 Kekoa Taparra
Kekoa Taparra Phuoc T. Tran1,2,3,4
Phuoc T. Tran1,2,3,4 Natasha E. Zachara
Natasha E. Zachara- 1Department of Radiation Oncology and Molecular Radiation Sciences, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 2Program in Cellular and Molecular Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 3Department of Oncology, Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 4Department of Urology, Johns Hopkins University School of Medicine, Baltimore, MD, USA
- 5Department of Biological Chemistry, Johns Hopkins University School of Medicine, Baltimore, MD, USA
The epithelial–mesenchymal transition (EMT) is a highly conserved program necessary for orchestrating distant cell migration during embryonic development. Multiple studies in cancer have demonstrated a critical role for EMT during the initial stages of tumorigenesis and later during tumor invasion. Transcription factors (TFs) such as SNAIL, TWIST, and ZEB are master EMT regulators that are aberrantly overexpressed in many malignancies. Recent evidence correlates EMT-related transcriptomic alterations with metabolic reprograming in cancer. Metabolic alterations may allow cancer to adapt to environmental stressors, supporting the irregular macromolecular demand of rapid proliferation. One potential metabolic pathway of increasing importance is the hexosamine biosynthesis pathway (HBP). The HBP utilizes glycolytic intermediates to generate the metabolite UDP–GlcNAc. This and other charged nucleotide sugars serve as the basis for biosynthesis of glycoproteins and other glycoconjugates. Recent reports in the field of glycobiology have cultivated great curiosity within the cancer research community. However, specific mechanistic relationships between the HBP and fundamental pathways of cancer, such as EMT, have yet to be elucidated. Altered protein glycosylation downstream of the HBP is well positioned to mediate many cellular changes associated with EMT including cell–cell adhesion, responsiveness to growth factors, immune system evasion, and signal transduction programs. Here, we outline some of the basics of the HBP and putative roles the HBP may have in driving EMT-related cancer processes. With novel appreciation of the HBP’s connection to EMT, we hope to illuminate the potential for new therapeutic targets of cancer.
Introduction
Since the time of Otto Warburg in the 1930s, scientists have been intrigued by the unique metabolic profile of cancer cells (1, 2). Current research corroborates Warburg’s original observation that cancer prefers glycolysis over mitochondrial oxidative phosphorylation (OXPHOS) (3). Initially, this metabolic reprograming appeared paradoxical due to the inefficiencies of glycolysis (i.e., ~38 ATP from OXPHOS versus 2 ATP from glycolysis). Despite early conflicting viewpoints on the Warburg Effect, aerobic glycolysis stands at the center of cancer metabolism demonstrating its importance as an “Emerging Hallmark of Cancer” (4).
Despite decades of research, the molecular advantages of the Warburg effect in cancer are still being interrogated (5). One popular explanation is the “Glycolytic Intermediate Diversion” hypothesis (6, 7). This hypothesis suggests that glycolysis is well positioned to support anabolic cell growth as it provides the metabolic intermediates (e.g., nucleosides, amino acids, and other carbon compounds) necessary for enzymatic reactions and organelle assembly. A second hypothesis involves the notion of “Cell Subpopulations” (8–10). This hypothesis posits that lactate from “Warburg-effect cells” is sent to neighboring cells, which utilize lactate through the citric acid cycle. The cell subpopulations symbiotically trade off waste for energy to support cancer progression. Interestingly, both hypotheses demonstrate the ability of the neoplastic state to commandeer normal biological processes observed in development and normal physiology (4).
The energetic demand required to survive adverse tumor environments is likely only a fraction of the functional significance underlying cancer metabolic reprograming. It is likely that glycolytic byproducts reinforce the cancer phenotype by modulating not just metabolic maintenance but also altering other cellular structures and functions. In particular, the role of post-translational modifications (PTM), such as glycosylation, are becoming of increasing importance as they provide rapid, reversible adaptations to the stressors that occur during early tumorigenesis. Recent studies have revealed new potential cancer treatment strategies specifically targeting these glycoconjugates (11).
Interestingly, one metabolic pathway with the potential of impacting functional macromolecular structures in cancer is an understudied pathway called the “hexosamine biosynthetic pathway” (HBP) (12–15). One downstream metabolite of this pathway, uridine diphosphate–N-acetylglucosamine (UDP–GlcNAc), serves as an essential building block for glycoconjugate biosynthesis. This pathway is well positioned to not only affect metabolic intermediates but also functional glycans that accelerate cancer progression (11, 16). The HBP has only recently gained traction in cancer biology and is becoming of increasing importance (17).
The epithelial–mesenchymal transition (EMT) is a conserved epithelial plasticity program capable of impacting cellular morphology, migration, stem cell-ness, among other malignant phenotypes (18). Moreover, the EMT is involved throughout the natural history of cancer from tumorigenesis to late metastatic progression (19–21). Master transcriptional regulators of EMT (i.e., TWIST, SNAIL, and ZEB) are elevated in a wide range of primary and metastatic tumors. Recent evidence demonstrates that the expression of key enzymes in the HBP is upregulated in cancer cells with a mesenchymal phenotype (22). Thus, in this review, we will highlight some of the relevant glycoconjugates downstream of the HBP and the implications this has on EMT-mediated cancer programs.
The Epithelial–Mesenchymal Transition
Epithelial–mesenchymal transition is an essential epithelial plasticity program deployed during development (23, 24), wound healing (25–27), and stem cell maintenance (28–31). The major characteristics of EMT include loss of cellular adhesion, reorganization of cytoskeleton, loss of cellular polarity, and a switch from epithelial to mesenchymal gene expression (18). Many of these EMT pathways are activated by extracellular signaling, highlighting the importance of the tumor microenvironment for the induction of EMT. Figure 1A outlines eight critical EMT-activating pathways: TGF-β, receptor tyrosine kinases (RTKs), integrin, WNT, NOTCH, Hedgehog (HH), hypoxia inducible factor 1α (HIF1α), and JAK/STAT.
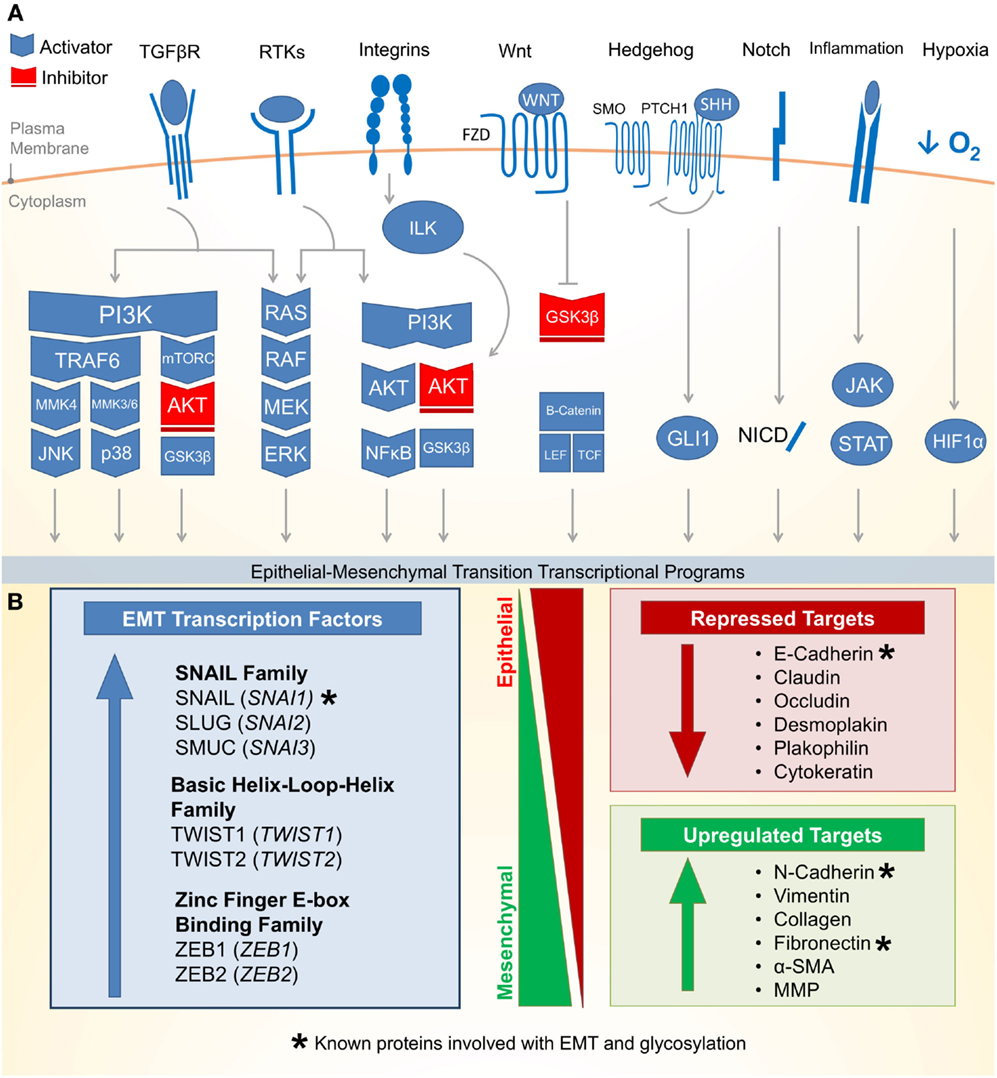
Figure 1. Molecular pathways and targets of the epithelial–mesenchymal transition. (A) (1) One of the most well characterized EMT-inducing pathways is the transforming growth factor-β (TGF-β) family receptors capable of inducing PI3K–AKT, ERK MAPK, p38 MAPK, and JUN N-terminal kinase (JNK) pathways (activating pathways in blue; inactivating pathways in red). (2) The RAS–RAF–MEK–ERK MAPK pathway lies downstream of a number of growth factor activated receptor tyrosine kinases (RTKs) and activates a number of major EMT transcription factors (TFs). (3) Integrin signaling can have a multipronged effect on EMT by both interrupting critical epithelial adhesion molecules (e.g., E-cadherin) and antagonizing GSK3-β via the integrin-linked kinase (ILK)-AKT signaling, thus promoting EMT. (4) WNT signaling can also interfere with GSK3-β, thus stabilizing β-catenin to promote EMT transcriptional programs in cooperation with lymphoid enhancer-binding factor 1 (LEF1) and T-cell factor (TCF). (5) The Hedgehog (HH)-glioma 1 (GLI1) and (6) NOTCH pathway both can promote transcription of the EMT regulators. (7) Recently, a number of inflammatory pathways downstream of interleukin (IL) signaling (e.g., IL-6) have demonstrated the activation of the Janus-kinase (JAK)-signal transducer and activator of transcription 3 (STAT3) pathway, which in turn promotes EMT transcription factors. (8) Hypoxia is capable of activating a number of key components of EMT through the hypoxia-induced factor 1 (HIF1α). (B) Downstream of these signal transduction pathways leading to EMT are a variety of transcription factors with the ability to alter epithelial gene expression. As an epithelial plasticity program, many of the target genes altered include adhesion molecules. Known glycosylated proteins involved with EMT are denoted with an asterisk (*).
