 Fabrizio Marcucci
Fabrizio Marcucci Cristiano Rumio
Cristiano Rumio François Lefoulon2
François Lefoulon2
- 1Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy
- 2Technologie Servier, Department of Synthesis, Orléans, France
Cancer stem-like cells (CSC) represent a subpopulation of tumor cells with elevated tumor-initiating potential. Upon differentiation, they replenish the bulk of the tumor cell population. Enhanced tumor-forming capacity, resistance to antitumor drugs, and metastasis-forming potential are the hallmark traits of CSCs. Given these properties, it is not surprising that CSCs have become a therapeutic target of prime interest in drug discovery. In fact, over the last few years, an enormous number of articles describing compounds endowed with anti-CSC activities have been published. In the meanwhile, several of these compounds and also approaches that are not based on the use of pharmacologically active compounds (e.g., vaccination, radiotherapy) have progressed into clinical studies. This article gives an overview of these compounds, proposes a tentative classification, and describes their biological properties and their developmental stage. Eventually, we discuss the optimal clinical setting for these compounds, the need for biomarkers allowing patient selection, the redundancy of CSC signaling pathways and the utility of employing combinations of anti-CSC compounds and the therapeutic limitations posed by the plasticity of CSCs.
Cancer Stem-Like Cells – Origin, Functional Properties, and Markers
Cancer stem-like cells (CSCs) represent a subpopulation of tumor cells. They are highly active in generating new tumors upon implantation in laboratory animals, while most tumor cells have low tumor-forming ability (1). CSCs are also resistant to conventional chemotherapeutics, radiation, and targeted therapies (2–4), and have enhanced metastasis-forming potential (5). Drug resistance allows CSCs to survive current therapies and to be ultimately responsible for relapse (4).
The first demonstration of the existence of CSCs was brought in acute myeloid leukemia (AML) (6). This initial observation was followed in ensuing year by similar observations in solid tumors (7–11).
The CSC population may not necessarily represent the original tumor-initiating cell (TIC) (12) and there is plasticity in the general tumor such that cells can loose and reacquire a CSC-like phenotype. Overall, it appears likely that, at any given time, CSCs are the result of the convergent action of two main events. The first is the genetic (mutations, rearrangements, and/or deregulation of genes) and the epigenetic (microRNAs, alterations of the methylation profile of genes, or gene promoters) instability of tumor cells and the products that are expressed, e.g., oncogenes (13, 14), or repressed as a consequence of this instability (15). The second event is represented by stimuli from the tumor microenvironment (TME). These stimuli are similar to those that promote the epithelial–mesenchymal transition (EMT) of tumor cells (16). Induction and maintenance of CSCs in response to these stimuli, however, is not a direct effect on tumor cells but, rather, the result of a cross-talk between different cell types within the TME that is largely effected by extracellular mediators released in response to the stimuli (17). Of note, genetic instability of tumor cells can also lead to overexpression of extracellular mediators (18, 19), which may add to those released in response to stimuli from the TME. The contribution of these two main events may differ quantitatively and qualitatively in different tumors and, over time, even within the same tumor, and this variability may underlie the plasticity and heterogeneity of CSCs. In fact, tumors may have a small number of CSCs, others a relatively large number, and still other tumors may even lack detectable CSCs (20, 21). Even within individual tumors, CSCs may express different, only partially overlapping phenotypes (22).
Given the multiplicity of genetic and environmental stimuli that are at the origin of CSCs, it is equally not surprising that a large number of signaling pathways have been reported being involved in the induction and maintenance of CSCs. Since CSCs and normal stem cells share a number of traits (4), it is logical that the role of signaling pathways involved in the physiology of normal stem cells, such as WNT, Notch, and Hedgehog (Hh), has been investigated with particular attention (4, 23).
The Relationship Between EMT and CSCs
CSCs can derive from bulk tumor cells that undergo an EMT, i.e., the conversion of tumor cells with an epithelial phenotype into cells with a mesenchymal phenotype (24, 25). EMT is critical for embryonic development and involves changes that lead to loss of cell–cell adhesion and cell polarity, with acquisition of migratory and invasive properties (26). In adults, EMT occurs during wound healing, tissue regeneration, organ fibrosis, and tumor progression (27). Tumor cells undergoing EMT are characterized by increased motility and invasiveness, resistance to antitumor drugs, and acquire tumor-initiating potential (28, 29). Reversal of EMT is accompanied by downregulation of CSC-associated traits (30). The question that obviously arises is whether tumor cells that have undergone an EMT are CSCs, i.e., if the two terms are interchangeable. The question is not of mere academic interest because, in case of coincidence, therapeutic approaches aimed at targeting CSCs would be identical to those addressing EMT (31). Overall, there is considerable evidence for the two cell types not being coincident. Thus, it has been proposed that cells that have undergone an EMT acquire a CSC phenotype by engaging additional programs, such as the WNT and Hippo pathways (32). Furthermore, under certain circumstances, EMT and stemness can be uncoupled (33). Similarly, Thiery and coworkers have recently proposed that it is not solely the acquisition of EMT but the EMT stem cell-like phenotype that engenders drug resistance (34). In accordance with this view, it has been observed that the anti-CSC compound salinomycin (see below for more details on this compound) can cause cell death and decrease stem cell properties despite activation of EMT (35). Eventually, in many instances CSCs have been reported to be in a quiescent, autophagic state, that is the greatly different from mesenchymal-type tumor cells with enhanced invasive and migratory potential (36, 37). Blockade of autophagy has even been reported to reduce CSC activity (38). Overall, it seems that CSCs represent a further developmental stage that ensues after tumor cells have undergone EMT (Figure 1), and that autophagy may be a specific trait of such CSCs. CSCs, however, may not loose mesenchymal traits, as has been shown for circulating tumor cells (39).
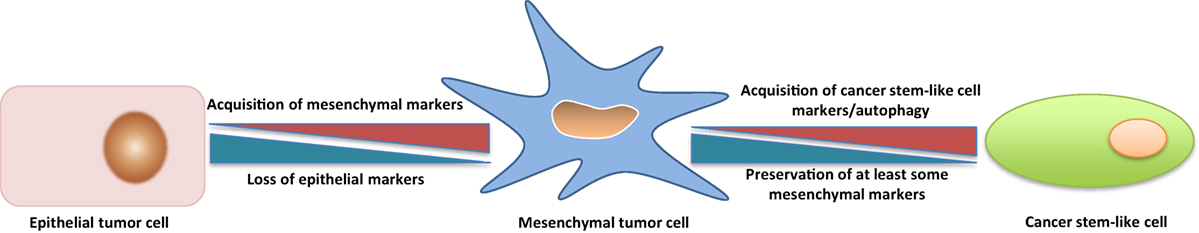
Figure 1. Transition from a Tumor Cell with a Predominantly Epithelial Phenotype to a Tumor Cell with a Predominantly Mesenchymal Phenotype and to a Cancer Stem-Like Cell (CSC). The transition from a predominantly epithelial to a predominantly mesenchymal phenotype is better understood in molecular and phenotypic terms, but most available data suggest that a tumor cell with a predominantly mesenchymal phenotype must undergo several changes in order to acquire properties of a CSC, including autophagy. The heterogeneity of CSC populations suggests also the possibility of different transition states between a predominantly mesenchymal tumor cell and a CSC, in analogy with what has been observed for the transition between predominantly epithelial and mesenchymal tumor cells. See text for discussion and references to this point.
Anti-CSC Compounds in Clinical Development
Search Criteria
A large number of compounds that have already received regulatory approval or are in clinical development have been tested for their anti-CSC activity. The goal of this article, however, is to address those compounds that are in clinical development for their anti-CSC activity. In order to identify these compounds, a search was performed in the ClinicalTrials.gov website using the key word CSC. Of the almost 3,500 clinical trials that were recovered, however, only a minute fraction were actually dealing with CSCs, while the vast majority were on stem cells used for transplantation. In the following, we realized that the few studies that were directly dealing with CSCs were only a fraction of those investigating anti-CSC compounds. Some other studies were recovered using the key word TIC. It became clear, however, that many studies that were investigating anti-CSC compounds did not use the terms CSC or TIC and, therefore, could not be recovered. For this reason, we expanded the study, including pharmaceutical companies known to be involved in anti-CSC drug discovery. Eventually, we included also antitumor compounds that are not or not yet being developed as anti-CSC compounds, but whose anti-CSC activities have been confirmed in numerous preclinical studies. On the other hand, we excluded compounds that have already received regulatory approval for antitumor indications unrelated to CSCs, but which have recently been found to possess some selectivity for CSCs [e.g., Ref. (40)]. Admittedly, this approach is somewhat arbitrary but it has allowed to keep the number of anti-CSC compounds to be discussed in a manageable size while, nevertheless, addressing all main classes of anti-CSC compounds with one or more compounds being discussed for each class.
Anti-CSC Compounds Targeting Extracellular Mediators or Cell Surface Molecules
In this section, we discuss compounds that target molecules that are extracellular mediators or are expressed on the surface of CSCs. Extracellular mediators and cell surface molecules that are part of complex signaling pathways involved in CSC biology and can be targeted at different levels of these pathways are discussed in later sections.
A large number of cell surface molecules that are expressed on CSCs of tumors or tumor subtypes of different tissue origin have been identified. CD44, CD47, CD33, CD133, CXC chemokine receptor (CXCR) 4, and CD26 are just some of these markers (41). Most of them are not CSC specific and in some cases may even be ubiquitously expressed, including blood cells (e.g., CD44, CD47) (42). Targeting these markers for therapeutic purposes may incur into severe side effects. Some CSC markers, however, have a more restricted expression and/or are overexpressed on CSCs, making them good potential targets for anti-CSC compounds. Recent approaches, such as the construction of bispecific antibodies, may offer, for the future, the opportunity to target also CSC markers that are broadly expressed (43).
Inhibitors of Clusterin
Clusterin is a stress-activated and apoptosis-associated molecular chaperone that protects cells from various stressors and is overexpressed in many human cancers (44). Antibodies targeting secreted clusterin inhibit transforming growth factor (TGF)-β-induced EMT in several tumor cell lines, without affecting the proliferation, and reduce lung metastasis in breast cancer and hepatocellular carcinoma (HCC) models (45, 46). Although no formal proof has been brought that anti-clusterin antibodies inhibit also CSCs, a humanized anti-clusterin mAb (AB-16B5) is being investigated in patients with solid tumors for clinical efficacy, and effects on EMT and CSC biomarkers (Table 1).
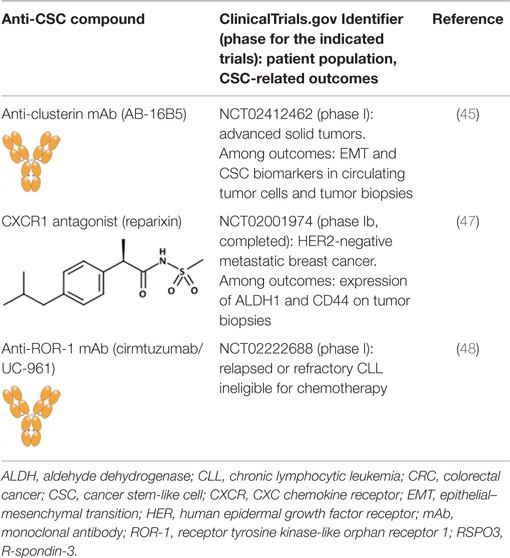
Table 1. Anti-CSC compounds targeting cell surface molecules.
Inhibitors of CXCR1
Gene expression profiling of tumor cell lines that had been enriched for aldehyde dehydrogenase (ALDH)-positive CSCs identified interleukin (IL)-8 and its receptor CXCR1 as constitutive of the CSC profile (49). IL-8 was also found to increase the number of ALDH-positive tumor cells and mammosphere formation in vitro, an assay that reflects the tumor-initiating potential of cancer cells. Later, the same group showed that CXCR1 blockade using either a CXCR1-specific blocking antibody or reparixin, a methanesulfonamide CXCR1 inhibitor, depleted the CSC population in two human breast cancer cell lines in vitro (50) and targeted the CSC population in breast cancer xenografts, retarding tumor growth and reducing metastasis. Another work showed that this compound, in combination with paclitaxel, inhibited formation of brain metastases in a breast cancer model (47). This was likely the result of the combined effect of the two drugs, the pro-apoptotic action of paclitaxel and the cytostatic and anti-migratory effects of reparixin. Reparixin has been investigated in a clinical study in patients with human epidermal growth factor receptor (HER) 2-negative metastatic breast cancer (Table 1).
Inhibitors of Receptor Tyrosine Kinase-Like Orphan Receptor 1
Receptor tyrosine kinase-like orphan receptor 1 (ROR1) is a type I orphan receptor, tyrosine kinase-like cell surface protein that is expressed during embryogenesis and is found on tumor cells of many different types of cancer, but not on normal adult tissues (51). ROR1 is preferentially expressed by less well-differentiated tumors with EMT-related markers that have high potential for relapse and metastasis. Silencing ROR1 in breast cancer cell lines attenuated expression of EMT-associated genes and impaired their metastatic potential in vivo (51). A recent study has reported that ROR1 is associated with ovarian cancer CSCs (48). Cirmtuzumab/UC-961, a humanized IgG1 mAb, binds with high-affinity ROR1, and inhibits migration in vitro and engraftment in mice of patient-derived tumor cells that had been treated with the antibody (48). Cirmtuzumab is currently being investigated in patients with chronic lymphocytic leukemia who are ineligible for chemotherapy (Table 1).
