 Katherine Karakasis
Katherine Karakasis Julia V. Burnier
Julia V. Burnier Amit M. Oza
Amit M. Oza Stephanie Lheureux
Stephanie Lheureux- Drug Development Program, Division of Medical Oncology and Hematology, Princess Margaret Cancer Centre, University of Toronto, Toronto, ON, Canada
Without prevention or screening options available, ovarian cancer is the most lethal malignancy of the female reproductive tract. High-grade serous ovarian cancer (HGSOC) is the most common histologic subtype, and the role of germline BRCA1/2 mutation in predisposition and prognosis is established. Given the targeted treatment opportunities with PARP inhibitors, a predictive role for BRCA1/2 mutation has emerged. Despite recommendations to provide BRCA1/2 testing to all women with histologically confirmed HGSOC, uniform implementation remains challenging. The opportunity to review and revise genetic screening and testing practices will identify opportunities, where universal adoption of BRCA1/2 mutation testing will impact and improve treatment of women with ovarian cancer. Improving education and awareness of genetic testing for women with cancer, as well as the broader general community, will help focus much-needed attention on opportunities to advance prevention and screening programs in ovarian cancer. This is imperative not only for women with cancer and those at risk of developing cancer but also for their first-degree relatives. In addition, BRCA1/2 testing may have direct implications for patients with other types of cancers, many of which are now being found to have BRCA1/2 involvement.
Introduction
Over the last four decades, there has been modest progress in the 5-year overall survival rates of women diagnosed with ovarian cancer, despite enhanced surgical efforts and introduction of doublet platinum/taxane chemotherapy. Worldwide, newly diagnosed cases of ovarian cancer have reached 239,000, positioning this malignancy as the seventh most common cancer in all women, with the highest incidence in Europe and North America (1). Typically diagnosed at an advanced stage (III/IV), high mortality rates for ovarian cancer continue to persist with almost 152,000 deaths per year (Figure 1) (2).
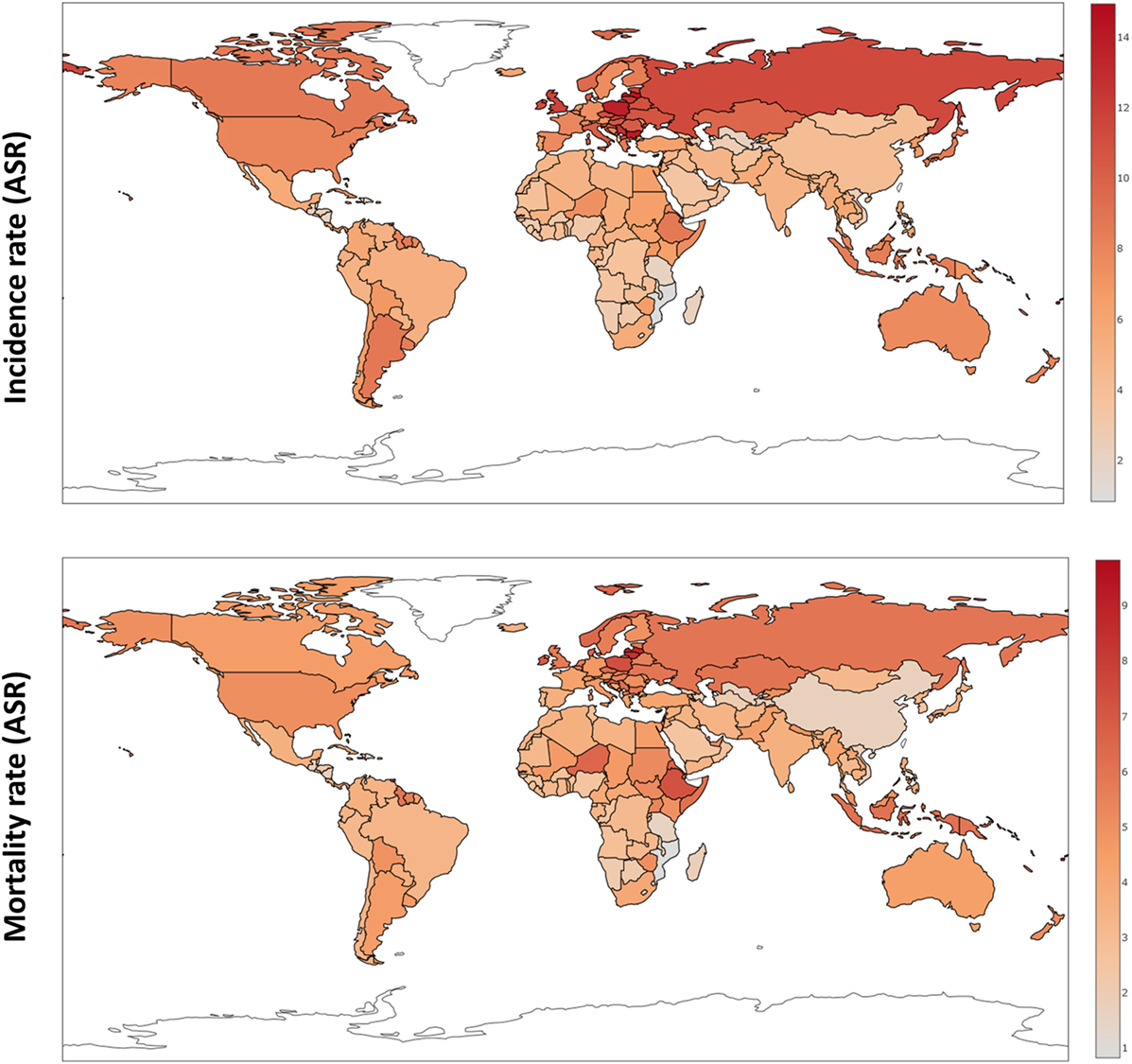
Figure 1. Ovarian cancer incidence and mortality rates worldwide (ASR) based on GloboCan data (1).
The lifetime risk of spontaneously developing and dying from ovarian cancer are 1.39 and 1.04%, respectively; however, the incidence of developing ovarian cancer significantly increases in carriers of germline mutations, mainly with either the breast cancer gene 1 (BRCA1) or 2 (BRCA2) genes. The lifetime risk of developing ovarian cancer is 40–60 and 11–27% for BRCA1 and BRCA2 mutation carriers, respectively (3). These particular mutations are implicated in 10–15% of all ovarian cancer cases and almost 20% of high-grade serous histology [high-grade serous ovarian cancer (HGSOC)] (4), including in women without a family history of breast or ovarian cancer. Approximately, one-third of patients with hereditary ovarian cancer have no close relatives with cancer (3). Family history-based testing for BRCA1/2 germline mutations has been shown to miss a significant proportion of women at risk for developing cancer (5), perhaps as a result of incomplete or incorrect family history reporting (6, 7) or potentially due to a lack of updating new family history information as it becomes available (8).
