PP2A–PR61β has been reported as a tumor suppressor that negatively regulates PIM1 protein kinase, enhancing the ability of c-MYC to induce lymphomas (21). PP2A–PR61γ plays a crucial role in cell proliferation (65), in part due to dephosphorylation of p53 (66). In accordance with this, it has been described that suppression of PR61γ expression contributes to the experimental transformation of human cells (31, 60). The function of PP2A–PR61γ in G2 is crucial to sustain normal G0/G1 control, and this G2 PP2A function involves modulation of endogenous RAS signaling (22). Therefore, loss of PR61β/B56β and PR61γ/B56γ could be playing a role in AML development, contributing to deregulate the correct PP2A function.
PR61ϵ/B56ϵ is recurrently downregulated at mRNA and protein level in AML patients (Table 1), contributing to cell proliferation (23). This regulatory subunit is involved in multiple signaling pathways and plays critical roles during early development (67–69). Moreover, PR61ϵ is an essential regulator of apoptosis (70), and acts as a negative regulator of MAP4K3, mediating its ability to signal to mTORC1 (71). In AML cells, PR61ϵ impairs cell proliferation, induces caspase-dependent apoptosis, affects the activation status of AKT, and reduces the colony-forming ability of the leukemic cells. Moreover, there is a good correlation between PR61ϵ downregulation and p53 levels, suggesting that the molecular effects of this B subunit in AML could occur, at least in part, via p53 (23). These results indicate that PR61ϵ downregulation has relevance in AML, and could allow distinguishing a subgroup of patients who could benefit from receiving future treatments with PP2A activators.
Aberrations of the other two families of regulatory PP2A-B subunits have been involved in several solid tumors (31, 72–75) but not in AML. Therefore, further studies are necessary to clarify the importance of these PP2A subunits in hematological malignancies.
SET/I2PP2A
The SET protein, also named I2PP2A (Inhibitor 2 of PP2A), TAF-1β or PHAP1, is a potent endogenous inhibitor of PP2A with an important role in myeloid leukemias (76). SET was first identified as an oncogene fused with the nucleoporin NUP214 (CAN) in acute undifferentiated leukemia (77), and soon after, it was described as a PP2A inhibitor (78). This protein is located mostly in the nucleus, and it is implicated in many cell processes, such as DNA replication, chromatin remodeling, gene transcription (79, 80), DNA repair (81), differentiation (82), migration (83), and cell-cycle regulation (84).
Protein phosphatase 2A is functionally inhibited as a consequence of the overexpression and/or post-translational modifications (e.g., phosphorylation) of SET, which results in an overall inhibition of PP2A phosphatase activity in both leukemic progenitors and stem cells (Figure 2) (13, 28, 29, 38, 85, 86). SET is upregulated in both hematological and solid tumors, including colorectal cancer (87) and breast cancer (88), and in most cases its effects as an oncogene are due to the concomitant inactivation of PP2A (87–90). The role of SET has been studied in depth in chronic myeloid leukemia (CML). SET is overexpressed in CML through BCR-ABL1, the constitutively active oncogenic tyrosine kinase that is essential for CML emergence, maintenance, and progression (85). Expression of BCR-ABL1 allows recruitment and activation of JAK2, which in turn, enhances β-catenin activity and induces SET-mediated inactivation of PP2A (91).
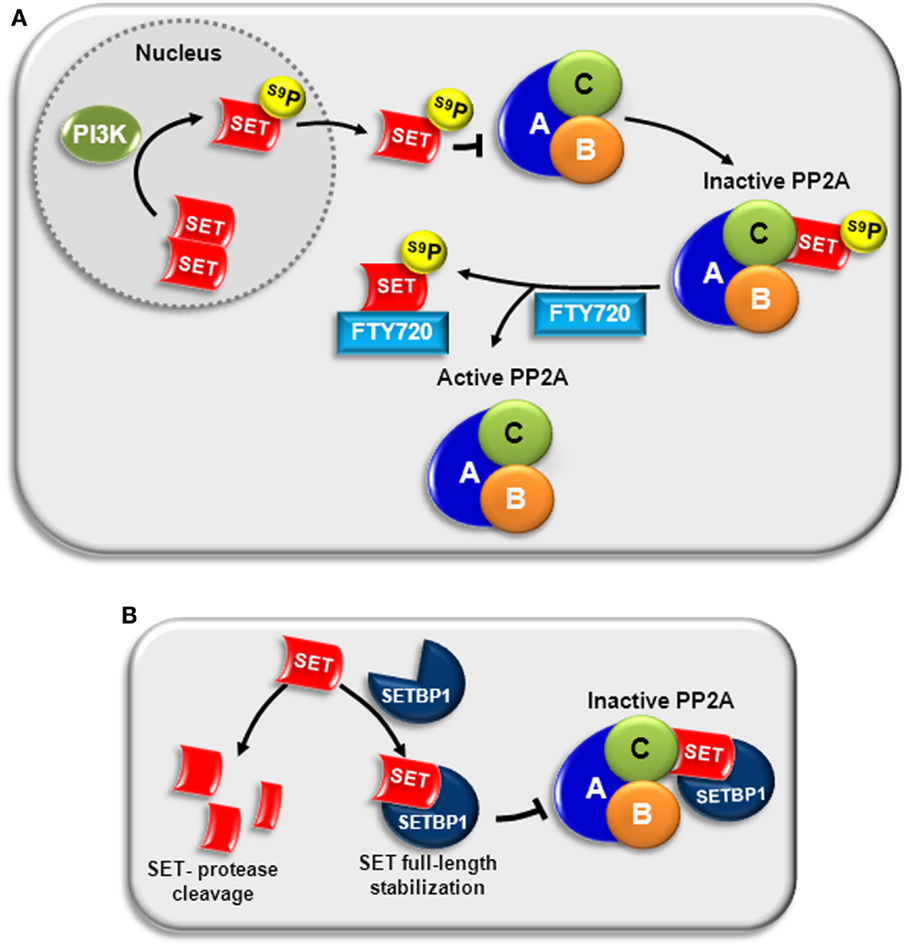
Figure 2. PP2A inactivation by SET in AML. (A) PI3K can phosphorylate SET at serine 9 (S9), located in the nuclear localization signal. This phosphorylation translocates SET to the cytosol and impairs its return to the nucleus, increasing its ability to bind to the catalytic subunit of PP2A (PP2A-C), and inactivating PP2A (92, 93). Treatment with FTY720 disrupts SET–PP2A interaction, allowing PP2A activation (39). (B) SETBP1 binds directly to SET, stabilizing full-length SET and protecting it from protease cleavage. The complex SETBP1–SET binds PP2A through SET, inhibiting PP2A activity (50).