Anti-CSC Compounds That Act on Ligand–Receptor Pairs and Their Signaling Pathways
Inhibitors of the TGF-β/TGF-β Receptor Pathway
The TGF-β/TGF-β receptor pathway is one of the most frequently involved in EMT and CSC biology. A recent study showed that blocking TGF-β signaling with a TGF-β type I receptor kinase inhibitor, EW-7197, suppressed paclitaxel-induced EMT and CSC functions, such as formation of mammospheres and ALDH activity, reduced the ratio of CD44+/CD24− CSCs, and CSC-associated transcription factors (52). Treatment with EW-7197 improved the efficacy of paclitaxel by decreasing the number of lung metastases and increasing survival time in vivo. The TGF-β pathway has also been shown to cross-talk with other signaling pathways involved in CSC biology, such as the Notch pathway (53). Thus, heightened Notch signaling in tumor cells magnified TGF-β-induced phosphorylation of signaling components and was required to sustain TGF-β-induced lung carcinoma cell growth. Conversely, Notch blockade reduced TGF-β signaling and limited lung carcinoma tumor progression. Overall, the TGF-β/TGF-β receptor signaling axis is involved in the CSC biology of several tumor types, such as breast cancer (54, 55), liver cancer (56), lung cancer (57), and head and neck cancer (53).
With regard to the clinical development of anti-TGF-β compounds, it must be considered that TGF-β is a multifunctional cytokine and its inhibition leads to effects that are likely unrelated to EMT or CSC inhibition, such as enhancement of adaptive antitumor immune responses or normalization of the tumor stroma (58, 59). Nevertheless, several inhibitors of this pathway are now in clinical development, both anti-ligand antibodies (60) and inhibitors of the TGF-β receptor tyrosine kinase (RTK). One of the most frequently investigated for its anti-CSC activity is the TGF-β type I RTK inhibitor galunisertib/LY2157299. In a preclinical study, this compound blocked paclitaxel-induced CSC expansion in triple-negative breast cancer (TNBC) cell lines and mouse xenografts where the chemotherapeutic drug paclitaxel increased autocrine TGF-β signaling and IL-8 expression and enriched for CSCs (61). Moreover, treatment with LY2157299 prevented reestablishment of tumors after paclitaxel treatment. The clinical development of this TGF-β inhibitor has been delayed owing to the detection of cardiovascular toxicity in preclinical studies (62, 63). This problem has now been bypassed through the application of judicious administration protocols (64). LY2157299 is now in several clinical trials although none of these trials explicitly refers to its anti-CSC activity (see Table 2).

Table 2. Anti-CSC compounds that act on ligand–receptor pairs and their signaling pathways.
Inhibitors of the Hedgehog Pathway
This signal transduction pathway, as well the WNT and Notch pathways are crucial for embryonic development. Therefore, compounds targeting these pathways may have negative consequences on embryonic patterning and child development. Furthermore, these pathways are utilized in the generation of stem cells and in regenerative processes, and compounds acting on these pathways may have untoward effects on these processes (82). In spite of these concerns, an inhibitor of this pathway, vismodegib, has received regulatory approval (83).
In adult tissues, Hh signaling is relatively quiescent except in tissue maintenance and repair. Aberrant activation of Hh signaling is implicated in multiple aspects of tumorigenesis, including maintenance of the CSC phenotype (84). Canonical Hh signaling is activated when a Hh ligand (Sonic Hh, Indian Hh, and Desert Hh) binds the transmembrane receptor Patched (PTCH) to relieve PTCH-mediated inhibition of the G-protein-coupled receptor-like protein Smoothened (SMO). SMO then drives a signaling cascade that results in nuclear translocation and activation of the glioma-associated oncogene transcription factors (GLI). GLI activate transcription of genes regulating self-renewal, cell fate, survival, angiogenesis, EMT, and cell invasion.
Both ligand-dependent and ligand-independent mechanisms (instabilities of genes encoding individual components that lead to constitutive activation of the pathway) result in aberrant Hh pathway activation in cancer. Hh signaling associates with CSC biology in several types of hematological malignancies and solid tumors, such as pancreatic cancer (85), prostate cancer (86), glioblastoma (87), lung cancer (88), breast cancer (89), colon cancer (90), chronic myeloid leukemia (91), and multiple myeloma (92). Preferential activation of Hh signaling in CSCs compared with bulk tumor cells has been reported (88–90, 92) as well as upregulation of CSC markers (65, 89, 93). Several inhibitors, mostly small-molecule inhibitors, of the Hh pathway are in clinical development for their antitumor effects. One of these, vismodegib, has already received regulatory approval. Some of these inhibitors, including vismodegib, are also being tested clinically for their anti-CSC activity.
The first small molecule that was found to inhibit the Hh pathway is cyclopamine, a naturally occurring compound that belongs to the group of jerveratum alkaloids (94). Pharmaceutical companies have set out to develop cyclopamine derivatives with improved pharmacologic properties or new molecules showing improved binding to SMO, so far the main target for Hh pathway inhibitors. Vismodegib has been approved for the treatment of advanced basal cell carcinoma, where it induces a high percentage of response rates (95), but inevitably incurs into acquired resistance (96). Vismodegib has demonstrated good efficacy also in medulloblastoma (97), but only limited activity in other tumor types. This lack of activity may be due to many factors, but the possibility of a tumor type-dependent redundancy of signaling pathways appears a likely possibility.
In preclinical studies, vismodegib inhibited cell viability and induced apoptosis in three pancreatic cancer cell lines and pancreatic CSCs (66). Suppression of both GLI1 plus GLI2 mimicked the changes in cell viability, spheroid formation, apoptosis, and gene expression observed in vismodegib-treated pancreatic CSCs. In another study, vismodegib decreased spheroid and colony formation of gastric cancer cell lines with upregulated CD44 and Hh pathway proteins (65). CD44-positive cells were more resistant to chemotherapeutics, showed enhanced migration, invasion, and anchorage-independent growth, and these properties were reversed by vismodegib. Vismodegib is being investigated in two phase II clinical studies for its anti-CSC activity. In both, vismodegib is studied in advanced pancreatic cancer in combination with chemotherapeutics (Table 2). Results of one of these studies have been published (67). Treatment for 3 weeks led to down-modulation of GL1 and PTCH1 and decreased fibrosis, but no significant changes in CSCs were observed, and combined treatment with vismodegib and gemcitabine was not superior to gemcitabine alone.
Saridegib/IPI-926 is another SMO inhibitor that is in clinical investigation (69). It is being studied in combination with the anti-epidermal growth factor receptor (EGFR) mAb cetuximab in recurrent head and neck cancer patients (Table 2). Preliminary results of this study have now been published (70). Among eight evaluable patients, the best responses were one partial response, four stable diseases, and three disease progressions. Decreases in tumor size were seen in both cetuximab-naïve patients. Toxicities were as expected. Tumor shrinkage and progression-free survival were associated with downregulation of intra-tumoral Hh pathway gene expression during therapy.
Inhibitors of the WNT Pathway
WNT proteins are a large family of secreted molecules that play a critical role in the development of various organisms (98). In the absence of extracellular WNT molecules, a destruction complex, including the proteins adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β) and AXIN, phosphorylates β-catenin, targeting it for ubiquitylation and degradation. The binding of WNTs to Frizzled receptors and the co-receptors low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6, transmits a signal through Dishevelled, which results in inhibition of the destruction complex and nuclear entry of β-catenin. In the nucleus, β-catenin acts as a bridge between members of the T cell factor (TCF) family of transcription factors and the basal transcriptional apparatus via co-activators [CREB-binding protein (CBP), E1A-associated protein p300, the co-activator Pygopus, B cell lymphoma 9, etc]. Aberrant activation of the WNT/β-catenin signaling pathway has recently been implicated in several types of human cancers (82), and shown to play a critical role in CSC biology (99, 100). Moreover, aberrant activation of the transcriptional activity of β-catenin, independently of upstream WNT signaling, has been associated with breast CSC amplification and tumorigenesis (101).
Inhibitors of the WNT pathway may act at different levels of the signaling chain. One approach is to act on ligands of this pathway. R-spondins (RSPOs) are secreted proteins that potentiate canonical WNT signaling (102). Translocations of RSPO genes are recurrent in a subset of colorectal tumors. PTPRK–RSPO3 is one of the fusions that can originate from these translocations. It has recently been shown that targeting RSPO3 in PTPRK–RSPO3-fusion-positive human tumor xenografts inhibits tumor growth and promotes differentiation (71). Genes expressed in the stem-cell compartment of the intestine were among those most sensitive to anti-RSPO3 treatment. A clinical study with the anti-RSPO3 mAb OMP-131R10 is ongoing in advanced solid tumors and in metastatic colorectal cancer (Table 1).
Another approach has been to raise inhibitory mAbs against Frizzled receptors. This is a challenging undertaking since there are 19 human WNTs and 10 Frizzled receptors. Nevertheless, a mAb, vantictumab/OMP-18R5, which binds to five distinct Frizzled receptors through a conserved epitope, has been obtained (72). In xenograft studies with minimally passaged human tumors, this antibody inhibited the growth of a range of tumor types, reduced tumor cell proliferation, and CSC frequency. Strong synergy was observed with several chemotherapeutics. Vantictumab is now being investigated in several phase I clinical trials in combination with chemotherapeutics in patients with advanced solid tumors (Table 2).
The fusion protein ipafricept/OMP-54F28 encompasses a portion of the Frizzled 8 receptor fused to the Fc portion of IgG1. This protein competes with the native Frizzled 8 receptor for its ligands and antagonizes WNT signaling. In preclinical models, OMP-54F28 reduced tumor growth and decreased CSC frequency as a single agent and in combination with chemotherapeutics (73). A phase Ia clinical study is nearing completion with this compound in advanced solid tumors and phase Ib studies have been started in combination with chemotherapeutics in ovarian, pancreatic, and hepatocellular cancers (Table 2).
Organic molecules have been synthesized that antagonize the binding of β-catenin to the TCF protein or to transcriptional co-activators. These molecules cause downregulation of β-catenin-responsive genes. CWP232228 inhibits the interaction of β-catenin with TCF (74), and this was shown to inhibit the growth of both breast CSCs and bulk tumor cells, but breast CSCs exhibited greater sensitivity. CWP232228 treatment blocked also secondary xenograft tumor development and inhibited metastasis formation. CWP232228 is not being investigated in clinical studies, but an undisclosed, but probably closely related molecule (CWP232291) is now in phase I clinical studies for the treatment of hematological malignancies (Table 2).
PRI-724 is a small molecule that inhibits the interaction between β-catenin and CBP. PRI-724 sensitized ovarian cancer cells to cisplatin and decreased tumor sphere formation (75). Importantly, CBP/β-catenin antagonists appear to have the ability to safely eliminate CSCs by taking advantage of an intrinsic differential preference in the way somatic stem cells and CSCs divide (76). PRI-724 is currently in phase I/II clinical trials for the treatment of solid tumors and hematological malignancies (Table 2).
Still another approach to interfere with this pathway is to inhibit the interaction between mucin 1 and β-catenin. Mucin 1 is a membrane-bound glycoprotein expressed by most glandular and ductal epithelial cells, and is overexpressed in many diverse human carcinomas. The mucin 1–β-catenin interaction is stabilized by extracellular matrix protein 1 and leads to the upregulation of EMT- and CSC-related genes (77, 78). The mucin 1 inhibitor GO-203-2C is a highly cationic peptide with a polyarginine tail at the N-terminus. It interacts with the mucin 1 C-terminus on the cell surface and inhibits cell–cell interactions and signaling. It is currently in a phase I/II trial in patients with relapsed or refractory AML (Table 2).
Several other small-molecule WNT/β-catenin signaling inhibitors have been developed and are in development [listed in Ref. (74)], but so far no direct evidence has been brought that these molecules inhibit CSCs, although this appears a likely possibility.
Inhibitors of the Notch Pathway
The Notch pathway regulates cell fate specification, tissue patterning, and cellular survival. In mammals, the Notch signaling system consists of five membrane-bound ligands, DLL1, 3, 4, and Jagged 1, 2, and four single-pass transmembrane receptors, Notch1–4. Ligand binding to Notch triggers the proteolytic activation of the receptor and translocation of its intracellular domain to the nucleus, where it interacts with the CSL transcription factor to regulate the expression of target genes (103). Notch signaling is involved in normal development and in most organs, including the hematopoietic system and the vasculature (103). Alterations in the Notch signaling pathway stimulate proliferation, restrict differentiation, promote cellular survival, and are associated with oncogenesis in several malignancies. Notch plays a critical role in CSC biology (86). Of the Notch ligands, DLL4 is commonly expressed in solid tumors and associated with chemoresistance. DLL4 binds all four Notch receptors, but the interaction with Notch 1 is preferred (103).
Several approaches are being pursued for inhibition of Notch signaling. A first has been to inhibit Notch receptor cleavage by γ-secretase inhibitors (104). The therapeutic utility of these compounds, however, is limited due to intestinal toxicity resulting from pan-Notch inhibition (105). One γ-secretase inhibitor (RO4929097) was being studied in clinical trials for its antitumor and anti-CSC activity in breast cancer, but its development has now been discontinued.