At present, a variety of selection criteria are used to determine the eligibility for BRCA1/2 testing, including family history, age at onset, tumor clinicopathological features, and computational risk prediction models (BRCAPRO, BOADICEA, Myriad, and Manchester scoring system) (9). The clinical criteria for risk assessment, genetic counseling, and genetic testing for BRCA-related cancers in women are based on personal and family history factors that may contribute to the disease (10) and are related to the likelihood of testing positive above a common testing threshold of 10% (11). These models often underestimate the probability of finding a mutation (12–14). It has been shown that the current family history approach does not identify 60% of Ashkenazi Jewish BRCA mutation carriers (15), thus creating a critical gap in practice that affects clinical treatment strategy and possibly patient outcome. As such, in light of advances in our understanding of BRCA-related breast and ovarian cancers – and the opportunity to directly impact therapeutic decision-making in these women – the recommendations to include universal germline BRCA1/2 testing to all women diagnosed with non-mucinous ovarian carcinoma (4) and women with triple-negative breast cancer (16) are growing in strength (17–19). Using next generation sequencing for 21 tumor suppressor genes of 360 subjects, ~24% carried germline loss-of-function mutations: 18% in BRCA1 or BRCA2 and 6% in BARD1, BRIP1, CHEK2, MRE11A, MSH6, NBN, PALB2, RAD50, RAD51C, or TP53 (20). The study also showed that 31% of women with an inherited mutation had no prior personal history of cancer or family history of breast or ovarian cancers (20). The National Comprehensive Cancer Network (NCCN) and Society of Gynecologic Oncology (SGO) guidelines suggest universal genetic counseling and testing of all women with ovarian cancer, including fallopian tube and peritoneal cancer (17, 19). Given the rate of BRCA1/2 mutation in HGSOC, germline BRCA1/2 testing is especially warranted in practice for this histology subtype. An immediate improvement to treatment opportunities would be to offer systematically genetic testing for BRCA1/2 mutation to all HGSOC, although it has been reported that 20% of women with ovarian cancer in community hospital settings were referred for genetic testing (21). While this was shown to be improved in academic centers, referral for germline BRCA1/2 testing was not systematic and did not reach the majority of patients (22). In clinical practice, there is a critical gap between the women eligible for BRCA1/2 counseling and those receiving testing (23, 24). With the recent approval of olaparib, a PARP inhibitor, it is likely that referral for genetic testing of BRCA1/2 status will improve.
Knowledge of BRCA1/2 Mutation Status Impacts Clinical Care of Women with Ovarian Cancer
Knowledge of BRCA1/2 status should be part of the standard of care at least for patients diagnosed with HGSOC. Indeed, there is a large body of evidence indicating benefits of targeting pathways involved in maintaining DNA integrity, including BRCA1 and BRCA2 signaling (25). Harboring a germline BRCA1/2 mutation is described as predictive of platinum sensitivity (26). Moreover, based on the synthetic lethality concept – the simultaneous promotion of DNA double-strand breaks (DSBs) and hindrance of DSB repair by inhibition of PARP protein expression (27, 28) – PARP inhibitors have been developed. This effect was shown clinically in the pivotal international, multicenter, randomized, phase II study that evaluated olaparib (a PARP inhibitor) as maintenance treatment in women with HGSOC who had responded to platinum-based chemotherapy (29). The preplanned retrospective analysis of outcomes by BRCA1/2 status in this study demonstrated that BRCA-mutated patients had better progression-free survival (PFS) with olaparib maintenance compared to those receiving placebo (11·2 versus 4·3 months; HR 0·18; p < 0·0001) (30). The PFS benefit was still observed when somatic BRCA-mutated patients were included in the analysis. Additional evidence supporting the role of olaparib as maintenance therapy was reported from an international, multicenter, randomized, open-label study in women with platinum-sensitive relapsed HGSOC (NCT01081951) (31). In this phase II, olaparib was given with carboplatin/paclitaxel chemotherapy and continued as maintenance monotherapy. Overall, study findings show a significant PFS improvement when compared to chemotherapy alone (12.2 and 9.6 median PFS, respectively; HR 0.51; 95% CI 0.34–0.77; p = 0.0012). A greater benefit was detected in patients with a BRCA1/2 mutation (PFS HR 0.21; 95% CI 0.08–0.55; p = 0.015) than in those without a BRCA1/2 mutation. Further, study analysis revealed strong evidence that olaparib maintenance is most likely a key contributor to the improvement in PFS in this patient population (31). There are numerous ongoing PARP inhibitor studies investigating women with BRCA1/2 mutations as well as mutations in other homologous recombination-deficient (HRD) genes, as data has shown HRD genes to exhibit BRCA-like behavior (32). To date, the use of olaparib maintenance has been approved in Europe after response to platinum-based chemotherapy in women with platinum-sensitive HGSOC who harbor a germline or somatic BRCA1/2 mutation (30) and in US, as single agent therapy after three lines of chemotherapy in patients with germline BRCA1/2 mutation HGSOC (33). Taken together, germline and somatic testing for BRCA1/2 provides important information for patients with ovarian cancer and this knowledge can directly impact clinical care.
Knowledge of Germline BRCA1/2 Mutation Status Impacts Ovarian Cancer Prevention
Germline BRCA1/2 status is not only relevant to women with ovarian cancer but also to women without cancer, who may be at an increased risk of developing the disease and could therefore benefit from prevention strategies. Currently, few prevention options are available for women with germline BRCA1/2 mutations. Women known to be at an increased genetic risk for developing OC, based on germline BRCA1/2 mutation carrier status, are offered risk-reducing salpingo–oophorectomy (RRSO), which reduces the risk of ovarian cancer by 71–96% (34–39). Surgery is usually performed after the completion of childbearing and while the woman is still pre-menopausal. Guidelines from the NCCN and the Society of Gynecologic Oncologists suggest that RRSO be completed by the age of 40 (19, 40); however, the majority of women who undergo RRSO do not do so by this age (41). This may be due to the potential side effects, such as premature surgical menopause (42), osteoporosis (43), cardiovascular disease (44, 45), cognitive impairments (46), symptoms of depression and anxiety (47), and consequences on quality of sleep, depression, and sexual dysfunction (48) associated with early RRSO. In light of these side effects – and the compelling evidence that high-grade serous epithelial ovarian cancer can be derived from the fallopian tube and not the ovary (49–53) – a recent committee opinion published by the American College of Obstetricians and Gynecologists outlines the opportunity for surgeon-led discussions with patients regarding the potential benefits of the removal of the fallopian tubes during hysterectomy in women at population risk of ovarian cancer who are not having an oophorectomy (54). Young BRCA1/2 mutation carriers can be counseled for risk-reducing bilateral salpingectomy initially, with subsequent bilateral oophorectomy after childbearing, although additional randomized controlled trials are warranted to support the validity of this approach. Further studies of associated hysterectomy are warranted in the population to provide appropriate family counseling guidance (55, 56). These discussions are important as data from nine countries have shown that preventative practices in women with germline BRCA1/2 mutations are varied despite guidelines (57). The study of 2677 women harboring germline BRCA1/2 mutations, who were an average of 3.9 years following genetic testing, showed that only 57.2% had undergone prophylactic surgery. This reveals differing uptake of preventative options by their country of residence (57). It also highlights the lack of effective alternate strategies for individuals identified to be at high risk, often for years before clinical development of disease or risk reduction procedures like surgery can be offered.
