 Pranshu Bansal
Pranshu Bansal Diaa Osman1,2
Diaa Osman1,2- 1Department of Internal Medicine, Division of Hematology/Oncology, University of New Mexico Comprehensive Cancer Center, University of New Mexico School of Medicine, Albuquerque, NM, USA
- 2Hematology/Oncology Fellowship Program, University of New Mexico Comprehensive Cancer Center, University of New Mexico School of Medicine, Albuquerque, NM, USA
- 3Section of Radiation Oncology, University of New Mexico Comprehensive Cancer Center, Albuquerque, NM, USA
- 4Department of Thoracic and Head/Neck Medical Oncology, Division of Cancer Medicine, University of Texas MD Anderson Cancer Center, Houston, TX, USA
- 5Cancer Genetics, Epigenetics, and Genomics Research Program, University of New Mexico Comprehensive Cancer Center, Albuquerque, NM, USA
Lung adenocarcinoma is the most common subtype of non-small cell lung cancer (NSCLC). With the discovery of epidermal growth factor receptor (EGFR) mutations, anaplastic lymphoma kinase (ALK) rearrangements, and effective targeted therapies, therapeutic options are expanding for patients with lung adenocarcinoma. Here, we review novel therapies in non-squamous NSCLC, which are directed against oncogenic targets, including EGFR, ALK, ROS1, BRAF, MET, human epidermal growth factor receptor 2 (HER2), vascular endothelial growth factor receptor 2 (VEGFR2), RET, and NTRK. With the rapidly evolving molecular testing and development of new targeted agents, our ability to further personalize therapy in non-squamous NSCLC is rapidly expanding.
Introduction
In recent years, advances in lung cancer are occurring at an accelerated pace. With several new targeted therapies approved last year for non-squamous non-small cell lung cancer (NSCLC) alone, it is now one of the most active areas in oncology. Approvals include one vascular endothelial growth factor receptor 2 (VEGFR2) inhibitor, two EGFR-targeting drugs, one anaplastic lymphoma kinase (ALK)-targeting drug, and one ROS1 inhibitor. This review summarizes breakthroughs that have already happened, and some promising ongoing areas of investigation where new drug approvals are anticipated.
Epidermal Growth Factor Receptor
Identification of oncogenic epidermal growth factor receptor (EGFR) mutations in NSCLC has ushered a new era of targeted therapies in metastatic NSCLC. These mutations lead to activation of EGFR, now effectively targeted by an ever-increasing list of tyrosine kinase inhibitors (TKIs). Exon 19 deletion and exon 21 L858R substitutions account for 90% of all EGFR + NSCLC (1). Activating EGFR mutation occur in ~10% Caucasians (2), higher in never smokers (30%) vs. ex-smokers (5%) and current smokers (3%) (2, 3).
Erlotinib and gefitinib are FDA-approved, first-generation reversible inhibitors of both wild-type and mutant EGFR. These are accepted first-line therapies in patients with known EGFR-sensitizing mutations in metastatic NSCLC and have shown overall response rate (ORR) up to 75% and advantage in progression-free survival (PFS) over chemotherapy in several clinical studies (4–6). Class side effects with these agents are primarily skin rash and diarrhea. Despite high ORR and improved PFS, long-term impact of such therapies is limited by the development of resistance.
Afatinib, a second-generation irreversible inhibitor of EGFR and human epidermal growth factor receptor 2 (HER2), also FDA-approved, is used as a frontline therapy in metastatic NSCLC patients with EGFR-sensitizing mutations. Side-effect profile is similar to the first-generation drugs. Two phase III trials, LUX-Lung 3 and LUX-Lung 6, have shown improved PFS and ORR with first-line afatinib compared to chemotherapy (6, 7). Recent subgroup analysis from LUX-Lung 3 and LUX-Lung 6 trials showed significantly improved OS in patients with exon 19 deletion treated with afatinib compared to chemotherapy: 33.3 vs. 21.1 months and 31.4 vs. 18.4 months (8). This OS difference was not seen in patients harboring L858R mutation (8). LUX-lung 7 is a phase II trial comparing afatinib with gefitinib in advanced NSCLC. Preliminary analysis showed that patients treated with afatinib had 27% reduction in risk of progression compared to patients treated with gefitinib. The benefits were seen in both L858R and exon 19 deletion subgroups (9).
The mechanisms of acquired resistance to first-generation EGFR inhibitors can be divided into three groups: secondary mutations, bypass signaling, and phenotypic alterations. The most common resistance mechanism seen in ~50% cases is the development of genetic alterations of EGFR, commonly a secondary mutation like T790M, a threonine to methionine substitution in exon 20. This mutation leads to an enhanced affinity for ATP and steric hindrance, reducing the ability of ATP-competitive, reversible EGFR TKIs to bind to the tyrosine kinase domain of EGFR (10–14). The second mechanism is the activation of additional signaling pathways, including MET, HER2, and CRKL amplification, AXL overexpression, and KRAS and BRAF mutations (15, 16). The third mechanism is histologic transformation into SCLC (17). In ~30% cases, resistance mechanisms are unknown.
One potential way to overcome T790M mutation is the use irreversible EGFR inhibitor afatinib. However, clinical studies with afatinib in patients progressing on the first-generation TKIs have shown a modest ORR of ~10% and PFS of ~4 months (18). Afatinib in combination with cetuximab showed an improved ORR of 30% in T790M+ patients after progression on first-generation TKIs; however, toxicity was a significant concern with this combination (19).