There are three major families of transcription factors (TFs) that contribute to EMT and may also be general drivers of cancer (Figure 1B): (1) the zinc finger protein SNAIL family (SNAI1, SNAI2, and SNAI3) (32), (2) the basic Helix-Loop-Helix (bHLH) proteins TWIST1 and TWIST2 (33), and (3) the zinc-finger E-box binding (ZEB) family of TFs (34). These TFs are evolutionarily conserved and critical for development. They bind short DNA segments called enhancer boxes (E-boxes) with the consensus sequence “CANNTG.” Like many TFs, they are able to modulate transcription by recruiting a variety of epigenetic regulators to alter the chromatin landscape of epithelial plasticity genes and interactions with transcriptional coactivators and corepressors (35).
The most well-established gene targets of EMT TFs are generally involved in epithelial cell adhesion (36–38). Cadherins represent a family of calcium-dependent cell–cell adhesion proteins particularly targeted by EMT (39–41). Loss of epithelial cadherin (E-cadherin) is a major hallmark of EMT (42–44). Thus, loss of E-cadherin has been used as a biomarker for many cancers. Additionally, loss of tight junctions (e.g., claudin and occludin), desmosomes (e.g., desmoplakin and plakophilin), and cytokeratins (intermediate filaments) are commonly observed during EMT (18). Conversely, while epithelial markers are repressed, mesenchymal markers are increased during EMT. These markers include N-cadherin, vimentin, and fibronectin (18). Following the transcriptional alterations of these adhesion molecules, protein degradation and endocytosis aid in recycling epithelial adhesion molecules to promote progress through EMT (45).
Altered gene expression of EMT targets, such as those involved in cellular adhesion, often facilitate biological and pathological functions such as migration and invasion (46–48). Upon detaching from the basal epithelium, epithelial cells undergoing EMT may alter their extracellular environment by expressing matrix metalloproteinases (MMPs) to promote directional migration (49–51). During migration, adhesion molecules are disproportionately redistributed between the leading and trailing edge of the cell, which allows the cell to coordinate directed migration leading to tumor dissemination and metastasis (24, 52). Beyond metastasis, EMT has recently been attributed to more fundamental roles in cancer biology including suppressing apoptosis and senescence (53). The EMT has also been implicated in immune evasion (54) and metabolic reprograming (22, 55) of cancer cells. Together, the data discussed above suggest that the EMT program promotes many cancer cell phenotypes leading to malignancy.
The Hexosamine Biosynthetic Pathway
Since the 1950s, cancer has been notorious for its addiction to glucose and glutamine (7, 56–58). Upon depletion of these carbon sources in cancer cell culture media, cellular growth is abrogated. Both glucose and glutamine (Gln) are essential for the first committed step and rate-limiting step of the HBP, the conversion of fructose-6-phosphate (Fru-6P) to glucosamine-6-phosphate. Approximately 2–5% of glucose (in adipocytes) is shunted through the HBP (59). Demonstrating the importance of extracellular glucose concentrations on the HBP, glucose starvation reduces UDP–GlcNAc levels (60, 61). Conversely, elevating extracellular glucose concentrations results in increased flux through the HBP (62). Figure 2A summarizes the four key enzymatic steps of the HBP:
(1) Glutamine:fructose-6-phosphate transaminase (GFAT; GFPT) utilizes glutamine in a transamination reaction, which converts fructose-6-phosphate (Fru-6-P) to glucosamine-6P (GlcN-6P);
(2) GlcN-6P is converted to N-acetylglucosamine-6-P (GlcNAc-6P) by Glucosamine-phosphate N-acetyltransferase (GNPNAT; GNPNAT), which requires acetyl-CoA;
(3) Phosphoglucomutase (PGM; PGM) isomerizes GlcNAc-6P to N-acetylglucosamine-1-phosphate (GlcNAc-1P);
(4) UDP–N-acetylglucosamine pyrophosphorylase (UAP1; UAP1) charges GlcNAc-1P with UDP to form uridine-5′-diphosphate–N-acetylglucosamine (UDP–GlcNAc).
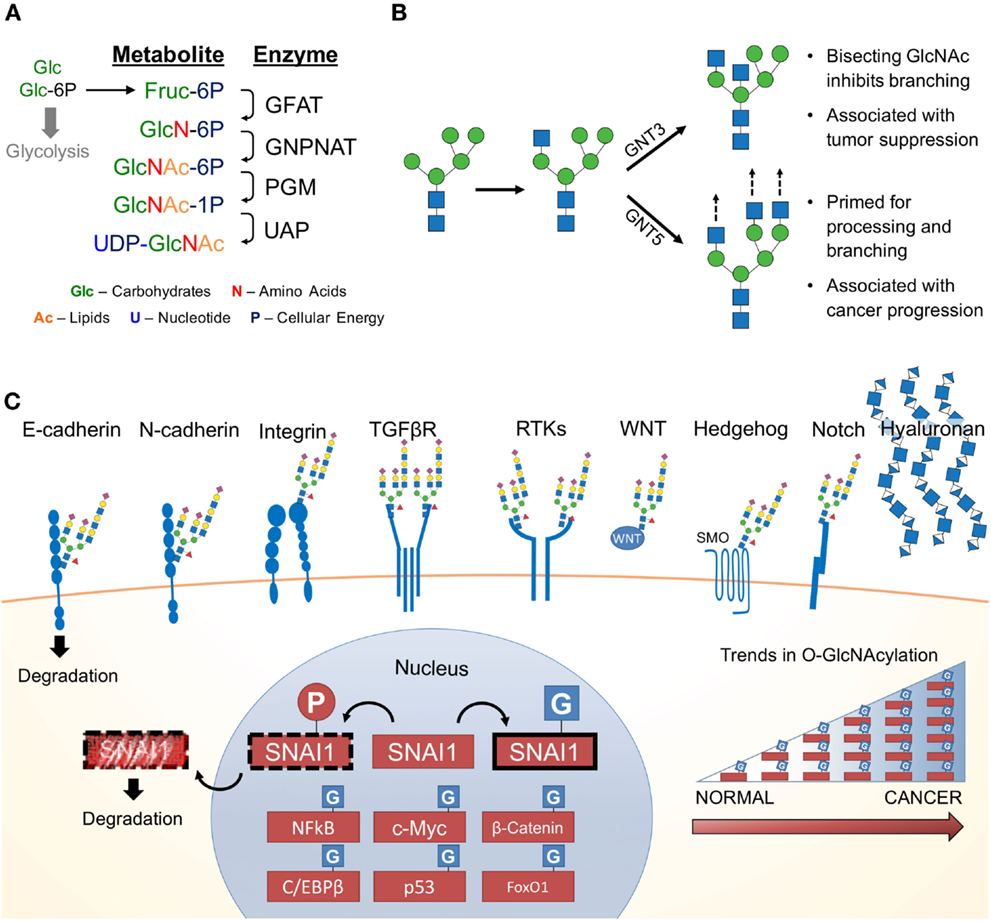
Figure 2. The hexosamine biosynthetic pathway (HBP) and glycosylated EMT targets. (A) First, the rate limiting enzyme of the HBP, glutamine:fructose-6-phosphate transaminase (GFAT), uses glutamine (Gln) as an amine donor to convert Fru-6P into glucosamine-6-P (GlcN-6P). Second, glucosamine-phosphate N-acetyltransferase (GNPNAT) N-acetylates GlcN-6P in an acetyl-CoA-mediated reaction to form N-acetylglucosamine-6-P (GlcNAc-6P). Third, phosphoglucomutase (PGM) isomerizes GlcNAc-6P to the highly active GlcNAc-1P. The final step is catalyzed by UDP–N-acetylglucosamine pyrophosphorylase (UAP1) and charges GlcNAc-1P with UDP to form uridine-5′-diphosphate-N-acetylglucosamine (UDP–GlcNAc). (B) UDP–GlcNAc (depicted as a blue square) is essential for N-glycosylation processing and elongation. One critical pivot point includes the branching of complex N-glycans. Inhibiting this process with a bisecting GlcNAc is associated with tumor suppressive phenotypes. In contrast, cancers have aberrant expression of glycosyltransferases responsible for branching and elongating complex N-glycans. (C) Many of the proteins commonly associated with promoting EMT are modified by glycans containing GlcNAc and are found on the cell surface. Hyaluronan, a glycosaminoglycan, is also found extracellularly and is a polymer of glucuronic acid and N-acetylglucosamine. Many nuclear, cytoplasmic and mitochondrial proteins are modified by monosaccharides of O-linked N-acetylglucosamine (O-GlcNAc), including many transcription factors, which appear to be stabilized by glycosylation (63). Numerous studies have identified various cancers with elevated levels of pan-O-GlcNAcylation (64).