We quantified the expression of SET in a series of patients with AML at diagnosis, observing that SET overexpression is a recurrent molecular event (60/214, 28%) associated with short overall survival in AML. Interestingly, overexpression of this oncogene also had prognostic impact in patients with normal karyotype, defining a subgroup of patients with a worse outcome. Although multivariate analysis confirmed SET overexpression as an independent prognostic marker in our series, it was associated with other adverse prognostic markers, such as monosomy 7, SETBP1 overexpression, and EVI1 overexpression. This observation suggests that SET deregulation could cooperate with other additional aberrations in the leukemogenesis program. Analysis by western blot confirmed high SET expression at protein level in both AML cell lines and patient samples. In addition, we observed that SET promotes cell viability by inhibiting the effect of PP2A in AML cells, contributing to malignancy progression (38). We also confirmed in AML cells that SET forms an inhibitory complex with PP2A-C, and that the whole structure of SET (amino-terminus and carboxy-terminus) is involved in the binding (39). Furthermore, the antileukemic effects of FTY720 and OP449, two recently discovered PADs, depend on the interaction/sequestration of SET, pointing out the importance of this oncogene in AML (37, 39). Nevertheless, despite the importance of SET overexpression and its prognostic impact in both hematological and solid tumors, little is known about the mechanisms involved in its transcriptional regulation. In addition, it has been reported that post-translational modification of SET can modulate SET affinity to PP2A. Phosphorylation at serine 9 (S9) in the nuclear localization signal, by either CKII (92) or PI3Kγ (93), impairs its returning to the nucleus, increasing its ability to inactivate PP2A (Figure 2A); whereas S171 phosphorylation by protein kinase D2 (PKD2) decreases its affinity for PP2A, lowering the inhibitory activity (94).
Our group found another mechanism that impairs PP2A activity via SET in AML: the overexpression of SETBP1 (SET binding protein 1) (50). SETBP1 upregulation leads to binding and stabilization of 39 kDa full-length SET, protecting from protease cleavage. The complex SETBP1–SET binds PP2A through SET, provoking PP2A activity inhibition and promoting cell proliferation. Interestingly, 27% of AML patients have SETBP1 overexpression, and shorter overall survival is predicted in older AML patients with this aberration (50) (Figure 2B). Later studies in other myeloid neoplasms have confirmed the important role of the SETBP1 oncogene in leukemogenesis.
Interestingly, SET is also implicated in natural killer (NK) cell cytotoxicity. After cytokine stimulation (Interleukin-12, -18, and -15), SET upregulation in human NK impairs IFN-γ production through PP2A inactivation, limiting the anti-tumor and/or anti-inflammatory effect of NK (95). Trotta et al. described a model where SET–PP2A regulates granzyme B expression at mRNA and protein levels, and therefore, determines NK cytotoxicity. They observed that SET knockdown inhibited the induction of granzyme B expression, normally induced by stimulation of NK cells with IL-2 or IL-15, limiting NK cytotoxicity (96). Other reported functions include inhibiting the DNase activity of the tumor-suppressor NM23-H1, increasing AP-1 activity, or activating MAPK signaling (97, 98). These data suggest the role of SET not only as a PP2A inhibitor but also contributing with other signaling pathways to promote tumor growth.
Cancerous Inhibitor of Protein Phosphatase 2A
Another PP2A endogenous inhibitor is CIP2A (cancerous inhibitor of PP2A) (99). CIP2A controls oncogenic cellular signals by inhibiting PP2A activity toward the oncogenic transcription factor c-MYC (8, 100, 101), which plays an important role in AML (102). CIP2A acts by impairing PP2A activity leading to the stabilization of c-MYC (99).
Cancerous inhibitor of protein phosphatase 2A is expressed in very few normal tissues but it is overexpressed in most human cancer types, where it is often associated with a clinically aggressive behavior (100, 101, 103–106). However, only a few studies have focused on AML. Wang et al., using conventional RT-PCR, found that 77.4% of AML cases expressed CIP2A (55 of 84), and confirmed their results at protein level; however, they provided no quantitative data (107). Recently, our group investigated its prevalence using quantitative real-time RT-PCR in a series of 203 normal karyotype AML patients (NK-AML) at diagnosis, and reported that high CIP2A expression is a is a recurrent event in this AML subgroup (51/203, 25%), with a poor prognostic impact in the overall survival of NK-AML cases (40). Our results indicate that CIP2A behaves as an oncoprotein in AML. CIP2A depletion downregulates c-MYC, leading to a reduction of cell proliferation, supporting the positive relationship between CIP2A and this oncogenic transcription factor in AML. Nevertheless, further studies are needed to elucidate the role of CIP2A in AML.
Cancerous inhibitor of protein phosphatase 2A has been extensively studied in CML. High levels of CIP2A at diagnosis are significantly associated with risk of progressing to blast crisis; therefore, the CIP2A protein level has been proposed as a prospective biomarker of disease progression in imatinib-treated CML patients (108). Moreover, high CIP2A levels are associated with high c-MYC and high BCR-ABL1 tyrosine kinase activity (108). In addition, second-generation tyrosine kinase inhibitors (TKI) disrupt the CIP2A/c-MYC/E2F1 positive feedback loop, leading to lower disease progression risk. These data support that CIP2A inhibits PP2Ac, stabilizing E2F1, and creating a CIP2A/c-MYC/E2F1 positive feedback loop, which imatinib cannot overcome (109).