Rociletinib and osimertinib are third-generation TKIs with specificity for EGFR T790M over wild-type EGFR. Osimertinib showed response rate of 60% in phase I/II studies in T790M+ tumors and ~30% in tumors negative for T790M in previously treated EGFR + NSCLC (20). This is a significant advancement over chemotherapy or afatinib/cetuximab combination, which leads to FDA approval of the drug for EGFR T790M+ patients progressing on first/second-generation TKIs (21–24). Rociletinib is another EGFR T790M selective TKI, awaiting FDA approval. Initial rociletinib phase I/II data reported an ORR of 60% and disease control rate (DCR) of 90%; however, initial data contained immature findings, including unconfirmed responses, and the revised ORR was significantly lower at 28–34% (25, 26). Both drugs have a lower incidence of grade 3/4 rash and diarrhea. The most common toxicity with rociletinib is hyperglycemia secondary to IGF1 inhibition (25). ASP8273 and HM61713 are third-generation EGFR TKIs currently in development (NCT02500927, NCT02485652, and NCT02588261).
As with the first- and second-generation EGFR TKIs, tumor evolution leads to resistance to third-generation TKI’s. So far, the data for resistance to third-generation EGFR TKI’s is mostly preclinical. Niederst et al. showed additional C797S EGFR mutations in EGFR T790M cell lines made resistant in vitro to third-generation TKIs. They showed resistant cell lines, harboring C797S and T790M mutations in cis-allelic conformation, were resistant to all types of TKIs; however, if these two mutations were expressed in trans conformation, the cells became resistant to third-generation TKIs but retained sensitivity to first-generation TKIs. Similarly, EGFR C797S mutant but T790M wild-type cell lines retained sensitivity to first-generation TKIs (27). In a separate study of 12 T790M+ patients who underwent tumor biopsy post-progression on rociletinib, 6 patients became T790M wild-type, 2 T790M wild-type cancers underwent SCLC transformation, 3 T790M-positive cancers acquired EGFR amplification, and 1 patient had T790 wild-type and mutant cell populations coexisting in the tumor (28). Other proposed mechanisms of third-generation TKI resistance include epithelial mesenchymal transition (EMT) (21), activation of the MAPK kinase pathway (22), and IGF1R bypass signaling (23).
Combination of an EGFR TKI with VEGF inhibitor, such as bevacizumab, has been studied in patients with EGFR-sensitizing mutations. In a phase II trial comparing erlotinib (E) to erlotinib plus bevacizumab (E + B), median PFS was 16 months compared to 9 months in the E arm (24). E + B combination has shown activity in T790M+ NSCLC, and a recent study showed a median PFS of 16 months in T790M+ NSCLC compared to 10.5 months in T790M wild-type patients (29).
MET amplification occurs in ~3–7% of untreated patients and ~21% patients previously treated with EGFR TKIs (30, 31). Tivantinib is a MET TKI, which was studied in combination with erlotinib compared to erlotinib alone. Combination showed improvement in PFS without advantage in OS; subgroup analysis showed a trend for OS advantage in high MET expression subgroup (32). INC280, another MET TKI, has shown promise in the initial dose escalation and combination phase I study with first-generation EGFR TKI, ongoing in patients who have progressed on EGFR TKI monotherapy (NCT01610336).
Anaplastic Lymphoma Kinase
The EML4–ALK fusion oncogene arises from an inversion on the short arm of chromosome 2 [Inv (2) (p21p23)] that joins exons 1–13 of EML4 to exons 20–29 of ALK (33). The EML4 fusion partner mediates ligand-independent dimerization and/or oligomerization of ALK, resulting in constitutive kinase activity. ALK+ NSCLC represents ~4–5% of NSCLC patients and is much higher among never/light smokers, up to 22% (34, 35).
Crizotinib is a small-molecule that competes with ATP tyrosine kinase activity. Initially developed as a c-MET inhibitor, it was later found to have potent inhibitory activity against ALK and ROS1 (36, 37). In phase I/II studies, crizotinib demonstrated ORR in ~60% of patients with ALK-positive NSCLC and PFS of 7–10 months (38–40). In a recent phase III study, crizotinib was compared to chemotherapy in untreated patients with ALK+ NSCLC, demonstrating a significant improvement in ORR and PFS. No significant OS benefit was shown since 70% of patients crossed over to crizotinib arm after progression on chemotherapy (41). Crizotinib is an FDA-approved, first-line therapy in ALK+ NSCLC metastatic patients.
In spite of the dramatic initial responses, patients develop resistance to crizotinib after a median of 8.9–10.5 months (42). In particular, central nervous system (CNS) is a common site of relapse in patients with ALK+ metastatic NSCLC (43). In a study of 28 patients treated with crizotinib, 13 developed CNS relapses (44). Nevertheless, recent update from the crizotinib phase III trial showed a delay in the onset of CNS relapse compared to chemotherapy (41).
Crizotinib resistance mechanisms can be ALK-dependent or -independent. ALK-dependent resistance includes ALK amplification/copy number gain and additional genetic alterations, including secondary mutations of the ALK kinase domain that preserve ALK signaling (45, 46). Of these, a secondary mutation L1196M, which interferes with binding of crizotinib, is the most common (47). This is very similar to T790M mutation in EGFR+ NSCLC. Additional resistance mutations that occur less frequently include G1269A, C1156Y, L1152R, G1202R, S1206Y, 1151Tins, F1174C, and D1203N.
The ALK-independent mechanisms of resistance involve activation of bypass signaling pathways, such as the EGFR, HSP90, MET, KRAS, or KIT. Analysis of 16 NSCLC patients treated with crizotinib and re-biopsied at progression showed ALK amplification in 13%, ALK mutations in 31%, and KRAS or EGFR pathway activation in 31%; 19% patients had HSP90, KIT, or HER2 activation (46).
In 2014, FDA approved ceritinib, a second-generation ALK inhibitor, when impressive results from a single-arm study of 163 patients with ALK+ positive metastatic NSCLC showed median ORR of 44%. Majority (91%) patients had progressed on prior crizoitinb. This trial also included patients with baseline brain metastasis (60%) (47). Two phase III trials comparing ceritinib to chemotherapy in treatment naïve and previously treated patients are ongoing (NCT01828099, NCT01828112).