A second approach has been to generate mAbs that inhibit Notch signaling. A humanized IgG2 anti-DLL4 mAb, demcizumab/OMP-21M18, is now in clinical investigation as anti-CSC compound (79) (Table 2). In human tumor models, both alone or in combination with chemotherapy, it reduced tumor growth, regrowth, and number of CSCs. A phase I clinical trial has allowed defining a recommended phase 2 dose. Moreover, 16 of 25 (64%) evaluable patients at one dose (10 mg/kg) had evidence of disease stabilization or response (79). Results suggested also that the antitumor effects of demcizumab might be the result of a combination of anti-CSC and anti-angiogenic effects, an observation consistent with the role of Notch signaling in normal angiogenesis (106) and tumor angiogenesis (107).
Given the role of Notch signaling in tumor angiogenesis, it is not surprising that other anti-DLL4 mAbs are in clinical investigation as inhibitors of tumor angiogenesis (e.g., enoticumab) (108). In fact, one compound in clinical development has been designed to optimally exploit both anti-angiogenic and anti-CSC activities. This is OMP-305B83, a bispecific anti-DLL4 and anti-vascular endothelial growth factor (VEGF) mAb that is currently in phase I clinical studies (Table 2).
In addition to inhibit the activity of Notch ligands, another possibility is to inhibit this pathway with anti-receptor (anti-Notch) antibodies. In order to avoid pitfalls deriving from pan-Notch inhibition, a novel mAb, tarextumab/OMP-59R5, has been generated that blocks the function of Notch 2 and Notch 3 (30). OMP-59R5 reduced CSC frequency in combination with chemotherapeutic agents in various cancer models, and the triple combination of anti-Notch2/3 with gemcitabine plus nab-paclitaxel produced striking tumor regression in pancreatic cancer xenografts. OMP-59R5 is now in phase I clinical trials (Table 2). Still another mAb that is in clinical trials binds only the Notch1 receptor (brontictuzumab/OMP-52M51). It is employed only for patients that demonstrate tumor overexpression of the activated form of Notch1. For this purpose, patients’ tumors are prescreened by immunohistochemistry to determine eligibility (Table 2).
A special role in the Notch pathway is played by DLL3. DLL3 predominantly localizes to the Golgi apparatus and is unable to activate Notch signaling (109). In the course of normal development, DLL3 inhibits Notch pathway activation by interacting with Notch and DLL1 and redirecting or retaining them to the late endosomal/lysosomal compartments or in the Golgi, respectively, thereby preventing their localization to the cell surface (110). DLL3 is overexpressed and relocalizes to the surface of small cell lung cancer (SCLC) and large cell neuroendocrine carcinoma cells (80). A DLL3-targeted antibody-drug conjugate (ADC), rovalpituzumab tesirine/SC16LD6.5, encompassing a humanized anti-DLL3 mAb conjugated to a DNA-damaging pyrrolobenzodiazepine dimer was synthesized and found to induce durable tumor regression in vivo across multiple patient-derived xenograft models. Lack of tumor recurrence resulted from effective targeting of DLL3-expressing CSCs. In vivo efficacy correlated with DLL3 expression, and responses were observed in xenograft models independently of their sensitivity to standard-of-care chemotherapy regimens. This ADC is now being investigated in a phase I/II clinical trial in patients with SCLC (Table 2).
Inhibitors of the Ephrin/Ephrin Receptor Pathway
Ephrin receptors comprise the largest family of RTKs in the human genome and modulate signaling pathways that impact cell fate decisions during embryogenesis and adult tissue homeostasis (111). Numerous therapeutics targeting this pathway are being tested in clinical trials; the majority are tyrosine kinase inhibitors. Vast functional redundancy within the Ephrin/Ephrin receptor pathway likely compromises the effect of blocking specific Ephrin ligands, while pan-Ephrin inhibition is toxic (111).
Gene expression analysis of breast and ovarian CSCs identified Ephrin A4 as a potential therapeutic target (81). An ADC targeting Ephrin A4 has been generated in order to induce cell death upon internalization, thereby avoiding redundancy of this highly diverse ligand–receptor family, and delaying the insurgence of resistance. This conjugate, labeled PF-06647263, encompasses a humanized anti-Ephrin A4 mAb conjugated to the DNA-damaging agent calicheamicin. It achieved sustained tumor regressions in both TNBC and ovarian cancer xenografts in vivo (81). Non-claudin low TNBC tumors exhibited higher expression and more robust responses than other breast cancer subtypes. PF-06647263 is currently being evaluated in a phase I clinical trial.
Anti-CSC Compounds That Inhibit Post-Receptor Signaling Pathways
Inhibitors of SRC and Focal Adhesion Kinase
SRC and focal adhesion kinase (FAK) are two interacting membrane-proximal tyrosine kinases. Both can be activated by adhesion receptors, RTKs, and cytokine receptors. Increased SRC and FAK phosphorylation and activity are upregulated in many cancers and have been implicated in several aspects of cancer progression. Both of these tyrosine kinases have been implicated in CSC biology. SRC has been reported being involved in CSC biology of, for example, hepatic cancer (112), pancreatic cancer (113), and breast cancer (114); FAK in breast cancer (115) and squamous cell carcinoma (116).
Several SRC and FAK inhibitors are in clinical studies. Some of these studies are centered on their anti-CSC activity. Dasatinib is a non-specific SRC inhibitor that inhibits also the Bcr/Abl tyrosine kinase. It has been approved for the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Dasatinib was shown in a clinical study to induce a long-standing decrease of leukemic stem cells in patients with CML (117). It is unclear, however, whether this effect is the result of Bcr/Abl inhibition in this patient population or whether the decrease is the result of inhibition of both tyrosine kinases. Dasatinib is being studied in a phase II clinical trial for the depletion of CSCs in CML patients (Table 3).
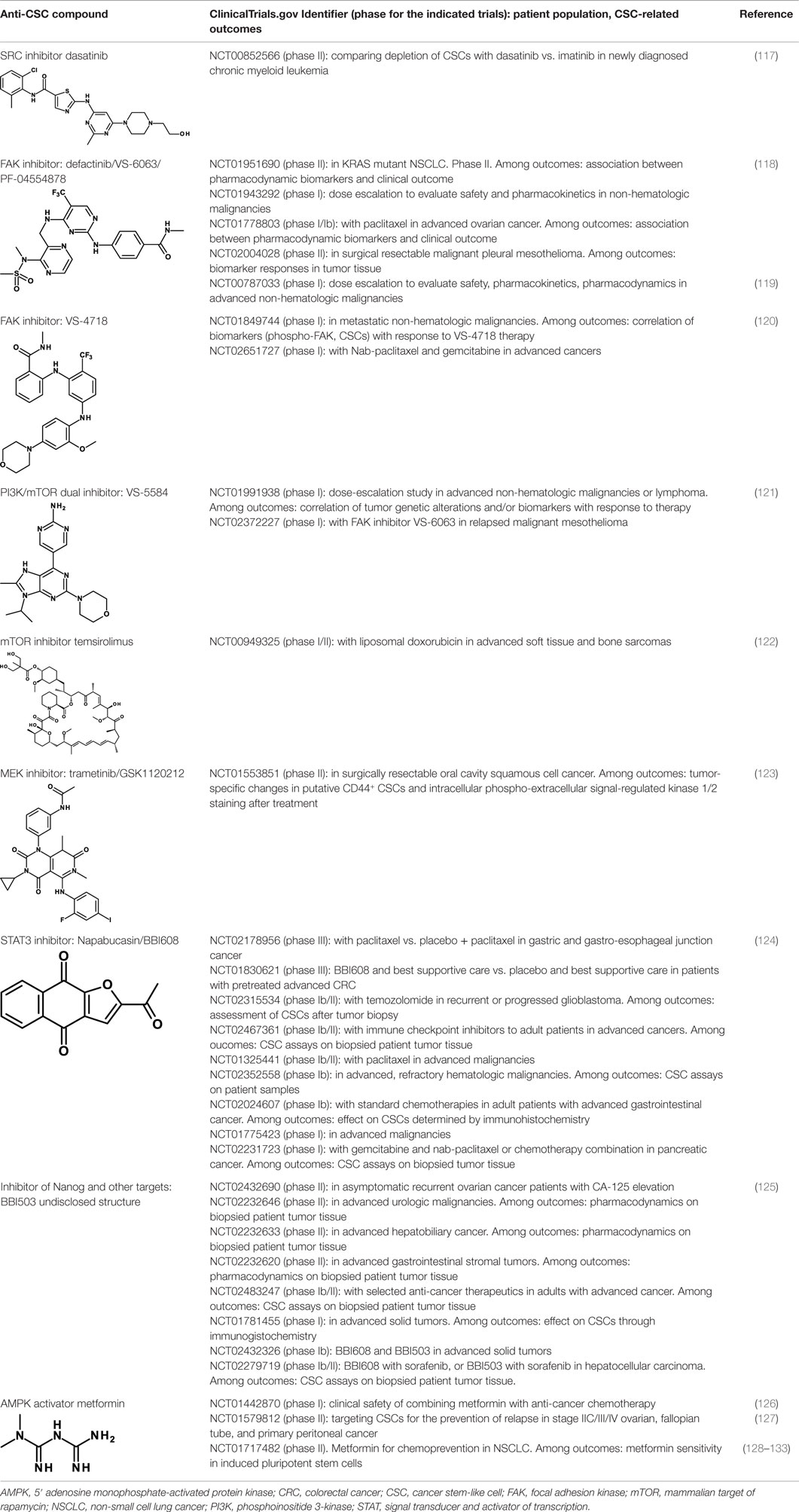
Table 3. Anti-CSC compounds that inhibit post-receptor signaling pathways.
Two FAK inhibitors that have been originated by the same company are now in clinical investigations for their antitumor and anti-CSC activity: defactinib/VS-6063/PF-04554878 and VS-4718. These are all small-molecule tyrosine kinase inhibitors. Results of a phase I study with defactinib have recently been reported (118). Disease stabilization at 12 weeks occurred in 6 of 37 patients receiving doses ≥200 mg/day. Treatment-related adverse events were mild to moderate, and reversible. A recommended phase II dose was defined. Of note, one clinical phase II study with defactinib in mesothelioma (NCT01870609) has been terminated due to lack of efficacy. Defactinib was also compared with a small-molecule FAK scaffolding inhibitor, Y15 (119). Cell viability was decreased in a dose-dependent manner in four thyroid cancer cell lines with Y15 and with higher doses of defactinib. Importantly, a combination of the two yielded synergistic effects, suggesting the possibility of enhancing efficacy by combining FAK inhibitors that work through different mechanisms of action. Defactinib is also being studied in a clinical trial in combination with VS-5584, a dual phosphoinositide 3-kinase (PI3K)-mammalian target of rapamycin (mTOR) inhibitor that will be discussed in the following. VS-4718 is in two clinical studies. In preclinical studies, VS-4718 showed that cells most sensitive to FAK inhibition lacked expression of the neurofibromatosis type 2 tumor suppressor gene product, Merlin (120). Merlin expression is often lost in malignant pleural mesothelioma. Low Merlin expression predicted for increased sensitivity of mesothelioma cells to VS-4718, in vitro and in vivo. Moreover, whereas pemetrexed and cisplatin, standard-of-care agents for mesothelioma, enrich for CSCs, FAK inhibitor treatment preferentially eliminated these cells.
PF-00562271 is another FAK inhibitor that was investigated in clinical studies, but its development has now been discontinued. In metastatic docetaxel-resistant prostate cancer cell lines, it reduced FAK phosphorylation in the resistant cells without affecting cell viability and overcame the chemoresistant phenotype (134).
Inhibitors of the PI3K–AKT–mTOR Pathway
The first component of this pathway, PI3K, encompasses three classes of molecules, with class IA PI3Ks being the most deeply investigated in cancer (135). Class IA PI3Ks are activated by RTKs, G-protein-coupled receptors, and some other post-receptor signaling molecules (e.g., RAS). Activated PI3K converts phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] into phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3]. PI(3,4,5)P3 binds and recruits the second pathway component, AKT, to the plasma membrane. This process is negatively regulated by the tumor suppressor phosphatase and tensin homolog (PTEN), which converts PI(3,4,5)P3 back to PI(4,5)P2. AKT is activated at the plasma membrane through phosphorylation. Activated AKT initiates a cascade of downstream signaling events, which promote cellular growth, metabolism, proliferation, survival, migration, apoptosis, and angiogenesis. A major downstream effector of AKT is mTOR complex (mTORC) 1; its downstream targets control protein synthesis. Another mTORC, mTORC2, participates in the phosphorylation and activation of AKT (136). The PI3K–AKT–mTOR pathway is one of the most frequently activated signaling pathways in cancer. The two most common mechanisms in human cancers are activation by RTKs and somatic mutations in specific components of the signaling pathway (135).
The PI3K–AKT–mTOR pathway is involved in CSC biology in several solid tumors and hematological malignancies: leukemias (122, 137), breast cancer (138, 139), colon cancer (140), pancreatic cancer (85), SCLC (141), glioblastoma (142), and bladder cancer (138).