Together, the four enzymes of the HBP orchestrate the de novo biosynthesis of the charged nucleotide sugar UDP–GlcNAc from glucose. This process can be manipulated by endogenous metabolites (i.e., glutamine) (65) as well as exogenous sugars (i.e., glucose, glucosamine, and N-acetylglucosamine) (66). Interestingly, this pathway is well positioned to sense the four macromolecules of life, coordinating carbohydrate, amino acid, lipid, and nucleotide donors through by Fru-6P, Gln, acetyl-CoA, and uridine, respectively (67). Despite the limited flux through the HBP, cellular UDP–GlcNAc levels can reach over 1 mM making it one of the most abundant high-energy cellular compounds (68). UDP–GlcNAc is utilized in the synthesis of numerous glycoconjugates and is interconverted into other nucleotide sugars (e.g., UDP–GalNAc, N-acetylmannosamine, CMP-neuraminic acid), which are incorporated into glycoconjugates (69). Together, the glycan structures downstream of the HBP metabolite, UDP–GlcNAc, influence a wide range of functional targets highly relevant to cancer and EMT.
Reinforcing the importance of UDP–GlcNAc incorporation, recent data suggest that the expression of multiple enzymes of the HBP and glycosyltransferases are altered in cancer, correlating with EMT, cancer progression, and metastasis. In a recent analysis using unsupervised hierarchical clustering of 1,704 metabolic genes and nearly 1,000 cancer cell lines, Shaul and colleagues identified a “mesenchymal metabolic signature” (MMS) (22). In this MMS, both GFPT2 and UAP1, key enzymes in the HBP, were found to be essential for the mesenchymal phenotype (22, 70). In other studies, metabolites of the HBP (e.g., UDP–GlcNAc) were reported to be elevated in cancer cells and this was linked to survival (60).
Glycosyltransferases consistently elevated in multiple cancers (e.g., stomach and pancreas cancer) include β-1,4-mannosyl-glycoprotein 4-β-N-acetylglucosaminyltransferase (GNT3), α-1,6-mannosylglycoprotein 6-β-N-acetylglucosaminyltransferase A (GNT5), core 2 β-1,3-galactosyl-O-glycosyl-glycoprotein β-1,6-N-acetylglucosaminyltransferase (Core 2 GNT; GCNT1), N-acetyllactosaminide β-1,6-N-acetylglucosaminyl-transferase-isoform A (GCNT2), and UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase (GPT1), encoded by the genes MGAT3, MGAT5, GCNT1, GCNT2, and DPAGT1, respectively (71). Notably, GNT5 is highly associated with breast, lung, and colon cancer metastasis (72–77), whereas GNT3 is associated with breast, skin, and colon cancer tumor suppression (78–80). GNT5 and GNT3 have antagonistic roles; GNT5 promotes complex N-linked glycan branching, whereas GNT3 suppresses branching (Figure 2B). It is thought that changes in glucose flux through the HBP impacts the function of GNT3 and GNT5 (66). The rate-limiting enzyme that forms the precursor for N-glycosylation, GPT1, has also been shown to drive proliferation, EMT, and cell morphology (81). As discussed below, flux through the HBP can alter the distribution patterns of glycosylation. To date, this has not been specifically studied in EMT. However, there are a number of glycoconjugates affected by changes in UDP–GlcNAc availability or changes in their biosynthesis. These glycoconjuagtes and their impact on EMT are discussed below.
External GlcNAc-Containing Glycoconjugates Observed during EMT
Accumulating evidence strongly suggest changes in protein glycosylation impact numerous cancers including melanoma (82), pancreas (83, 84), colon (85), ovarian and breast (86), brain and lung (87), liver (88), and prostate (89) cancers. Generally, alterations in N-glycan structure profoundly affect cellular adhesion and epithelial morphology in vitro (90). Figures 2B,C show that many glycoproteins utilizing UDP–GlcNAc in their biosynthesis occur on key EMT adhesion molecules (e.g., E- and N-cadherin). E-cadherin has four putative N-linked glycosylation sites (91), which are modified by complex N-linked glycans. The number of “antennae” on these glycans is regulated by the competing activities of GNT3 and GNT5. The introduction of a bisecting GlcNAc by GNT3 (Figure 2B) reduces the number of antennae and thus complexity of the N-linked glycans. Epigenetic regulation of the gene encoding GNT3, MGAT3, stabilizes E-cadherin and inhibits EMT (92). In contrast, elevated activity or expression of GNT5 results in more complex N-glycans, which impairs E-cadherin localization and cellular aggregation in mice (93). Additional studies in mice have revealed that MGAT5 knockdown leads to a reduction of N-glycosylated E-cadherin, which increases E-cadherin cis-dimerization, catenin recruitment, and cell membrane localization (94, 95). Importantly, aberrantly N-glycosyated E-cadherin is found in gastric cancer patients and correlates with poor patient survival (94).
Mesenchymal N-cadherin is also modified by N-linked glycans, and the modification of these glycans with GlcNAc by GNT5 promotes cell migration, MAPK signaling, and reduced adhesion (96, 97). Furthermore, N-cadherin N-glycans attract galectin-3, forming highly organized lipid rafts on the cell surface, which stabilizes the galectin lattice and enhances cancer cell mobility (98). This galectin lattice structure also recruits several major signaling receptors such as epidermal growth factor (EGF) receptor (EGFR) and TGF-β to promote oncogenic signaling (99, 100).
Integrins are heterodimeric glycoproteins responsible for cell–cell and cell–extracellular matrix interactions (101). The α5β1 integrin serves as the receptor for fibronectin, and their interaction is critical for cellular migration in development (102–104). While both integrin and fibronectin are N-glycosylated, the activity of GNT3 is associated with shorter less complex N-glycans, which is thought to result in reduced integrin-mediated EMT signaling (105, 106). Additionally, without N-linked glycans, integrins show significantly decreased heterodimerization, cell surface localization, and promotion of migration in vitro (107).
Receptor tyrosine kinases are vital to transducing external stimuli into internal signals for induction of EMT in many cancer (e.g., carcinomas). Interestingly, RTKs involved in growth and proliferation (e.g., EGFR) have approximately five times more N-glycosylation sites than receptors involved with organogenesis, differentiation, or cell cycle arrest (108). The HBP has been shown to drive changes in EGFR N-glycosylation; feeding both GNT5 wild type and GNT5 null tumor cells with N-acetylglucosamine elevated UDP–GlcNAc levels and the number of terminal GlcNAc residues on cell surface proteins. Analysis of the N-linked glycans demonstrated increased flux through the HBP results in increased triantennary structures in GNT5 null cells (twofold) and a smaller increase in both tri- and tetra-antennary N-glycans in GNT5 wild-type cells. Functionally, increased flux through the HBP-altered EGFR plasma membrane retention, active conformation, EGF ligand binding, and inhibition of endocytosis mediated degradation (109, 110). N-glycosylated EGFR recruits N-glycosylated TGF-βR to the galectin lattice thereby promoting TGF-β and SMAD autocrine signaling. TGF-βR with highly branched glycans, a result of increased GNT5 activity, localizes to the plasma membrane, binds galectin-3, inhibits receptor endocytosis, enhances TGF-βR heterodimerization, increases tumor metastasis, and promotes EMT-mediated cell migration (100, 111). TGF-β itself upregulates GCNT1, a critical GlcNAc branching enzyme, producing similar effects in prostate, colorectal, pancreatic, testicular, and breast cancers (112).