Alectinib is another FDA-approved, second-generation ALK inhibitor with activity in crizotinib-resistant ALK+ metastatic NSCLC. A phase II study with 138 patients progressing on crizotinib had 50% ORR and DCR of 79% (48). In a frontline, phase I/II study of crizotinib-naive patients with ALK+ NSCLC treated with alectinib showed an impressive ORR of 93.5% (49). Alectinib has shown activity in patients with CNS metastasis; one study showed an intracranial ORR of 52% in patients progressing on crizotinib (50). A phase III clinical trial comparing alectinib and crizotinib upfront was stopped early as it met its primary endpoint (ALEX trial, NCT02075840).
Mechanisms of resistance to ceritinib and alectinib are undergoing active investigation. G1202R ALK mutation is pan-resistant to crizotinib, ceritinib, and alectinib (43, 51). Multiple ALK and ROS1 inhibitors are in development. Lorlatinib (PF-06463922) is an ALK/ROS1 novel CNS-penetrant inhibitor with preclinical activity against the G1202R mutation that demonstrated encouraging activity in resistant patients: 40% PRs, including patients progressing following crizotinib ± ceritinib; intracranial responses were also observed (52, 53). Brigatinib (AP26113) is an exciting TKI with dual activity against EGFR T790M and ALK L1196M mutations. The results from phase I/II showed significant activity in ALK+ patients. Of 72 evaluable pts, 72% responded: 45/65 (69%) with prior crizotinib and all 7 crizotinib-naive pts. Median duration of response (DOR) was 49 weeks (54).
ROS1
The ROS1 oncogene encodes an orphan receptor tyrosine kinase related to ALK. ROS1 rearrangements occur in ~1–2% of patients with NSCLC, mainly non-smokers or light smokers (55). Phase 1 expansion trial with crizotinib in ROS1 rearrangement-positive NSCLC patients demonstrated marked antitumor activity with 72% ORR and 17.6 months median DOR, which led to its FDA approval in 2016 (56). Similar to experience with EGFR and ALK, crizotinib resistance develops with acquired mutations, such as G2032R and L2155S, in ROS1 kinase domain (57, 58). Preclinical studies with lorlatinib look encouraging, as it has shown activity against novel mutations in both ALK and ROS1, which cause resistance to first- and second-generation TKIs (52). In phase I/II trial, 22 patients with ALK+ or ROS1+ NSCLC received lorlatinib, which was well tolerated with encouraging clinical activity (59). Phase II part of this trial is ongoing (NCT01970865).
BRAF
Oncogenic BRAF mutation is found in ~3–4% of NSCLC, usually non-overlapping with other oncogenic driver mutations with ~50% cases harboring the characteristic V600E mutation (60, 61). Unlike EGFR and ALK, BRAF mutations commonly occur in smokers (60–62). Vemurafenib and dabrafenib are BRAF-targeting TKIs, approved in BRAF-mutated metastatic melanoma. They have shown promise in early-phase trials in NSCLC. A basket phase II trial looking into the efficacy of vemurafenib in different tumors harboring BRAF mutations included 20 patients with advanced NSCLC. The ORR was 42%, superior to other cancers types (63). Dabrafenib showed a ORR of 32% in 78 patients with BRAF-mutated NSCLC; 2-stage phase II study with the second phase evaluating the combination of MEK inhibitor trametinib and dabrafenib is ongoing. Interim analysis showed an encouraging ORR of 63% in 24 patients (64).
c-MET Inhibitors
The MET amplification is oncogenic in 3–7% of NSCLC and confers resistance to EGFR in ~21% cases (31, 32). In addition to amplification, MET gene alterations, namely exon 14 internal deletions and mutations, are oncogenic in a small fraction of NSCLC patients (65). MET receptor tyrosine kinase and its ligand hepatocyte growth factor (HGF) are implicated in tumor cell proliferation, invasion, and angiogenesis in NSCLC (66). Responses with anti-MET TKIs, such as crizotinib and cabozantinib, in MET mutant and amplified NSCLC have been reported (67, 68).
Glesatinib (MGCD265, Mirati) is an oral TKI targeting MET and AXL. A phase I/II trial has been initiated in MET mutant or MET-amplified NSCLC. Early data from phase Ib part had shown confirmed PRs and significant tumor reductions; phase II part is ongoing (NCT02544633).
INC280 is a highly selective small-molecule MET inhibitor with preclinical activity in human tumor models; early-phase trial is initiated of INC280 alone or in combination with chemotherapy or erlotinib (NCT02468661).
Vascular Endothelial Growth Factor Receptor 2
Blockade of VEGFR2 signaling inhibits formation, proliferation, and migration of new blood vessels (69). Ramucirumab is a human IgG1 monoclonal antibody that targets the extracellular domain of VEGFR2, FDA-approved in metasatic NSCLC, in combination with docetaxel, after a phase III trial demonstrated OS benefit over docetaxel alone (69).
Cabozantinib is a VEGFR2/MET inhibitor assessed in a phase II trials in pretreated patients with NSCLC (n = 125). Cabozantinib alone or in combination with erlotinib significantly improved PFS over erlotinib in pts with EGFR wild-type NSCLC (70).