Germline BRCA1/2 Testing Strategy
The current germline BRCA1/2 testing strategy is mainly based on patients diagnosed with cancer. As previously discussed, as a minimum, all patients with HGSOC should be approached for BRCA1/2 testing as well as those patients diagnosed with non-mucinous ovarian cancer (Figure 2). Furthermore, knowledge of germline BRCA1/2 status in women living with ovarian cancer directly impacts first-degree relatives (FDRs), who have a 50% probability of carrying the same mutation and are yet to be diagnosed, and therefore, could also benefit from risk-reducing prevention strategies (58).
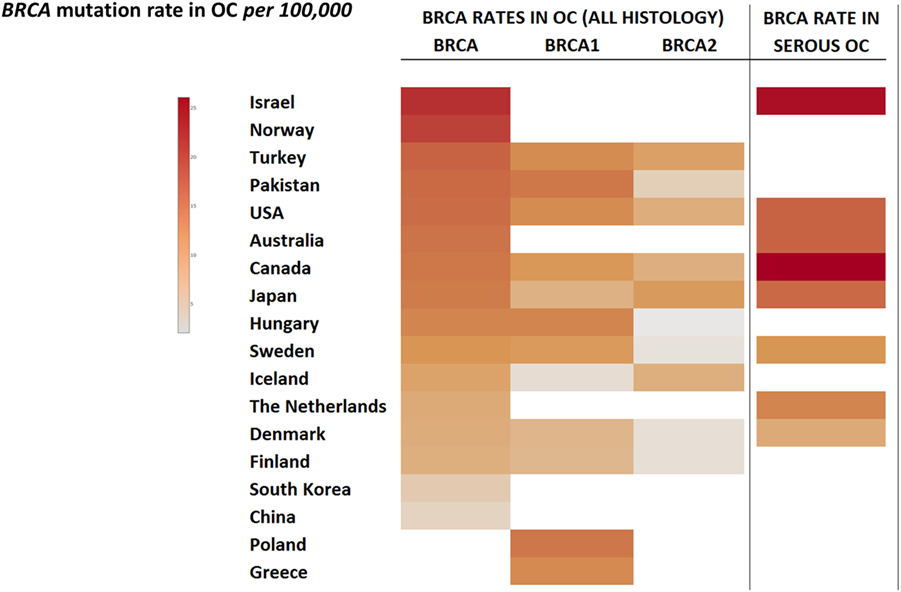
Figure 2. Germline BRCA1/2 mutational frequencies worldwide – examples.
While there has been much debate regarding the concept of population-based germline BRCA1/2 screening (59), this targeted approach within the Ashkenazi Jewish community has been shown to be more effective than family history-based testing and cost-effective. A Canadian-led study comparing the detection of BRCA1/2 mutation carriers through Jewish population-based genetic testing versus clinic-based genetic testing found that more unaffected women with a BRCA1 or 2 mutation were identified as a result of a genetic testing program targeting all Jewish women (60). This evidence supports the provision of genetic testing to all Jewish women (60). Conducted between 2008 and 2012, around 6179 Jewish women were tested through the population-based program, which identified 93 mutation carriers (92 unaffected with cancer) in comparison to 38 female carriers identified through 487 referrals to the genetics center (29 unaffected with cancer). Study findings showed that population genetic testing does not contribute to increased genetic counseling time but in fact decreases the overall time required when utilizing a population-based approach. Of particular importance, the 38% of women identified as having a BRCA1/2 mutation would have qualified for genetic testing but were either unaware of the recommendation or had not been referred by their health-care provider (60). Examining a similar approach, a randomized controlled trial of germline BRCA1/2 gene mutation testing in Ashkenazi Jewish women that compared family based testing to population screening, successfully enrolled and randomized 1034 participants (691 women, 343 men), of which 1017 were eligible for analysis. Similarly, findings showed that overall 56% of carriers did not fulfill clinical criteria for genetic testing, and germline BRCA1/2 prevalence was 2.45%. The fact that more than half of participants did not fulfill testing criteria is in agreement with previous data (61, 62), in which carriers lacked a strong family history of cancer. Moreover, the study also provided evidence that population-based genetic testing of Ashkenazi Jewish women does not adversely affect short-term psychological or quality of life outcomes (63). Cost-effective analyses conducted in parallel to the above study show that even when incorporating BRCA1/2 prevalence in family history negative individuals and genetic counseling costs, this specific population-based screening for germline BRCA1/2 mutations is highly cost-effective compared to family history-based approaches in Ashkenazi Jewish women aged 30 years and older (15). Screening based on founder mutations is feasible if the type of mutation is well known and allows for population-based screening approaches, such as in the Ashkenazi Jewish population, where two founder mutations in BRCA1 (185delAG and 5382insC) and one in BRCA2 (6174delT) account for 98–99% of identified mutations (64–67). This population-based screening approach is cost-effective, as previously described, given that 2.5% of this population carry one of these three mutations (64), and these mutations account for 40% of ovarian cancer (68, 69).
Worldwide, variation in the distribution of BRCA1 and BRCA2 mutations is well recognized, and in certain countries and ethnic communities the germline BRCA1/2 mutation spectrum is limited to a few founder mutations (70). However, both the number and frequency of germline BRCA1 and BRCA2 mutations vary among populations (Figures 3 and 4) (71–73). Findings from an international observational study of 19,581 BRCA1 and 11,900 BRCA2 carriers from 55 centers in 33 countries on 6 continents provide strong evidence that breast and ovarian cancer risks vary by type and location of BRCA1/2 mutation (73). As such, much research is moving toward characterizing the functional significance of specific mutations or mutation locations (74, 75).