The WNT, NOTCH, and HH pathways are also critical for EMT and are modified by glycans that utilize GlcNAc for modulation of pathway activity. All 19 known WNT ligands contain at least one N-linked glycosylation site, and these sites are critical for ligand maturation, lipid processing, secretion, and β-catenin signal transduction (113, 114). WNT also regulates transcription of DPAGT1 to promote EMT through E-cadherin glycosylation (81).
The Notch signaling pathway regulates cell proliferation, survival, and differentiation while glycosylation of components in this pathway are associated with poor prognosis and metastasis in numerous cancers (115, 116). Over two decades of research demonstrates the extracellular domain of Notch receptor is glycosylated with N-linked (117), O-fucose (117, 118), O-GlcNAc (119), and O-glucose (117, 120) glycans. Extension of O-fucose with GlcNAc [catalyzed by O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase (Fringe in Drosophila)] alters Notch ligand–receptor specificity. In Drosophila, extended O-fucose glycans are associated with increase sensitization of Notch to the Delta ligands and reduced sensitivity to the Serrate/Jagged ligands (116). Little is known about the impact of altered HBP flux on the Notch receptor, although one might postulate that changes in UDP–GlcNAc levels may alter Notch glycosylation and thus signaling downstream of this receptor. In the Sonic HH pathway, the G protein-couple receptor (GPCR), smoothened (SMO), is activated to promote cell proliferation and migration (121). Recently, critical N-glycans on SMO were found to abrogate HH induced cell migration due to blunted small heterotrimeric Gαi protein signaling (122).
Beyond the suite of GlcNAc-modified adhesion molecules and receptors, hyaluronic acid (hyaluronan or HA) is an oligomer found ubiquitously in the extracellular space particularly of connective, epithelial, and neural tissues (123). Human HA is a massive (0.5–2 MDa), unbranched glycosaminoglycan composed of the repeating disaccharide consisting of GlcNAc and glucuronic Acid (GlcNAcβ1–4GlcAβ1–3) (124). It is synthesized by HA synthase (HAS) and is extruded through the plasma membrane as it is synthesized. Recent reports suggest hyaluronan synthesis and catabolism is controlled by UDP–GlcNAc concentrations, with hyaluronan serving as a sink for excess UDP–GlcNAc (125). Recent studies have demonstrated that modulating levels of UDP–GlcNAc and glucuronic acid alter the localization of the HAS enzymes (126). Low levels of UDP–GlcNAc are associated with an inhibition of HA synthesis, whereas elevated levels of UDP–GlcNAc are associated with HA synthesis and melanoma progression (126). Consistent with these data, several studies have demonstrated patients with higher extracellular HA or HAS expression have a worse prognosis and survival with more aggressive and metastatic cancers including breast (127–129), prostate (130, 131), lung (132, 133), pancreatic (134), colorectal (135), and ovarian (136) cancers. With respect to EMT, high levels of HA are sufficient to induce the EMT in kidney and mammary epithelial cells (137). Taken together, HA synthesis is in part driven by the HBP, has been associated with EMT, and is found at high levels in many cancers.
Nuclear, Cytoplasmic, and Mitochondrial Glycosylation Observed during EMT
Uridine diphosphate–N-acetylglucosamine can also be utilized for the synthesis of O-linked β-N-acetylglucosamine (O-GlcNAc), an essential PTM of metazoans (138). O-GlcNAc is found on more than 3,000 cytoplasmic, nuclear, and mitochondrial proteins (67). O-GlcNAcylation is thought to regulate protein function in a manner analogous to phosphorylation. O-GlcNAc has been demonstrated to regulate cellular processes such as epigenetics, transcription, translation, protein degradation, metabolism, ribosomal bioenergentics, and cytokinesis (139).
Unlike N-glycans, the O-GlcNAc modification (or O-GlcNAcylation) consists of a monosaccharide of GlcNAc covalently attached to serine and threonine residues through an O-glycosidic bond (138). Where N-linked glycan synthesis and processing is regulated by upwards of 18 enzymes (depending on the structure formed), the dynamic cycling of O-GlcNAc on proteins is regulated by just two enzymes: the O-GlcNAc transferase (OGT) and the O-GlcNAcase (OGA), which add and remove O-GlcNAc, respectively (140). OGT activity and substrate specificity are regulated by changes in UDP–GlcNAc concentrations, and this has led many to suggest that OGT may regulate cell function in a manner dependent on extracellular glucose concentrations (140). Cancer cells which are dependent on glucose and glutamine have been demonstrated to have high UDP–GlcNAc levels (discussed above), high O-GlcNAc levels, and in some cases increased expression of OGT (140). In sum, elevated protein O-GlcNAcylation and OGT expression have been reported in numerous malignancies including breast (16, 63, 64, 141, 142), prostate (143–145), lung (146), pancreas (147), liver (148), and colon (146, 149, 150) cancers. Importantly, levels of O-GlcNAc, OGT, and OGA have correlated with aggressiveness (e.g., Gleason score for prostate cancer) in a number of patient tumor samples including prostate (144), breast (64), endometrial (151), and bladder (152) cancers.
One important class of proteins heavily O-GlcNAcylated are TFs (Figure 2C). Early analyses suggested that over 25% of known O-GlcNAcylated proteins were TFs (14). For many of these TFs, O-GlcNAcylation serves as a direct or indirect competitor of key phosphorylation sites (140). Particularly relevant to EMT is the O-GlcNAcylation and regulation of SNAI1. Upon serial phosphorylation by CK1 and glycogen synthase kinase (GSK)-3β, SNAI1 is primed for nuclear export, β-TrCP ubiquitination, and subsequent proteosomal degradation (153, 154). Interestingly, SNAI1 is O-GlcNAcylated in hyperglycemic conditions preventing GSK-3β phosphorylation, which results in SNAI1 stabilization (155). O-GlcNAcylated SNAI1 is associated with enhanced EMT and migration, which is linked to a repression of E-cadherin. Whether other EMT-inducing TFs are similarly regulated by O-GlcNAcylation is yet to be determined. Beyond SNAI1, O-GlcNAcylation occurs on other TFs generally relevant to cancer including c-Myc (156, 157), β-catenin (158), C/EBPβ (159), p53 (160), and FoxO1 (161), NF-kB (162, 163). Thus, while more experimentation is needed to demonstrate causality between EMT and O-GlcNAcylation, O-GlcNAcylation has demonstrated to be a key regulator of cancer biology.
Previous studies from our lab and others have elucidated the role of the EMT TF, TWIST1, in suppression of oncogene-induced senescence (OIS) (20, 21, 164). While normal cells respond to oncogene activation with p53-p19ARF, p16-Rb, and Atf4-p27KIP-dependent OIS (165, 166), suppression of these pathways through EMT TFs provide an alternative route for cancer to maintain cell cycle progression and proceed along a tumorigenic path. Due to the metabolic regulation of the cell cycle, it is not surprising many of these proteins orchestrating cellular division are also O-GlcNAcylated. Knockdown of OGT results in elevated expression of p27Kip (63), a reduction of cyclin D1 and B1, and diminished PI3K/AKT signaling (167), suggesting that OGT/O-GlcNAc plays key roles during cell cycle progression. Furthermore, OGT is thought to control cytokinesis as it is localized to the mitotic spindle where it interacts with Polo-like kinase. Disrupting O-GlcNAcylation results in defects in cytokinesis and multinucleated cells (168). Overall, global O-GlcNAc levels have numerous effects on the cell cycle, indicative of yet another link to advancing the neoplastic phenotype.
Conclusion
The data discussed here highlight alterations in intracellular and extracellular glycoconjugates that impact different EMT tumorigenic pathways and associated proteins/biomolecules. With recent controversies of EMT transcription programs continuing to unfold (169, 170), it is likely that the role of EMT may extend beyond cancer development and metastasis, including cancer treatment resistance. Thus, understanding how changes in metabolic pathways observed in cancer (e.g., the HBP) impact the distribution and composition of glycoconjugates may provide deeper insights into mechanisms of cancer biology. While most of the research discussed here demonstrates the potential for glycoconjugates to regulate EMT, it may be interesting to see in the future how EMT reciprocally promotes metabolic reprograming and the HBP.