Human Epidermal Growth Factor Receptor 2
Human epidermal growth factor receptor 2 (ERBB2) belongs to ErbB receptor tyrosine kinase family along with EGFR (HER1), HER3, and HER4. Ligand binding and subsequent homo and heterodimerization of these receptors activate EGFR, HER3, and HER4. In this regard, HER2 is unique, as no HER2 ligand has been identified. HER2 is the preferred binding partner of ERBB receptors, in particular EGFR. HER2/EGFR heterodimers have an increased potential for signaling than EGFR homodimers (71). HER2 overexpression (~35%) and amplification (~20%) have been reported in NSCLC (72–74). Clinical results with HER2-directed therapy in these patients have not been convincing (75–77). HER2 mutations, including exon 20 insertions, are oncogenic in breast and lung cancer (77–81). HER2 mutations occur in ~2–5% of NSCLC, more commonly in Asian, non-smoker, and female populations (80, 81). HER2-targeted therapies in HER2-mutant population are an ongoing area of research. The largest retrospective study looking at HER2-targeted agents in this population demonstrated 50% ORR among 16 HER2-mutant patients, primarily treated with trastuzumab and afatinib (82). A trial (NCT02369484) initiated with afatinib in HER2 mutation-positive NSCLC is ongoing.
Neratinib is another irreversible pan-HER inhibitor, which has shown activity in trastuzumab-resistant breast cancer (83). It was evaluated in a phase I study in combination with temsirolimus (mTOR inhibitor) and showed activity in 2/6 HER2-mutant NSCLC (84). Based on these results, a phase II study of neratinib with and without temsirolimus is ongoing (NCT01827267).
Dacomitinib, an irreversible pan-EGFR inhibitor, demonstrated ORR of 13% in HER2-mutant NSCLC patients in a phase II study (85).
RET
The RET gene encodes a RET family receptor tyrosine kinase. Activating somatic point mutations in RET occur in medullary thyroid cancer (86). Recurrent translocations between RET and various fusion partners occur in ~12% NSCLC (87–89). The prevalence is higher among non-smokers, negative for other driver mutations (90).
Cabozantinib showed encouraging results in a phase II study of 16 patients with RET fusion-positive disease, 7/16 had PR with median PFS of 7 months and OS 10 months (91). Studies in RET+ NSCLC with lenvatinib (NCT01877083), apatinib (NCT02540824), vandetinib (NCT01823068), and ponatinib (NCT01813734) are ongoing.
NTRK
NTRK gene encodes for tropomyosin receptor kinase (Trk) protein. Vaishnavi et al. in their pioneering work have shown MPRIP–NTrk1 and CD74–NTrk1 fusions leading to constitutive TrkA activity in 3.3% patients with NSCLC (92). In the same study, authors reported oncogenic TPM3–NTRK1 fusion that has also been reported in a small fraction of colon cancer (93). Stransky et al. reported TRIM24–NTRK2 gene fusion in a NSCLC patient (94).
Entrectinib (RXDX-101) is a highly potent inhibitor of TRK as well as ROS1 and ALK. In a phase I trial, entrectinib has demonstrated clinical activity in TRK-fusion-positive advanced solid malignancies. Trials with this drug and other novel NTRK inhibitors are ongoing [(95), NCT02576431, NCT01639508].
Conclusion
Advances in targeted therapy for metastatic non-squamous NSCLC have now expanded from EGFR and ALK to additional oncogenic targets, including ROS1, BRAF, RET, HER2, NTRK, and MET. Testing for these genes is now standard in many centers and is recommended by the NCCN (96). Identification of new drivers leading to effective personalized therapy is an exciting but challenging task in today’s world. As we know, most targetable mutations are rare, and therefore development of standardized therapies calls for innovative ways to improve our clinical and translational knowledge. In basket trials, patients are included based on molecular aberration regardless of histology, whereas umbrella trials include patients of single tumor types (97). The latter involves a group of two or more enrichment designs, or sub-studies, connected through a central infrastructure overseeing screening and identification of patients and centralized tissue analysis for standardized genotyping (98).
The other major issue we face with molecularly targeted agents is the inevitable emergence of resistance. We are now venturing into the era of resistance to third-generation TKI’s in EGFR/ALK therapies, and the treatment paradigm changes with every successive generation of inhibitors. Patients progressing on first-generation EGFR inhibitors are expected to undergo tumor re-biopsies; and tumor heterogeneity and false-negative results make future treatments more challenging.
Table 1 summarizes ongoing trials in NSCLC, and Figure 1 describes mechanisms of action. Novel drug testing and development of rational drug combination in frontline and recurrent settings in NSCLC remains one of the most exciting, rapidly evolving areas in oncology, with hopes to dramatically increase the numbers of long-term survivors with stage IV disease.
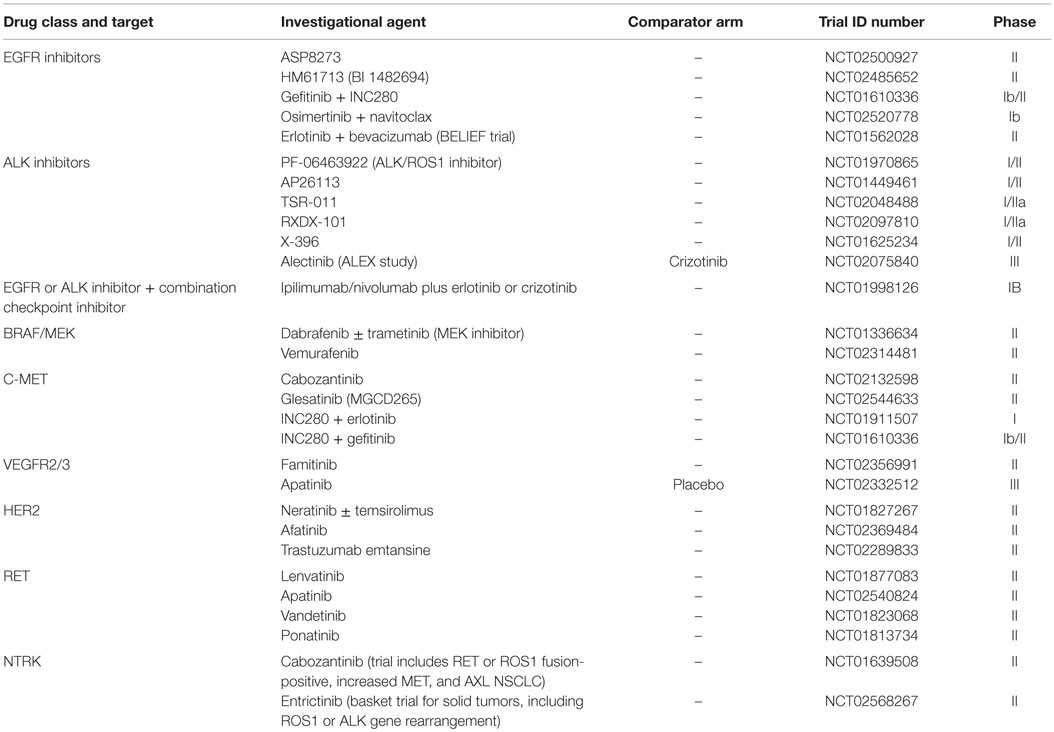
Table 1. Selective ongoing non-squamous NSCLC targeted therapy trials.