Given the relevance of this pathway in tumorigenesis, it is not surprising that the number of compounds that inhibit one or more components of these pathways and that are in clinical studies is enormous (135). Yet, few of them are being investigated as anti-CSC compounds. One is VS-5584, a PI3K/mTOR dual inhibitor. It exhibits approximately equal low nanomolar potency against mTOR and all PI3K class I isoforms (121). VS-5584 is up to 30-fold more potent in inhibiting the proliferation and survival of CSCs compared with non-CSCs in solid tumor cell populations. It preferentially diminishes CSC levels in mouse xenograft models and, ex vivo, in surgically resected breast and ovarian patient tumors. VS-5584 delayed tumor regrowth following chemotherapy in xenograft models of SCLC. The preferential activity on CSCs compared to non-CSCs may explain the limited efficacy of PI3K inhibitors used as monotherapies in trials on patients with tumors harboring PI3K pathway activation (143). VS-5584 is currently in phase I clinical trials in hematological and non-hematological malignancies, alone or in combination with the FAK inhibitor VS-6063 (Table 3).
BEZ235 is another PI3K/mTOR dual inhibitor that has prominent anti-CSC activity (144). Thus, combination of BEZ235 with radiotherapy effectively increased radiosensitivity of radioresistant prostate cancer cell lines, induced more apoptosis in radioresistant cells, reduced the expression of EMT and CSC markers and of the PI3K–AKT–mTOR pathway compared with radiotherapy alone. The PI3K–AKT–mTOR signaling pathway is highly activated also in colon CSCs and inhibition with BEZ235 suppressed their proliferation (140). BEZ235 has been investigated in numerous clinical studies for its antitumor activity, but the development has now been discontinued.
Eventually, also the mTOR inhibitor temsirolimus, which has already been approved for the therapy of renal cell carcinoma is being evaluated in a phase I/II clinical trial in combination with liposomal doxorubicin in patients with advanced soft tissue and bone sarcomas. Clinical efficacy and proportion of CSCs before and after therapy are some of the outcomes of this study (Table 3).
Inhibitors of the RAS–RAF–MEK–ERK Pathway
The RAS–RAF–MEK-extracellular signal-regulated kinase (ERK) signaling pathway is, together with the PI3K–AKT–mTOR pathway, one of the most frequently involved in cancer biology. In fact, this pathway is hyperactivated in a high percentage of tumors, mostly because of the presence of activating mutations of the RAS genes (145). Recently, several compounds targeting components of this pathway have been approved for the therapy of metastatic melanoma and have shown promising clinical activity in other tumor types (68, 146). Several preclinical studies showed an involvement of this signaling pathway in the CSC biology of several tumor types. Thus, deficient expression of a negative regulator, dual specificity phosphatase-4, leads to aberrant activation of the pathway with consequent resistance to chemotherapeutics, increased mammosphere formation of CD44+/CD24− tumor cells in basal-like breast cancer (147). This pathway is also involved in the biology of CSCs of several tumor types, such as bladder cancer (148), colon cancer (149), and leiomyosarcoma (150). In some cases, a coordinate activation of the PI3K–AKT–mTOR and RAS–RAF–MEK–ERK has been demonstrated (150, 151). This finding is not surprising, given the intimate relationship between these two pathways, with RAS being an activating signaling node for both pathways (152).
A MEK inhibitor, trametinib (GSK1120212), is being tested in a phase II clinical trial in surgically resectable oral cavity squamous cell cancer (Table 3). Measurement of changes of CD44+ CSCs before and after treatment is one of the outcomes of this study. In fact, a preclinical study in a model of head and neck cancer had shown that CD44 is a critical target of ERK in promoting tumor aggressiveness and proposed this pathway as a target to treat head and neck cancer (123).
Inhibitors of Signal Transducer and Activator of Transcription 3 and Other Transcription Factors
Signal transducer and activator of transcription (STAT) 3 is a transcription factor downstream of several cytokine (e.g., IL-6) and growth factor receptors and non-RTKs, such as SRC (153, 154). STAT3 is frequently overexpressed in carcinomas secondary to the activation of upstream kinases (155). It modulates the expression of a broad range of downstream genes (156), and plays a crucial role in CSC biology in several tumor types, such as breast cancer (139), colon cancer (157), endometrial cancer (158), prostate cancer (159), lung cancer (160), pancreatic cancer (161), and glioblastoma (162).
Napabucasin/BBI608 is a synthetic napthofurandione natural product, which was originally extracted from Bignoniaceae tabeluia. It inhibits gene transcription driven by STAT3, blocks spherogenesis of CSCs, kills CSCs, and downregulates CSC-associated genes (124). In vivo, BBI608 prevents cancer relapse in a pancreatic cancer model and metastasis but completely spares hematopoietic stem cells. BBI608 is well tolerated with no signs of adverse events in preclinical toxicology studies. BBI608 is in phase III clinical trials (Table 3). While BBI608 is the most advanced anti-CSC compound in clinical development, there are concerns about the specificity of this compound, given its quinone structure and potential to exert pleiotropic effects inside cells. Risks and opportunities of such promiscuous compounds have been recently discussed (163, 164).
The same company is developing also another small-molecule inhibitor, BBI503. While no publication is available about this inhibitor, it is claimed to inhibit Nanog and other CSC pathways by targeting kinases. Nanog is a transcription factor that acts as a key regulator of embryonic stem cell maintenance. Nanog is dysregulated in cancer and is involved in the maintenance of CSCs (125). BBI503 is in several phase I–II clinical studies, and two clinical studies are also investigating the combined use of BBI608 and BBI503 (Table 3).
Activators of 5′ Adenosine Monophosphate-Activated Protein Kinase
While most anti-CSC compounds are inhibitors of pathways that are conducive to CSCs, 5′ adenosine monophosphate-activated protein kinase (AMPK) acts as an endogenous inhibitor of CSCs and, consequently, activators of AMPK are being investigated as anti-CSC compounds. The anti-diabetic drug metformin is the prototypic member of this class. It inhibits preferentially the growth of CSCs compared to the bulk of tumor cells and combinatorial therapy with standard chemotherapeutics (doxorubicin, paclitaxel, and cisplatin) increases tumor regression and prolongs remission in mouse xenografts (126). In addition, metformin can decrease the chemotherapeutic dose for prolonging tumor remission in xenografts of multiple cancer types. Phenformin, a related biguanide, is generally considered to represent a stronger version of metformin. While the most broadly accepted mechanism of action whereby metformin inhibits CSCs is through AMPK activation and consequent inhibition of PI3K–AKT–mTOR signaling (127), other AMPK-independent mechanisms may contribute to the antitumor activity of metformin (128, 165). Metformin has also been shown to inhibit and reverse EMT in cell lines resistant to EGFR tyrosine kinase inhibitors through inhibition of TGF-β-induced EMT (131) and of the IL-6/STAT3 axis (132). Recent results in a non-tumor system have shown that metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation (133). These observations establish a link between AMPK and another crucial pathway in CSC biology, STAT3, in addition to PI3K–AKT–mTOR. Metformin is now being studied in a large number of clinical trials for its antitumor activity, and some of these trials investigate also the anti-CSC activity (Table 3).
Inducers of CSC Differentiation
The possibility of inducing the differentiation of CSCs into non-CSC tumor cells appears a logical approach for anti-CSC therapy. It is speculated that some of the anti-CSC compounds discussed so far act by inducing differentiation of CSCs (98). In the following, we discuss some other compounds that act by inducing CSC differentiation.
All-trans-retinoic acid/tretinoin is currently used for the therapy of acute promyelocytic leukemia (APL), a subtype of AML, where it induces differentiation of leukemic blasts. However, among patients with non-APL AML, tretinoin-based treatment has not been effective. It has now been shown that, through epigenetic reprograming, inhibitors of lysine-specific demethylase 1, including tranylcypromine, unlock the tretinoin-driven therapeutic response in non-APL AML (166). Treatment with tretinoin plus tranylcypromine markedly diminished the engraftment of primary human AML cells in vivo in immunodeficient mice, suggesting that tretinoin in combination with tranylcypromine may target leukemia CSCs. This therapeutic combination is now being studied in a phase I clinical trial in AML patients (Table 4).
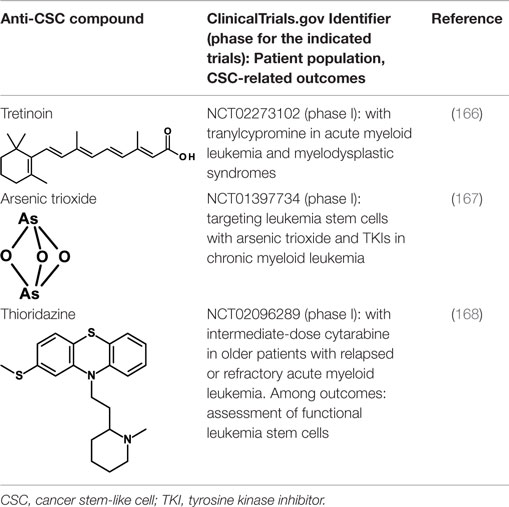
Table 4. Inducers of CSC differentiation.
Also arsenic trioxide is used for the therapy of APL where it induces differentiation and apoptosis of leukemic cells. Arsenic trioxide has now been reported to induce differentiation of HCC CSCs by downregulating the expression of CD133 and other CSC markers (167). The self-renewal ability and tumorigenic capacity, but not the proliferation in vitro, were inhibited. In vivo, arsenic trioxide decreased recurrence rates after radical resection and prolonged survival in a mouse model. Arsenic trioxide is being studied, in combination with tyrosine kinase inhibitors in a clinical trial for its capacity to target the CML stem cell population (Table 4).
Thioridazine is an antipsychotic drug that antagonizes dopamine receptors. It was identified using a discovery platform that reveals differences between neoplastic and normal human stem cells in undergoing differentiation in response to molecules from libraries of known compounds (168). Thioridazine was found to selectively target neoplastic cells, and to impair human somatic CSCs capable of in vivo leukemic disease initiation while having no effect on normal blood stem cells. Thioridazione is now undergoing a phase I trial in combination with cytarabine in patients with AML. Objective response rates and effects on leukemic stem cells are among the outcomes of this study (see Table 4).
Radiotherapeutic Targeting of CSC-Enriched Domains
CSCs are resistant to radiation because of enhanced self-renewal capacity, DNA-repair capacity, and enhanced reactive oxygen species defenses. In spite of this radioresistance, new radiotherapeutic approaches appear to be promising treatments for the targeting of CSCs (169). Moreover, it is currently believed that glioblastoma CSCs derive from the transformation of normal tissue stem cells residing in the subventricular zones of the brain (170). This finite anatomic location of the glioblastoma CSC pool has suggested the possibility of targeting these cells with radiotherapeutic approaches. One clinical trial investigating the effects of radiotherapy to brain areas containing CSCs in combination with temozolomide in glioblastoma patients is ongoing (NCT02039778, phase I/II).
Anti-CSC Compounds with Undefined Mechanism(s) of Action or Targeting Diverse Signaling Pathways
Salinomycin
Salinomycin is the first molecule that was found to have anti-CSC activities. It was identified in a chemical screen that had been designed to discover compounds showing selective toxicity for breast CSCs (171). Salinomycin has been widely used as an anticoccidiosis agent in chickens. It is a natural, fused polypyran ionophore able to shield charge of ions and to cross cell lipid membranes, thereby interfering with transmembrane potassium potential and promoting mitochondrial and cellular potassium flux. Salinomycin is a complex natural molecule produced by fermentation with many stereocenters. Efforts are underway to develop synthetic salinomycin analogs. Salinomycin reduced the proportion of CSCs by >100-fold relative to paclitaxel, inhibited mammary tumor growth in vivo and reduced expression of breast CSC genes. Ionic changes induced by salinomycin were found to inhibit proximal WNT signaling by interfering with phosphorylation of the WNT coreceptor LPR6 (172). In the following, salinomycin has also been found to modulate Hh signaling (173), to induce accumulation of reactive oxygen species (174), and inhibit STAT3 activation (175). In general, compounds endowed with a pleiotropic array of mechanisms of action are, clinically, of little promise. Nevertheless, salinomycin has been reported being investigated in clinical trials (176), but these trials are not reported under the clinicaltrials.gov website.
Disulfiram
This is a dithiocarbamate used for the treatment of chronic alcoholism where it produces acute sensitivity to ethanol due to its inhibitory activity on ALDH (177). Disulfiram was found to possess antitumor activity in early clinical studies (178) and, in recent years, to possess anti-CSC activities. Thus, disulfiram inhibits TGF-β-induced EMT and CSC-like features in breast cancer cells (179). It can also inhibit the proliferation and self-renewal of glioblastoma CSCs (180). Regarding the mechanism of action, disulfiram can act as a proteasome inhibitor and this, in turn, inhibits nuclear translocation and DNA-binding activity of NF-κB (130). Moreover, being an irreversible inhibitor of ALDH, disulfiram might also act as an inhibitor of ALDH-positive CSCs (129). Disulfiram is now being investigated in clinical studies, and the results of a preliminary clinical trial appear encouraging (181). In one study, it is used in combination with temozolomide in newly diagnosed glioblastoma (Table 5). This study refers explicitly to the anti-CSC activity of disulfiram.