PP2A-Activating Drugs
The increased number of studies showing that PP2A is frequently inactivated in cancer and has raised interest in developing new drugs that could act as PP2A activators (12, 110). The most widely studied drugs are FTY720 and OP449.
FTY720 is an oral sphingosine analog derived from myriocin, a metabolite isolated from fungus Isaria Sinclairii that has been approved for the treatment of patients with relapsing multiple sclerosis (111). After phosphorylation by sphingosine kinase 2 (SPHK2), FTY720 binds to one of the sphingosine-1-phosphate receptor (S1P1, S1P3, S1P4, or S1P5). Phosphorylated FTY720 does not impair T-lymphocyte or B-lymphocyte activation, but does interfere with immune cell trafficking between lymphoid organs and peripheral blood (112).
FTY720 is also a potent inhibitor of tumor growth and angiogenesis, pointing to the use of this drug in the treatment of both solid and hematological tumors. The anticancer activity of FTY720 depends on its ability, at least in leukemias, to act as a potent PP2A activator [reviewed in Ref. (86)]. FTY720-induced PP2A activity induces apoptosis by interfering with BCL2; and suppresses mitogenic and survival signals by inhibiting the ERK and PI3K/AKT pathways (13, 28, 32, 39, 41, 113–115).
The effects induced by FTY720 are well characterized in both Ph positive and negative leukemias. Several reports highlight the efficacy of FTY720 in vitro and in vivo models of AML, reporting restored PP2A activity, decreased clonogenicity, and suppression of disease (12, 38–41). In CML and Ph-positive B-ALL progenitors, FTY720 promotes BCR-ABL1 inactivation and degradation. This leads to inhibition of survival factors (such as JAK2, AKT, and ERK1/2), which result in apoptosis of CD34+ progenitors in patients with TKI sensitive and TKI-resistant CML (12, 28, 85). These findings are promising in CML and in other myeloproliferative neoplasms, suggesting the possibility that patients could be brought into remission by TKIs and then treated with FTY720 or its derivatives (12).
Mechanistically, as indicated above, FTY720 prevents SET/PP2A-C binding through its interaction with SET in a C-terminal hydrophobic pocket that contains a globular amphipathic domain (116). Our group confirmed these results in AML (39). FTY720, which binds SET within the last 100 amino acids of the C-terminal fragment (39, 117), produces a destabilization of the SET/PP2A-C complex, leading to the reactivation of PP2A function and a reduction of AML cell viability (39). Interestingly, FTY720 not only disrupts complex formation between SET and PP2A-C but it also induces increased translocation of SET to the nucleus (39), possibly by reducing the phosphorylation of SET S9 without affecting the protein levels (86) (Figure 2A). Moreover, apart from inducing apoptosis of AML cells by reactivation of PP2A activity, FTY720 treatment could perturb the sphingolipid metabolism pathway. This disruption leads to the accumulation of ceramide, a pro-apoptotic second messenger, mostly in the mitochondria membrane, contributing to the death of AML cells (115). In addition, in a recent study in in vitro and in vivo models of AML, we found that FTY720 lipid nanoparticles were more efficient at inducing cell growth arrest and apoptosis than FTY720 in solution in AML cells. Interestingly, the use of lipid nanoparticles containing FTY720 significantly increased oral bioavailability of the free drug. These results provide the first evidence for the potential use of FTY720 lipid nanoparticles as an oral therapeutic agent in AML (41).
Since the anticancer activity of FTY720 does not require SPHK2 phosphorylation or S1PR1 interaction, and FTY720-P seems to have pro-proliferative properties [reviewed in Ref. (12)], it has been proposed that FTY720 analogs that are not targets for phosphorylation by SPHK2, as [S]-FTY720-OMe, [S]-FTY720-regioisomer, and OSU-2S, may have less toxicity and be more useful as anticancer drugs. Of note, these FTY720 derivatives do not induce lymphopenia, undergo phosphorylation, or interact with the S1PR1 receptor [(13), reviewed in Ref. (12)].
Other novel molecules have been tested to activate PP2A in AML, such as the small peptide OP449 (118). OP449 was reported as a novel, physiologically stable, cell-penetrating peptide, which binds specifically to SET and antagonizes the inhibition of PP2A. Furthermore, OP449 treatment suppresses growth, enhances apoptosis, and impairs clonogenicity of CML and AML cell lines and primary patient cells, leading to the activation of the PP2A function. It has been reported that the SET binding peptides COG112 and OP449 reactivate PP2A upon interaction with SET, preventing SET–PP2Ac interaction and, therefore, the inhibition of PP2A activity (90, 118). Furthermore, the combination of OP449 with specific TKI or chemotherapy in treatment of CML and AML cell lines and primary patient samples have synergistic effects (37). These findings open new possibilities to establish innovative strategies for combined therapy that targets PP2A and tyrosine kinase signaling pathways in order to improve therapeutic options in AML patients.
Conclusion
Despite progressive advances in our understanding of the molecular biology of AML, the general therapeutic strategy in patients with this leukemia has not changed substantially, and the outcome for most patients is poor. New compounds targeting a variety of cellular processes have been developed for the treatment of AML, although few have been translated into clinical practice. Nevertheless, it is unlikely that any of these compounds, when used as single agents, will cure the disease, which suggests the need for combinatorial therapy (7). Furthermore, the results of the Cancer Genome Atlas Research Network confirm the molecular heterogeneity of this disease, and show that genes that are significantly mutated in AML are organized into several functional categories, suggesting the importance of developing treatments directed at target pathways (48). In this regard, the tumor-suppressor PP2A has emerged as a promising therapeutic target in AML, since it is a negative regulator of several survival and proliferation pathways that are frequently activated in AML as a result of aberrant activation of oncogenic kinases.