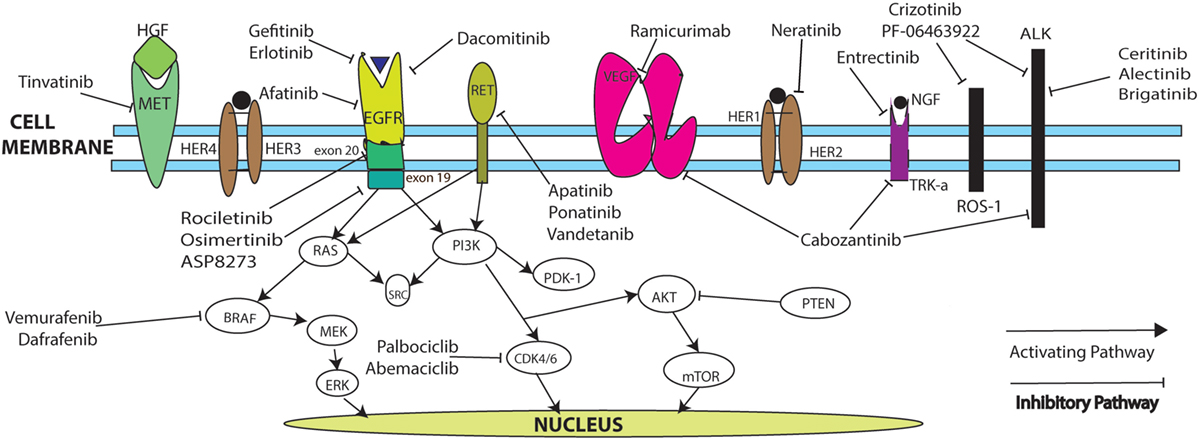
Figure 1. Targeted pathways for NSCLC.
Author Contributions
First author PB – prepared the main manuscript and table. Corresponding author YB – provided guidance in preparing the manuscript, contributed to literature review and vital modifications, and also contributed to the main manuscript and table. Coauthor DO – prepared part of the manuscript and pathway diagram. Coauthor GG – helped with review, edition, and vital updates to the article. Coauthor GS – helped with editing and review of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
NCI P30 CA118100 Core Grant Funding to UNM Comprehensive Cancer Center (PB, DO, YB, and GG) and NCI P30 Core Grant CA016672 to MD Anderson Cancer Center (GS).
References
1. Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol (2008) 21(Suppl 2):S16–22. doi: 10.1038/modpathol.2008.12
2. Skov BG, Høgdall E, Clementsen P, Krasnik M, Larsen KR, Sørensen JB, et al. The prevalence of EGFR mutations in non-small cell lung cancer in an unselected Caucasian population. APMIS (2015) 123(2):108–15. doi:10.1111/apm.12328
3. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer (2007) 7(3):169–81. doi:10.1038/nrc2088
4. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol (2012) 13(3):239–46. doi:10.1016/S1470-2045(12)70227-9
5. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) 361(10):947–57. doi:10.1056/NEJMoa0810699
6. Sequist LV, Yang JC, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol (2013) 31(27):3327–34. doi:10.1200/JCO.2012.44.2806
7. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol (2014) 15(2):213–22. doi:10.1016/S1470-2045(13)70604-1
8. Yang JC, Wu YL, Schuler M, Sebastian M, Popat S, Yamamoto N, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol (2015) 16(2):141–51. doi:10.1016/S1470-2045(15)00026-1
9. Park K, Tan E-H, Zhang L, Hirsh V, O’Byrne K, Boyer M, et al. Afatinib versus gefitinib as first-line treatment for patients with advanced non-small-cell lung cancer harboring activating EGFR mutations: LUX-Lung 7 a phase 2b, open label, randomized controlled trial. Lancet Oncol (2016). doi:10.1016/S1470-2045(16)30033-X
10. Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med (2005) 352(8):786–92. doi:10.1056/NEJMoa044238
11. Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med (2011) 3(75):75ra26. doi:10.1126/scitranslmed.3002003
12. Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res (2011) 17(5):1169–80. doi:10.1158/1078-0432.CCR-10-2277
13. Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res (2006) 66(16):7854–8. doi:10.1158/0008-5472.CAN-06-1951
14. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science (2007) 316(5827):1039–43. doi:10.1126/science.1145015
15. Sierra JR, Cepero V, Giordano S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol Cancer (2010) 9:75. doi:10.1186/1476-4598-9-75
16. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet (2012) 44(8):852–60. doi:10.1038/ng.2412
17. Morinaga R, Okamoto I, Furuta K, Kawano Y, Sekijima M, Dote K, et al. Sequential occurrence of non-small cell and small cell lung cancer with the same EGFR mutation. Lung Cancer (2007) 58(3):411–3. doi:10.1016/j.lungcan.2007.05.014
18. Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol (2012) 13(5):528–38. doi:10.1016/S1470-2045(12)70087-6
19. Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov (2014) 4(9):1036–45. doi:10.1158/2159-8290.CD-14-0326
20. Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor – resistant non-small-cell lung cancer. N Engl J Med (2015) 372(18):1689–99. doi:10.1056/NEJMoa1411817
21. Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov (2013) 3(12):1404–15. doi:10.1158/2159-8290.CD-13-0314
22. Ercan D, Xu C, Yanagita M, Monast CS, Pratilas CA, Montero J, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov (2012) 2(10):934–47. doi:10.1158/2159-8290.CD-12-0103
23. Cortot AB, Repellin CE, Shimamura T, Capelletti M, Zejnullahu K, Ercan D, et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res (2013) 73(2):834–43. doi:10.1158/1538-7445.AM2013-834
24. Seto T, Kato T, Nishio M, Goto K, Atagi S, Hosomi Y, et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomised, multicentre, phase 2 study. Lancet Oncol (2014) 15(11):1236–44. doi:10.1016/S1470-2045(14)70381-X
25. Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med (2015) 372(18):1700–9. doi:10.1056/NEJMoa1413654
26. Sequist LV, Goldman JW, Wakelee HA, Camidge R, Yu HA, Varga A, et al. Efficacy of rociletinib (CO-1686) in plasma-genotyped T790M-positive non-small cell lung cancer (NSCLC) patients (pts). J Clin Oncol (2015) 33(Suppl):abstr8001.