Table 5. Anti-CSC compounds with undefined mechanism of action or targeting diverse signaling pathways.
Chloroquine
Chloroquine has anti-CSC activity (182), and this effect is considered to be the consequence of the lysosomotropic action of chloroquine and consequent inhibition of autophagy. Balic et al. (183), however, have found that chloroquine targets pancreatic CSCs through a dual mechanism of action: first, inhibition of CXC ligand 12/CXCR4 signaling, resulting in reduced phosphorylation of ERK and STAT3. Second, chloroquine inhibited Hh signaling by decreasing the production of SMO, translating into downregulation of downstream targets in CSCs and the surrounding stroma. Chloroquine-induced inhibition of STAT3 signaling as a mechanism for the anti-CSC activity of chloroquine has been reported also by Choi et al. (185). Chloroquine is currently being investigated in clinical studies for its anti-CSC activity (see Table 5).
Oncolytic Adenovirus
The tumor-selective oncolytic adenovirus Delta-24-RGD has antiglioma effects (186) and overcomes temozolomide resistance by silencing the O6-methylguanine-DNA methyltransferase promoter (187). Delta-24-RGD infects, replicates, and induces autophagic cell death in brain CSCs (184). It is being studied in combination with temozolomide in a phase I clinical trial in glioblastoma patients (Table 5).
Vaccination Against CSCs
Vaccination against CSCs or individual proteins overexpressed in CSCs is another anti-CSC approach. In preclinical studies, syngeneic hosts have been immunized with CSC-enriched populations (188). This source was more effective than unselected tumor cells in inducing protective antitumor immunity. Immune sera from vaccinated hosts contained lytic anti-CSC IgG antibodies, and cytotoxic T-lymphocytes that were capable of killing CSCs in vitro.
Recently, anti-CSC vaccination against individual proteins has been proposed. Sox2 is a transcription factor required for the maintenance of normal neural stem cells and for the biology of oligodendroglioma CSCs. Immunization of mice with Sox2 peptides delayed tumor development and prolonged survival (189). In another study, xCT, the functional subunit of the cystine/glutamate antiporter system xc-, has been found to be upregulated in mammospheres derived from murine human EGFR2-positive breast tumor cells (190). Downregulation of xCT impaired mammosphere generation and altered CSC intracellular redox balance in vitro. DNA vaccination-based immunotargeting of xCT in mice delayed subcutaneous growth of mammophere-derived cells, impaired pulmonary metastasis formation, and increased CSC chemosensitivity to doxorubicin in vivo. Table 6 lists ongoing clinical trials aimed at vaccinating tumor-bearing patients against CSCs.

Table 6. Anti-CSC vaccines.
Looking Ahead – 1. Epigenetic Drugs for Anti-CSC Therapy
Epigenetic reprograming is a characteristic trait of tumorigenesis and CSC biology. Reprograming has been demonstrated for every component of the epigenetic machinery, including DNA methylation, histone modifications, nucleosome positioning, and non-coding RNAs, specifically microRNA expression (194). The possibility of therapeutically manipulating these events is attractive because cancer-associated epigenetic aberrations are reversible. Not surprisingly, drug discovery in epigenetics is being widely pursued and four epigenetic drugs have already received FDA approval (azacitidine, decitabine, vorinostat, and romidepsin). Inhibition of CSCs with epigenetic drugs has been demonstrated in several instances (195–197), and this is likely to become a rapidly progressing domain. A note of caution to be sounded for these drugs comes from the knowledge that some epigenetic drugs have been reported to have tumor-promoting or tumor-inhibiting activities in a context-dependent manner (31, 194).
Looking Ahead – 2. Improving Tumor Delivery of Anti-CSC Medicines
CSCs are often reported to be concealed in hypoxic tumor areas (198), and these areas pose significant problems for drug penetration (199). Therefore, administration of anti-CSC compounds may not achieve complete depletion of CSCs present at any given time, because of inadequate tumor penetration. Moreover, inadequate penetration of antitumor drugs may contribute to the induction of drug resistance (200). Therefore, improving the delivery of anti-CSC compounds to their target cells has become an area of active research (201). In general, one can envisage two main approaches. First, through the use of passively or actively targeted nanoparticles (202). Second, through coadministration of promoter drugs that facilitate the penetration of compounds endowed with direct antitumor effects (the effector drug) (199, 203). While none of these approaches has yet made its way to the clinic, several preclinical studies suggest their feasibility. Thus, PEGylated polymeric micelles were loaded with salinomycin to effectively target breast CSCs and shown to be more effective than salinomycin in vivo (204). Others demonstrated that nanoparticles can penetrate, upon convection-enhanced delivery, large intracranial volumes in experimental animals. When loaded into these nanoparticles, a compound that had been identified possessing anti-CSC activity, ditiazanine iodide, significantly increased survival in rats bearing brain CSC-derived xenografts (205).
Regarding the possibility of improving the penetration of effector drugs through the coadministration of a promoter drug, it is interesting to note that an anti-CSC compound that has been discussed above, IPI-926, a Hh pathway inhibitor, has been shown to improve penetration of an effector drug (gemcitabine) into pancreatic tumors through depletion of tumor-associated stromal tissue (206). In fact, paracrine Hh signaling from neoplastic cells promotes stromal desmoplasia (207). Combined with their anti-CSC activity, compounds of this class may be of particular interest because they have the potential to combine in a single molecule anti-CSC activity and promotion of tumor penetration.
Anti-CSC Compounds in Clinical Development – A Critical Appraisal
There are several crucial points to address when taking an overall look to anti-CSC compounds in current clinical investigation.
The first, most important and most difficult point to answer is how to evidence clinical benefit that may derive from anti-CSC compounds. Currently used antitumor drugs target mainly proliferating tumor cells, while sparing CSCs and even inducing the generation of new CSCs. On the other hand, anti-CSC compounds are inactive against the bulk of proliferating tumor cells because they target markers or pathways that are overexpressed or selectively expressed on CSCs compared to bulk tumor cells. Consequently, the vast majority of ongoing clinical trials with anti-CSC compounds are performed in combination with other antitumor drugs belonging to different classes of compounds. Therefore, the easiest way to answer the question as to how evidence clinical benefit it to say that anti-CSC compounds should improve the efficacy of anti-cancer therapies that are given in combination, i.e., higher percentages of patients with clinical responses or stable disease for longer periods of time than patients treated with standard-of-care therapies. This, however, is the goal of any anti-cancer therapy, whether or not targeting CSCs. So, the question arises as to how it is possible to recognize bona fide anti-CSC activity compared to generic antitumor effects.
A first possibility, and probably also the most realistic, is to state that any clinical benefit deriving from anti-CSC therapy should be accompanied by a decline of tumor tissue CSCs or circulating CSCs (208). Not surprisingly, many clinical trials with anti-CSC compounds include, among the study outcomes, the determination of tumor tissue or circulating CSCs (see the tables throughout this article). In fact, it is not obvious that anti-CSC compounds exert antitumor effects through their anti-CSC activity, because most, if not all of them, are not CSC-specific. One example is represented by anti-DLL4 antibodies. While one anti-DLL4 antibody is being developed as an anti-CSC compound (79), a second antibody is being developed as an anti-angiogenic compound (108), and anti-DLL4 antibodies have even been shown to exert antitumor effects by acting on cells of the tumor stroma (107).
In spite of such a pharmacodynamic approach it would be, nevertheless, desirable to identify the clinical setting that would profit most from anti-CSC therapy. If we consider that CSCs replenish the bulk of proliferating tumor cells, then anti-CSC compounds should be most efficacious after debulking of tumor cells, i.e., in an adjuvant setting. In fact, in tumors that do not display HER2 gene amplification, HER2 may be selectively expressed on CSCs, and an anti-HER2 antibody blocked growth of these HER2-negative tumors in mouse models when administered in the adjuvant setting but had no effect on established tumors (209). Given that, it is somehow surprising to note that there are no ongoing clinical trials studying anti-CSC compounds in an adjuvant setting. Only one study evaluates the expression of a CSC marker as a predictor of adjuvant chemotherapy response in breast cancers of high-risk women (NCT00949013). A likely reason for this attitude is that the execution of clinical trials in an adjuvant setting is challenging for most tumor types, both in terms of duration and costs.
Another question is as to which molecular targets should be addressed in order to have the greatest chances of success. Although, also in this case, only results from clinical practice will give definitive answers, some guidelines may help in taking the most promising roads. First, going through the pathways that are involved in CSC biology and are targets for anti-CSC compounds, it can be appreciated that none of these pathways or targets have been clearly attributed to a single tumor type or subtype. This implies that in each tumor several of these pathways can be active, i.e., the system appears highly redundant and prone to native and acquired resistance. There are several possibilities to mitigate these consequences.
First, using a given anti-CSC compound only when aberrant activation of the relevant pathway is clearly demonstrated. This is important not only for avoiding native drug resistance and delaying insurgence of acquired drug resistance, but also because some of the pathways that are being targeted for anti-CSC therapy play a context-dependent tumor- and CSC-promoting role, while, in a different context, they may also exert tumor-suppressive effects. There are several examples of these opposite, context-dependent effects, for example, the Notch (210, 211), TGF-β (212), WNT (82, 213), and STAT3 (214) pathways.
Second, using anti-CSC compounds that are directly cytotoxic, such as ADCs or native antibodies that act through antibody-dependent cellular cytotoxicity or complement-dependent cytotoxicity. Use of these compounds does not exclude the possibility of selecting for cells that are mutated in the epitope recognized by the targeting moiety, as has been elegantly demonstrated in a recent work (215). Nevertheless, these compounds are expected to be less conducive to resistance than anti-CSC compounds that inhibit signaling pathways without being directly cytotoxic. In fact, these latter compounds may themselves promote acquisition of drug resistance when acting on cells at suboptimal concentrations like those that are achieved in hypoperfused tumor domains (200).
A further turn of complexity is given by the fact that, in any given tumor and at any given time, CSCs may represent a heterogeneous population, both functionally and phenotypically (116, 216). This fact, taken together with the redundancy of the signaling systems underlying CSC biology, suggests that the combined use of anti-CSC compounds may represent still another approach to mitigate untoward consequences. Thus, combined use of different anti-CSC compounds may embrace a larger part of the whole CSC population than a single compound. Preclinical studies suggest that, indeed, combined use of anti-CSC compounds is more efficacious than their individual use (85, 217), and, as already mentioned before in this article, clinical studies with combinations of anti-CSC compounds are ongoing.
Another last point to consider is the plasticity of CSCs. We have already discussed the role of stimuli from the tumor micronevironment in CSC biology. These stimuli are the result of unlimited tumor cell proliferation that causes the generation of different forms of stressors, which, in combination with the inherent genetic instability of tumor cells, shapes the CSC landscape. The implicit consequence of this is that once tumor cells resume growth after drug-induced depletion, the same conditions that generated the CSC-inducing stimuli will be recreated and give rise again to a CSC population. Therefore, anti-CSC therapy should not be expected to act as a device that leads, in combination with other antitumor therapies, to tumor eradication but, rather, as a tool to improve the clinical efficacy of existing drugs. The ongoing clinical trials will tell us in the next years if this is a feasible goal to achieve.
Author Contributions
FM, CR, and FL contributed to the conception of the work, drafted or revisited it critically, approved the final version, and agreed to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the conception and preparation of this article was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature (2001) 414:105–11. doi: 10.1038/35102167
2. Ghisolfi L, Keates AC, Hu X, Lee DK, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS One (2012) 7:e43628. doi:10.1371/journal.pone.0043628
3. Hu X, Ghisolfi L, Keates AC, Zhang J, Xiang S, Lee DK, et al. Induction of cancer cell stemness by chemotherapy. Cell Cycle (2012) 11:2691–8. doi:10.4161/cc.21021
4. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells – what challenges do they pose? Nat Rev Drug Discov (2014) 13:497–512. doi:10.1038/nrd4253
5. Sun S, Liu S, Duan SZ, Zhang L, Zhou H, Hu Y, et al. Targeting the c-Met/FZD8 signaling axis eliminates patient-derived cancer stem-like cells in head and neck squamous carcinomas. Cancer Res (2014) 74:7546–59. doi:10.1158/0008-5472.CAN-14-0826
6. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature (1994) 367:645–8. doi:10.1038/367645a0
7. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A (2003) 100:3983–8. doi:10.1073/pnas.0530291100
8. Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res (2005) 65:3025–9. doi:10.1158/0008-5472.CAN-04-3931
9. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res (2005) 65:10946–51. doi:10.1158/0008-5472.CAN-05-2018
10. Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature (2007) 445:111–5. doi:10.1038/nature05384
11. Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res (2003) 63:5821–8.