Author Contributions
KT drafted the manuscript and figures based on discussions with Drs NZ and PT. Drs PT and NZ reviewed the manuscript for accuracy and provided constructive criticism while editing the manuscript for flow and content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
NZ acknowledges support from the National Institutes of Health (NHLBI P01HL107153, NCI CA199806) and the Mizutani Foundations for the Glycosciences. KT was funded by the NIH (F31CA189588). PT was funded by the Keeling Family, the DoD (W81XWH-11-1-0272 and W81XWH-13-1-0182), a Kimmel Translational Science Award (SKF-13-021), an ACS Scholar award (122688-RSG-12-196-01-TBG), and the NIH (R01CA166348).
References
1. Warburg O. On the origin of cancer cells. Science (1956) 123:309–14. doi:10.1126/science.123.3191.309
2. Warburg O. Injuring of respiration the origin of cancer cells. Science (1956) 123:309–14. doi:10.1126/science.123.3191.309
3. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer (2011) 11:325–37. doi:10.1038/nrc3108
4. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
5. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi:10.1038/nrc2981
7. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324:1029–33. doi:10.1126/science.1160809
8. Kennedy KM, Dewhirst MW. Tumor metabolism of lactate: the influence and therapeutic potential for MCT and CD147 regulation. Future Oncol (2010) 6:127–48. doi:10.2217/fon.09.145
9. Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol (2009) 92:329–33. doi:10.1016/j.radonc.2009.06.025
10. Semenza GL. Tumor metabolism: cancer cells give and take lactate. J Clin Invest (2008) 118:3835–7. doi:10.1172/JCI37373
11. Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science (2012) 337:975–80. doi:10.1126/science.1222278
12. Lau KS, Dennis JW. N-glycans in cancer progression. Glycobiology (2008) 18:750–60. doi:10.1093/glycob/cwn071
13. Taniguchi N, Kizuka Y. Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv Cancer Res (2015) 126:11–51. doi:10.1016/bs.acr.2014.11.001
14. Love D, Hanover J. The hexosamine signaling pathway: deciphering the “O-GlcNAc code.” Sci STKE (2005) 2005:re13. doi:10.1126/stke.3122005re13
15. Hanover JA, Krause MW, Love DC. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim Biophys Acta (2010) 1800:80–95. doi:10.1016/j.bbagen.2009.07.017
16. Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell (2014) 54:820–31. doi:10.1016/j.molcel.2014.04.026
17. Vasconcelos-dos-Santos A, Oliveira I, Lucena MC, Mantuano NR, Whelan S, Dias WB, et al. Biosynthetic machinery involved in aberrant glycosylation: promising targets for developing of drugs against cancer. Front Oncol (2015) 5:1–23. doi:10.3389/fonc.2015.00138
18. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol (2014) 15:178–96. doi:10.1038/nrm3758
19. Smit MA, Peeper DS. Deregulating EMT and senescence: double impact by a single twist. Cancer Cell (2008) 14:5–7. doi:10.1016/j.ccr.2008.06.012
20. Tran PT, Shroff EH, Burns TF, Thiyagarajan S, Das ST, Zabuawala T, et al. Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis. PLoS Genet (2012) 8:e1002650. doi:10.1371/journal.pgen.1002650
21. Ansieau S, Bastid J, Doreau A, Morel A-P, Bouchet BP, Thomas C, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell (2008) 14:79–89. doi:10.1016/j.ccr.2008.06.005
22. Shaul YD, Freinkman E, Comb WC, Cantor JR, Tam WL, Thiru P, et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell (2014) 158:1094–109. doi:10.1016/j.cell.2014.07.032
23. Hay ED. An overview of epithelio-mesenchymal transformation. Acta Anat (Basel) (1995) 154:8–20. doi:10.1159/000147748
24. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139:871–90. doi:10.1016/j.cell.2009.11.007
25. Schnaper HW, Hayashida T, Hubchak SC, Poncelet A-C. TGF-β signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol (2003) 284:F243–52. doi:10.1152/ajprenal.00300.2002
26. Margadant C, Sonnenberg A. Integrin-TGF-beta crosstalk in fibrosis, cancer and wound healing. EMBO Rep (2010) 11:97–105. doi:10.1038/embor.2009.276
27. Yan C, Grimm WA, Garner WL, Qin L, Travis T, Tan N, et al. Epithelial to mesenchymal transition in human skin wound healing is induced by tumor necrosis factor-alpha through bone morphogenic protein-2. Am J Pathol (2010) 176:2247–58. doi:10.2353/ajpath.2010.090048
28. Brabletz T. EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell (2012) 22:699–701. doi:10.1016/j.ccr.2012.11.009
29. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene (2010) 29:4741–51. doi:10.1038/onc.2010.215
30. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
31. Fuxe J, Vincent T, De Herreros AG. Transcriptional crosstalk between TGFβ and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle (2010) 9:2363–74. doi:10.4161/cc.9.12.12050
32. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol (2002) 3:155–66. doi:10.1038/nrm757
33. Kang Y, Massagué J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell (2004) 118:277–9. doi:10.1016/j.cell.2004.07.011
34. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer (2007) 7:415–28. doi:10.1038/nrc2131
35. Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med (2013) 19:1438–49. doi:10.1038/nm.3336
36. Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene (2008) 27:6958–69. doi:10.1038/onc.2008.346
37. Díaz V, Viñas-Castells R, García de Herreros A. Regulation of the protein stability of EMT transcription factors. Cell Adh Migr (2014) 8:418–28. doi:10.4161/19336918.2014.969998
38. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev (2009) 28:15–33. doi:10.1007/s10555-008-9169-0
39. van Roy F. Beyond E-cadherin: roles of other cadherin superfamily members in cancer. Nat Rev Cancer (2014) 14:121–34. doi:10.1038/nrc3647
40. Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, et al. E/N-cadherin switch mediates cancer progression via TGF-β-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer (2011) 105:1885–93. doi:10.1038/bjc.2011.452
41. Gheldof A, Berx G. Cadherins and epithelial-to-mesenchymal transition. Prog Mol Biol Transl Sci (2013) 116:317–36. doi:10.1016/B978-0-12-394311-8.00014-5
42. Vesuna F, van Diest P, Chen JH, Raman V. Twist is a transcriptional repressor of E-cadherin gene expression in breast cancer. Biochem Biophys Res Commun (2008) 367:235–41. doi:10.1016/j.bbrc.2007.11.151
43. Sánchez-Tilló E, Lázaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene (2010) 29:3490–500. doi:10.1038/onc.2010.102
44. Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol (2000) 2:84–9. doi:10.1038/35000034
45. Corallino S, Malabarba MG, Zobel M, Di Fiore PP, Scita G. Epithelial-to-mesenchymal plasticity harnesses endocytic circuitries. Front Oncol (2015) 5:45. doi:10.3389/fonc.2015.00045
46. Nakaya Y, Sheng G. EMT in developmental morphogenesis. Cancer Lett (2013) 341:9–15. doi:10.1016/j.canlet.2013.02.037
47. Revenu C, Gilmour D. EMT 2.0: shaping epithelia through collective migration. Curr Opin Genet Dev (2009) 19:338–42. doi:10.1016/j.gde.2009.04.007
48. Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol (2009) 10:445–57. doi:10.1038/nrm2720
49. Stallings-Mann ML, Waldmann J, Zhang Y, Miller E, Gauthier ML, Visscher DW, et al. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci Transl Med (2012) 4:ra95–142. doi:10.1126/scitranslmed.3004062
50. Zheng G, Lyons JG, Tan TK, Wang Y, Hsu T-T, Min D, et al. Disruption of E-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol (2009) 175:580–91. doi:10.2353/ajpath.2009.080983
51. Nelson CM, Khauv D, Bissell MJ, Radisky DC. Change in cell shape is required for matrix metalloproteinase-induced epithelial-mesenchymal transition of mammary epithelial cells. J Cell Biochem (2008) 105:25–33. doi:10.1002/jcb.21821
52. Szabova L, Chrysovergis K, Yamada SS, Holmbeck K. MT1-MMP is required for efficient tumor dissemination in experimental metastatic disease. Oncogene (2008) 27:3274–81. doi:10.1038/sj.onc.1210982
54. López-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med (2009) 1:303–14. doi:10.1002/emmm.200900043
55. Cha YH, Yook JI, Kim HS, Kim NH. Catabolic metabolism during cancer EMT. Arch Pharm Res (2015) 38:313–20. doi:10.1007/s12272-015-0567-x
56. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang X-Y, Pfeiffer HK, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A (2008) 105:18782–7. doi:10.1073/pnas.0810199105
57. DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A (2007) 104:19345–50. doi:10.1073/pnas.0709747104
58. Eagle H. Nutrition needs of mammalian cells in tissue culture. Science (1955) 122:501–14. doi:10.1126/science.122.3168.501
59. Marshall S, Bacote V, Traxinger RR. Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system: role of hexosamine biosynthesis in the induction of insulin resistance. J Biol Chem (1991) 266:4706–12.
