 Beth Walters
Beth Walters Sunnie R. Thompson
Sunnie R. Thompson- Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, USA
Translational regulation has been shown to play an important role in cancer and tumor progression. Despite this fact, the role of translational control in cancer is an understudied and under appreciated field, most likely due to the technological hurdles and paucity of methods available to establish that changes in protein levels are due to translational regulation. Tumors are subjected to many adverse stress conditions such as hypoxia or starvation. Under stress conditions, translation is globally downregulated through several different pathways in order to conserve energy and nutrients. Many of the proteins that are synthesized during stress in order to cope with the stress use a non-canonical or cap-independent mechanism of initiation. Tumor cells have utilized these alternative mechanisms of translation initiation to promote survival during tumor progression. This review will specifically discuss the role of cap-independent translation initiation, which relies on an internal ribosome entry site (IRES) to recruit the ribosomal subunits internally to the messenger RNA. We will provide an overview of the role of IRES-mediated translation in cancer by discussing the types of genes that use IRESs and the conditions under which these mechanisms of initiation are used. We will specifically focus on three well-studied examples: Apaf-1, p53, and c-Jun, where IRES-mediated translation has been demonstrated to play an important role in tumorigenesis or tumor progression.
Introduction
Gene expression is regulated at multiple levels: DNA, transcription, translation, messenger RNA (mRNA) turnover, mRNA or protein localization, and protein stability. Dysregulation of any one of these steps results in aberrant gene expression, which can be detrimental to the cell or organism. The sheer complexity of these processes and the energy that is invested in them demonstrates the importance of accurate regulation of gene expression. The major research focus in aberrant gene expression in cancer has been on alterations in DNA or transcription. However, the improved methods for proteomics and transcriptomics have revealed that there is a remarkably low correlation between mRNA transcript and protein levels (1), suggesting that protein expression is extensively regulated at the posttranscriptional or posttranslational level. This review focuses on translational control mechanisms during cellular stress. Global mechanisms of translational control in cancer were reviewed elsewhere (2).
The vast majority (>90%) of mRNAs are translated using a cap-dependent mechanism of initiation. Briefly, the eukaryotic initiation factor 4F (eIF4F) complex consisting of eIF4E, eIF4A, eIF4G recognizes and binds to the 5′ cap structure on the mRNA. The 40S subunit is recruited to the 5′ end of the mRNA as a 43S complex (40S, eIF5, eIF3, eIF1, eIF1A, eIF2, and Met-tRNAi). The 40S subunit scans down the mRNA in a 5′–3′ direction until a start codon is recognized, the 60S subunit joins, forming an 80S ribosome, and protein synthesis begins (3). Translation is a very energy-demanding process (4); therefore, under conditions of cellular stress, cap-dependent translation is globally downregulated by several mechanisms, depending on the type and extent of the stress. Protein synthesis can be inhibited globally by phosphorylating eIF2α, which delivers the initiator met-tRNAi for each round of initiation (5). There are four kinases that sense distinct cellular stresses and phosphorylate eIF2α. This globally downregulates protein synthesis while simultaneously enhances translation of certain mRNAs that encode for proteins involved in cell adaptation to cellular stress. Some IRESs are less sensitive to eIF2α phosphorylation during global inhibition of translation (6–8).
Mammalian target of rapamycin (mTOR) is a conserved protein kinase that regulates a multitude of cellular processes in response to growth factors or nutrients (9, 10). mTOR forms complexes with other proteins to form two distinct complexes, mTORC1 and mTORC2 (11). The mTORC1 complex is best understood and senses both intracellular and extracellular cues: growth factors, stress, oxygen, energy status, and amino acids. Signals that are pro-growth, such as growth factors, and nutrients stimulate mTOR, whereas stresses inhibit mTOR activity. Inhibition of mTOR results in de-repression of eIF4E-binding protein (4E-BP), which sequesters the cap-binding protein [eukaryotic initiation factor 4E (eIF4E)] that is required for cap-dependent initiation (12). 4E-BP inhibits the most predominant mechanism of translation initiation, cap-dependent initiation. Proteins synthesized from mRNAs under these conditions use a non-canonical, cap-independent mechanism of initiation from an IRES located in the 5′ untranslated region (5′UTR). Initiation of protein synthesis by an IRES involves internal recruitment of the 40S ribosome either upstream or directly at the start codon by an unknown mechanism that does not require a 5′ cap structure, eIF4E cap-binding protein, or a free 5′ end. IRES-containing mRNAs encode for proteins that are involved in cell growth, proliferation, apoptosis, and angiogenesis. There are several examples in the literature that demonstrate that IRES-mediated translation of certain mRNAs is required for tumor growth or vascularization (13–24) or resistance to apoptosis (20, 25, 26).
Viral IRESs were the first IRESs discovered, followed by the identification of cellular IRESs (27–29). IRESs are structurally and functionally diverse from one another and must be identified using a properly controlled functional assay, thus making identification of IRESs difficult. A commonly used functional assay has been the bicistronic reporter, whereby translation of an upstream open reading frame (ORF) is translated by a cap-dependent mechanism and translation of the downstream ORF relies on an IRES in the intergenic region for translation. The bicistronic reporter is generally transfected into cells as DNA, depending on the cellular machinery to transcribe it into RNA, which is transported to the cytoplasm for translation. However, if there are cryptic splice sites or promoters that are introduced into the intergenic region (either in the IRES or instead of the IRES), then this can result in a transcript that encodes for a chimeric protein or a monocistronic message, respectively. Either way translation of the downstream reporter would be cap-dependent. Therefore, it is important to show not only that the bicistronic reporter yields expression of the downstream ORF but also that translation of the downstream ORF is independent of the first ORF and not from a monocistronic mRNA. Another potential false positive is if both ORFs are in the same reading frame, then there can be readthrough of the upstream stop codon, resulting in a chimeric protein that is translated cap-dependently. While readthrough is easy to control for, the others are difficult since this requires proving a negative that monocistronic mRNAs or alternatively spliced mRNAs are not present even at low levels (30). Since most of these artifacts arise from transfecting DNA, the obvious solution is to transfect RNA to bypass cellular splicing and transcription; however, while this works fine for viral IRESs, many cellular IRESs may require a “nuclear experience,” to bind IRES-transacting factors that are important for stabilizing the IRES structure (31, 32). This may be due to the fact that unlike the highly structured viral IRESs, mRNAs that contain cellular IRESs may switch between cap-dependent and cap-independent translation, depending on the cellular conditions. Stable RNA structures would be detrimental for cap-dependent translation, which involves scanning of the 40S subunit from the 5′ end of the mRNA to the start codon. These challenges have led to the false reporting of some IRESs in the literature. This has resulted in a lot of controversies around cellular IRESs and which one have been correctly identified (33, 34). Nevertheless, there are compelling data demonstrating internal initiation of both cellular and viral IRESs (35–37), as well as their role in cancer development and gene expression during growth and development (16, 17, 24, 38–40).