13. Martinez-Climent JA, Andreu EJ, Prosper F. Somatic stem cells and the origin of cancer. Clin Transl Oncol (2006) 8:647–63. doi:10.1007/s12094-006-0035-7
14. Vaz AP, Deb S, Rachagani S, Dey P, Muniyan S, Lakshmanan I, et al. Overexpression of PD2 leads to increased tumorigenicity and metastasis in pancreatic ductal adenocarcinoma. Oncotarget (2016) 7:3317–31. doi:10.18632/oncotarget.6580
15. Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res (2014) 74:4922–36. doi:10.1158/0008-5472.CAN-14-1022
16. Marcucci F, Bellone M, Caserta CA, Corti A. Pushing tumor cells towards a malignant phenotype: stimuli from the microenvironment, intercellular communications and alternative roads. Int J Cancer (2014) 135:1265–76. doi:10.1002/ijc.28572
17. Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest (2011) 121:3804–9. doi:10.1172/JCI57099
18. Lu T, Stark GR. Cytokine overexpression and constitutive NF-κB in cancer. Cell Cycle (2004) 3:1114–7. doi:10.4161/cc.3.9.1130
19. Catanzaro JM, Sheshadri N, Pan JA, Sun Y, Shi C, Li J, et al. Oncogenic Ras induces inflammatory cytokine production by upregulating the squamous cell carcinoma antigens SerpinB3/B4. Nat Commun (2014) 5:3729. doi:10.1038/ncomms4729
20. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer (2008) 8:755–68. doi:10.1038/nrc2499
21. Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol (2012) 13:e83–9. doi:10.1016/S1470-2045(11)70257-1
22. Poleszczuk J, Hahnfeldt P, Enderling H. Evolution and phenotypic selection of cancer stem cells. PLoS Comput Biol (2015) 11:e1004025. doi:10.1371/journal.pcbi.1004025
23. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol (2015) 12:445–64. doi:10.1038/nrclinonc.2015.61
24. Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
25. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell (2012) 148:349–61. doi:10.1016/j.cell.2011.11.025
26. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial−mesenchymal transitions in development and disease. Cell (2009) 139:871–90. doi:10.1016/j.cell.2009.11.007
27. Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest (2009) 119:1417–9. doi:10.1172/JCI39675
28. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer (2009) 9:265–73. doi:10.1038/nrc2620
29. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene (2010) 29:4741–51. doi:10.1038/onc.2010.215
30. Yen WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res (2015) 21:2084–95. doi:10.1158/1078-0432.CCR-14-2808
31. Marcucci F, Stassi G, De Maria R. Epithelial-mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov (2016). doi:10.1038/nrd.2015.13
32. Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci (2013) 126:21–9. doi:10.1242/jcs.120907
33. Ocaña OH, Córcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell (2012) 22:709–24. doi:10.1016/j.ccr.2012.10.012
34. Tan TZ, Miow QH, Miki Y, Noda T, Mori S, Huang RY, et al. Epithelial-mesenchymal transition spectrum quantification and its efficacy in deciphering survival and drug responses of cancer patients. EMBO Mol Med (2014) 6:1279–93. doi:10.15252/emmm.201404208
35. Kuo SZ, Blair KJ, Rahimy E, Kiang A, Abhold E, Fan JB, et al. Salinomycin induces cell death and differentiation in head and neck squamous cell carcinoma stem cells despite activation of epithelial-mesenchymal transition and Akt. BMC Cancer (2012) 12:556. doi:10.1186/1471-2407-12-556
36. Wiener Z, Högström J, Hyvönen V, Band AM, Kallio P, Holopainen T, et al. Prox1 promotes expansion of the colorectal cancer stem cell population to fuel tumor growth and ischemia resistance. Cell Rep (2014) 8:1943–56. doi:10.1016/j.celrep.2014.08.034
37. Ojha R, Bhattacharyya S, Singh SK. Autophagy in cancer stem cells: a potential link between chemoresistance, recurrence, and metastasis. Biores Open Access (2015) 4:97–108. doi:10.1089/biores.2014.0035
38. Yang MC, Wang HC, Hou YC, Tung HL, Chiu TJ, Shan YS. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer (2015) 14:179. doi:10.1186/s12943-015-0449-3
39. Papadaki MA, Kallergi G, Zafeiriou Z, Manouras L, Theodoropoulos PA, Mavroudis D, et al. Co-expression of putative stemness and epithelial-to-mesenchymal transition markers on single circulating tumour cells from patients with early and metastatic breast cancer. BMC Cancer (2014) 14:651. doi:10.1186/1471-2407-14-651
40. Diessner J, Bruttel V, Stein RG, Horn E, Häusler SF, Dietl J, et al. Targeting of preexisting and induced breast cancer stem cells with trastuzumab and trastuzumab emtansine (T-DM1). Cell Death Dis (2014) 5:e1149. doi:10.1038/cddis.2014.115
41. Herrmann H, Sadovnik I, Cerny-Reiterer S, Rülicke T, Stefanzl G, Willmann M, et al. Dipeptidylpeptidase IV (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood (2014) 123:3951–62. doi:10.1182/blood-2013-10-536078
42. Cioffi M, Trabulo S, Hidalgo M, Costello E, Greenhalf W, Erkan M, et al. Inhibition of CD47 effectively targets pancreatic cancer stem cells via dual mechanisms. Clin Cancer Res (2015) 21:2325–37. doi:10.1158/1078-0432.CCR-14-1399
43. Emlet DR, Gupta P, Holgado-Madruga M, Del Vecchio CA, Mitra SS, Han SY, et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res (2014) 74:1238–49. doi:10.1158/0008-5472.CAN-13-1407
44. Shiota M, Zardan A, Takeuchi A, Kumano M, Beraldi E, Naito S, et al. Clusterin mediates TGF-β-induced epithelial-mesenchymal transition and metastasis via Twist1 in prostate cancer cells. Cancer Res (2012) 72:5261–72. doi:10.1158/0008-5472.CAN-12-0254
45. Lenferink AE, Cantin C, Nantel A, Wang E, Durocher Y, Banville M, et al. Transcriptome profiling of a TGF-β-induced epithelial-to-mesenchymal transition reveals extracellular clusterin as a target for therapeutic antibodies. Oncogene (2010) 29:831–44. doi:10.1038/onc.2009.399
46. Wang C, Jiang K, Kang X, Gao D, Sun C, Li Y, et al. Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int J Biochem Cell Biol (2012) 44:2308–20. doi:10.1016/j.biocel.2012.09.012
47. Brandolini L, Cristiano L, Fidoamore A, De Pizzol M, Di Giacomo E, Florio TM, et al. Targeting CXCR1 on breast cancer stem cells: signaling pathways and clinical application modelling. Oncotarget (2015) 6:43375–94. doi:10.18632/oncotarget.6234
48. Zhang S, Cui B, Lai H, Liu G, Ghia EM, Widhopf GF II, et al. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc Natl Acad Sci U S A (2014) 111:17266–71. doi:10.1073/pnas.1419599111
49. Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res (2009) 69:1302–13. doi:10.1158/0008-5472.CAN-08-2741
50. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, et al. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J Clin Invest (2010) 120:485–97. doi:10.1172/JCI39397
51. Cui B, Zhang S, Chen L, Yu J, Widhopf GF II, Fecteau J-F, et al. Targeting ROR1 inhibits epithelial-mesenchymal transition and metastasis. Cancer Res (2013) 73:3649–60. doi:10.1158/0008-5472.CAN-12-3832
52. Park SY, Kim MJ, Park SA, Kim JS, Min KN, Kim DK, et al. Combinatorial TGF-β attenuation with paclitaxel inhibits the epithelial-to-mesenchymal transition and breast cancer stem-like cells. Oncotarget (2015) 6:37526–43. doi:10.18632/oncotarget.6063
53. Ohnuki H, Jiang K, Wang D, Salvucci O, Kwak H, Sánchez-Martín D, et al. Tumor-infiltrating myeloid cells activate Dll4/Notch/TGF-β signaling to drive malignant progression. Cancer Res (2014) 74:2038–49. doi:10.1158/0008-5472.CAN-13-3118
54. Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFβ/TNFα-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res (2011) 71:4707–19. doi:10.1158/0008-5472.CAN-10-4554
55. Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell (2011) 145:926–40. doi:10.1016/j.cell.2011.04.029
56. Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett (2014) 352:160–8. doi:10.1016/j.canlet.2014.05.008
57. Park CY, Min KN, Son JY, Park SY, Nam JS, Kim DK, et al. An novel inhibitor of TGF-β type I receptor, IN-1130, blocks breast cancer lung metastasis through inhibition of epithelial-mesenchymal transition. Cancer Lett (2014) 351:72–80. doi:10.1016/j.canlet.2014.05.006
58. Zhong Z, Driscoll Carroll K, Policarpio D, Osborn C, Gregory M, Bassi R, et al. Anti-transforming growth factor β receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin Cancer Res (2010) 16:1191–205. doi:10.1158/1078-0432.CCR-09-1634
59. Liu J, Liao S, Diop-Frimpong B, Chen W, Goel S, Naxerova K, et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A (2012) 109:16618–23. doi:10.1073/pnas.1117610109
60. Morris JC, Tan AR, Olencki TE, Shapiro GI, Dezube BJ, Reiss M, et al. Phase I study of GC1008 (fresolimumab): a human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS One (2014) 9:e90353. doi:10.1371/journal.pone.0090353
61. Bhola NE, Balko JM, Dugger TC, Kuba MG, Sánchez V, Sanders M, et al. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J Clin Invest (2013) 123:1348–58. doi:10.1172/JCI65416
62. Akhurst RJ, Hata A. Targeting the TGF-β signalling pathway. Nat Rev Drug Discov (2012) 11:790–811. doi:10.1038/nrd3810
63. Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol (2011) 39:916–24. doi:10.1177/0192623311416259
64. Gueorguieva I, Cleverly AL, Stauber A, Sada Pillay N, Rodon JA, Miles CP, et al. Defining a therapeutic window for the novel TGF-β inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br J Clin Pharmacol (2014) 77:796–807. doi:10.1111/bcp.12256
65. Yoon C, Park do J, Schmidt B, Thomas NJ, Lee HJ, Kim TS, et al. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin Cancer Res (2014) 20:3974–88. doi:10.1158/1078-0432.CCR-14-0011
66. Singh BN, Fu J, Srivastava RK, Shankar S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One (2011) 6:e27306. doi:10.1371/journal.pone.0027306
67. Kim EJ, Sahai V, Abel EV, Griffith KA, Greenson JK, Takebe N, et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin Cancer Res (2014) 20:5937–45. doi:10.1158/1078-0432.CCR-14-1269
68. Kim G, McKee AE, Ning YM, Hazarika M, Theoret M, Johnson JR, et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res (2014) 20:4994–5000. doi:10.1158/1078-0432.CCR-14-0776
69. Jimeno A, Weiss GJ, Miller WH Jr, Gettinger S, Eigl BJ, Chang AL, et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin Cancer Res (2013) 19:2766–74. doi:10.1158/1078-0432.CCR-12-3654
70. Bowles DW, Keysar SB, Eagles JR, Wang G, Glogowska MJ, McDermott JD, et al. A pilot study of cetuximab and the hedgehog inhibitor IPI-926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol (2016) 53:74–9. doi:10.1016/j.oraloncology.2015.11.014
71. Storm EE, Durinck S, de Sousa e Melo F, Tremayne J, Kljavin N, Tan C, et al. Targeting PTPRK-RSPO3 colon tumours promotes differentiation and loss of stem-cell function. Nature (2016) 529:97–100. doi:10.1038/nature16466
72. Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A (2012) 109:11717–22. doi:10.1073/pnas.1120068109
73. Le PN, McDermott JD, Jimeno A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol Ther (2015) 146:1–11. doi:10.1016/j.pharmthera.2014.08.005
74. Jang G-B, Hong I-S, Kim R-J, Lee S-Y, Park S-J, Lee E-S, et al. Wnt/β-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res (2015) 75:1691–702. doi:10.1158/0008-5472.CAN-14-2041
75. Nagaraj AB, Joseph P, Kovalenko O, Singh S, Armstrong A, Redline R, et al. Critical role of Wnt/β-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget (2015) 6:23720–34. doi:10.18632/oncotarget.4690
76. Lenz H-J, Kahn M. Safely targeting cancer stem cells via selective catenin coactivator antagonism. Cancer Sci (2014) 105:1087–92. doi:10.1111/cas.12471
77. Lee K-M, Nam K, Oh S, Lim J, Kim RK, Shim D, et al. ECM1 regulates tumor metastasis and CSC-like property through stabilization of β-catenin. Oncogene (2015) 34:6055–65. doi:10.1038/onc.2015.54
78. Ponnusamy MP, Seshacharyulu P, Lakshmanan I, Vaz AP, Chugh S, Batra SK. Emerging role of mucins in epithelial to mesenchymal transition. Curr Cancer Drug Targets (2013) 13:945–56. doi:10.2174/15680096113136660100
79. Smith DC, Eisenberg PD, Manikhas G, Chugh R, Gubens MA, Stagg RJ, et al. A phase I dose escalation and expansion study of the anticancer stem cell agent demcizumab (anti-DLL4) in patients with previously treated solid tumors. Clin Cancer Res (2014) 20:6295–303. doi:10.1158/1078-0432.CCR-14-1373
80. Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med (2015) 7:302ra136. doi:10.1126/scitranslmed.aac9459