27. Niederst MJ, Hu H, Mulvey HE, Lockerman EL, Garcia AR, Piotrowska Z, et al. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin Cancer Res (2015) 21(17):3924–33. doi:10.1158/1078-0432.CCR-15-0560
28. Piotrowska Z, Niederst MJ, Karlovich CA, Wakelee HA, Neal JW, Mino-Kenudson M, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov (2015) 5(7):713–22. doi:10.1158/2159-8290.CD-15-0399
29. Stahel RA, Dafni U, Gautschi O, Felip E, Curioni-Fontecedro A, Peters S, et al. A phase II trial of erlotinib (E) and bevacizumab (B) in patients with advanced non-small-cell lung cancer (NSCLC) with activating epidermal growth factor receptor (EGFR) mutations with and without T790M mutation. The Spanish Lung Cancer Group (SLCG) and the European Thoracic Oncology Platform (ETOP) BELIEF trial (Abstract 3BA). Presented at 2015 European Cancer Congress; 2015 Sep 25–29; Vienna, Austria. (2015)
30. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A (2007) 104(52):20932–7. doi:10.1073/pnas.0710370104
31. Cappuzzo F, Jänne PA, Skokan M, Finocchiaro G, Rossi E, Ligorio C, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol (2009) 20(2):298–304. doi:10.1093/annonc/mdn635
32. Scagliotti G, von Pawel J, Novello S, Ramlau R, Favaretto A, Barlesi F, et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol (2015) 33(24):2667–74. doi:10.1200/JCO.2014.60.7317
33. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature (2007) 448(7153):561–6. doi:10.1038/nature05945
34. Shaw AT, Yeap BY, Mino-Kenudson M, Digumarthy SR, Costa DB, Heist RS, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol (2009) 27(26):4247–53. doi:10.1200/JCO.2009.22.6993
35. Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res (2008) 14(13):4275–83. doi:10.1158/1078-0432.CCR-08-0168
36. Cui JJ, Tran-DubeÌ M, Shen H, Nambu M, Kung PP, Pairish M, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem (2011) 54(18):6342–63. doi:10.1021/jm2007613
37. Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther (2007) 6(12 Pt 1):3314–22. doi:10.1158/1535-7163.MCT-07-0365
38. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med (2010) 363:1693–703. doi:10.1056/NEJMoa1006448
39. Camidge DR, Bang YJ, Kwak EL, Iafrate AJ, Varella-Garcia M, Fox SB, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol (2012) 13:1011–9. doi:10.1016/S1470-2045(12)70344-3
40. Kim DW, Ahn M-J, Shi Y, Shi Y, De Pass TM, Yang P-C, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). J Clin Oncol (2012) 30. doi:10.1200/JCO.2011.39.9766
41. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med (2014) 371(23):2167–77. doi:10.1056/NEJMoa1408440
42. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med (2012) 4(120):120ra17. doi:10.1126/scitranslmed.3003316
43. Otterson GA, Riely GJ, Shaw AT, Crinò L, Kim D-W, Martins R, et al. Clinical characteristics of ALK+ NSCLC patients (pts) treated with crizotinib beyond disease progression (PD): potential implications for management. J Clin Oncol (2012) 30(15 Suppl).
44. Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A (2011) 108(18):7535–40. doi:10.1073/pnas.1019559108
45. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res (2012) 18(5):1472–82. doi:10.1158/1078-0432.CCR-11-2906
46. Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med (2010) 363(18):1734–9. doi:10.1056/NEJMoa1007478
47. Kim D-W, Mehra R, Tan DSW, Felip E, Chow LQM, Camidge DR, et al. Activity and safety of ceritinib in patients with ALK-rearranged non-small-cell lung cancer (ASCEND-1): updated results from the multicentre, open-label, phase 1 trial. Lancet Oncol (2016). doi:10.1016/S1470-2045(15)00614-2
48. Ou SH, Ahn JS, De Petris L, Govindan R, Yang JC, Hughes B, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol (2016) 34(7):661–8. doi:10.1200/JCO.2015.63.9443
49. Seto T, Kiura K, Nishio M, Nakagawa K, Maemondo M, Inoue A, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1-2 study. Lancet Oncol (2013) 14(7):590–8. doi:10.1016/S1470-2045(13)70142-6
50. Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol (2014) 15(10):1119–28. doi:10.1016/S1470-2045(14)70362-6
51. Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov (2014) 4(6):662–73. doi:10.1158/2159-8290.CD-13-0846
52. Zou HY, Li Q, Engstrom LD, West M, Appleman V, Wong KA, et al. PF-06463922 is a potent and selective next-generation ROS1/ALK inhibitor capable of blocking crizotinib-resistant ROS1 mutations. Proc Natl Acad Sci U S A (2015) 112(11):3493–8. doi:10.1073/pnas.1420785112
53. Shaw TA, Bauer MT, Felip E, Besse B, James LP, Clancy JS, et al. Clinical activity and safety of PF-06463922 from a dose escalation study in patients with advanced ALK+ or ROS1+ NSCLC. J Clin Oncol (2015) 33(Suppl):abstr8018.