In this review, we will describe the basic stress conditions that are known to upregulate IRES-mediated translation during tumorigenesis. In order to demonstrate the significance of the role that cellular IRESs have in cancer, we have compiled a table of validated, bona fide IRES-containing cellular mRNAs with important roles in tumorigenesis (Table 1). In addition, we provide a detailed review of three cellular mRNAs that contain IRESs and discuss how their translational regulation impacts oncogenesis, cancer progression, and survival of the tumor cells.
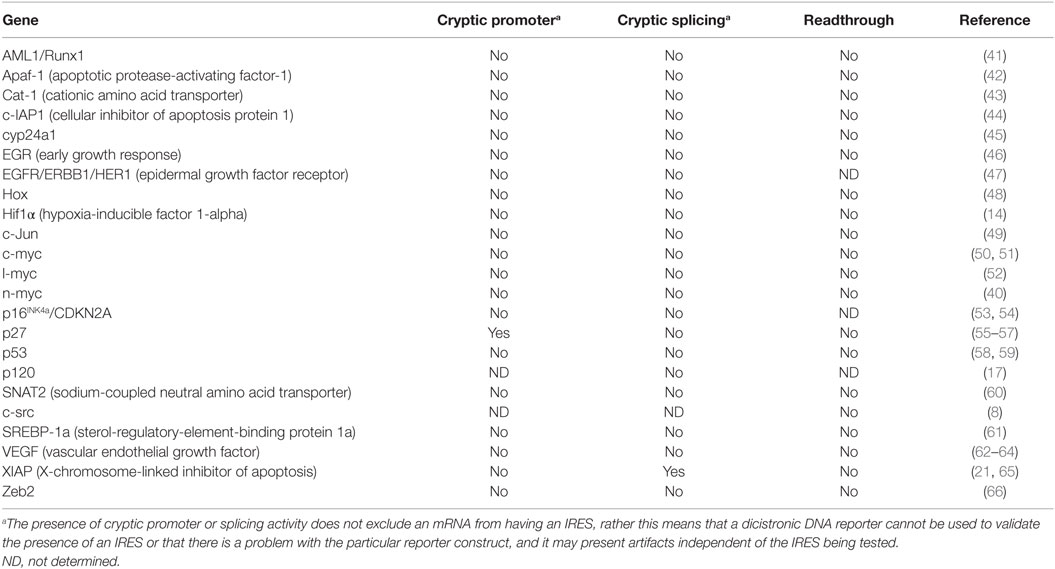
Table 1. IRES-containing cellular mRNAs with important roles in tumorigenesis.
IRES-Mediated Translation During Cellular Stress
mRNAs Encoding Proteins Involved in Tumorigenesis and Cell Survival Are IRES-Dependent
Translation is globally downregulated under a variety of cellular conditions, such as mitosis, heat shock, cold shock, hypoxia, DNA damage, osmotic shock, starvation, and apoptosis (67–70). Many of these conditions accompany tumor progression, metastasis, and cancer treatments. In order for the tumor cells to survive and for the tumor to progress, proteins must be synthesized to cope with these stresses. Many of the mRNAs that encode for these proteins contain an IRES (Table 1), suggesting that IRES-mediated translation must play an important role in tumor progression and survival. Direct evidence supporting this model comes from several studies (15–17, 71). IRES-mediated translation was shown to promote cell survival and the formation of tumor emboli in inflammatory breast cancer (16, 17). Another study showed that treatment of a 3D ovarian cell culture with a PI3K/mTOR inhibitor (to globally downregulate cap-dependent protein synthesis) resulted in apoptosis of the majority of the cancer cells. Importantly, a sub-population of the cells that were resistant to treatment overexpressed survival proteins that contained IRESs (15). These data demonstrate that IRES-mediated translation may be required for cancer progression and survival.
Additional support for the role of IRES-driven translation in tumorigenesis comes from studies of a genetic disorder called X-linked dyskeratosis (X-DC). Patients with mutations in the DKC1 gene, which causes X-DC, exhibit an increased susceptibility to cancer among other abnormalities (72). DKC1 encodes for dyskerin, a pseudouridine synthase that isomerizes uridines on rRNA to pseudouridines in a sequence-specific manner. Biochemical evidence demonstrated that ribosomes with decreased pseudouridylation displayed a reduced affinity for IRES-containing mRNAs (73). Importantly, when rRNA pseudouridylation is reduced, there is a specific decrease in translation of some IRES-containing mRNAs: p27, XIAP, and Bcl-xL (74), while another IRES-containing mRNA, vascular endothelial growth factor (VEGF), is translationally induced (75). Reduced levels of p27, a tumor suppressor, could at least, in part, explain why patients with mutations in DKC1 have an increased susceptibility to cancer (76). Altogether, these studies reveal a significant role for posttranscriptional control of gene expression during tumorigenesis that requires IRES-mediated initiation.