Protein phosphatase 2A inactivation is a recurrent event in AML, and restoration of its activity by PADs has antileukemic effects in both KIT-positive and KIT-negative AML cells. Preclinical studies show that pharmacological restoration of PP2A tumor-suppressor activity by PADs (e.g., FTY720, FTY720 analogs, or OP499) effectively antagonizes leukemogenesis, and that these drugs have synergistic cytotoxic effects with both conventional chemotherapy and TKIs, opening new possibilities for precision medicine, or personalized treatment, in AML patients (30, 37, 41). Interestingly, the anticancer activity of several PADs depends on interaction/sequestration of its endogenous inhibitor SET, an oncogene overexpressed in 28% of AML patients (38).
These results indicate that the combination of kinase inhibitors and PADs may be a valid therapeutic option for AML, especially for treating leukemias characterized by SET-dependent inactivation of PP2A. Therefore, PADs might be clinically relevant anticancer drugs that could be introduced into therapeutic protocols for patients with hematopoietic and non-hematopoietic malignancies characterized by functional loss of the PP2A tumor suppressor (13, 37).
Author Contributions
All authors listed, have made substantial, direct, and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by Ministerio de Economía y Competitividad (PI14/02073), integrado en el Plan Estatal de I + D + I 2013-2016 y cofinanciado por el ISCIII-Subdirección General de Evaluación y Fomento de la investigación y el Fondo Europeo de Desarrollo Regional (FEDER); Departamento de Salud del Gobierno de Navarra (29/2015); and ISCIII-RTICC (RD12/0036/0063).
Abbreviations
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; ATM, ataxia-telangiectasia mutated; CIP2A, cancerous inhibitor of protein phosphatase 2A; CML, chronic myeloid leukemia; LCMT1, leucine carboxyl methyltransferase 1; NK, natural killer; OA, okadaic acid; PADs, PP2A activating drugs; PKR, interferon-inducible dsRNA-activated protein kinase; PP2A, protein phosphatase 2A; SETBP1, SET binding protein 1; SPHK2, sphingosine kinase 2.
References
1. Marcucci G, Haferlach T, Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol (2011) 29:475–86. doi: 10.1200/JCO.2010.30.2554
2. Yohe S. Molecular genetic markers in acute myeloid leukemia. J Clin Med (2015) 4:460–78. doi:10.3390/jcm4030460
3. Ofran Y, Rowe JM. Treatment for relapsed acute myeloid leukemia: what is new? Curr Opin Hematol (2012) 19:89–94. doi:10.1097/MOH.0b013e32834ff4e1
4. Seval GC, Ozcan M. Treatment of acute myeloid leukemia in adolescent and young adult patients. J Clin Med (2015) 4:441–59. doi:10.3390/jcm4030441
5. Rowe JM. Optimal induction and post-remission therapy for AML in first remission. ASH Hematol Educ Program (2009) 2009(1):396–405. doi:10.1182/asheducation-2009.1.396
6. Bryan JC, Jabbour EJ. Management of relapsed/refractory acute myeloid leukemia in the elderly: current strategies and developments. Drugs Aging (2015) 32:623–37. doi:10.1007/s40266-015-0285-6
7. Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med (2015) 373:1136–52. doi:10.1056/NEJMra1406184
8. Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell (2007) 130:21–4. doi:10.1016/j.cell.2007.06.034
9. Bononi A, Agnoletto C, De Marchi E, Marchi S, Patergnani S, Bonora M, et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res (2011) 2011:329098. doi:10.4061/2011/329098
10. Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med (2008) 14:152–60. doi:10.1016/j.molmed.2008.02.001
11. Low IC, Loh T, Huang Y, Virshup DM, Pervaiz S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56delta stabilizes its antiapoptotic activity. Blood (2014) 124:2223–34. doi:10.1182/blood-2014-03-563296
12. Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol (2013) 14:e229–38. doi:10.1016/S1470-2045(12)70558-2
13. Neviani P, Harb JG, Oaks JJ, Santhanam R, Walker CJ, Ellis JJ, et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J Clin Invest (2013) 123:4144–57. doi:10.1172/JCI68951
14. Schonthal AH. Role of serine/threonine protein phosphatase 2A in cancer. Cancer Lett (2001) 170:1–13. doi:10.1016/S0304-3835(01)00561-4
15. Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem (2008) 283:1882–92. doi:10.1074/jbc.M709585200
16. Ruvolo PP, Qui YH, Coombes KR, Zhang N, Ruvolo VR, Borthakur G, et al. Low expression of PP2A regulatory subunit B55alpha is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia (2011) 25:1711–7. doi:10.1038/leu.2011.146
17. Ruvolo PP. The interplay between PP2A and microRNAs in leukemia. Front Oncol (2015) 5:43. doi:10.3389/fonc.2015.00043
18. Cheng X, Byrne M, Brown KD, Konopleva MY, Kornblau SM, Bennett RL, et al. PKR inhibits the DNA damage response, and is associated with poor survival in AML and accelerated leukemia in NHD13 mice. Blood (2015) 126:1585–94. doi:10.1182/blood-2015-03-635227
19. Ruvolo PP, Ruvolo VR, Jacamo R, Burks JK, Zeng Z, Duvvuri SR, et al. The protein phosphatase 2A regulatory subunit B55alpha is a modulator of signaling and microRNA expression in acute myeloid leukemia cells. Biochim Biophys Acta (2014) 1843:1969–77. doi:10.1016/j.bbamcr.2014.05.006