60. Abdel Rahman AM, Ryczko M, Pawling J, Dennis JW. Probing the hexosamine biosynthetic pathway in human tumor cells by multitargeted tandem mass spectrometry. ACS Chem Biol (2013) 8:2053–62. doi:10.1021/cb4004173
61. Nakajima K, Kitazume S, Angata T, Fujinawa R, Ohtsubo K, Miyoshi E, et al. Simultaneous determination of nucleotide sugars with ion-pair reversed-phase HPLC. Glycobiology (2010) 20:865–71. doi:10.1093/glycob/cwq044
62. Marshall S, Nadeau O, Yamasaki K. Dynamic actions of glucose and glucosamine on hexosamine biosynthesis in isolated adipocytes: differential effects on glucosamine 6-phosphate, UDP-N-acetylglucosamine, and ATP levels. J Biol Chem (2004) 279:35313–9. doi:10.1074/jbc.M404133200
63. Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene (2010) 29:2831–42. doi:10.1038/onc.2010.41
64. Krzeslak A, Forma E, Bernaciak M, Romanowicz H, Brys M. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin Exp Med (2012) 12:61–5. doi:10.1007/s10238-011-0138-5
65. Broschat KO, Gorka C, Page JD, Martin-Berger CL, Davies MS, Huang HC, et al. Kinetic characterization of human glutamine-fructose-6-phosphate amidotransferase I. Potent feedback inhibition by glucosamine 6-phosphate. J Biol Chem (2002) 277:14764–70. doi:10.1074/jbc.M201056200
66. Wellen KE, Lu C, Mancuso A, Lemons JMS, Ryczko M, Dennis JW, et al. The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev (2010) 24:2784–99. doi:10.1101/gad.1985910
67. Wells L, Vosseller K, Hart GW. A role for N-acetylglucosamine as a nutrient sensor and mediator of insulin resistance. Cell Mol Life Sci (2003) 60:222–8. doi:10.1007/s000180300017
68. Sasai K, Ikeda Y, Fujii T, Tsuda T, Taniguchi N. UDP-GlcNAc concentration is an important factor in the biosynthesis of beta1,6-branched oligosaccharides: regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology (2002) 12:119–27. doi:10.1093/glycob/12.2.119
69. Freeze HH, Elbein AD. Glycosylation precursors. 2nd ed. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Source Essentials of Glycobiology, Chapter 4. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press (2009). p. 47–61.
70. Itkonen HM, Engedal N, Babaie E, Luhr M, Guldvik IJ, Minner S, et al. UAP1 is overexpressed in prostate cancer and is protective against inhibitors of N-linked glycosylation. Oncogene (2014) 34:1–7. doi:10.1038/onc.2014.307
71. Narimatsu H. Human glycogene cloning: focus on beta 3-glycosyltransferase and beta 4-glycosyltransferase families. Curr Opin Struct Biol (2006) 16:567–75. doi:10.1016/j.sbi.2006.09.001
72. Granovsky M, Fata J, Pawling J, Muller WJ, Khokha R, Dennis JW. Suppression of tumor growth and metastasis in Mgat5-deficient mice. Nat Med (2000) 6:306–12. doi:10.1038/73163
73. Li D, Li Y, Wu X, Li Q, Yu J, Gen J, et al. Knockdown of Mgat5 inhibits breast cancer cell growth with activation of CD4+ T cells and macrophages. J Immunol (2008) 180:3158–65. doi:10.4049/jimmunol.180.5.3158
74. Zhou X, Chen H, Wang Q, Zhang L, Zhao J. Knockdown of Mgat5 inhibits CD133+ human pulmonary adenocarcinoma cell growth in vitro and in vivo. Clin Invest Med (2011) 34:E155–62.
75. Fernandes B, Sagman U, Auger M, Demetrio M, Dennis JW. Beta 1-6 branched oligosaccharides as a marker of tumor progression in human breast and colon neoplasia. Cancer Res (1991) 51:718–23.
76. Seelentag WKF, Li W, Schmitz SH, Carcinoma C. Prognostic value of β 1, 6-branched oligosaccharides in human colorectal carcinoma. Cancer Res (1998) 58:5559–64.
77. Murata K, Miyoshi E, Kameyama M, Ishikawa O, Kabuto T, Sasaki Y, et al. Expression of N-acetylglucosaminyltransferase V in colorectal cancer correlates with metastasis and poor prognosis. Clin Cancer Res (2000) 6:1772–7.
78. Sethi MK, Thaysen-Andersen M, Smith JT, Baker MS, Packer NH, Hancock WS, et al. Comparative N-glycan profiling of colorectal cancer cell lines reveals unique bisecting GlcNAc and α-2,3-linked sialic acid determinants are associated with membrane proteins of the more metastatic/aggressive cell lines. J Proteome Res (2014) 13:277–88. doi:10.1021/pr400861m
79. Bubka M, Link-Lenczowski P, Janik M, Pocheć E, Lityńska A. Overexpression of N-acetylglucosaminyltransferases III and V in human melanoma cells. Implications for MCAM N-glycosylation. Biochimie (2014) 103:37–49. doi:10.1016/j.biochi.2014.04.003
80. Song Y, Aglipay JA, Bernstein JD, Goswami S, Stanley P. The bisecting GlcNAc on N-glycans inhibits growth factor signaling and retards mammary tumor progression. Cancer Res (2010) 70:3361–71. doi:10.1158/0008-5472.CAN-09-2719
81. Sengupta PK, Bouchie MP, Kukuruzinska MA. N-glycosylation gene DPAGT1 is a target of the Wnt/beta-catenin signaling pathway. J Biol Chem (2010) 285:31164–73. doi:10.1074/jbc.M110.149195
82. Pocheć E, Janik M, Hoja-Łukowicz D, Link-Lenczowski P, Przybyło M, Lityńska A. Expression of integrins α3β1 and α5β1 and GlcNAc β1,6 glycan branching influences metastatic melanoma cell migration on fibronectin. Eur J Cell Biol (2013) 92:355–62. doi:10.1016/j.ejcb.2013.10.007
83. McCarter C, Kletter D, Tang H, Partyka K, Ma Y, Singh S, et al. Prediction of glycan motifs using quantitative analysis of multi-lectin binding: motifs on MUC1 produced by cultured pancreatic cancer cells. Proteomics Clin Appl (2013) 7:632–41. doi:10.1002/prca.201300069
84. Tang H, Hsueh P, Kletter D, Bern M, Haab B. The detection and discovery of glycan motifs in biological samples using lectins and antibodies: new methods and opportunities. Adv Cancer Res (2015) 126:167–202. doi:10.1016/bs.acr.2014.11.003
85. Holst S, Wuhrer M, Rombouts Y. Glycosylation characteristics of colorectal cancer. Adv Cancer Res (2015) 126:203–56. doi:10.1016/bs.acr.2014.11.004
86. Guo H, Abbott KL. Functional impact of tumor-specific N-linked glycan changes in breast and ovarian cancers. Adv Cancer Res (2015) 126:281–303. doi:10.1016/bs.acr.2014.11.006
87. Lemjabbar-Alaoui H, McKinney A, Yang Y, Tran V, Phillips J. Glycosylation alterations in lung and brain cancer. Adv Cancer Res (2015) 126:305–44. doi:10.1016/bs.acr.2014.11.007
88. Mehta A, Herrera H, Block T. Glycosylation and liver cancer. Adv Cancer Res (2015) 126:257–79. doi:10.1016/bs.acr.2014.11.005
89. Drake RR, Jones EE, Powers TW, Nyalwidhe JO. Altered glycosylation in prostate cancer. Adv Cancer Res (2015) 126:345–82. doi:10.1016/bs.acr.2014.12.001
90. Liwosz A, Lei T, Kukuruzinska MA. N-glycosylation affects the molecular organization and stability of e-cadherin junctions. J Biol Chem (2006) 281:23138–49. doi:10.1074/jbc.M512621200
91. Pinho SS, Seruca R, Gärtner F, Yamaguchi Y, Gu J, Taniguchi N, et al. Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol Life Sci (2011) 68:1011–20. doi:10.1007/s00018-010-0595-0
92. Pinho SS, Oliveira P, Cabral J, Carvalho S, Huntsman D, Gärtner F, et al. Loss and recovery of Mgat3 and GnT-III mediated E-cadherin N-glycosylation is a mechanism involved in epithelial-mesenchymal-epithelial transitions. PLoS One (2012) 7:1–9. doi:10.1371/journal.pone.0033191
93. Carvalho S, Catarino TA, Dias AM, Kato M, Almeida A, Hessling B, et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene (2016) 35:1619–31. doi:10.1038/onc.2015.225
94. Zhou F, Su J, Fu L, Yang Y, Zhang L, Wang L, et al. Unglycosylation at Asn-633 made extracellular domain of E-cadherin folded incorrectly and arrested in endoplasmic reticulum, then sequentially degraded by ERAD. Glycoconj J (2008) 25:727–40. doi:10.1007/s10719-008-9133-9
95. Pinho SS, Osório H, Nita-Lazar M, Gomes J, Lopes C, Gärtner F, et al. Role of E-cadherin N-glycosylation profile in a mammary tumor model. Biochem Biophys Res Commun (2009) 379:1091–6. doi:10.1016/j.bbrc.2009.01.024
96. Guo H-B, Lee I, Kamar M, Pierce M. N-acetylglucosaminyltransferase V expression levels regulate cadherin-associated homotypic cell-cell adhesion and intracellular signaling pathways. J Biol Chem (2003) 278:52412–24. doi:10.1074/jbc.M308837200
97. Guo HB, Johnson H, Randolph M, Pierce M. Regulation of homotypic cell-cell adhesion by branched N-glycosylation of N-cadherin extracellular EC2 and EC3 domains. J Biol Chem (2009) 284:34986–97. doi:10.1074/jbc.M109.060806
98. Boscher C, Zheng YZ, Lakshminarayan R, Johannes L, Dennis JW, Foster LJ, et al. Galectin-3 protein regulates mobility of N-cadherin and GM1 ganglioside at cell-cell junctions of mammary carcinoma cells. J Biol Chem (2012) 287:32940–52. doi:10.1074/jbc.M112.353334
99. Lajoie P, Partridge EA, Guay G, Goetz JG, Pawling J, Lagana A, et al. Plasma membrane domain organization regulates EGFR signaling in tumor cells. J Cell Biol (2007) 179:341–56. doi:10.1083/jcb.200611106
100. Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, et al. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science (2004) 306:120–4. doi:10.1126/science.1102109
101. Gu JG, Isaji T, Sato Y, Kariya Y, Fukuda T. Importance of N-glycosylation on alpha 5 beta 1 integrin for its biological functions. Biol Pharm Bull (2009) 32:780–5. doi:10.1248/bpb.32.780