Cap-Independent Translation in Apoptosis
p53 IRES-Mediated Translation Is Required for p53 Induction of Cellular Senescence and Apoptosis
The p53 tumor suppressor protein is dysregulated in over half of all cancers. It is a transcription factor that controls the expression of protein coding genes as well as micro-RNAs (miRNAs) (77, 78). It plays a critical role in cellular responses to DNA damage and other stresses by inducing cell-cycle arrest and programed cell death (79). Failure to induce senescence or apoptosis following DNA damage results in genetic instability or inappropriate survival of damaged cells. Thus, inherited or spontaneous mutations in p53 contribute to tumorigenesis.
Since p53 plays a vital role in controlling cellular functions, its activity is highly regulated. Optimal induction of growth arrest or apoptosis following DNA damage requires an increase in the intracellular p53 protein levels. Under normal growth conditions, the cell maintains a low level of p53 protein due to proteasomal targeting of p53 protein by the E3 ubiquitin ligase mouse double minute 2 (Mdm2) (80–82) (Figure 1, left). In addition, when Mdm2 and/or Mdm4 are bound to p53, they mask the transactivation domain. Following DNA damage or cell stress, the level of p53 protein in the cell increases, while mRNA levels remain constant (83). The increase in p53 protein level is controlled by two distinct mechanisms: stabilization of the p53 protein caused by the loss of Mdm2 recruitment and an increase in translation of the p53 mRNA (20, 84–86) (Figure 1, right). The TP53 gene can express 12–13 different isoforms of the p53 protein with the major transcriptional start site generating a transcript with a 147 nucleotides long 5′UTR that contains an IRES (58, 87). IRES-mediated translation of the p53 mRNA generates ΔNp53 (also known as p54/47, p47, and Δ40p53) (25, 58, 88). Interestingly, IRES-mediated translation of p53 was shown to be important for increasing p53 protein levels following DNA damage in order to induce senescence (25, 26, 71).
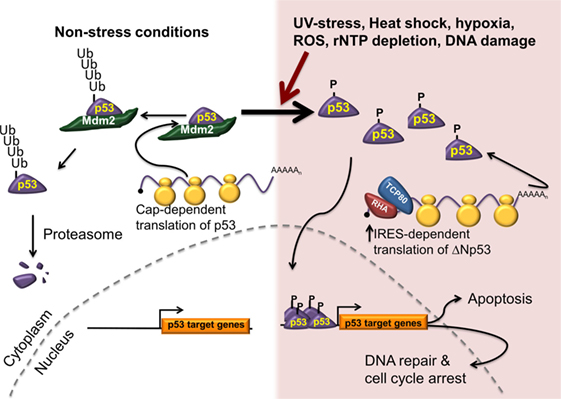
Figure 1. IRES-mediated translation is required for cell survival and tumor progression. Under normal conditions (left), p53 protein is synthesized predominately using a cap-dependent mechanism of initiation. The newly synthesized p53 protein is recognized by an E3 ubiquitin ligase (Mdm2), ubiquitinated, and degraded by the proteasome. In response to multiple stresses, p53 is phosphorylated, thus stabilizing it by preventing its interaction with Mdm2. In addition, IRES-mediated translation of the p53 mRNA is upregulated generating ΔNp53 and resulting in increased p53 levels. The phosphorylated p53 translocates to the nucleus to promote transcription of a number of genes involved in cell cycle arrest, DNA repair, and apoptosis.
The p53 IRES binds two IRES transacting factors, translational control protein 80 (TCP80), and RNA helicase (RHA) (25, 86), which were shown to modulate p53 IRES activity. If these proteins were either knocked down or overexpressed in cells, p53 proteins levels decreased or increased, respectively (25). This suggests that defective p53 regulation may be observed in cells with wild-type p53, but with dysregulated TCP80 or RHA. Thus, it is conceivable that many cancer cells that have wild-type p53 may still be unable to induce p53 due to defective p53 IRES-mediated translation (20, 25, 26).
Apoptotic Protease-Activating Factor-1 Regulation Requires IRES-Mediated Translation during Apoptosis
Apaf-1 plays a central role in initiating the intrinsic or mitochondrial apoptotic pathway following cellular stresses, such as DNA damage (89–91). Under normal conditions, Apaf-1 is present in the cytoplasm as a monomer (92, 93). However, early apoptosis signals induce Apaf-1 oligomerization, which commits the cell to apoptosis. Briefly, exposure to proapoptotic stimuli triggers release of cytochrome c from the outer mitochondrial membrane into the cytoplasm. Binding of cytochrome c to dATP stimulates Apaf-1 oligomerization to form an apoptosome, which recruits and activates caspase 9 (91), which in turn induces the caspase cascade and commits the cell to apoptosis (91). Interestingly, Apaf-1 is expressed in all tissues except for muscle, which could be to avoid inappropriate activation of apoptosis when the mitochondria swell or degenerate following strenuous exercise (42, 94). Apaf-1 knockout mice are embryonic lethal due to reduced apoptosis resulting in an accumulation of neurons in the central nervous system causing many morphological abnormalities (95, 96). Overexpression of Apaf-1 increases the sensitivity of cells to proapoptotic stimuli (97). Cell lines that have Apaf-1 knocked out exhibit an increased propensity toward oncogenic transformation when c-Myc is overexpressed in cells (98). Significantly, a reduction in Apaf-1 expression is a negative prognostic marker for a variety of cancers including melanoma, cervical carcinoma, colon cancer, and acute myeloid leukemia (99, 100).