20. Ruvolo VR, Kurinna SM, Karanjeet KB, Schuster TF, Martelli AM, McCubrey JA, et al. PKR regulates B56(alpha)-mediated BCL2 phosphatase activity in acute lymphoblastic leukemia-derived REH cells. J Biol Chem (2008) 283:35474–85. doi:10.1074/jbc.M800951200
21. Ma J, Arnold HK, Lilly MB, Sears RC, Kraft AS. Negative regulation of Pim-1 protein kinase levels by the B56beta subunit of PP2A. Oncogene (2007) 26:5145–53. doi:10.1038/sj.onc.1210323
22. Naetar N, Soundarapandian V, Litovchick L, Goguen KL, Sablina AA, Bowman-Colin C, et al. PP2A-mediated regulation of Ras signaling in G2 is essential for stable quiescence and normal G1 length. Mol Cell (2014) 54:932–45. doi:10.1016/j.molcel.2014.04.023
23. Cristobal I, Cirauqui C, Castello-Cros R, Garcia-Orti L, Calasanz MJ, Odero MD. Downregulation of PPP2R5E is a common event in acute myeloid leukemia that affects the oncogenic potential of leukemic cells. Haematologica (2013) 98:e103–4. doi:10.3324/haematol.2013.084731
24. Haesen D, Sents W, Lemaire K, Hoorne Y, Janssens V. The basic biology of PP2A in hematologic cells and malignancies. Front Oncol (2014) 4:347. doi:10.3389/fonc.2014.00347
25. Price NE, Mumby MC. Effects of regulatory subunits on the kinetics of protein phosphatase 2A. Biochemistry (2000) 39:11312–8. doi:10.1021/bi0008478
26. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta (2009) 1795:1–15. doi:10.1016/j.bbcan.2008.05.005
27. Lambrecht C, Haesen D, Sents W, Ivanova E, Janssens V. Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol Biol (2013) 1053:283–305. doi:10.1007/978-1-62703-562-0_17
28. Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest (2007) 117:2408–21. doi:10.1172/JCI31095
29. Roberts KG, Smith AM, McDougall F, Carpenter H, Horan M, Neviani P, et al. Essential requirement for PP2A inhibition by the oncogenic receptor c-KIT suggests PP2A reactivation as a strategy to treat c-KIT+ cancers. Cancer Res (2010) 70:5438–47. doi:10.1158/0008-5472.CAN-09-2544
30. Cristobal I, Garcia-Orti L, Cirauqui C, Alonso MM, Calasanz MJ, Odero MD. PP2A impaired activity is a common event in acute myeloid leukemia and its activation by forskolin has a potent anti-leukemic effect. Leukemia (2011) 25:606–14. doi:10.1038/leu.2010.294
31. Sablina AA, Hector M, Colpaert N, Hahn WC. Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res (2010) 70:10474–84. doi:10.1158/0008-5472.CAN-10-2855
32. Yang Y, Huang Q, Lu Y, Li X, Huang S. Reactivating PP2A by FTY720 as a novel therapy for AML with C-KIT tyrosine kinase domain mutation. J Cell Biochem (2012) 113:1314–22. doi:10.1002/jcb.24003
33. Ramaswamy K, Spitzer B, Kentsis A. Therapeutic re-activation of protein phosphatase 2A in acute myeloid leukemia. Front Oncol (2015) 5:16. doi:10.3389/fonc.2015.00016
34. Janssens V, Longin S, Goris J. PP2A holoenzyme assembly: in cauda venenum (the sting is in the tail). Trends Biochem Sci (2008) 33:113–21. doi:10.1016/j.tibs.2007.12.004
35. Leulliot N, Quevillon-Cheruel S, Sorel I, Li de La Sierra-Gallay I, Collinet B, Graille M, et al. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J Biol Chem (2004) 279:8351–8. doi:10.1074/jbc.M311484200
36. Longin S, Zwaenepoel K, Louis JV, Dilworth S, Goris J, Janssens V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic subunit. J Biol Chem (2007) 282:26971–80. doi:10.1074/jbc.M704059200
37. Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res (2014) 20:2092–103. doi:10.1158/1078-0432.CCR-13-2575
38. Cristobal I, Garcia-Orti L, Cirauqui C, Cortes-Lavaud X, Garcia-Sanchez MA, Calasanz MJ, et al. Overexpression of SET is a recurrent event associated with poor outcome and contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica (2012) 97:543–50. doi:10.3324/haematol.2011.050542
39. Pippa R, Dominguez A, Christensen DJ, Moreno-Miralles I, Blanco-Prieto MJ, Vitek MP, et al. Effect of FTY720 on the SET-PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia (2014) 28:1915–8. doi:10.1038/leu.2014.141
40. Barragan E, Chillon MC, Castello-Cros R, Marcotegui N, Prieto MI, Hoyos M, et al. CIP2A high expression is a poor prognostic factor in normal karyotype acute myeloid leukemia. Haematologica (2015) 100:e183–5. doi:10.3324/haematol.2014.118117
41. Estella-Hermoso de Mendoza A, Castello-Cros R, Imbuluzqueta E, Cirauqui C, Pippa R, Odero MD, et al. Lipid nanosystems enhance the bioavailability and the therapeutic efficacy of FTY720 in acute myeloid leukemia. J Biomed Nanotechnol (2015) 11:691–701. doi:10.1166/jbn.2015.1944
42. Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J, et al. Alterations of the PPP2R1B gene in human lung and colon cancer. Science (1998) 282:284–7. doi:10.1126/science.282.5387.284
43. Calin GA, di Iasio MG, Caprini E, Vorechovsky I, Natali PG, Sozzi G, et al. Low frequency of alterations of the alpha (PPP2R1A) and beta (PPP2R1B) isoforms of the subunit A of the serine-threonine phosphatase 2A in human neoplasms. Oncogene (2000) 19:1191–5. doi:10.1038/sj.onc.1203389
44. Ruediger R, Pham HT, Walter G. Alterations in protein phosphatase 2A subunit interaction in human carcinomas of the lung and colon with mutations in the A beta subunit gene. Oncogene (2001) 20:1892–9. doi:10.1038/sj.onc.1204279