54. Camidge RD, Bazhenova L, Salgia R, Langer CJ, Gold KA, Rosell R, et al. Safety and efficacy of brigatinib (AP26113) in advanced malignancies, including ALK+ non-small cell lung cancer (NSCLC). J Clin Oncol (2015) 33(Suppl):abstr8062.
55. Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol (2012) 30(8):863–70. doi:10.1200/JCO.2011.35.6345
56. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med (2014) 371(21):1963–71. doi:10.1056/NEJMoa1406766
57. Engelman JA, Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr Opin Genet Dev (2008) 18(1):73–9. doi:10.1016/j.gde.2008.01.004
58. Song A, Kim TM, Kim DW, Kim S, Keam B, Lee SH, et al. Molecular changes associated with acquired resistance to crizotinib in ROS1-rearranged non-small cell lung cancer. Clin Cancer Res (2015) 21(10):2379–87. doi:10.1158/1078-0432.CCR-14-2191
59. Bauer MT, Shaw TA, Solomon B, Besse B, James LP, Clancy JS, et al. Phase I/II study of PF-06463922, an ALK/ROS1 tyrosine kinase inhibitor, in patients with advanced non-small-cell lung cancer harboring specific molecular alterations. J Clin Oncol (2015) 33(Suppl):abstrTS2620.
60. Paik PK, Arcila ME, Fara M, Sima CS, Miller VA, Kris MG, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol (2011) 29(15):2046–51. doi:10.1200/JCO.2011.36.9025
61. Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C, et al. Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res (2013) 19(16):4532–40. doi:10.1158/1078-0432.CCR-13-0657
62. Pratilas CA, Hanrahan AJ, Halilovic E, Persaud Y, Soh J, Chitale D, et al. Genetic predictors of MEK dependence in non-small cell lung cancer. Cancer Res (2008) 68(22):9375–83. doi:10.1158/0008-5472.CAN-08-2223
63. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med (2015) 373(8):726–36. doi:10.1056/NEJMoa1502309
64. Planchard D, Groen HJM, Kim TM, Rigas JR, Souquet PJ, Baik CS, et al. Interim results of a phase II study of the BRAF inhibitor (BRAFi) dabrafenib (D) in combination with the MEK inhibitor trametinib (T) in patients (pts) with BRAF V600E mutated (mut) metastatic non-small cell lung cancer (NSCLC). J Clin Oncol (2015) 33(Suppl):abstr8006.
65. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov (2015) 5(8):850–9. doi:10.1158/2159-8290.CD-15-0285
66. Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer (2006) 6(8):637–45. doi:10.1038/nrc1912
67. Camidge RD, Ou IH-S, Shapiro G, Otterson AG, Villaruz CL, Villalona-Calero AM, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). J Clin Oncol (2014) 32(Suppl):abstr8001.
68. Paik KP, Drilon EA, Yu AH, Krug ML, Rekhtman N, Borsu L, et al. Response to crizotinib and cabozantinib in stage IV lung adenocarcinoma patients with mutations that cause MET exon 14 skipping. J Clin Oncol (2015) 33(Suppl):abstr8021.
69. Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet (2015) 384(9944):665–73. doi:10.1016/S0140-6736(14)60845-X
70. Neal JW, Dahlberg ES, Wakelee HA, Aisner SC, Bowden M, Carbone DP, et al. Cabozantinib (C), erlotinib (E) or the combination (E+C) as second- or third-line therapy in patients with EGFR wild-type (wt) non-small cell lung cancer (NSCLC): a randomized phase 2 trial of the ECOG-ACRIN Cancer Research Group (E1512). J Clin Oncol (2015) 33(Suppl):abstr8003.
71. Lenferink AE, Pinkas-Kramarski R, van de Poll ML, van Vugt MJ, Klapper LN, Tzahar E, et al. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J (1998) 17(12):3385–97. doi:10.1093/emboj/17.12.3385
72. Ménard S, Casalini P, Campiglio M, Pupa S, Agresti R, Tagliabue E. HER2 overexpression in various tumor types, focussing on its relationship to the development of invasive breast cancer. Ann Oncol (2001) 12(Suppl 1):S15–9. doi:10.1093/annonc/12.suppl_3.S15
73. Heinmöller P, Gross C, Beyser K, Schmidtgen C, Maass G, Pedrocchi M, et al. HER2 status in non-small cell lung cancer: results from patient screening for enrollment to a phase II study of herceptin. Clin Cancer Res (2003) 9(14):5238–43.
74. Pellegrini C, Falleni M, Marchetti A, Cassani B, Miozzo M, Buttitta F, et al. HER-2/Neu alterations in non-small cell lung cancer: a comprehensive evaluation by real time reverse transcription-PCR, fluorescence in situ hybridization, and immunohistochemistry. Clin Cancer Res (2003) 9(10 Pt 1):3645–52.