The Apaf-1 5′UTR is long, G-C rich, contains upstream start codons, has 56% homology between human and mouse, and was inhibitory to 40S scanning (42). This suggests that Apaf-1 is translationally regulated through a conserved mechanism. Indeed, the 5′UTR of Apaf-1 contains an IRES, and any possibility of cryptic promoter, cryptic splicing, or readthrough was ruled out (42). Furthermore, the Apaf-1 IRES was shown to be dependent on the ribosomal protein S25 (RPS25/eS25), which has been shown to be required for IRES-mediated translation, but has no affect on cap-dependent initiation (59, 101). Additionally, the Apaf-1 IRES is active if it is transfected into the cell as an RNA (102), which is highly significant since many cellular IRESs are only functional when they are transfected as a DNA. RNA transfection bypasses cellular splicing and transcription machinery, which are the major sources of false positives in the bicistronic reporter assay. Lastly, in response to ultraviolet C irradiation, an increase in Apaf-1 IRES activity correlates with an increase in Apaf-1 protein levels without a corresponding increase in mRNA levels (89), indicating Apaf-1 expression is translationally regulated. Furthermore, Apaf-1 protein levels are associated with increased sensitivity to ultraviolet-induced apoptosis (95, 103, 104).
Since proapoptotic stimuli triggers downregulation of cap-dependent translation (105, 106), IRES-mediated initiation of Apaf-1 ensures that Apaf-1 protein levels are maintained during apoptosis, which is necessary to ensure propagation of the caspase cascade (89, 107). Moreover, while many mRNAs that have cellular IRESs can be translated by both cap-dependent and cap-independent mechanisms, the 5′UTR of Apaf-1 is inhibitory for the scanning mechanism of initiation (42, 102). Therefore, the Apaf-1 mRNA represents an exquisite example of gene expression control in which mRNA is translationally repressed when cap-dependent translation predominates and is translationally upregulated upon stresses that downregulate cap-dependent translation such as apoptosis (108, 109). A possible mechanism for such regulation of Apaf-1 during apoptosis comes from studies of death-associated protein 5 (DAP5) protein. DAP5 is an eIF4G family member that gets activated, while other eIF4G proteins are inactivated during apoptosis by caspase cleavage (109, 110). Upon cleavage, DAP5 can stimulate IRES-mediated translation of Apaf-1 but not cap-dependent translation because it lacks the domain that binds eIF4E (the cap-binding protein).
Many cellular IRESs are cell type specific and Apaf-1 is no exception (111, 112). The Apaf-1 IRES activity is highest in neuronal cells (111). This cell type specificity is consistent with the fact that Apaf-1 protein is highly expressed in neurons, and the knockout mice exhibit defects in brain formation (95, 96). In fact, it was reported that the Apaf-1 IRES is significantly more active in neuronal cell lines compared to HeLa or HEK293T cells due to a stronger binding preference for the neuronal pyrimidine tract binding (nPTB) over the PTB1 that is expressed in non-neuronal cell types (111).
IRES-Mediated Translation in Tumor Progression
Cap-Independent Translation of c-Jun Is Required for Tumor Progression
The oncoprotein, c-Jun, is a component of the activator protein 1 (AP-1) transcription factor, which is involved in regulating proliferation, differentiation, growth, apoptosis, cell-migration, and transformation (113). AP-1 is regulated at many levels, which include dimer composition, transcriptional, translational, and posttranslational regulation (49, 114, 115). The c-Jun protein is known to stimulate transcription of components of the cell cycle, repress transcription of tumor suppressor genes such as TP53, and induce expression of metalloproteinases, which are proteolytic enzymes that promote growth, invasion, and metastasis of cancer cells (116). Surprisingly, c-Jun mRNA expression levels are only elevated in a few cancers (115, 117, 118). However, high c-Jun protein levels have been observed in glioblastoma, malignant melanoma, invasive breast cancer, and colorectal cancers without corresponding increases in mRNA levels or changes in protein stability. Likewise, the c-Jun protein levels are low in normal cells, but high in glioblastoma and melanoma cell lines (49, 115). Together, these data support a model whereby c-Jun expression is translationally controlled (49, 114, 115, 119).
The 5′UTR of c-Jun is 974 nucleotides long, conserved across species, and contains an IRES (49). Importantly, IRES-mediated translation of c-Jun mRNA resulted in accumulation of c-Jun protein without an increase in mRNA levels or changes in protein stability (120). IRES-mediated translation of c-Jun can be induced by loss of cell–cell contacts, such as when there is a loss of E-cadherin, which causes a disruption or restructuring of the cytoskeletal network (114, 120, 121). Disruption of the cytoskeletal network activates a signaling pathway that upregulates IRES-mediated translation of c-Jun and induces an invasive program (122, 123). Thus, IRES-mediated translation of c-Jun likely plays an important role in tumor progression, following the loss of adhesion molecules and/or restructuring of the cytoskeleton.
Conclusion
Cap-dependent translation is the primary mechanism of initiating translation for the majority of mRNAs; however, cancer cells must adapt to many stresses that downregulate cap-dependent translation, including, but not limited to, hypoxia, nutrient deprivation, and DNA damage. In order for the cancer to progress, it must rely on other translational mechanisms, such as IRES-mediated translation, to support survival, growth, angiogenesis, and metastasis. Many genes that have been shown to be important in tumorigenesis have also had IRESs identified in their 5′UTR (Table 1). Thus, there is a need to better understand how IRES-mediated translation contributes to proteome changes in both healthy and tumor cells. This information will be critical for prognosis and development of more effective anticancer therapeutics.
Author Contributions
SRT and BW contributed to the development, writing, and editing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Robert Curtis Hendrickson and Olga Viktorovskaya for comments, and our support from the DoD Congressionally Directed Medical Research Programs Prostate Cancer Research Program (25W81XWH-15-1-0483) and the UAB Breast Spore (P50CA089019).