81. Damelin M, Bankovich A, Park A, Aguilar J, Anderson W, Santaguida M, et al. Anti-EFNA4 calicheamicin conjugates effectively target triple-negative breast and ovarian tumor-initiating cells to result in sustained tumor regressions. Clin Cancer Res (2015) 21:4165–73. doi:10.1158/1078-0432.CCR-15-0695
82. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer (2013) 13:11–26. doi:10.1038/nrc3419
84. Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin Cancer Res (2015) 21:505–13. doi:10.1158/1078-0432.CCR-14-0507
85. Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP, Shankar S, et al. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget (2015) 6:32039–60. doi:10.18632/oncotarget.5055
86. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn A, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of Notch and Hedgehog-dependent tumor-initiating cells. Cancer Cell (2012) 22:373–88. doi:10.1016/j.ccr.2012.07.016
87. Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells (2007) 25:2524–33. doi:10.1634/stemcells.2007-0166
88. Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell (2014) 25:139–51. doi:10.1016/j.ccr.2014.01.008
89. Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res (2006) 66:6063–71. doi:10.1158/0008-5472.CAN-06-0054
90. Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med (2009) 1:338–51. doi:10.1002/emmm.200900039
91. Dierks C, Beigi R, Guo GR, Zirlik K, Stegert MR, Manley P, et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell (2008) 14:238–49. doi:10.1016/j.ccr.2008.08.003
92. Peacock CD, Wang Q, Gesell GS, Corcoran-Schwartz IM, Jones E, Kim J, et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc Natl Acad Sci U S A (2007) 104:4048–53. doi:10.1073/pnas.0611682104
93. Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature (2008) 455:406–10. doi:10.1038/nature07275
94. Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature (2000) 406:1005–9. doi:10.1038/35023008
95. Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med (2012) 366:2171–9. doi:10.1056/NEJMoa1113713
96. Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science (2009) 326:572–4. doi:10.1126/science.1179386
97. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med (2009) 361:1173–8. doi:10.1056/NEJMoa0902903
98. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov (2014) 13:513–32. doi:10.1038/nrd4233
99. Eaves CJ, Humphries RK. Acute myeloid leukemia and the Wnt pathway. N Engl J Med (2010) 362:2326–7. doi:10.1056/NEJMcibr1003522
100. Monteiro J, Gaspar C, Richer W, Franken PF, Sacchetti A, Joosten R, et al. Cancer stemness in Wnt-driven mammary tumorigenesis. Carcinogenesis (2014) 35:2–13. doi:10.1093/carcin/bgt279
101. White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology (2012) 142:219–32. doi:10.1053/j.gastro.2011.12.001
102. Yoon JK, Lee JS. Cellular signaling and biological functions of R-spondins. Cell Signal (2012) 24:369–77. doi:10.1016/j.cellsig.2011.09.023
103. Andersson ER, Ledahl U. Therapeutic modulation of Notch signalling – are we there yet? Nat Rev Drug Discov (2014) 13:357–78. doi:10.1038/nrd4252
104. Huynh C, Poliseno L, Segura MF, Medicherla R, Haimovic A, Menendez S, et al. The novel gamma secretase inhibitor RO4929097 reduces the tumor initiating potential of melanoma. PLoS One (2011) 6:e25264. doi:10.1371/journal.pone.0025264
105. Takebe N, Nguyen D, Yang SX. Targeting Notch pathway in cancer: clinical development advances and challenges. Pharmacol Ther (2014) 141:140–9. doi:10.1016/j.pharmthera.2013.09.005
106. Hellström M, Phng L-K, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature (2007) 445:776–80. doi:10.1038/nature05571
107. Kuhnert F, Chen G, Coetzee S, Thambi N, Hickey C, Shan J, et al. Dll4 blockade in stromal cells mediates antitumor effects in preclinical models of ovarian cancer. Cancer Res (2015) 75:4086–96. doi:10.1158/0008-5472.CAN-14-3773
108. Chiorean EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA, et al. A phase I first-in-human study of enoticumab (REGN421), a fully human Delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin Cancer Res (2015) 21:2695–703. doi:10.1158/1078-0432.CCR-14-2797
109. Geffers I, Serth K, Chapman G, Jaekel R, Schuster-Gossler K, Cordes R, et al. Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J Cell Biol (2007) 178:465–76. doi:10.1083/jcb.200702009
110. Chapman G, Sparrow DB, Kremmer E, Dunwoodie SL. Notch inhibition by the ligand Delta-like 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Hum Mol Genet (2011) 20:905–16. doi:10.1093/hmg/ddq529
111. Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat Rev Drug Discov (2014) 13:39–62. doi:10.1038/nrd4175
112. Marquardt JU, Raggi C, Andersen JB, Seo D, Avital I, Geller D, et al. Human hepatic cancer stem cells are characterized by common stemness traits and diverse oncogenic pathways. Hepatology (2011) 54:1031–42. doi:10.1002/hep.24454
113. Duong HQ, Yi YW, Kang HJ, Bae I, Jang YJ, Kwak SJ, et al. Combination of dasatinib and gemcitabine reduces the ALDH1A1 expression and the proliferation of gemcitabine-resistant pancreatic cancer MIA PaCa-2 cells. Int J Oncol (2014) 44:2132–8. doi:10.3892/ijo.2014.2357
114. Thakur R, Trivedi R, Rastogi N, Singh M, Mishra DP. Inhibition of STAT3, FAK and Src mediated signaling reduces cancer stem cell load, tumorigenic potential and metastasis in breast cancer. Sci Rep (2015) 5:10194. doi:10.1038/srep10194
115. Williams KE, Bundred NJ, Landberg G, Clarke RB, Farnie G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells (2015) 33:327–41. doi:10.1002/stem.1843
116. Schober M, Fuchs E. Tumor-initiating stem cells of squamous cell carcinomas and their control by TGF-β and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci U S A (2011) 108:10544–9. doi:10.1073/pnas.1107807108
117. Mustjoki S, Richter J, Barbany G, Ehrencrona H, Fioretos T, Gedde-Dahl T, et al. Impact of malignant stem cell burden on therapy outcome in newly diagnosed chronic myeloid leukemia patients. Leukemia (2013) 27:1520–6. doi:10.1038/leu.2013.19
118. Jones SF, Siu LL, Bendell JC, Cleary JM, Razak AR, Infante JR, et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs (2015) 33:1100–7. doi:10.1007/s10637-015-0282-y
119. O’Brien S, Golubovskaya VM, Conroy J, Liu S, Wang D, Liu B, et al. FAK inhibition with small molecule inhibitor Y15 decreases viability, clonogenicity, and cell attachment in thyroid cancer cell lines and synergizes with targeted therapeutics. Oncotarget (2014) 5:7945–59. doi:10.18632/oncotarget.2381
120. Shapiro IM, Kolev VN, Vidal CM, Kadariya Y, Ring JE, Wright Q, et al. Merlin deficiency predicts FAK inhibitor sensitivity: a synthetic lethal relationship. Sci Transl Med (2014) 6:237ra68. doi:10.1126/scitranslmed.3008639
121. Kolev VN, Wright QG, Vidal CM, Ring JE, Shapiro IM, Ricono J, et al. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res (2015) 75:446–55. doi:10.1158/0008-5472.CAN-14-1223
122. Chiarini F, Lonetti A, Teti G, Orsini E, Bressanin D, Cappellini A, et al. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget (2012) 3:1615–28. doi:10.18632/oncotarget.762
123. Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS Jr, et al. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer Res (2012) 72:365–74. doi:10.1158/0008-5472.CAN-11-1831
124. Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A (2015) 112:1839–44. doi:10.1073/pnas.1424171112
125. Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer (2014) 135:2741–8. doi:10.1002/ijc.28690
126. Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res (2009) 69:7507–11. doi:10.1158/0008-5472.CAN-09-2994
127. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer (2009) 9:563–75. doi:10.1038/nrc2676
128. Janzer A, German NJ, Gonzalez-Herrera KN, Asara JM, Haigis MC, Struhl K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc Natl Acad Sci U S A (2014) 111:10574–9. doi:10.1073/pnas.1409844111
129. Liu P, Brown S, Goktug T, Channathodiyil P, Kannappan V, Hugnot JP, et al. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br J Cancer (2012) 107:1488–97. doi:10.1038/bjc.2012.442
130. Liu P, Wang Z, Brown S, Kannappan V, Tawari PE, Jiang W, et al. Liposome encapsulated Disulfiram inhibits NF-κB pathway and targets breast cancer stem cells in vitro and in vivo. Oncotarget (2014) 5:7471–85. doi:10.18632/oncotarget.2166
131. Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFβ-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle (2010) 9:4461–8. doi:10.4161/cc.9.22.14048
132. Li L, Han R, Xiao H, Lin C, Wang Y, Liu H, et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res (2014) 20:2714–26. doi:10.1158/1078-0432.CCR-13-2613
133. Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes (2015) 64:2028–41. doi:10.2337/db14-1225
134. Lee BY, Hochgräfe F, Lin HM, Castillo L, Wu J, Raftery MJ, et al. Phosphoproteomic profiling identifies focal adhesion kinase as a mediator of docetaxel resistance in castrate-resistant prostate cancer. Mol Cancer Ther (2014) 13:190–201. doi:10.1158/1535-7163.MCT-13-0225-T
135. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer (2015) 15:7–24. doi:10.1038/nrc3860
136. Slomovitz BM, Coleman RL. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin Cancer Res (2012) 18:5856–64. doi:10.1158/1078-0432.CCR-12-0662
137. Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature (2006) 441:475–82. doi:10.1038/nature04703
138. He K, Xu T, Xu Y, Ring A, Kahn M, Goldkorn A. Cancer cells acquire a drug resistant, highly tumorigenic, cancer stem-like phenotype through modulation of the PI3K/Akt/β-catenin/CBP pathway. Int J Cancer (2014) 134:43–54. doi:10.1002/ijc.28341
139. Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A (2007) 104:16158–63. doi:10.1073/pnas.0702596104
140. Chen J, Shao R, Li F, Monteiro M, Liu JP, Xu ZP, et al. PI3K/Akt/mTOR pathway dual inhibitor BEZ235 suppresses the stemness of colon cancer stem cells. Clin Exp Pharmacol Physiol (2015) 42:1317–26. doi:10.1111/1440-1681.12493
141. Sarvi S, Mackinnon AC, Avlonitis N, Bradley M, Rintoul RC, Rassl DM, et al. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res (2014) 74:1554–65. doi:10.1158/0008-5472.CAN-13-1541
142. Luchman HA, Stechishin OD, Nguyen SA, Lun XQ, Cairncross JG, Weiss S. Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin Cancer Res (2014) 20:5756–67. doi:10.1158/1078-0432.CCR-13-3389
143. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol (2013) 10:143–53. doi:10.1038/nrclinonc.2013.10
144. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis (2013) 4:e875. doi:10.1038/cddis.2013.407
145. Samatar AA, Pulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov (2014) 13:928–42. doi:10.1038/nrd4281
146. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (2012) 380:358–65. doi:10.1016/S0140-6736(12)60868-X
147. Balko JM, Schwarz LJ, Bhola NE, Kurupi R, Owens P, Miller TW, et al. Activation of MAPK pathways due to DUSP4 loss promotes cancer stem cell-like phenotypes in basal-like breast cancer. Cancer Res (2013) 73:6346–58. doi:10.1158/0008-5472.CAN-13-1385
148. Hepburn AC, Veeratterapillay R, Williamson SC, El-Sherif A, Sahay N, Thomas HD, et al. Side population in human non-muscle invasive bladder cancer enriches for cancer stem cells that are maintained by MAPK signalling. PLoS One (2012) 7:e50690. doi:10.1371/journal.pone.0050690
149. Fischer M, Yen WC, Kapoun AM, Wang M, O’Young G, Lewicki J, et al. Anti-DLL4 inhibits growth and reduces tumor-initiating cell frequency in colorectal tumors with oncogenic KRAS mutations. Cancer Res (2011) 71:1520–5. doi:10.1158/0008-5472.CAN-10-2817
150. Sette G, Salvati V, Memeo L, Fecchi K, Colarossi C, Di Matteo P, et al. EGFR inhibition abrogates leiomyosarcoma cell chemoresistance through inactivation of survival pathways and impairment of CSC potential. PLoS One (2012) 7:e46891. doi:10.1371/journal.pone.0046891
151. Wang YK, Zhu YL, Qiu FM, Zhang T, Chen ZG, Zheng S, et al. Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis (2010) 31:1376–80. doi:10.1093/carcin/bgq120
152. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget (2011) 2:135–64. doi:10.18632/oncotarget.240
153. Zhong Z, Wen Z, Darnell JE Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin–6. Science (1994) 264:95–8.
154. Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, et al. Stat3–mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A (2001) 98:7319–24.
155. Yu CL, Meyer DJ, Campbell GS, Larner AC, Carter-Su C, Schwartz J, et al. Enhanced DNA-binding activity of a Stat3–related protein in cells transformed by the Src oncoprotein. Science (1995) 269:81–3.
156. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer (2014) 14:736–46. doi:10.1038/nrc3818
157. Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, et al. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res (2011) 71:7226–37. doi:10.1158/0008-5472.CAN-10-4660
158. van der Zee M, Sacchetti A, Cansoy M, Joosten R, Teeuwssen M, Heijmans-Antonissen C, et al. IL6/JAK1/STAT3 signaling blockade in endometrial cancer affects the ALDHhi/CD126+ stem-like component and reduces tumor burden. Cancer Res (2015) 75:3608–22. doi:10.1158/0008-5472.CAN-14-2498
159. Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, et al. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res (2014) 74:1227–37. doi:10.1158/0008-5472.CAN-13-0594
160. Shao C, Sullivan JP, Girard L, Augustyn A, Yenerall P, Rodriguez-Canales J, et al. Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway. Clin Cancer Res (2014) 20:4154–66. doi:10.1158/1078-0432.CCR-13-3292
161. Li L, Tang W, Wu X, Karnak D, Meng X, Thompson R, et al. HAb18G/CD147 promotes pSTAT3-mediated pancreatic cancer development via CD44s. Clin Cancer Res (2013) 19:6703–15. doi:10.1158/1078-0432.CCR-13-0621
162. Moon SH, Kim DK, Cha Y, Jeon I, Song J, Park KS. PI3K/Akt and Stat3 signaling regulated by PTEN control of the cancer stem cell population, proliferation and senescence in a glioblastoma cell line. Int J Oncol (2013) 42:921–8. doi:10.3892/ijo.2013.1765
163. Baell JB. Feeling nature’s PAINS: natural products, natural product drugs, and pan assay interference compounds (PAINS). J Nat Prod (2016) 79(3):616–28. doi:10.1021/acs.jnatprod.5b00947
164. Senger MR, Fraga CA, Dantas RF, Silva FP. Filtering promiscuous compounds in early drug discovery: is it a good idea? Drug Discov Today (2016). doi:10.1016/j.drudis.2016.02.004
165. Liu X, Chhipa RR, Pooya S, Wortman M, Yachyshin S, Chow LM, et al. Discrete mechanisms of mTOR and cell cycle regulation by AMPK agonists independent of AMPK. Proc Natl Acad Sci U S A (2014) 111:E435–44. doi:10.1073/pnas.1311121111
166. Schenk T, Chen WC, Göllner S, Howell L, Jin L, Hebestreit K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med (2012) 18:605–11. doi:10.1038/nm.2661
167. Zhang KZ, Zhang QB, Zhang QB, Sun HC, Ao JY, Chai ZT, et al. Arsenic trioxide induces differentiation of CD133+ hepatocellular carcinoma cells and prolongs posthepatectomy survival by targeting GLI1 expression in a mouse model. J Hematol Oncol (2014) 7:28. doi:10.1186/1756-8722-7-28
168. Sachlos E, Risueño EM, Laronde S, Shapovalova Z, Lee J-H, Russell J, et al. Identification of drugs including a dopamine receptor antagonist that selectively target cancer stem cells. Cell (2012) 149:1284–97. doi:10.1016/j.cell.2012.03.049
169. Ogawa K, Yoshioka Y, Isohashi F, Seo Y, Yoshida K, Yamazaki H. Radiotherapy targeting cancer stem cells: current views and future perspectives. Anticancer Res (2013) 33:747–54.
170. Gupta T, Nair V, Paul SN, Kannan S, Moiyadi A, Epari S, et al. Can irradiation of potential cancer stem-cell niche in the subventricular zone influence survival in patients with newly diagnosed glioblastoma? J Neurooncol (2012) 109:195–203. doi:10.1007/s11060-012-0887-3
171. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell (2009) 138:645–59. doi:10.1016/j.cell.2009.06.034
172. Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci U S A (2011) 108:13253–7. doi:10.1073/pnas.1110431108
173. Lu Y, Ma W, Mao J, Yu X, Hou Z, Fan S, et al. Salinomycin exerts anticancer effects on human breast carcinoma MCF-7 cancer stem cells via modulation of Hedgehog signaling. Chem Biol Interact (2015) 228:100–7. doi:10.1016/j.cbi.2014.12.002
174. Zhou J, Li P, Xue X, He S, Kuang Y, Zhao H, et al. Salinomycin induces apoptosis in cisplatin-resistant colorectal cancer cells by accumulation of reactive oxygen species. Toxicol Lett (2013) 222:139–45. doi:10.1016/j.toxlet.2013.07.022
175. An H, Kim JY, Oh E, Lee N, Cho Y, Seo JH. Salinomycin promotes anoikis and decreases the CD44+/CD24- stem-like population via inhibition of STAT3 activation in MDA-MB-231 Cells. PLoS One (2015) 10:e0141919. doi:10.1371/journal.pone.0141919
176. Naujokat C, Steinhart R. Salinomycin as a drug for targeting human cancer stem cells. J Biomed Biotechnol (2012) 2012:950658. doi:10.1155/2012/950658
177. Wright C, Moore R. Disulfiram treatment of alcoholism. Am J Med (1990) 88:647–55. doi:10.1016/0002-9343(90)90534-K
178. Dufour P, Lang JM, Giron C, Duclos B, Haehnel P, Jaeck D, et al. Sodium dithiocarb as adjuvant immunotherapy for high risk breast cancer: a randomized study. Biotherapy (1993) 6:9–12. doi:10.1007/BF01877380
179. Han D, Wu G, Chang C, Zhu F, Xiao Y, Li Q, et al. Disulfiram inhibits TGF-β-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-κB/Snail pathway. Oncotarget (2015) 6:40907–19. doi:10.18632/oncotarget.5723
180. Hothi P, Martins TJ, Chen L, Deleyrolle L, Yoon JG, Reynolds B, et al. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget (2012) 3:1124–36. doi:10.18632/oncotarget.707
181. Nechushtan H, Hamamreh Y, Nidal S, Gotfried M, Baron A, Shalev YI, et al. A phase IIb trial assessing the addition of disulfiram to chemotherapy for the treatment of metastatic non-small cell lung cancer. Oncologist (2015) 20:366–7. doi:10.1634/theoncologist.2014-0424
182. Firat E, Weyerbrock A, Gaedicke S, Grosu AL, Niedermann G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PLoS One (2012) 7:e47357. doi:10.1371/journal.pone.0047357
183. Balic A, Sørensen MD, Trabulo SM, Sainz B Jr, Cioffi M, Vieira CR, et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol Cancer Ther (2014) 13:1758–71. doi:10.1158/1535-7163.MCT-13-0948
184. Jiang H, Gomez-Manzano C, Lang FF, Alemany R, Fueyo J. Oncolytic adenovirus: preclinical and clinical studies in patients with human malignant gliomas. Curr Gene Ther (2009) 9:422–7. doi:10.2174/156652309789753356
185. Choi DS, Blanco E, Kim YS, Rodriguez AA, Zhao H, Huang TH, et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells (2014) 32:2309–23. doi:10.1002/stem.1746
186. Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene (2000) 19:2–12. doi:10.1038/sj.onc.1203251
187. Alonso MM, Gomez-Manzano C, Bekele BN, Yung WK, Fueyo J. Adenovirus-based strategies overcome temozolomide resistance by silencing the O6-methylguanine-DNA methyltransferase promoter. Cancer Res (2007) 67:11499–504. doi:10.1158/0008-5472.CAN-07-5312
188. Ning N, Pan Q, Zheng F, Teitz-Tennenbaum S, Egenti M, Yet J, et al. Cancer stem cell vaccination confers significant antitumor immunity. Cancer Res (2012) 72:1853–64. doi:10.1158/0008-5472.CAN-11-1400
189. Favaro R, Appolloni I, Pellegatta S, Sanga AB, Pagella P, Gambini E, et al. Sox2 is required to maintain cancer stem cells in a mouse model of high-grade oligodendroglioma. Cancer Res (2014) 74:1833–44. doi:10.1158/0008-5472.CAN-13-1942
190. Lanzardo S, Conti L, Rooke R, Ruiu R, Accart N, Bolli E, et al. Immunotargeting of antigen xCT attenuates stem-like cell behavior and metastatic progression in breast cancer. Cancer Res (2016) 76:62–72. doi:10.1158/0008-5472.CAN-15-1208
191. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tønnesen P, Suso EM, et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol Immunother (2013) 62:1499–509. doi:10.1007/s00262-013-1453-3
192. Xu M, Yao Y, Hua W, Wu Z, Zhong P, Mao Y, et al. Mouse glioma immunotherapy mediated by A2B5+ GL261 cell lysate-pulsed dendritic cells. J Neurooncol (2014) 116:497–504. doi:10.1007/s11060-013-1334-9
193. Rezvani K, Yong AS, Mielke S, Jafarpour B, Savani BN, Le RQ, et al. Repeated PR1 and WT1 peptide vaccination in Montanide-adjuvant fails to induce sustained high-avidity, epitope-specific CD8+ T cells in myeloid malignancies. Haematologica (2011) 96:432–40. doi:10.3324/haematol.2010.031674
194. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis (2010) 31:27–36. doi:10.1093/carcin/bgp220
195. Lu S, Labhasetwar V. Drug resistant breast cancer cell line displays cancer stem cell phenotype and responds sensitively to epigenetic drug SAHA. Drug Deliv Transl Res (2013) 3:183–94. doi:10.1007/s13346-012-0113-z
196. Nalls D, Tang S-N, Rodova M, Srivastava RK, Shankar S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One (2011) 6:e24099. doi:10.1371/journal.pone.0024099
197. Peitzsch C, Cojoc M, Hein L, Kurth I, Mäbert K, Trautmann F, et al. An epigenetic reprogramming strategy to re-sensitize radioresistant prostate cancer cells. Cancer Res (2016). doi:10.1158/0008-5472
198. Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, et al. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta (2012) 1826:272–96. doi:10.1016/j.bbcan.2012.04.008
199. Marcucci F, Corti A. How to improve exposure of tumor cells to drugs – promoter drugs increase tumor uptake and penetration of effector drugs. Adv Drug Deliv Rev (2012) 64:53–68. doi:10.1016/j.addr.2011.09.007
200. Marcucci F, Corti A. Improving drug penetration to curb tumor drug resistance. Drug Discov Today (2012) 17:1139–47. doi:10.1016/j.drudis.2012.06.004
201. Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev (2013) 65:1866–79. doi:10.1016/j.addr.2013.09.019
202. Marcucci F, Lefoulon F. Active targeting with particulate drug carriers in tumor therapy: fundamentals and recent progresses. Drug Discov Today (2004) 9:219–28. doi:10.1016/S1359-6446(03)02988-X
203. Marcucci F, Bellone M, Rumio C, Corti A. Approaches to improve tumor accumulation and interactions between monoclonal antibodies and immune cells. MAbs (2013) 5:34–46. doi:10.4161/mabs.22775
204. Zhang Y, Zhang H, Wang X, Wang J, Zhang X, Zhang Q. The eradication of breast cancer and cancer stem cells using octreotide modified paclitaxel active targeting micelles and salinomycin passive targeting micelles. Biomaterials (2012) 33:679–91. doi:10.1016/j.biomaterials.2011.09.072
205. Zhou J, Patel TR, Sirianni RW, Strohbehn G, Zheng MQ, Duong N, et al. Highly penetrative, drug-loaded nanocarriers improve treatment of glioblastoma. Proc Natl Acad Sci U S A (2013) 110:11751–6. doi:10.1073/pnas.1304504110
206. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science (2009) 324:1457–61. doi:10.1126/science.1171362
207. Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res (2008) 14:5995–6004. doi:10.1158/1078-0432.CCR-08-0291
208. Krawczyk N, Meier-Stiegen F, Banys M, Neubauer H, Ruckhaeberle E, Fehm T. Expression of stem cell and epithelial-mesenchymal transition markers in circulating tumor cells of breast cancer patients. Biomed Res Int (2014) 2014:415721. doi:10.1155/2014/415721
209. Korkaya H, Wicha MS. HER2 and breast cancer stem cells: more than meets the eye. Cancer Res (2013) 73:3489–93. doi:10.1158/0008-5472.CAN-13-0260
210. Lobry C, Oh P, Mansour MR, Look AT, Aifantis I. Notch signaling: switching an oncogene to a tumor suppressor. Blood (2014) 123:2451–9. doi:10.1182/blood-2013-08-355818
211. Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, Valavanis C, et al. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med (2014) 20:1199–207. doi:10.1038/nm.3678
212. Massagué J. TGF-β signalling in context. Nat Rev Mol Cell Biol (2012) 13:616–30. doi:10.1038/nrm3434
213. Chen EY, DeRan MT, Ignatius MS, Grandinetti KB, Clagg R, McCarthy KM, et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/β-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc Natl Acad Sci U S A (2014) 111:5349–54. doi:10.1073/pnas.1317731111
214. Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simões M, et al. STAT3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci U S A (2012) 109:E2361–70. doi:10.1073/pnas.1201232109
215. Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov (2015) 5:1282–9. doi:10.1158/2159-8290.CD-15-1020
216. He Y-C, Zhou F-L, Shen Y, Liao D-F, Cao D. Apoptotic death of cancer stem cells for cancer therapy. Int J Mol Sci (2014) 15:8335–51. doi:10.3390/ijms15058335
Keywords: cancer stem-like cells, epithelial–mesenchymal transition, therapy, biomarkers, classification, clinical development
Citation: Marcucci F, Rumio C and Lefoulon F (2016) Anti-Cancer Stem-like Cell Compounds in Clinical Development – An Overview and Critical Appraisal. Front. Oncol. 6:115. doi: 10.3389/fonc.2016.00115
Received: 03 March 2016; Accepted: 25 April 2016;
Published: 10 May 2016
Edited by:
Julie Gavard, University Nantes, FranceCopyright: © 2016 Marcucci, Rumio and Lefoulon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabrizio Marcucci, ZmFibWFyY3VAZ21haWwuY29t