45. Ruediger R, Pham HT, Walter G. Disruption of protein phosphatase 2A subunit interaction in human cancers with mutations in the A alpha subunit gene. Oncogene (2001) 20:10–5. doi:10.1038/sj.onc.1204279
46. Esplin ED, Ramos P, Martinez B, Tomlinson GE, Mumby MC, Evans GA. The glycine 90 to aspartate alteration in the Abeta subunit of PP2A (PPP2R1B) associates with breast cancer and causes a deficit in protein function. Genes Chromosomes Cancer (2006) 45:182–90. doi:10.1002/gcc.20284
47. Kalla C, Scheuermann MO, Kube I, Schlotter M, Mertens D, Dohner H, et al. Analysis of 11q22-q23 deletion target genes in B-cell chronic lymphocytic leukaemia: evidence for a pathogenic role of NPAT, CUL5, and PPP2R1B. Eur J Cancer (2007) 43:1328–35. doi:10.1016/j.ejca.2007.02.005
48. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368:2059–74. doi:10.1056/NEJMoa1301689
49. Seshacharyulu P, Pandey P, Datta K, Batra SK. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett (2013) 335:9–18. doi:10.1016/j.canlet.2013.02.036
50. Cristobal I, Blanco FJ, Garcia-Orti L, Marcotegui N, Vicente C, Rifon J, et al. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood (2010) 115:615–25. doi:10.1182/blood-2009-06-227363
51. Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, Bulmer SE, et al. The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell (2007) 129:969–82. doi:10.1016/j.cell.2007.03.047
52. Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res (2005) 65:8183–92. doi:10.1158/0008-5472.CAN-05-1103
53. Gao X, Lin J, Gao L, Deng A, Lu X, Li Y, et al. High expression of c-kit mRNA predicts unfavorable outcome in adult patients with t(8;21) acute myeloid leukemia. PLoS One (2015) 10:e0124241. doi:10.1371/journal.pone.0124241
54. Turowski P, Myles T, Hemmings BA, Fernandez A, Lamb NJ. Vimentin dephosphorylation by protein phosphatase 2A is modulated by the targeting subunit B55. Mol Biol Cell (1999) 10:1997–2015. doi:10.1091/mbc.10.6.1997
55. Wang P, Malumbres M, Archambault V. The Greatwall-PP2A axis in cell cycle control. Methods Mol Biol (2014) 1170:99–111. doi:10.1007/978-1-4939-0888-2_6
56. Grallert A, Boke E, Hagting A, Hodgson B, Connolly Y, Griffiths JR, et al. A PP1-PP2A phosphatase relay controls mitotic progression. Nature (2015) 517:94–8. doi:10.1038/nature14019
57. Gallay N, Dos Santos C, Cuzin L, Bousquet M, Simmonet Gouy V, Chaussade C, et al. The level of AKT phosphorylation on threonine 308 but not on serine 473 is associated with high-risk cytogenetics and predicts poor overall survival in acute myeloid leukaemia. Leukemia (2009) 23:1029–38. doi:10.1038/leu.2008.395
58. Blalock WL, Grimaldi C, Fala F, Follo M, Horn S, Basecke J, et al. PKR activity is required for acute leukemic cell maintenance and growth: a role for PKR-mediated phosphatase activity to regulate GSK-3 phosphorylation. J Cell Physiol (2009) 221:232–41. doi:10.1002/jcp.21848
59. Blalock WL, Bavelloni A, Piazzi M, Tagliavini F, Faenza I, Martelli AM, et al. Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia (2011) 25:236–45. doi:10.1038/leu.2010.264
60. Chen W, Possemato R, Campbell KT, Plattner CA, Pallas DC, Hahn WC. Identification of specific PP2A complexes involved in human cell transformation. Cancer Cell (2004) 5:127–36. doi:10.1016/S1535-6108(04)00026-1
61. Margolis SS, Perry JA, Forester CM, Nutt LK, Guo Y, Jardim MJ, et al. Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell (2006) 127:759–73. doi:10.1016/j.cell.2006.10.035
62. Letourneux C, Rocher G, Porteu F. B56-containing PP2A dephosphorylate ERK and their activity is controlled by the early gene IEX-1 and ERK. EMBO J (2006) 25:727–38. doi:10.1038/sj.emboj.7600980
63. Arnold HK, Sears RC. Protein phosphatase 2A regulatory subunit B56alpha associates with c-myc and negatively regulates c-myc accumulation. Mol Cell Biol (2006) 26:2832–44. doi:10.1128/MCB.26.7.2832-2844.2006
64. Xu Z, Williams BR. The B56alpha regulatory subunit of protein phosphatase 2A is a target for regulation by double-stranded RNA-dependent protein kinase PKR. Mol Cell Biol (2000) 20:5285–99. doi:10.1128/MCB.20.14.5285-5299.2000
65. Shouse GP, Nobumori Y, Panowicz MJ, Liu X. ATM-mediated phosphorylation activates the tumor-suppressive function of B56gamma-PP2A. Oncogene (2011) 30:3755–65. doi:10.1038/onc.2011.95
66. Shouse GP, Cai X, Liu X. Serine 15 phosphorylation of p53 directs its interaction with B56gamma and the tumor suppressor activity of B56gamma-specific protein phosphatase 2A. Mol Cell Biol (2008) 28:448–56. doi:10.1128/MCB.00983-07
67. Seeling JM, Miller JR, Gil R, Moon RT, White R, Virshup DM. Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science (1999) 283:2089–91. doi:10.1126/science.283.5410.2089
68. Yang J, Wu J, Tan C, Klein PS. PP2A:B56epsilon is required for Wnt/beta-catenin signaling during embryonic development. Development (2003) 130:5569–78. doi:10.1242/dev.00762
69. Rorick AM, Mei W, Liette NL, Phiel C, El-Hodiri HM, Yang J. PP2A:B56epsilon is required for eye induction and eye field separation. Dev Biol (2007) 302:477–93. doi:10.1016/j.ydbio.2006.10.011