References
1. Ghazalpour A, Bennett B, Petyuk VA, Orozco L, Hagopian R, Mungrue IN, et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet (2011) 7(6):e1001393. doi:10.1371/journal.pgen.1001393
2. Leprivier G, Rotblat B, Khan D, Jan E, Sorensen PH. Stress-mediated translational control in cancer cells. Biochim Biophys Acta (2015) 1849(7):845–60. doi:10.1016/j.bbagrm.2014.11.002
3. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol (2010) 11(2):113–27. doi:10.1038/nrm2838
4. Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem J (1995) 312(Pt 1):163–7. doi:10.1042/bj3120163
5. Hinnebusch AG. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem (2014) 83:779–812. doi:10.1146/annurev-biochem-060713-035802
6. Kim JH, Park SM, Park JH, Keum SJ, Jang SK. eIF2A mediates translation of hepatitis C viral mRNA under stress conditions. EMBO J (2011) 30(12):2454–64. doi:10.1038/emboj.2011.146
7. Thompson SR, Gulyas KD, Sarnow P. Internal initiation in Saccharomyces cerevisiae mediated by an initiator tRNA/eIF2-independent internal ribosome entry site element. Proc Natl Acad Sci U S A (2001) 98(23):12972–7. doi:10.1073/pnas.241286698
8. Allam H, Ali N. Initiation factor eIF2-independent mode of c-Src mRNA translation occurs via an internal ribosome entry site. J Biol Chem (2010) 285(8):5713–25. doi:10.1074/jbc.M109.029462
9. Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci (2013) 38(5):233–42. doi:10.1016/j.tibs.2013.01.004
10. Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle (2011) 10(14):2305–16. doi:10.4161/cc.10.14.16586
11. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell (2012) 149(2):274–93. doi:10.1016/j.cell.2012.03.017
12. Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev (1998) 12(4):502–13. doi:10.1101/gad.12.4.502
13. Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature (1992) 359(6398):845–8. doi:10.1038/359845a0
14. Lang KJ, Kappel A, Goodall GJ. Hypoxia-inducible factor-1alpha mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol Biol Cell (2002) 13(5):1792–801. doi:10.1091/mbc.02-02-0017
15. Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, et al. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell (2012) 21(2):227–39. doi:10.1016/j.ccr.2011.12.024
16. Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H, et al. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell (2007) 28(3):501–12. doi:10.1016/j.molcel.2007.10.019
17. Silvera D, Arju R, Darvishian F, Levine PH, Zolfaghari L, Goldberg J, et al. Essential role for eIF4GI overexpression in the pathogenesis of inflammatory breast cancer. Nat Cell Biol (2009) 11(7):903–8. doi:10.1038/ncb1900
18. Silvera D, Schneider RJ. Inflammatory breast cancer cells are constitutively adapted to hypoxia. Cell Cycle (2009) 8(19):3091–6. doi:10.4161/cc.8.19.9637
19. Le Quesne JP, Spriggs KA, Bushell M, Willis AE. Dysregulation of protein synthesis and disease. J Pathol (2010) 220(2):140–51. doi:10.1002/path.2627
20. Halaby MJ, Yang DQ. p53 translational control: a new facet of p53 regulation and its implication for tumorigenesis and cancer therapeutics. Gene (2007) 395(1–2):1–7. doi:10.1016/j.gene.2007.01.029
21. Holcik M, Lefebvre C, Yeh C, Chow T, Korneluk RG. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat Cell Biol (1999) 1(3):190–2. doi:10.1038/11109
22. Holcik M. Targeting translation for treatment of cancer – a novel role for IRES? Curr Cancer Drug Targets (2004) 4(3):299–311. doi:10.2174/1568009043333005
23. Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol (2005) 6(4):318–27. doi:10.1038/nrm1618
24. Holcik M, Yeh C, Korneluk RG, Chow T. Translational upregulation of X-linked inhibitor of apoptosis (XIAP) increases resistance to radiation induced cell death. Oncogene (2000) 19(36):4174–7. doi:10.1038/sj.onc.1203765
25. Halaby MJ, Harris BR, Miskimins WK, Cleary MP, Yang DQ. Deregulation of IRES-mediated p53 translation in cancer cells with defective p53 response to DNA damage. Mol Cell Biol (2015) 35(23):4006–17. doi:10.1128/MCB.00365-15
26. Bellodi C, Kopmar N, Ruggero D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J (2010) 29(11):1865–76. doi:10.1038/emboj.2010.83
27. Jang SK, Krausslich HG, Nicklin MJ, Duke GM, Palmenberg AC, Wimmer E. A segment of the 5’ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J Virol (1988) 62(8):2636–43.
28. Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature (1988) 334(6180):320–5. doi:10.1038/334320a0
29. Yang Q, Sarnow P. Location of the internal ribosome entry site in the 5’ non-coding region of the immunoglobulin heavy-chain binding protein (BiP) mRNA: evidence for specific RNA-protein interactions. Nucleic Acids Res (1997) 25(14):2800–7. doi:10.1093/nar/25.14.2800
30. Thompson SR. So you want to know if your message has an IRES? Wiley Interdiscip Rev RNA (2012) 3(5):697–705. doi:10.1002/wrna.1129
31. Stoneley M, Subkhankulova T, Le Quesne JP, Coldwell MJ, Jopling CL, Belsham GJ, et al. Analysis of the c-myc IRES; a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res (2000) 28(3):687–94. doi:10.1093/nar/28.3.687
32. Semler BL, Waterman ML. IRES-mediated pathways to polysomes: nuclear versus cytoplasmic routes. Trends Microbiol (2008) 16(1):1–5. doi:10.1016/j.tim.2007.11.001
33. Jackson RJ. The current status of vertebrate cellular mRNA IRESs. Cold Spring Harb Perspect Biol (2013) 5(2): 1–20. doi:10.1101/cshperspect.a011569
34. Andreev DE, Dmitriev SE, Terenin IM, Shatsky IN. Cap-independent translation initiation of apaf-1 mRNA based on a scanning mechanism is determined by some features of the secondary structure of its 5’ untranslated region. Biochemistry (Mosc) (2013) 78(2):157–65. doi:10.1134/S0006297913020041
35. Schneider R, Agol VI, Andino R, Bayard F, Cavener DR, Chappell SA, et al. New ways of initiating translation in eukaryotes. Mol Cell Biol (2001) 21(23):8238–46. doi:10.1128/MCB.21.23.8238-8246.2001
36. Schneider R, Kozak M. New ways of initiating translation in eukaryotes? Mol Cell Biol (2001) 21(23):8238–46. doi:10.1128/mcb.21.23.8238-8246.2001
37. Kozak M. New ways of initiating translation in eukaryotes? Mol Cell Biol (2001) 21(6):1899–907. doi:10.1128/MCB.21.6.1899-1907.2001
38. Kondrashov N, Pusic A, Stumpf CR, Shimizu K, Hsieh AC, Xue S, et al. Ribosome-mediated specificity in Hox mRNA translation and vertebrate tissue patterning. Cell (2011) 145(3):383–97. doi:10.1016/j.cell.2011.03.028
39. Desplanques G, Giuliani N, Delsignore R, Rizzoli V, Bataille R, Barille-Nion S. Impact of XIAP protein levels on the survival of myeloma cells. Haematologica (2009) 94(1):87–93. doi:10.3324/haematol.13483
40. Jopling CL, Willis AE. N-myc translation is initiated via an internal ribosome entry segment that displays enhanced activity in neuronal cells. Oncogene (2001) 20(21):2664–70. doi:10.1038/sj.onc.1204404
41. Pozner A, Goldenberg D, Negreanu V, Le SY, Elroy-Stein O, Levanon D, et al. Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms. Mol Cell Biol (2000) 20(7):2297–307. doi:10.1128/MCB.20.7.2297-2307.2000
42. Coldwell MJ, Mitchell SA, Stoneley M, MacFarlane M, Willis AE. Initiation of Apaf-1 translation by internal ribosome entry. Oncogene (2000) 19(7):899–905. doi:10.1038/sj.onc.1203407
43. Fernandez J, Yaman I, Mishra R, Merrick WC, Snider MD, Lamers WH, et al. Internal ribosome entry site-mediated translation of a mammalian mRNA is regulated by amino acid availability. J Biol Chem (2001) 276(15):12285–91. doi:10.1074/jbc.M009714200
44. Van Eden ME, Byrd MP, Sherrill KW, Lloyd RE. Translation of cellular inhibitor of apoptosis protein 1 (c-IAP1) mRNA is IRES mediated and regulated during cell stress. RNA (2004) 10(3):469–81. doi:10.1261/rna.5156804
45. Rubsamen D, Kunze MM, Buderus V, Brauss TF, Bajer MM, Brune B, et al. Inflammatory conditions induce IRES-dependent translation of cyp24a1. PLoS One (2014) 9(1):e85314. doi:10.1371/journal.pone.0085314
46. Rubsamen D, Blees JS, Schulz K, Doring C, Hansmann ML, Heide H, et al. IRES-dependent translation of egr2 is induced under inflammatory conditions. RNA (2012) 18(10):1910–20. doi:10.1261/rna.033019.112
47. Webb TE, Hughes A, Smalley DS, Spriggs KA. An internal ribosome entry site in the 5’ untranslated region of epidermal growth factor receptor allows hypoxic expression. Oncogenesis (2015) 4:e134. doi:10.1038/oncsis.2014.43
48. Xue S, Tian S, Fujii K, Kladwang W, Das R, Barna M. RNA regulons in Hox 5’ UTRs confer ribosome specificity to gene regulation. Nature (2015) 517(7532):33–8. doi:10.1038/nature14010
49. Blau L, Knirsh R, Ben-Dror I, Oren S, Kuphal S, Hau P, et al. Aberrant expression of c-Jun in glioblastoma by internal ribosome entry site (IRES)-mediated translational activation. Proc Natl Acad Sci U S A (2012) 109(42):E2875–84. doi:10.1073/pnas.1203659109
50. Stoneley M, Paulin FE, Le Quesne JP, Chappell SA, Willis AE. C-Myc 5’ untranslated region contains an internal ribosome entry segment. Oncogene (1998) 16(3):423–8. doi:10.1038/sj.onc.1201763
51. Nanbru C, Lafon I, Audigier S, Gensac MC, Vagner S, Huez G, et al. Alternative translation of the proto-oncogene c-myc by an internal ribosome entry site. J Biol Chem (1997) 272(51):32061–6. doi:10.1074/jbc.272.51.32061
52. Jopling CL, Spriggs KA, Mitchell SA, Stoneley M, Willis AE. L-Myc protein synthesis is initiated by internal ribosome entry. RNA (2004) 10(2):287–98. doi:10.1261/rna.5138804
53. Bisio A, Latorre E, Andreotti V, Bressac-de Paillerets B, Harland M, Scarra GB, et al. The 5’-untranslated region of p16INK4a melanoma tumor suppressor acts as a cellular IRES, controlling mRNA translation under hypoxia through YBX1 binding. Oncotarget (2015) 6(37):39980–94. doi:10.18632/oncotarget.5387