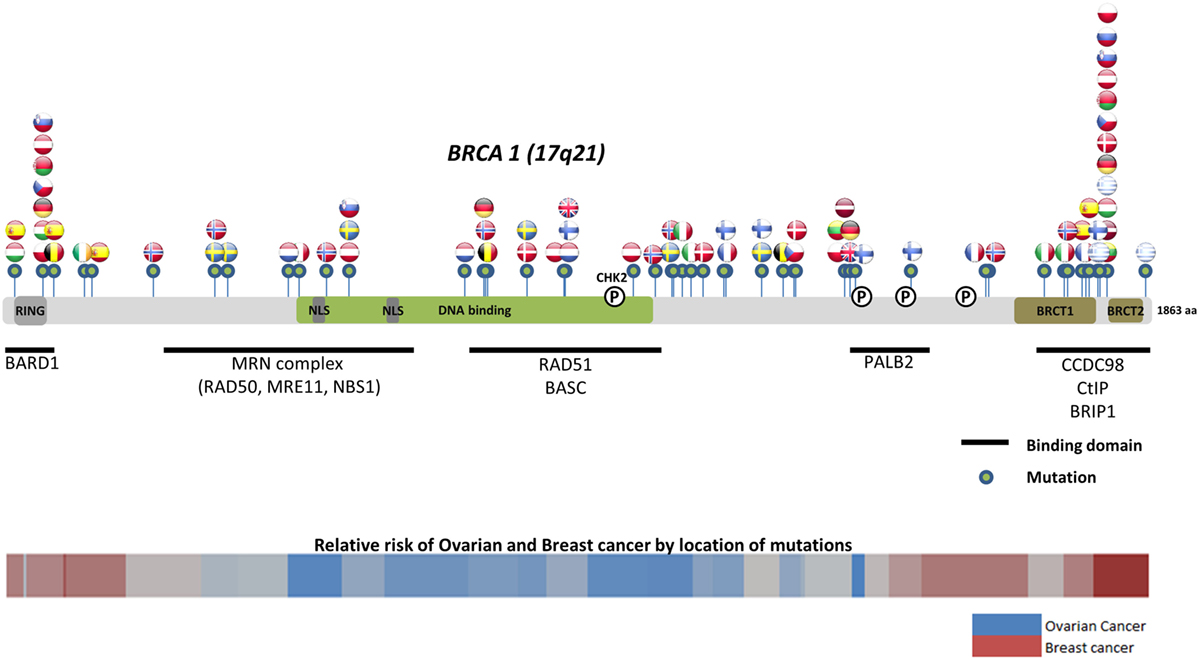
Figure 3. Location of common and founder BRCA1 mutations by country. Schematic of common and founder mutations by country modified from Janavičius (71) and Ramus and Gayther (72). Relative risk data taken from Rebbeck et al. (73).
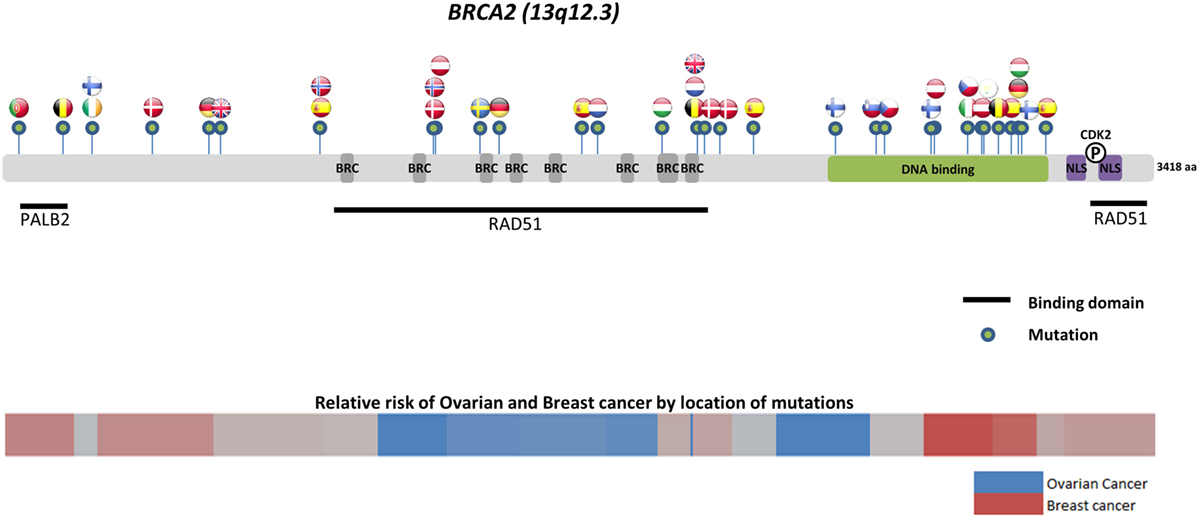
Figure 4. Location of common and founder BRCA2 mutations by country. Schematic of common and founder mutations by country modified from Janavičius (71) and Ramus and Gayther (72). Relative risk data taken from Rebbeck et al. (73).
Located on the long arm of chromosome 17, BRCA1 (MIM#113705) comprises 22 coding exons spanning 80 kb of genomic DNA and has a 7.8-kb transcript coding for an 1863-amino-acid protein (76). BRCA2 (MIM#600185) is located on chromosome 13 and comprises 26 coding exons spanning 70 kb of genomic DNA and gives an 11.4-kb transcript that encodes a protein of 3418 amino acids (77). Multifunctional in nature, BRCA proteins play important control functions in homologous recombination, the DNA DSB repair pathway, and early cellular response to DNA damage. BRCA1 also has a transcriptional activator or repressor function and possesses a central role in chromatin remodeling and centrosome regulation. BRCA1 and BRCA2 appear to behave as tumor suppressor genes, and mutations in either of these genes have been found throughout the entire coding region and at splice sites (78). In light of the structural and interactive complexity of BRCA1/2, international collaborations will not only continue to improve our understanding of BRCA1/2 mutations and how mutation type and location influence breast and ovarian cancer risks (Figures 3 and 4) (71–73) but also help devise novel, targeted testing panels that can potentially support specific population-based genetic testing, similar to the Ashkenazi Jewish population.
Translating Knowledge into Practice
To ensure successful uptake of germline BRCA1/2 testing or preventative strategies, wide community engagement and education regarding ovarian cancer are imperative. Following Angelina Jolie’s announcement that she carried a genetic mutation that increased her odds of developing breast and ovarian cancer, referrals for genetic counseling and BRCA1/2 testing appeared to have increased the awareness of cancer, particularly for breast cancer (79–81). While celebrities can bring heightened awareness to health issues, there is a need for these messages to be accompanied by more purposeful communication efforts to assist the public in understanding and using the complex diagnostic and treatment information that these stories convey (82).
In a small US study, data show that despite a significant proportion of primary care patients requiring genetic counseling, there is compelling evidence that few are actually receiving these services (23). Data from the same study also indicate that while overall perceived cancer risk was higher among women with familial cancer risk, 27% of women with familial breast/ovarian cancer felt their risk was “low” and 32% felt their risk was lower than average – highlighting the need for educational interventions for patients as well as providers (23). This highlights the importance of considering the potential psychological impacts that may be associated with BRCA1/2 testing over time. Employing qualitative interviews (N = 49) and reflective diaries, a study of 33 patients showed that the short-term impact of a positive BRCA1/2 test result differs prior to, immediately following, and up to 24 months after having received test results (83). Conducted from December 2006 to March 2010, data show that while women with cancer initially undergo genetic testing for their children, on confirmation of a positive test, the focus temporarily shifts to decision-making around their personal health needs. In fact, the threat of further disease caused anxiety around nurturing children and personal survival, which remained unresolved until women underwent risk-reducing surgery and in many continued as cancer worry (83). Here, findings help to illustrate where additional support for women during the testing process may be most beneficial. The long-term effects of a positive BRCA1/2 test result are also of relevance. A prospective single US centre study evaluating the long-term psychosocial effects of BRCA1/2 testing in a cohort of 464 women who had undergone genetic testing found that at long-term follow up (median 5 years; range 3.4–9.1 years), when assessing cancer-specific and genetic testing distress, perceived stress, and perceived cancer risk, there is modest increased distress in BRCA1/2 carriers compared to those women who received uninformative or negative test results (84). Despite the modest increase in distress, the group found no evidence of clinically significant dysfunction or impact of long-term psychological dysfunction due to testing (84). Data indicate that when patients receive counseling both before and after testing, they have more knowledge and experience less uncertainty and anxiety after learning the results of BRCA1/2 test. Although, patient experiences may vary with test results (85). Therefore, when taken together, it is imperative that appropriate multidisciplinary, supportive structures are in place that women eligible for testing can rely upon, leading up to and following a positive test result, including at the time of risk-reducing surgery and during surveillance.