70. Jin Z, Wallace L, Harper SQ, Yang J. PP2A:B56{epsilon}, a substrate of caspase-3, regulates p53-dependent and p53-independent apoptosis during development. J Biol Chem (2010) 285:34493–502. doi:10.1074/jbc.M110.169581
71. Yan L, Mieulet V, Burgess D, Findlay GM, Sully K, Procter J, et al. PP2A T61 epsilon is an inhibitor of MAP4K3 in nutrient signaling to mTOR. Mol Cell (2010) 37:633–42. doi:10.1016/j.molcel.2010.01.031
72. Zwaenepoel K, Goris J, Erneux C, Parker PJ, Janssens V. Protein phosphatase 2A PR130/B''alpha1 subunit binds to the SH2 domain-containing inositol polyphosphate 5-phosphatase 2 and prevents epidermal growth factor (EGF)-induced EGF receptor degradation sustaining EGF-mediated signaling. FASEB J (2010) 24:538–47. doi:10.1096/fj.09-140228
73. Perot G, Soubeyran I, Ribeiro A, Bonhomme B, Savagner F, Boutet-Bouzamondo N, et al. Identification of a recurrent STRN/ALK fusion in thyroid carcinomas. PLoS One (2014) 9:e87170. doi:10.1371/journal.pone.0087170
74. Kelly LM, Barila G, Liu P, Evdokimova VN, Trivedi S, Panebianco F, et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc Natl Acad Sci U S A (2014) 111:4233–8. doi:10.1073/pnas.1321937111
75. Wong M, Hyodo T, Asano E, Funasaka K, Miyahara R, Hirooka Y, et al. Silencing of STRN4 suppresses the malignant characteristics of cancer cells. Cancer Sci (2014) 105:1526–32. doi:10.1111/cas.12541
76. Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem (1996) 271:11059–62. doi:10.1074/jbc.271.19.11059
77. Adachi Y, Pavlakis GN, Copeland TD. Identification and characterization of SET, a nuclear phosphoprotein encoded by the translocation break point in acute undifferentiated leukemia. J Biol Chem (1994) 269:2258–62.
78. Saito S, Miyaji-Yamaguchi M, Nagata K. Aberrant intracellular localization of SET-CAN fusion protein, associated with a leukemia, disorganizes nuclear export. Int J Cancer (2004) 111:501–7. doi:10.1002/ijc.20296
79. Seo SB, McNamara P, Heo S, Turner A, Lane WS, Chakravarti D. Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell (2001) 104:119–30. doi:10.1016/S0092-8674(01)00196-9
80. Kutney SN, Hong R, Macfarlan T, Chakravarti D. A signaling role of histone-binding proteins and INHAT subunits pp32 and Set/TAF-Ibeta in integrating chromatin hypoacetylation and transcriptional repression. J Biol Chem (2004) 279:30850–5. doi:10.1074/jbc.M404969200
81. Kalousi A, Hoffbeck AS, Selemenakis PN, Pinder J, Savage KI, Khanna KK, et al. The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep (2015) 11:149–63. doi:10.1016/j.celrep.2015.03.005
82. Kandilci A, Mientjes E, Grosveld G. Effects of SET and SET-CAN on the differentiation of the human promonocytic cell line U937. Leukemia (2004) 18:337–40. doi:10.1038/sj.leu.2403227
83. ten Klooster JP, Leeuwen I, Scheres N, Anthony EC, Hordijk PL. Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J (2007) 26:336–45. doi:10.1038/sj.emboj.7601518
84. Canela N, Rodriguez-Vilarrupla A, Estanyol JM, Diaz C, Pujol MJ, Agell N, et al. The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J Biol Chem (2003) 278:1158–64. doi:10.1074/jbc.M207497200
85. Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell (2005) 8:355–68. doi:10.1016/j.ccr.2005.10.015
86. Oaks JJ, Santhanam R, Walker CJ, Roof S, Harb JG, Ferenchak G, et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood (2013) 122:1923–34. doi:10.1182/blood-2013-03-492181
87. Cristobal I, Rincon R, Manso R, Carames C, Zazo S, Madoz-Gurpide J, et al. Deregulation of the PP2A inhibitor SET shows promising therapeutic implications and determines poor clinical outcome in patients with metastatic colorectal cancer. Clin Cancer Res (2015) 21:347–56. doi:10.1158/1078-0432.CCR-14-0724
88. Janghorban M, Farrell AS, Allen-Petersen BL, Pelz C, Daniel CJ, Oddo J, et al. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc Natl Acad Sci U S A (2014) 111:9157–62. doi:10.1073/pnas.1317630111
89. Cervoni N, Detich N, Seo SB, Chakravarti D, Szyf M. The oncoprotein Set/TAF-1beta, an inhibitor of histone acetyltransferase, inhibits active demethylation of DNA, integrating DNA methylation and transcriptional silencing. J Biol Chem (2002) 277:25026–31. doi:10.1074/jbc.M202256200
90. Switzer CH, Cheng RY, Vitek TM, Christensen DJ, Wink DA, Vitek MP. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene (2011) 30:2504–13. doi:10.1038/onc.2010.622
91. Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest (2010) 120:2254–64. doi:10.1172/JCI41246
92. Yu G, Yan T, Feng Y, Liu X, Xia Y, Luo H, et al. Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol Aging (2013) 34:1748–58. doi:10.1016/j.neurobiolaging.2012.12.025
93. Vasudevan NT, Mohan ML, Gupta MK, Hussain AK, Naga Prasad SV. Inhibition of protein phosphatase 2A activity by PI3Kgamma regulates beta-adrenergic receptor function. Mol Cell (2011) 41:636–48. doi:10.1016/j.molcel.2011.02.025
94. Irie A, Harada K, Araki N, Nishimura Y. Phosphorylation of SET protein at Ser171 by protein kinase D2 diminishes its inhibitory effect on protein phosphatase 2A. PLoS One (2012) 7:e51242. doi:10.1371/journal.pone.0051242