75. Gatzemeier U, Groth G, Butts C, Van Zandwijk N, Shepherd F, Ardizzoni A, et al. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann Oncol (2004) 15(1):19–27. doi:10.1093/annonc/mdh031
76. Lara PN Jr, Laptalo L, Longmate J, Lau DH, Gandour-Edwards R, Gumerlock PH, et al. Trastuzumab plus docetaxel in HER2/neu-positive non-small-cell lung cancer: a California Cancer Consortium screening and phase II trial. Clin Lung Cancer (2004) 5(4):231–6. doi:10.3816/CLC.2004.n.004
77. Bose R, Kavuri SM, Searleman AC, Shen W, Shen D, Koboldt DC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov (2013) 3(2):224–37. doi:10.1158/2159-8290.CD-12-0349
78. Shih J, Bashir B, Gustafson KS, Andrake M, Dunbrack RL, Goldstein LJ, et al. Cancer signature investigation: ERBB2 (HER2)-activating mutation and amplification-positive breast carcinoma mimicking lung primary. J Natl Compr Canc Netw (2015) 13(8):947–52.
79. Minami Y, Shimamura T, Shah K, LaFramboise T, Glatt KA, Liniker E, et al. The major lung cancer-derived mutants of ERBB2 are oncogenic and are associated with sensitivity to the irreversible EGFR/ERBB2 inhibitor HKI-272. Oncogene (2007) 26(34):5023–7. doi:10.1038/sj.onc.1210292
80. Arcila ME, Chaft JE, Nafa K, Roy-Chowdhuri S, Lau C, Zaidinski M, et al. Prevalence, clinicopathologic associations, and molecular spectrum of ERBB2 (HER2) tyrosine kinase mutations in lung adenocarcinomas. Clin Cancer Res (2012) 18(18):4910–8. doi:10.1158/1078-0432.CCR-12-0912
81. Buttitta F, Barassi F, Fresu G, Felicioni L, Chella A, Paolizzi D, et al. Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer (2006) 119(11):2586–91. doi:10.1002/ijc.22143
82. Mazières J, Peters S, Lepage B, Cortot AB, Barlesi F, Beau-Faller M, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol (2013) 31(16):1997–2003. doi:10.1200/JCO.2012.45.6095
83. Burstein HJ, Sun Y, Dirix LY, Jiang Z, Paridaens R, Tan AR, et al. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol (2010) 28(8):1301–7. doi:10.1200/JCO.2009.26.3756
84. Gandhi L, Bahleda R, Tolaney SM, Kwak EL, Cleary JM, Pandya SS, et al. Phase I study of neratinib in combination with temsirolimus in patients with human epidermal growth factor receptor 2-dependent and other solid tumors. J Clin Oncol (2014) 32(2):68–75. doi:10.1200/JCO.2012.47.2787
85. Kris MG, Camidge DR, Giaccone G, Hida T, Li BT, O’Connell J, et al. Targeting HER2 aberrations as actionable drivers in lung cancers: phase II trial of the pan-HER tyrosine kinase inhibitor dacomitinib in patients with HER2-mutant or amplified tumors. Ann Oncol (2015) 26(7):1421–7. doi:10.1093/annonc/mdv532.48
86. Salvatore D, Barone MV, Salvatore G, Melillo RM, Chiappetta G, Mineo A, et al. Tyrosines 1015 and 1062 are in vivo autophosphorylation sites in ret and ret-derived oncoproteins. J Clin Endocrinol Metab (2000) 85(10):3898–907. doi:10.1210/jc.85.10.3898
87. Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med (2012) 18(3):382–4. doi:10.1038/nm.2673
88. Wang R, Hu H, Pan Y, Li Y, Ye T, Li C, et al. RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J Clin Oncol (2012) 30(35):4352–9. doi:10.1200/JCO.2011.39.5855
89. Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med (2012) 18(3):375–7. doi:10.1038/nm.2644
90. Drilon A, Wang L, Hasanovic A, Suehara Y, Lipson D, Stephens P, et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov (2013) 3(6):630–5. doi:10.1158/2159-8290.CD-13-0035
91. Drilon AE, Sima CS, Somwar R, Smith R, Ginsberg MS, Riely GJ, et al. Phase II study of cabozantinib for patients with advanced RET-rearranged lung cancers. J Clin Oncol (2015) 33(Suppl):abstr8007.
92. Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med (2013) 19(11):1469–72. doi:10.1038/nm.3352
93. Créancier L, Vandenberghe I, Gomes B, Dejean C, Blanchet JC, Meilleroux J, et al. Chromosomal rearrangements involving the NTRK1 gene in colorectal carcinoma. Cancer Lett (2015) 365(1):107–11. doi:10.1016/j.canlet.2015.05.013
94. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun (2014) 5:4846. doi:10.1038/ncomms5846
95. De Braud FG, Pilla L, Niger M, Damian S, Bardazza B, Martinetti A, et al. Phase 1 open label, dose escalation study of RXDX101, an oral pan-trk, ROS1, and ALK inhibitor, in patients with advanced solid tumors with relevant molecular alterations. J Clin Oncol (2014) 2014(32):2502.
96. National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Non-Small Cell Lung Cancer. Fort Washington, PA: NCCN (2015).
97. Redig AJ, Janne PA. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J Clin Oncol (2015) 33(9):975–7. doi:10.1200/JCO.2014.59.8433
Keywords: NSCLC, EGFR, ALK, ROS1, HER2, BRAF, c-MET, VEGFR2
Citation: Bansal P, Osman D, Gan GN, Simon GR and Boumber Y (2016) Recent Advances in Targetable Therapeutics in Metastatic Non-Squamous NSCLC. Front. Oncol. 6:112. doi: 10.3389/fonc.2016.00112
Received: 29 February 2016; Accepted: 19 April 2016;
Published: 04 May 2016
Edited by:
Taofeek K. Owonikoko, Emory University, USAReviewed by:
Matthew A. Gubens, University of California San Francisco, USAK. Shilo, The Ohio State University, USA
Copyright: © 2016 Bansal, Osman, Gan, Simon and Boumber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanis Boumber, eWJvdW1iZXJAc2FsdWQudW5tLmVkdQ==