Testing positive for a germline BRCA1/2 mutation goes beyond the patient herself potentially impacting her children and other members of their family by allowing cascade testing to proceed, if warranted (86). Accurate communication of test results is therefore critical for subsequent members to be tested. Research suggests fractured information dissemination among families when a positive germline BRCA1/2 test is communicated. In a systematic review of 29 publications from 26 studies, family communication regarding genetic risk is described as a deliberative process whereby the individual’s personal risk is determined, within the context of family dynamics, family vulnerability and receptivity is assessed, which mediates what information will be conveyed, and ultimately, the appropriate time to disclose information (87). Numerous studies provide complementary data illustrating that issues impacting the communication of test results within families includes an individual’s responsibility to inform, emotional and developmental readiness – such as when parents disclose BRCA1/2 results to children (88) – and again, communicating in the context of the existing family culture (89, 90). A retrospective study highlighted many errors in the transmission of DNA-test results in families from early stages of probands recalling information directly from genetic counselors, to the interpretation of information by family members (91). Therefore, support provided by genetic counselors could improve the overall process, not only during communication to family members but also during the education of physicians regarding family centered genetic testing for the physicians who may have referred the patient for testing (92).
BRCA1/2 Mutation Impacts More than Ovarian Cancer Treatment and Prevention
While the most described cancers driven by germline mutations in BRCA1/2 have been breast and ovarian, there is also mounting evidence to support the role of germline BRCA1/2 mutations contributing to other solid tumors, such as in prostate (93) and pancreatic (94, 95) cancers. In a United Kingdom study, Kote-Jarai et al. screened 1864 men with prostate cancer between 36 and 88 years of age and following analysis of the BRCA2 gene, findings show that all carriers of truncating mutations developed prostate cancer at ≤65 years (93). In this study, the prevalence of BRCA2 mutations was 1.27% (8/632) for cases diagnosed ≤55 years, 1.20% (19/1589) for cases diagnosed ≤65 years, and 0% (0/243) for cases diagnosed >65 years; p = 0.14 (81). It is estimated that germline mutations in the BRCA2 gene confer an ~8.6× increased risk of prostate cancer by 65 years of age, corresponding to an absolute risk of ~15% by age 65. A higher risk is perhaps conferred due to mutations in the BRCA2 ovarian cancer cluster region (OCCR) (96). Data suggest that routine testing of early onset prostate cancer cases for germline BRCA2 mutations would further help refine the prevalence of risk associated with BRCA2 mutations (93). A study examining other cancers in 268 BRCA1 and 222 BRCA2 families in the United Kingdom from 1975 to 2005 using person-years at risk analysis showed BRCA2 mutation increased risks for pancreatic cancers (RR 4.1, 95% CI 1.9–7.8) and uveal melanoma (RR 99.4 95% CI 11.1–359.8). Study data also showed possible novel associations with upper gastrointestinal malignancies and BRCA1 mutations, although this requires confirmation in future large prospective studies (96). Recently, a study provided evidence supporting current recommendations for hereditary breast and/or ovarian cancer screening of cancers other than breast and ovarian by the NCCN. In the study of 1072 patients who tested positive for a deleterious BRCA1/2 mutation, 1177 cancers comprising 30 different cancer types were detected (97). Findings show that while individuals harboring BRCA1 mutation did not have a significant increase in the development of cancers other than breast and ovarian, a trend in melanoma was observed. In addition, patients harboring a BRCA2 mutation had a significantly higher number of observed cases compared to expected cases for pancreatic cancer (SIR 21.7, 95% CI = 13.1–34.0; p < 0.001) in both men and women and prostate cancer in men (SIR 4.9, 95% CI = 2.0–10.1; p < 0.002) (97). Taken together, germline BRCA1/2 mutations bear significance in more than just breast and ovarian cancers. Future studies are warranted to provide evidence of access to BRCA1/2 testing and counseling for these cancers as well.
Conclusion
Worldwide, give the high incidence of ovarian cancer, the opportunity to identify BRCA1/2 carriers at the time of their cancer diagnosis – and those at risk for developing disease – can impact therapeutic interventions. Therefore, it also provides compelling evidence to improve and standardize BRCA1/2 testing practices. This becomes further punctuated when the opportunity to prevent or diagnose disease early in FDRs is also considered. In appropriate settings, population-based testing may be effective in identifying individuals at risk, who, with current criteria, would otherwise be missed. Future research should strive to build novel, targeted testing panels that will facilitate treatment/prevention-based decision-making. Therefore, it will be important to invest in resources and approaches that will change how ovarian cancer and other solid tumors with BRCA1/2 involvement are managed and prevented, to improve the current paradigm of care.
Author Contributions
KK wrote the draft manuscript and reviewed the article. JB did the figures and reviewed the article. VB and AO reviewed the manuscript. SL worked on the concept, the manuscript writing, and the review of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.1, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet]. Lyon, France: International Agency for Research on Cancer (2014). Available from: http://globocan.iarc.fr
2. Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, et al., editors. SEER Cancer Statistics Review, 1975-2012. Bethesda, MD: National Cancer Institute (2015). Available from: http://seer.cancer.gov/csr/1975_2012/
3. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol (2007) 25(11):1329–33. doi: 10.1200/JCO.2006.09.1066
4. Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol (2012) 30(21):2654–63. doi:10.1200/JCO.2011.39.8545 Erratum in: J Clin Oncol (2012) 30(33):4180
5. Møller P, Hagen AI, Apold J, Maehle L, Clark N, Fiane B, et al. Genetic epidemiology of BRCA mutations – family history detects less than 50% of the mutation carriers. Eur J Cancer (2007) 43(11):1713–7. doi:10.1016/j.ejca.2007.04.023
6. Murff HJ, Spigel DR, Syngal S. Does this patient have a family history of cancer? An evidence-based analysis of the accuracy of family cancer history. JAMA (2004) 292(12):1480–9. doi:10.1001/jama.292.12.1480
7. ASHG statement. Professional disclosure of familial genetic information. The American Society of Human Genetics Social Issues Subcommittee on Familial Disclosure. Am J Hum Genet (1998) 62(2):474–83.
8. Riley BD, Culver JO, Skrzynia C, Senter LA, Peters JA, Costalas JW, et al. Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns (2012) 21(2):151–61. doi:10.1007/s10897-011-9462-x
9. Lheureux S, Karakasis K, Harter P, Scott C, Bacon M, Bryce J, et al. Germline BRCA1/2 testing practices in ovarian cancer: current state and opportunities for new directions. Gynecol Oncol (2016) 140(1):90–4. doi:10.1016/j.ygyno.2015.10.010
10. Moyer VA; U.S. Preventive Services Task Force. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med (2014) 160(4):271–81. doi:10.7326/M13-2771
11. Daly MB, Axilbund JE, Buys S, Crawford B, Farrell CD, Friedman S, et al. Genetic/familial high-risk assessment: breast and ovarian. J Natl Compr Canc Netw (2010) 8(5):562–94.