102. Yang JT, Rayburn H, Hynes RO. Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development (1993) 119:1093–105.
103. Goh KL, Yang JT, Hynes RO. Mesodermal defects and cranial neural crest apoptosis in alpha5 integrin-null embryos. Development (1997) 124:4309–19.
104. George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development (1993) 119:1079–91.
105. Sato Y, Isaji T, Tajiri M, Yoshida-Yamamoto S, Yoshinaka T, Somehara T, et al. An N-glycosylation site on the beta-propeller domain of the integrin alpha5 subunit plays key roles in both its function and site-specific modification by beta1,4-N-acetylglucosaminyltransferase III. J Biol Chem (2009) 284:11873–81. doi:10.1074/jbc.M807660200
106. Isaji T, Gu J, Nishiuchi R, Zhao Y, Takahashi M, Miyoshi E, et al. Introduction of bisecting GlcNAc into integrin alpha5beta11 reduces ligand binding and down-regulates cell adhesion and cell migration. J Biol Chem (2004) 279:19747–54. doi:10.1074/jbc.M311627200
107. Isaji T, Sato Y, Zhao Y, Miyoshi E, Wada Y, Taniguchi N, et al. N-glycosylation of the β-propeller domain of the integrin α5 subunit is essential for α5β1 heterodimerization, expression on the cell surface, and its biological function. J Biol Chem (2006) 281:33258–67. doi:10.1074/jbc.M607771200
108. Nabi IR, Shankar J, Dennis JW. The galectin lattice at a glance. J Cell Sci (2015) 128(13):2213–9. doi:10.1242/jcs.151159
109. Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell (2007) 129:123–34. doi:10.1016/j.cell.2007.01.049
110. Kaszuba K, Grzybek M, Orłowski A, Danne R, Róg T, Simons K, et al. N-glycosylation as determinant of epidermal growth factor receptor conformation in membranes. Proc Natl Acad Sci U S A (2015) 112:2–7. doi:10.1073/pnas.1503262112
111. Kim Y-W, Park J, Lee H-J, Lee S-Y, Kim S-J. TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem J (2012) 445:403–11. doi:10.1042/BJ20111923
112. Zhang H, Meng F, Wu S, Kreike B, Sethi S, Chen W, et al. Engagement of I-branching {beta}-1, 6-N-acetylglucosaminyltransferase 2 in breast cancer metastasis and TGF-{beta} signaling. Cancer Res (2011) 71:4846–56. doi:10.1158/0008-5472.CAN-11-0414
113. Komekado H, Yamamoto H, Chiba T, Kikuchi A. Glycosylation and palmitoylation of Wnt-3a are coupled to produce an active form of Wnt-3a. Genes Cells (2007) 12:521–34. doi:10.1111/j.1365-2443.2007.01068.x
114. Anagnostou SH, Shepherd PR. Glucose induces an autocrine activation of the Wnt/beta-catenin pathway in macrophage cell lines. Biochem J (2008) 416:211–8. doi:10.1042/BJ20081426
116. Kopan R, Ilagan MX. The canonical notch signaling pathway: unfolding the activation mechanism. Cell (2009) 137:216–33. doi:10.1016/j.cell.2009.03.045
117. Moloney DJ, Shair LH, Lu FM, Xia J, Locke R, Matta KL, et al. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J Biol Chem (2000) 275:9604–11. doi:10.1074/jbc.275.13.9604
118. Shao L, Moloney DJ, Haltiwanger R. Fringe modifies o-fucose on mouse Notch1 at epidermal growth factor-like repeats within the ligand-binding site and the Abruptex region. J Biol Chem (2003) 278:7775–82. doi:10.1074/jbc.M212221200
119. Matsuura A, Ito M, Sakaidani Y, Kondo T, Murakami K, Furukawa K, et al. O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem (2008) 283:35486–95. doi:10.1074/jbc.M806202200
120. Rana NA, Nita-Lazar A, Takeuchi H, Kakuda S, Luther KB, Haltiwanger RS. O-glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. J Biol Chem (2011) 286:31623–37. doi:10.1074/jbc.M111.268243
121. Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem (2011) 286:19589–96. doi:10.1074/jbc.M110.197111
122. Marada S, Navarro G, Truong A, Stewart DP, Arensdorf AM, Nachtergaele S, et al. Functional divergence in the role of N-linked glycosylation in smoothened signaling. PLoS Genet (2015) 11:e1005473. doi:10.1371/journal.pgen.1005473
123. Vigetti D, Karousou E, Viola M, Deleonibus S, De Luca G, Passi A. Hyaluronan: biosynthesis and signaling. Biochim Biophys Acta (2014) 1840:2452–9. doi:10.1016/j.bbagen.2014.02.001
124. Holmes MW, Bayliss MT, Muir H. Hyaluronic acid in human articular cartilage. Age-related changes in content and size. Biochem J (1988) 250:435–41. doi:10.1042/bj2500435
125. Hascall VC, Wang A, Tammi M, Oikari S, Tammi R, Passi A, et al. The dynamic metabolism of hyaluronan regulates the cytosolic concentration of UDP-GlcNAc. Matrix Biol (2014) 35:14–7. doi:10.1016/j.matbio.2014.01.014
126. Deen AJ, Arasu UT, Pasonen-Seppänen S, Hassinen A, Takabe P, Wojciechowski S, et al. UDP-sugar substrates of HAS3 regulate its O-GlcNAcylation, intracellular traffic, extracellular shedding and correlate with melanoma progression. Cell Mol Life Sci (2016). doi:10.1007/s00018-016-2158-5
127. Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, Johansson R, et al. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol (2000) 156:529–36. doi:10.1016/S0002-9440(10)64757-8
128. Auvinen P, Tammi R, Kosma V-M, Sironen R, Soini Y, Mannermaa A, et al. Increased hyaluronan content and stromal cell CD44 associate with HER2 positivity and poor prognosis in human breast cancer. Int J Cancer (2013) 132:531–9. doi:10.1002/ijc.27707
129. Auvinen P, Rilla K, Tumelius R, Tammi M, Sironen R, Soini Y, et al. Hyaluronan synthases (HAS1-3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival. Breast Cancer Res Treat (2014) 143:277–86. doi:10.1007/s10549-013-2804-7
130. Lipponen P, Aaltomaa S, Tammi R, Tammi M, Agren U, Kosma VM. High stromal hyaluronan level is associated with poor differentiation and metastasis in prostate cancer. Eur J Cancer (2001) 37:849–56. doi:10.1016/S0959-8049(00)00448-2
131. Aaltomaa S, Lipponen P, Tammi R, Tammi M, Viitanen J, Kankkunen J-P, et al. Strong stromal hyaluronan expression is associated with PSA recurrence in local prostate cancer. Urol Int (2002) 69:266–72. doi:10.1159/000066123
132. Sá VK, Rocha TP, Moreira A, Soares FA, Takagaki T, Carvalho L, et al. Hyaluronidases and hyaluronan synthases expression is inversely correlated with malignancy in lung/bronchial pre-neoplastic and neoplastic lesions, affecting prognosis. Brazilian J Med Biol Res (2015) 48:1039–47. doi:10.1590/1414-431X20154693
133. Pirinen R, Leinonen T, Böhm J, Johansson R, Ropponen K, Kumpulainen E, et al. Versican in nonsmall cell lung cancer: relation to hyaluronan, clinicopathologic factors, and prognosis. Hum Pathol (2005) 36:44–50. doi:10.1016/j.humpath.2004.10.010
134. Cheng X-B, Sato N, Kohi S, Yamaguchi K. Prognostic impact of hyaluronan and its regulators in pancreatic ductal adenocarcinoma. PLoS One (2013) 8:e80765. doi:10.1371/journal.pone.0080765
135. Ropponen K, Tammi M, Parkkinen J, Eskelinen M, Tammi R, Lipponen P, et al. Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res (1998) 58:342–7.