54. Andreotti V, Bisio A, Bressac-de Paillerets B, Harland M, Cabaret O, Newton-Bishop J, et al. The CDKN2A/p16 5’UTR sequence and translational regulation: impact of novel variants predisposing to melanoma. Pigment Cell Melanoma Res (2015) 29(2):210–21. doi:10.1111/pcmr.12444
55. Miskimins WK, Wang G, Hawkinson M, Miskimins R. Control of cyclin-dependent kinase inhibitor p27 expression by cap-independent translation. Mol Cell Biol (2001) 21(15):4960–7. doi:10.1128/MCB.21.15.4960-4967.2001
56. Jiang H, Coleman J, Miskimins R, Srinivasan R, Miskimins WK. Cap-independent translation through the p27 5’-UTR. Nucleic Acids Res (2007) 35(14):4767–78. doi:10.1093/nar/gkm512
57. Cuesta R, Martinez-Sanchez A, Gebauer F. miR-181a regulates cap-dependent translation of p27kip1 mRNA in myeloid cells. Mol Cell Biol (2009) 29(10):2841–51. doi:10.1128/MCB.01971-08
58. Ray PS, Grover R, Das S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep (2006) 7(4):404–10. doi:10.1038/sj.embor.7400623
59. Hertz MI, Landry DM, Willis AE, Luo G, Thompson SR. Ribosomal protein S25 dependency reveals a common mechanism for diverse internal ribosome entry sites and ribosome shunting. Mol Cell Biol (2013) 33(5):1016–26. doi:10.1128/MCB.00879-12
60. Gaccioli F, Huang CC, Wang C, Bevilacqua E, Franchi-Gazzola R, Gazzola GC, et al. Amino acid starvation induces the SNAT2 neutral amino acid transporter by a mechanism that involves eukaryotic initiation factor 2alpha phosphorylation and cap-independent translation. J Biol Chem (2006) 281(26):17929–40. doi:10.1074/jbc.M600341200
61. Damiano F, Alemanno S, Gnoni GV, Siculella L. Translational control of the sterol-regulatory transcription factor SREBP-1 mRNA in response to serum starvation or ER stress is mediated by an internal ribosome entry site. Biochem J (2010) 429(3):603–12. doi:10.1042/BJ20091827
62. Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E. Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol (1998) 18(6):3112–9. doi:10.1128/MCB.18.6.3112
63. Huez I, Creancier L, Audigier S, Gensac MC, Prats AC, Prats H. Two independent internal ribosome entry sites are involved in translation initiation of vascular endothelial growth factor mRNA. Mol Cell Biol (1998) 18(11):6178–90. doi:10.1128/MCB.18.11.6178
64. Akiri G, Nahari D, Finkelstein Y, Le SY, Elroy-Stein O, Levi BZ. Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene (1998) 17(2):227–36. doi:10.1038/sj.onc.1202019
65. Saffran HA, Smiley JR. The XIAP IRES activates 3’ cistron expression by inducing production of monocistronic mRNA in the betagal/CAT bicistronic reporter system. RNA (2009) 15(11):1980–5. doi:10.1261/rna.1557809
66. Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, Pena R, et al. A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev (2008) 22(6):756–69. doi:10.1101/gad.455708
67. Bonneau AM, Sonenberg N. Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J Biol Chem (1987) 262(23):11134–9.
68. Panniers R. Translational control during heat shock. Biochimie (1994) 76(8):737–47. doi:10.1016/0300-9084(94)90078-7
69. Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell (2010) 40(2):228–37. doi:10.1016/j.molcel.2010.09.028
70. Wang X, Proud CG. Nutrient control of TORC1, a cell-cycle regulator. Trends Cell Biol (2009) 19(6):260–7. doi:10.1016/j.tcb.2009.03.005
71. Bellodi C, Krasnykh O, Haynes N, Theodoropoulou M, Peng G, Montanaro L, et al. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res (2010) 70(14):6026–35. doi:10.1158/0008-5472.CAN-09-4730
72. Dokal I. Dyskeratosis congenita in all its forms. Br J Haematol (2000) 110(4):768–79. doi:10.1046/j.1365-2141.2000.02109.x
73. Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell (2011) 44(4):660–6. doi:10.1016/j.molcel.2011.09.017
74. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science (2006) 312(5775):902–6. doi:10.1126/science.1123835
75. Rocchi L, Pacilli A, Sethi R, Penzo M, Schneider RJ, Trere D, et al. Dyskerin depletion increases VEGF mRNA internal ribosome entry site-mediated translation. Nucleic Acids Res (2013) 41(17):8308–18. doi:10.1093/nar/gkt587
76. Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature (1998) 396(6707):177–80. doi:10.1038/24179
77. Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer (2012) 12(9):613–26. doi:10.1038/nrc3318
78. Hoffman Y, Pilpel Y, Oren M. microRNAs and Alu elements in the p53-Mdm2-Mdm4 regulatory network. J Mol Cell Biol (2014) 6(3):192–7. doi:10.1093/jmcb/mju020
79. Hainaut P, Hernandez T, Robinson A, Rodriguez-Tome P, Flores T, Hollstein M, et al. IARC database of p53 gene mutations in human tumors and cell lines: updated compilation, revised formats and new visualisation tools. Nucleic Acids Res (1998) 26(1):205–13. doi:10.1093/nar/26.1.205
80. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature (1997) 387(6630):296–9. doi:10.1038/387296a0
81. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett (1997) 420(1):25–7. doi:10.1016/S0014-5793(97)01480-4
82. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature (1997) 387(6630):299–303. doi:10.1038/387299a0
83. Giaccia AJ, Kastan MB. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev (1998) 12(19):2973–83. doi:10.1101/gad.12.19.2973
84. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res (1991) 51(23 Pt 1):6304–11.