12. Kwong A, Wong CH, Suen DT, Co M, Kurian AW, West DW, et al. Accuracy of BRCA1/2 mutation prediction models for different ethnicities and genders: experience in a southern Chinese cohort. World J Surg (2012) 36(4):702–13. doi:10.1007/s00268-011-1406-y
13. Varesco L, Viassolo V, Viel A, Gismondi V, Radice P, Montagna M, et al. Performance of BOADICEA and BRCAPRO genetic models and of empirical criteria based on cancer family history for predicting BRCA mutation carrier probabilities: a retrospective study in a sample of Italian cancer genetics clinics. Breast (2013) 22(6):1130–5. doi:10.1016/j.breast.2013.07.053
14. Kurian AW, Gong GD, John EM, Miron A, Felberg A, Phipps AI, et al. Performance of prediction models for BRCA mutation carriage in three racial/ethnic groups: findings from the Northern California Breast Cancer Family Registry. Cancer Epidemiol Biomarkers Prev (2009) 18(4):1084–91. doi:10.1158/1055-9965.EPI-08-1090
15. Manchanda R, Legood R, Burnell M, McGuire A, Raikou M, Loggenberg K, et al. Cost-effectiveness of population screening for BRCA mutations in Ashkenazi Jewish women compared with family history-based testing. J Natl Cancer Inst (2014) 107(1):380. doi:10.1093/jnci/dju380
16. Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol (2015) 33(4):304–11. doi:10.1200/JCO.2014.57.1414
17. Lancaster JM, Powell CB, Chen LM, Richardson DL; SGO Clinical Practice Committee. Society of Gynecologic Oncology statement on risk assessment for inherited gynecologic cancer predispositions. Gynecol Oncol (2015) 136(1):3–7. doi:10.1016/j.ygyno.2014.09.009 Erratum in: Gynecol Oncol (2015) 138(3):765
18. Walker JL, Powell CB, Chen LM, Carter J, Bae Jump VL, Parker LP, et al. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer (2015) 121(13):2108–20. doi:10.1002/cncr.29321
19. Daly MB, Pilarski R, Axilbund JE, Berry M, Buys SS, Crawford B, et al. Genetic/familial high-risk assessment: breast and ovarian, version 2.2015. J Natl Compr Canc Netw (2016) 14(2):153–62.
20. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A (2011) 108(44):18032–7. doi:10.1073/pnas.1115052108
21. Beattie MS, Copeland K, Fehniger J, Cheung E, Joseph G, Lee R, et al. Genetic counseling, cancer screening, breast cancer characteristics, and general health among a diverse population of BRCA genetic testers. J Health Care Poor Underserved (2013) 24(3):1150–66. doi:10.1353/hpu.2013.0151
22. Meyer LA, Anderson ME, Lacour RA, Suri A, Daniels MS, Urbauer DL, et al. Evaluating women with ovarian cancer for BRCA1 and BRCA2 mutations: missed opportunities. Obstet Gynecol (2010) 115(5):945–52. doi:10.1097/AOG.0b013e3181da08d7
23. Quillin JM, Krist AH, Gyure M, Corona R, Rodriguez V, Borzelleca J, et al. Patient-reported hereditary breast and ovarian cancer in a primary care practice. J Community Genet (2014) 5(2):179–83. doi:10.1007/s12687-013-0161-1
24. Wright JD, Chen L, Tergas AI, Accordino M, Ananth CV, Neugut AI, et al. Underuse of BRCA testing in patients with breast and ovarian cancer. Am J Obstet Gynecol (2016). doi:10.1016/j.ajog.2016.02.011
25. McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res (2006) 66(16):8109–15. doi:10.1158/0008-5472.CAN-06-0140
26. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res (2014) 20(3):764–75. doi:10.1158/1078-0432.CCR-13-2287
27. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol (2008) 26(22):3785–90. doi:10.1200/JCO.2008.16.0812
28. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) 434(7035):917–21. doi:10.1038/nature03445
29. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med (2012) 366(15):1382–92. doi:10.1056/NEJMoa1105535
30. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol (2014) 15(8):852–61. doi:10.1016/S1470-2045(14)70228-1 Erratum in: Lancet Oncol (2015) 16(4):e158
31. Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RH, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol (2015) 16(1):87–97. doi:10.1016/S1470-2045(14)71135-0 Erratum in: Lancet Oncol (2015) 16(1):e6. Lancet Oncol (2015) 16(2):e55
32. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474(7353):609–15. doi:10.1038/nature10166 Erratum in: Nature (2012) 490(7419):298
33. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol (2015) 33(3):244–50. doi:10.1200/JCO.2014.56.2728
34. Finch A, Beiner M, Lubinski J, Lynch HT, Moller P, Rosen B, et al. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 mutation. JAMA (2006) 296(2):185–92. doi:10.1001/jama.296.2.185
35. Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van’t Veer L, Garber JE, et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med (2002) 346(21):1616–22. doi:10.1056/NEJMoa012158
36. Kauff ND, Satagopan JM, Robson ME, Scheuer L, Hensley M, Hudis CA, et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med (2002) 346(21):1609–15. doi:10.1056/NEJMoa020119
37. Domchek SM, Friebel TM, Neuhausen SL, Wagner T, Evans G, Isaacs C, et al. Mortality after bilateral salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers: a prospective cohort study. Lancet Oncol (2006) 7(3):223–9. doi:10.1016/S1470-2045(06)70585-X
38. Kauff ND, Domchek SM, Friebel TM, Robson ME, Lee J, Garber JE, et al. Risk-reducing salpingo-oophorectomy for the prevention of BRCA1- and BRCA2-associated breast and gynecologic cancer: a multicenter, prospective study. J Clin Oncol (2008) 26(8):1331–7. doi:10.1200/JCO.2007.13.9626
39. Rutter JL, Wacholder S, Chetrit A, Lubin F, Menczer J, Ebbers S, et al. Gynecologic surgeries and risk of ovarian cancer in women with BRCA1 and BRCA2 Ashkenazi founder mutations: an Israeli population-based case-control study. J Natl Cancer Inst (2003) 95(14):1072–8. doi:10.1093/jnci/95.14.1072
40. American College of Obstetricians and Gynecologists, ACOG Committee on Practice Bulletins – Gynecology, ACOG Committee on Genetics, Society of Gynecologic Oncologists. ACOG Practice Bulletin No. 103: hereditary breast and ovarian cancer syndrome. Obstet Gynecol (2009) 113(4):957–66. doi:10.1097/AOG.0b013e3181a106d4
41. Garcia C, Wendt J, Lyon L, Jones J, Littell RD, Armstrong MA, et al. Risk management options elected by women after testing positive for a BRCA mutation. Gynecol Oncol (2014) 132(2):428–33. doi:10.1016/j.ygyno.2013.12.014
42. Stan DL, Shuster LT, Wick MJ, Swanson CL, Pruthi S, Bakkum-Gamez JN. Challenging and complex decisions in the management of the BRCA mutation carrier. J Womens Health (Larchmt) (2013) 22(10):825–34. doi:10.1089/jwh.2013.4407
43. Garcia C, Lyon L, Conell C, Littell RD, Powell CB. Osteoporosis risk and management in BRCA1 and BRCA2 carriers who undergo risk-reducing salpingo-oophorectomy. Gynecol Oncol (2015) 138(3):723–6. doi:10.1016/j.ygyno.2015.06.020
44. Parker WH, Jacoby V, Shoupe D, Rocca W. Effect of bilateral oophorectomy on women’s long-term health. Womens Health (Lond Engl) (2009) 5(5):565–76. doi:10.2217/whe.09.42
45. Rivera CM, Grossardt BR, Rhodes DJ, Brown RD Jr, Roger VL, Melton LJ III, et al. Increased cardiovascular mortality after early bilateral oophorectomy. Menopause (2009) 16(1):15–23. doi:10.1097/gme.0b013e31818888f7
46. Rocca WA, Bower JH, Maraganore DM, Ahlskog JE, Grossardt BR, de Andrade M, et al. Increased risk of cognitive impairment or dementia in women who underwent oophorectomy before menopause. Neurology (2007) 69(11):1074–83. doi:10.1212/01.wnl.0000276984.19542.e6
47. Rocca WA, Grossardt BR, Geda YE, Gostout BS, Bower JH, Maraganore DM, et al. Long-term risk of depressive and anxiety symptoms after early bilateral oophorectomy. Menopause (2008) 15(6):1050–9. doi:10.1097/gme.0b013e318174f155
48. Domchek SM, Li J, Digiovanni L, Voong C, Mueller R, Johnson L, et al. Quality of life in BRCA1 and BRCA2 mutation carriers (B1/2) following risk-reducing salpingo-oophorectomy (RRSO). J Clin Oncol (2014) 32:5s.