136. Anttila MA, Tammi RH, Tammi MI, Syrjanen KJ, Saarikoski SV, Kosma VM. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res (2000) 60:150–5.
137. Zoltan-Jones A, Huang L, Ghatak S, Toole BP. Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem (2003) 278:45801–10. doi:10.1074/jbc.M308168200
138. Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem (1984) 259:3308–17.
139. Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer (2011) 11:678–84. doi:10.1038/nrc3114
140. Iyer SPN, Hart GW. Dynamic nuclear and cytoplasmic glycosylation: enzymes of O-GlcNAc cycling. Biochemistry (2003) 42:2493–9. doi:10.1021/bi020685a
141. Champattanachai V, Netsirisawan P, Chaiyawat P, Phueaouan T, Charoenwattanasatien R, Chokchaichamnankit D, et al. Proteomic analysis and abrogated expression of O-GlcNAcylated proteins associated with primary breast cancer. Proteomics (2013) 13:2088–99. doi:10.1002/pmic.201200126
142. Gu Y, Mi W, Ge Y, Liu H, Fan Q, Han C, et al. GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res (2010) 70:6344–51. doi:10.1158/0008-5472.CAN-09-1887
143. Gu Y, Gao J, Han C, Zhang X, Liu H, Ma L, et al. O-GlcNAcylation is increased in prostate cancer tissues and enhances malignancy of prostate cancer cells. Mol Med Rep (2014) 10:897–904. doi:10.3892/mmr.2014.2269
144. Kamigaito T, Okaneya T, Kawakubo M, Shimojo H, Nishizawa O, Nakayama J. Overexpression of O-GlcNAc by prostate cancer cells is significantly associated with poor prognosis of patients. Prostate Cancer Prostatic Dis (2014) 17:18–22. doi:10.1038/pcan.2013.56
145. Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, Reginato MJ. Critical role of O-Linked β-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem (2012) 287:11070–81. doi:10.1074/jbc.M111.302547
146. Mi W, Gu Y, Han C, Liu H, Fan Q, Zhang X, et al. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim Biophys Acta (2011) 1812:514–9. doi:10.1016/j.bbadis.2011.01.009
147. Ma Z, Vocadlo DJ, Vosseller K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-κB activity in pancreatic cancer cells. J Biol Chem (2013) 288:15121–30. doi:10.1074/jbc.M113.470047
148. Zhu Q, Zhou L, Yang Z, Lai M, Xie H, Wu L, et al. O-GlcNAcylation plays a role in tumor recurrence of hepatocellular carcinoma following liver transplantation. Med Oncol (2012) 29:985–93. doi:10.1007/s12032-011-9912-1
149. Phueaouan T, Chaiyawat P, Netsirisawan P, Chokchaichamnankit D, Punyarit P, Srisomsap C, et al. Aberrant O-GlcNAc-modified proteins expressed in primary colorectal cancer. Oncol Rep (2013) 30:2929–36. doi:10.3892/or.2013.2794
150. Yehezkel G, Cohen L, Kliger A, Manor E, Khalaila I. O-linked β-N-acetylglucosaminylation (O-GlcNAcylation) in primary and metastatic colorectal cancer clones and effect of N-acetyl-β-D-glucosaminidase silencing on cell phenotype and transcriptome. J Biol Chem (2012) 287:28755–69. doi:10.1074/jbc.M112.345546
151. Krześlak A, Wójcik-Krowiranda K, Forma E, Bieńkiewicz A, Bryś M. Expression of genes encoding for enzymes associated with O-GlcNAcylation in endometrial carcinomas: clinicopathologic correlations. Ginekol Pol (2012) 83:22–6.
152. Rozanski W, Krzeslak A, Forma E, Brys M, Blewniewski M, Wozniak P, et al. Prediction of bladder cancer based on urinary content of MGEA5 and OGT mRNA level. Clin Lab (2012) 58:579–83.
153. Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol (2004) 6:931–40. doi:10.1038/ncb1173
154. Xu Y, Lee S-H, Kim HS, Kim NH, Piao S, Park S-H, et al. Role of CK1 in GSK3beta-mediated phosphorylation and degradation of snail. Oncogene (2010) 29:3124–33. doi:10.1038/onc.2010.77
155. Park SY, Kim HS, Kim NH, Ji S, Cha SY, Kang JG, et al. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J (2010) 29:3787–96. doi:10.1038/emboj.2010.254
156. Chou TY, Dang CV, Hart GW. Glycosylation of the c-Myc transactivation domain. Proc Natl Acad Sci U S A (1995) 92:4417–21. doi:10.1073/pnas.92.10.4417
157. Chou TY, Hart GW, Dang CV. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J Biol Chem (1995) 270:18961–5. doi:10.1074/jbc.270.32.18961
158. Olivier-Van Stichelen S, Dehennaut V, Buzy A, Zachayus J-L, Guinez C, Mir A-M, et al. O-GlcNAcylation stabilizes β-catenin through direct competition with phosphorylation at threonine 41. FASEB J (2014) 28:3325–38. doi:10.1096/fj.13-243535
159. Li X, Molina H, Huang H, Zhang Y-Y, Liu M, Qian S-W, et al. O-linked N-acetylglucosamine modification on CCAAT enhancer-binding protein beta: role during adipocyte differentiation. J Biol Chem (2009) 284:19248–54. doi:10.1074/jbc.M109.005678
160. Yang WH, Kim JE, Nam HW, Ju JW, Kim HS, Kim YS, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nat Cell Biol (2006) 8:1074–83. doi:10.1038/ncb1470
161. Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, et al. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem (2008) 283:16283–92. doi:10.1074/jbc.M802240200
162. Yang WH, Park SY, Nam HW, Kim DH, Kang JG, Kang ES, et al. NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci U S A (2008) 105:17345–50. doi:10.1073/pnas.0806198105
163. Allison DF, Wamsley JJ, Kumar M, Li D, Gray LG, Hart GW, et al. Modification of RelA by O-linked N-acetylglucosamine links glucose metabolism to NF-κB acetylation and transcription. Proc Natl Acad Sci U S A (2012) 109:16888–93. doi:10.1073/pnas.1208468109
164. Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell (2010) 18:448–58. doi:10.1016/j.ccr.2010.10.020
165. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell (1997) 88:593–602. doi:10.1016/S0092-8674(00)81902-9
166. Lin H-K, Chen Z, Wang G, Nardella C, Lee S-W, Chan C-H, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature (2010) 464:374–9. doi:10.1038/nature08815
167. Olivier-Van Stichelen S, Drougat L, Dehennaut V, El Yazidi-Belkoura I, Guinez C, Mir A-M, et al. Serum-stimulated cell cycle entry promotes ncOGT synthesis required for cyclin D expression. Oncogenesis (2012) 1:e36. doi:10.1038/oncsis.2012.36
168. Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem (2005) 280:32944–56. doi:10.1074/jbc.M503396200
169. Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, et al. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature (2015) 527:525–30. doi:10.1038/nature16064
Keywords: glycoproteins, glycosylation, O-GlcNAcylation, O-GlcNAc, EMT, cancer, nucleotide sugar, metabolism
Citation: Taparra K, Tran PT and Zachara NE (2016) Hijacking the Hexosamine Biosynthetic Pathway to Promote EMT-Mediated Neoplastic Phenotypes. Front. Oncol. 6:85. doi: 10.3389/fonc.2016.00085
Received: 22 February 2016; Accepted: 27 March 2016;
Published: 18 April 2016
Edited by:
Leonardo Freire-de-Lima, Federal University of Rio de Janeiro, BrazilReviewed by:
Michaela Ruth Reagan, Maine Medical Center Research Institute, USALuciana Boffoni Gentile, Universidade Federal do Rio de Janeiro, Brazil
Copyright: © 2016 Taparra, Tran and Zachara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Natasha E. Zachara, bnphY2hhcmFAamhtaS5lZHU=