95. Trotta R, Ciarlariello D, Dal Col J, Allard J II, Neviani P, Santhanam R, et al. The PP2A inhibitor SET regulates natural killer cell IFN-gamma production. J Exp Med (2007) 204:2397–405. doi:10.1084/jem.20070419
96. Trotta R, Ciarlariello D, Dal Col J, Mao H, Chen L, Briercheck E, et al. The PP2A inhibitor SET regulates granzyme B expression in human natural killer cells. Blood (2011) 117:2378–84. doi:10.1182/blood-2010-05-285130
97. Al-Murrani SW, Woodgett JR, Damuni Z. Expression of I2PP2A, an inhibitor of protein phosphatase 2A, induces c-Jun and AP-1 activity. Biochem J (1999) 341(Pt 2):293–8. doi:10.1042/bj3410293
98. Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell (2003) 112:659–72. doi:10.1016/S0092-8674(03)00150-8
99. Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, et al. CIP2A inhibits PP2A in human malignancies. Cell (2007) 130:51–62. doi:10.1016/j.cell.2007.04.044
100. Niemela M, Kauko O, Sihto H, Mpindi JP, Nicorici D, Pernila P, et al. CIP2A signature reveals the MYC dependency of CIP2A-regulated phenotypes and its clinical association with breast cancer subtypes. Oncogene (2012) 31:4266–78. doi:10.1038/onc.2011.599
101. Khanna A, Pimanda JE, Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res (2013) 73:6548–53. doi:10.1158/0008-5472.CAN-13-1994
102. Luo H, Li Q, O’Neal J, Kreisel F, Le Beau MM, Tomasson MH. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood (2005) 106:2452–61. doi:10.1182/blood-2005-02-0734
103. Come C, Laine A, Chanrion M, Edgren H, Mattila E, Liu X, et al. CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res (2009) 15:5092–100. doi:10.1158/1078-0432.CCR-08-3283
104. Khanna A, Bockelman C, Hemmes A, Junttila MR, Wiksten JP, Lundin M, et al. MYC-dependent regulation and prognostic role of CIP2A in gastric cancer. J Natl Cancer Inst (2009) 101:793–805. doi:10.1093/jnci/djp103
105. Dong QZ, Wang Y, Dong XJ, Li ZX, Tang ZP, Cui QZ, et al. CIP2A is overexpressed in non-small cell lung cancer and correlates with poor prognosis. Ann Surg Oncol (2011) 18:857–65. doi:10.1245/s10434-010-1313-8
106. Xue Y, Wu G, Wang X, Zou X, Zhang G, Xiao R, et al. CIP2A is a predictor of survival and a novel therapeutic target in bladder urothelial cell carcinoma. Med Oncol (2013) 30:406. doi:10.1007/s12032-012-0406-6
107. Wang J, Li W, Li L, Yu X, Jia J, Chen C. CIP2A is over-expressed in acute myeloid leukaemia and associated with HL60 cells proliferation and differentiation. Int J Lab Hematol (2011) 33:290–8. doi:10.1111/j.1751-553X.2010.01288.x
108. Lucas CM, Harris RJ, Giannoudis A, Copland M, Slupsky JR, Clark RE. Cancerous inhibitor of PP2A (CIP2A) at diagnosis of chronic myeloid leukemia is a critical determinant of disease progression. Blood (2011) 117:6660–8. doi:10.1182/blood-2010-08-304477
109. Lucas CM, Harris RJ, Holcroft AK, Scott LJ, Carmell N, McDonald E, et al. Second generation tyrosine kinase inhibitors prevent disease progression in high-risk (high CIP2A) chronic myeloid leukaemia patients. Leukemia (2015) 29:1514–23. doi:10.1038/leu.2015.71
110. Ciccone M, Calin GA, Perrotti D. From the biology of PP2A to the PADs for therapy of hematologic malignancies. Front Oncol (2015) 5:21. doi:10.3389/fonc.2015.00021
111. Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med (2010) 362:402–15. doi:10.1056/NEJMoa0907839
112. Ingwersen J, Aktas O, Kuery P, Kieseier B, Boyko A, Hartung HP. Fingolimod in multiple sclerosis: mechanisms of action and clinical efficacy. Clin Immunol (2012) 142:15–24. doi:10.1016/j.clim.2011.10.008
113. Liu Q, Zhao X, Frissora F, Ma Y, Santhanam R, Jarjoura D, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood (2008) 111:275–84. doi:10.1182/blood-2006-10-053884
114. Shah MV, Zhang R, Irby R, Kothapalli R, Liu X, Arrington T, et al. Molecular profiling of LGL leukemia reveals role of sphingolipid signaling in survival of cytotoxic lymphocytes. Blood (2008) 112:770–81. doi:10.1182/blood-2007-11-121871
115. Chen L, Luo LF, Lu J, Li L, Liu YF, Wang J, et al. FTY720 induces apoptosis of M2 subtype acute myeloid leukemia cells by targeting sphingolipid metabolism and increasing endogenous ceramide levels. PLoS One (2014) 9:e103033. doi:10.1371/journal.pone.0103033
116. Arnaud L, Chen S, Liu F, Li B, Khatoon S, Grundke-Iqbal I, et al. Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett (2011) 585:2653–9. doi:10.1016/j.febslet.2011.07.020
117. Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med (2013) 5:105–21. doi:10.1002/emmm.201201283
Keywords: PP2A, SET, AML, FTY720, OP449
Citation: Arriazu E, Pippa R and Odero MD (2016) Protein Phosphatase 2A as a Therapeutic Target in Acute Myeloid Leukemia. Front. Oncol. 6:78. doi: 10.3389/fonc.2016.00078
Received: 02 February 2016; Accepted: 21 March 2016;
Published: 06 April 2016
Edited by:
Peter Ruvolo, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Alex Kentsis, Memorial Sloan Kettering Cancer Center, USAVeerle Janssens, University of Leuven (KU Leuven), Belgium
Copyright: © 2016 Arriazu, Pippa and Odero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María D. Odero, bW9kZXJvQHVuYXYuZXM=