49. Kurman RJ, Shih IeM. The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol (2010) 34(3):433–43. doi:10.1097/PAS.0b013e3181cf3d79
50. Erickson BK, Conner MG, Landen CN Jr. The role of the fallopian tube in the origin of ovarian cancer. Am J Obstet Gynecol (2013) 209(5):409–14. doi:10.1016/j.ajog.2013.04.019
51. Crum CP. Intercepting pelvic cancer in the distal fallopian tube: theories and realities. Mol Oncol (2009) 3(2):165–70. doi:10.1016/j.molonc.2009.01.004
52. Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, et al. Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am J Surg Pathol (2007) 31(2):161–9. doi:10.1097/01.pas.0000213335.40358.47
53. Kamran MW, Vaughan D, Crosby D, Wahab NA, Saadeh FA, Gleeson N. Opportunistic and interventional salpingectomy in women at risk: a strategy for preventing pelvic serous cancer (PSC). Eur J Obstet Gynecol Reprod Biol (2013) 170(1):251–4. doi:10.1016/j.ejogrb.2013.06.030
54. Committee on Gynecologic Practice. Committee opinion no. 620: salpingectomy for ovarian cancer prevention. Obstet Gynecol (2015) 125(1):279–81. doi:10.1097/01.AOG.0000459871.88564.09
55. Pennington KP, Walsh T, Lee M, Pennil C, Novetsky AP, Agnew KJ, et al. BRCA1, TP53, and CHEK2 germline mutations in uterine serous carcinoma. Cancer (2013) 119(2):332–8. doi:10.1002/cncr.27720
56. Hiramatsu K, Yoshino K, Serada S, Yoshihara K, Hori Y, Fujimoto M, et al. Similar protein expression profiles of ovarian and endometrial high-grade serous carcinomas. Br J Cancer (2016) 114(5):554–61. doi:10.1038/bjc.2016.27
57. Metcalfe KA, Birenbaum-Carmeli D, Lubinski J, Gronwald J, Lynch H, Moller P, et al. International variation in rates of uptake of preventive options in BRCA1 and BRCA2 mutation carriers. Int J Cancer (2008) 122(9):2017–22. doi:10.1002/ijc.23340
58. Kwon JS, Daniels MS, Sun CC, Lu KH. Preventing future cancers by testing women with ovarian cancer for BRCA mutations. J Clin Oncol (2010) 28(4):675–82. doi:10.1200/JCO.2008.21.4684
59. King MC, Levy-Lahad E, Lahad A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA (2014) 312(11):1091–2. doi:10.1001/jama.2014.12483
60. Metcalfe KA, Poll A, Royer R, Nanda S, Llacuachaqui M, Sun P, et al. A comparison of the detection of BRCA mutation carriers through the provision of Jewish population-based genetic testing compared with clinic-based genetic testing. Br J Cancer (2013) 109(3):777–9. doi:10.1038/bjc.2013.309
61. Metcalfe KA, Poll A, Royer R, Llacuachaqui M, Tulman A, Sun P, et al. Screening for founder mutations in BRCA1 and BRCA2 in unselected Jewish women. J Clin Oncol (2010) 28(3):387–91. doi:10.1200/JCO.2009.25.0712
62. Levy-Lahad E, Gabai-Kapara E, Kaufman B, Catane R, Segev S, Renbaum P, et al. Idenfication of BRCA1/BRCA2 carriers by screening in the healthy population and its implications. J Clin Oncol (2011) 29(Suppl):abstr 1513.
63. Manchanda R, Loggenberg K, Sanderson S, Burnell M, Wardle J, Gessler S, et al. Population testing for cancer predisposing BRCA1/BRCA2 mutations in the Ashkenazi-Jewish community: a randomized controlled trial. J Natl Cancer Inst (2014) 107(1):379. doi:10.1093/jnci/dju379
64. Roa BB, Boyd AA, Volcik K, Richards CS. Ashkenazi Jewish population frequencies for common mutations in BRCA1 and BRCA2. Nat Genet (1996) 14(2):185–7. doi:10.1038/ng1096-185
65. Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, et al. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med (1997) 336(20):1401–8. doi:10.1056/NEJM199705153362001
66. Kauff ND, Perez-Segura P, Robson ME, Scheuer L, Siegel B, Schluger A, et al. Incidence of non-founder BRCA1 and BRCA2 mutations in high risk Ashkenazi breast and ovarian cancer families. J Med Genet (2002) 39(8):611–4. doi:10.1136/jmg.39.8.611
67. Phelan CM, Kwan E, Jack E, Li S, Morgan C, Aubé J, et al. A low frequency of non-founder BRCA1 mutations in Ashkenazi Jewish breast-ovarian cancer families. Hum Mutat (2002) 20(5):352–7. doi:10.1002/humu.10123
68. King MC, Marks JH, Mandell JB; New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science (2003) 302(5645):643–6. doi:10.1126/science.1088759
69. Hirsh-Yechezkel G, Chetrit A, Lubin F, Friedman E, Peretz T, Gershoni R, et al. Population attributes affecting the prevalence of BRCA mutation carriers in epithelial ovarian cancer cases in Israel. Gynecol Oncol (2003) 89(3):494–8. doi:10.1016/S0090-8258(03)00152-5
70. Shanmughapriya S, Nachiappan V, Natarajaseenivasan K. BRCA1 and BRCA2 mutations in the ovarian cancer population across race and ethnicity: special reference to Asia. Oncology (2013) 84(4):226–32. doi:10.1159/000346593
71. Janavičius R. Founder BRCA1/2 mutations in the Europe: implications for hereditary breast-ovarian cancer prevention and control. EPMA J (2010) 1(3):397–412. doi:10.1007/s13167-010-0037-y
72. Ramus SJ, Gayther SA. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol Oncol (2009) 3(2):138–50. doi:10.1016/j.molonc.2009.02.001
73. Rebbeck TR, Mitra N, Wan F, Sinilnikova OM, Healey S, McGuffog L, et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA (2015) 313(13):1347–61. doi:10.1001/jama.2014.5985 Erratum in: JAMA (2015) 314(6):628
74. Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, et al. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging (Albany NY) (2011) 3(5):515–32. doi:10.18632/aging.100325
75. Clark SL, Rodriguez AM, Snyder RR, Hankins GD, Boehning D. Structure-function of the tumor suppressor BRCA1. Comput Struct Biotechnol J (2012) 1(1):pii:e201204005. doi:10.5936/csbj.201204005
76. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (1994) 266(5182):66–71. doi:10.1126/science.7545954
77. Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature (1995) 378(6559):789–92. doi:10.1038/378789a0 Erratum in: Nature (1996) 379(6567):749
78. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell (2002) 108(2):171–82. doi:10.1016/S0092-8674(02)00615-3
79. Lebo PB, Quehenberger F, Kamolz LP, Lumenta DB. The Angelina effect revisited: exploring a media-related impact on public awareness. Cancer (2015) 121(22):3959–64. doi:10.1002/cncr.29461
80. Dean M. Celebrity health announcements and online health information seeking: an analysis of Angelina Jolie’s preventative health decision. Health Commun (2016) 31(6):752–61. doi:10.1080/10410236.2014.995866
81. Juthe RH, Zaharchuk A, Wang C. Celebrity disclosures and information seeking: the case of Angelina Jolie. Genet Med (2015) 17(7):545–53. doi:10.1038/gim.2014.141
82. Borzekowski DL, Guan Y, Smith KC, Erby LH, Roter DL. The Angelina effect: immediate reach, grasp, and impact of going public. Genet Med (2014) 16(7):516–21. doi:10.1038/gim.2013.181
83. Jeffers L, Morrison PJ, McCaughan E, Fitzsimons D. Maximising survival: the main concern of women with hereditary breast and ovarian cancer who undergo genetic testing for BRCA1/2. Eur J Oncol Nurs (2014) 18(4):411–8. doi:10.1016/j.ejon.2014.03.007
84. Graves KD, Vegella P, Poggi EA, Peshkin BN, Tong A, Isaacs C, et al. Long-term psychosocial outcomes of BRCA1/BRCA2 testing: differences across affected status and risk-reducing surgery choice. Cancer Epidemiol Biomarkers Prev (2012) 21(3):445–55. doi:10.1158/1055-9965.EPI-11-0991
85. Printz C. BRCA1/2-negative patients who receive counseling after genetic testing have lower anxiety. Cancer (2016) 122(8):1149. doi:10.1002/cncr.30002
86. Tercyak KP, Peshkin BN, Demarco TA, Patenaude AF, Schneider KA, Garber JE, et al. Information needs of mothers regarding communicating BRCA1/2 cancer genetic test results to their children. Genet Test (2007) 11(3):249–55. doi:10.1089/gte.2006.0534
87. Gaff CL, Clarke AJ, Atkinson P, Sivell S, Elwyn G, Iredale R, et al. Process and outcome in communication of genetic information within families: a systematic review. Eur J Hum Genet (2007) 15(10):999–1011. doi:10.1038/sj.ejhg.5201883 Erratum in: Eur J Hum Genet (2008) 16(3):402
88. Bradbury AR, Patrick-Miller L, Egleston BL, Olopade OI, Daly MB, Moore CW, et al. When parents disclose BRCA1/2 test results: their communication and perceptions of offspring response. Cancer (2012) 118(13):3417–25. doi:10.1002/cncr.26471
89. Dancyger C, Wiseman M, Jacobs C, Smith JA, Wallace M, Michie S. Communicating BRCA1/2 genetic test results within the family: a qualitative analysis. Psychol Health (2011) 26(8):1018–35. doi:10.1080/08870446.2010.525640
90. Patenaude AF, Dorval M, DiGianni LS, Schneider KA, Chittenden A, Garber JE. Sharing BRCA1/2 test results with first-degree relatives: factors predicting who women tell. J Clin Oncol (2006) 24(4):700–6. doi:10.1200/JCO.2005.01.7541
91. Vos J, Menko F, Jansen AM, van Asperen CJ, Stiggelbout AM, Tibben A. A whisper-game perspective on the family communication of DNA-test results: a retrospective study on the communication process of BRCA1/2-test results between proband and relatives. Fam Cancer (2011) 10(1):87–96. doi:10.1007/s10689-010-9385-y
92. Plon SE, Cooper HP, Parks B, Dhar SU, Kelly PA, Weinberg AD, et al. Genetic testing and cancer risk management recommendations by physicians for at-risk relatives. Genet Med (2011) 13(2):148–54. doi:10.1097/GIM.0b013e318207f564
93. Kote-Jarai Z, Leongamornlert D, Saunders E, Tymrakiewicz M, Castro E, Mahmud N, et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: implications for genetic testing in prostate cancer patients. Br J Cancer (2011) 105(8):1230–4. doi:10.1038/bjc.2011.383
94. Iqbal J, Ragone A, Lubinski J, Lynch HT, Moller P, Ghadirian P, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer (2012) 107(12):2005–9. doi:10.1038/bjc.2012.483
95. Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol (2015) 33(28):3124–9. doi:10.1200/JCO.2014.59.7401
96. Moran A, O’Hara C, Khan S, Shack L, Woodward E, Maher ER, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer (2012) 11(2):235–42. doi:10.1007/s10689-011-9506-2
Keywords: ovarian cancer, BRCA1/2, testing, treatment, prevention
Citation: Karakasis K, Burnier JV, Bowering V, Oza AM and Lheureux S (2016) Ovarian Cancer and BRCA1/2 Testing: Opportunities to Improve Clinical Care and Disease Prevention. Front. Oncol. 6:119. doi: 10.3389/fonc.2016.00119
Received: 26 February 2016; Accepted: 28 April 2016;
Published: 11 May 2016
Edited by:
Sarah M. Temkin, National Cancer Institute, USAReviewed by:
Tamara Louise Kalir, The Mount Sinai School of Medicine, USAPaola Gehrig, University of North Carolina, USA
Copyright: © 2016 Karakasis, Burnier, Bowering, Oza and Lheureux. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie Lheureux, c3RlcGhhbmllLmxoZXVyZXV4QHVobi5jYQ==