85. Fu L, Minden MD, Benchimol S. Translational regulation of human p53 gene expression. EMBO J (1996) 15(16):4392–401.
86. Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell (2005) 123(1):49–63. doi:10.1016/j.cell.2005.07.034
87. Yang DQ, Halaby MJ, Zhang Y. The identification of an internal ribosomal entry site in the 5’-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene (2006) 25(33):4613–9. doi:10.1038/sj.onc.1209483
88. Candeias MM, Powell DJ, Roubalova E, Apcher S, Bourougaa K, Vojtesek B, et al. Expression of p53 and p53/47 are controlled by alternative mechanisms of messenger RNA translation initiation. Oncogene (2006) 25(52):6936–47. doi:10.1038/sj.onc.1209996
89. Ungureanu NH, Cloutier M, Lewis SM, de Silva N, Blais JD, Bell JC, et al. Internal ribosome entry site-mediated translation of Apaf-1, but not XIAP, is regulated during UV-induced cell death. J Biol Chem (2006) 281(22):15155–63. doi:10.1074/jbc.M511319200
90. Jagot-Lacoussiere L, Faye A, Bruzzoni-Giovanelli H, Villoutreix BO, Rain JC, Poyet JL. DNA damage-induced nuclear translocation of Apaf-1 is mediated by nucleoporin Nup107. Cell Cycle (2015) 14(8):1242–51. doi:10.1080/15384101.2015.1014148
91. Reubold TF, Eschenburg S. A molecular view on signal transduction by the apoptosome. Cell Signal (2012) 24(7):1420–5. doi:10.1016/j.cellsig.2012.03.007
92. Haraguchi M, Torii S, Matsuzawa S, Xie Z, Kitada S, Krajewski S, et al. Apoptotic protease activating factor 1 (Apaf-1)-independent cell death suppression by Bcl-2. J Exp Med (2000) 191(10):1709–20. doi:10.1084/jem.191.10.1709
94. Zamora AJ, Tessier F, Marconnet P, Margaritis I, Marini JF. Mitochondria changes in human muscle after prolonged exercise, endurance training and selenium supplementation. Eur J Appl Physiol Occup Physiol (1995) 71(6):505–11. doi:10.1007/BF00238552
95. Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell (1998) 94(6):739–50. doi:10.1016/S0092-8674(00)81733-X
96. Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell (1998) 94(6):727–37. doi:10.1016/S0092-8674(00)81732-8
97. Perkins C, Kim CN, Fang G, Bhalla KN. Overexpression of Apaf-1 promotes apoptosis of untreated and paclitaxel- or etoposide-treated HL-60 cells. Cancer Res (1998) 58(20):4561–6.
98. Soengas MS, Alarcon RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW, et al. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science (1999) 284(5411):156–9. doi:10.1126/science.284.5411.156
99. Furukawa Y, Sutheesophon K, Wada T, Nishimura M, Saito Y, Ishii H, et al. Methylation silencing of the Apaf-1 gene in acute leukemia. Mol Cancer Res (2005) 3(6):325–34. doi:10.1158/1541-7786.MCR-04-0105
100. Krajewska M, Kim H, Kim C, Kang H, Welsh K, Matsuzawa S, et al. Analysis of apoptosis protein expression in early-stage colorectal cancer suggests opportunities for new prognostic biomarkers. Clin Cancer Res (2005) 11(15):5451–61. doi:10.1158/1078-0432.CCR-05-0094
101. Landry DM, Hertz MI, Thompson SR. RPS25 is essential for translation initiation by the Dicistroviridae and hepatitis C viral IRESs. Genes Dev (2009) 23(23):2753–64. doi:10.1101/gad.1832209
102. Andreev DE, Dmitriev SE, Terenin IM, Prassolov VS, Merrick WC, Shatsky IN. Differential contribution of the m7G-cap to the 5’ end-dependent translation initiation of mammalian mRNAs. Nucleic Acids Res (2009) 37(18):6135–47. doi:10.1093/nar/gkp665
103. Jia L, Srinivasula SM, Liu FT, Newland AC, Fernandes-Alnemri T, Alnemri ES, et al. Apaf-1 protein deficiency confers resistance to cytochrome c-dependent apoptosis in human leukemic cells. Blood (2001) 98(2):414–21. doi:10.1182/blood.V98.2.414
104. Fu WN, Bertoni F, Kelsey SM, McElwaine SM, Cotter FE, Newland AC, et al. Role of DNA methylation in the suppression of Apaf-1 protein in human leukaemia. Oncogene (2003) 22(3):451–5. doi:10.1038/sj.onc.1206147
105. Clemens MJ, Bushell M, Morley SJ. Degradation of eukaryotic polypeptide chain initiation factor (eIF) 4G in response to induction of apoptosis in human lymphoma cell lines. Oncogene (1998) 17(22):2921–31. doi:10.1038/sj.onc.1202227
106. Morley SJ, McKendrick L, Bushell M. Cleavage of translation initiation factor 4G (eIF4G) during anti-Fas IgM-induced apoptosis does not require signalling through the p38 mitogen-activated protein (MAP) kinase. FEBS Lett (1998) 438(1–2):41–8. doi:10.1016/S0014-5793(98)01269-1
107. Holcik M, Sonenberg N, Korneluk RG. Internal ribosome initiation of translation and the control of cell death. Trends Genet (2000) 16(10):469–73. doi:10.1016/S0168-9525(00)02106-5
108. Deckwerth TL, Johnson EM Jr. Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol (1993) 123(5):1207–22. doi:10.1083/jcb.123.5.1207
109. Clemens MJ, Bushell M, Jeffrey IW, Pain VM, Morley SJ. Translation initiation factor modifications and the regulation of protein synthesis in apoptotic cells. Cell Death Differ (2000) 7(7):603–15. doi:10.1038/sj.cdd.4400695
110. Henis-Korenblit S, Shani G, Sines T, Marash L, Shohat G, Kimchi A. The caspase-cleaved DAP5 protein supports internal ribosome entry site-mediated translation of death proteins. Proc Natl Acad Sci U S A (2002) 99(8):5400–5. doi:10.1073/pnas.082102499
111. Mitchell SA, Spriggs KA, Coldwell MJ, Jackson RJ, Willis AE. The Apaf-1 internal ribosome entry segment attains the correct structural conformation for function via interactions with PTB and unr. Mol Cell (2003) 11(3):757–71. doi:10.1016/S1097-2765(03)00093-5
112. Mitchell SA, Brown EC, Coldwell MJ, Jackson RJ, Willis AE. Protein factor requirements of the Apaf-1 internal ribosome entry segment: roles of polypyrimidine tract binding protein and upstream of N-ras. Mol Cell Biol (2001) 21(10):3364–74. doi:10.1128/MCB.21.10.3364-3374.2001
113. Vesely PW, Staber PB, Hoefler G, Kenner L. Translational regulation mechanisms of AP-1 proteins. Mutat Res (2009) 682(1):7–12. doi:10.1016/j.mrrev.2009.01.001
114. Polak P, Oren A, Ben-Dror I, Steinberg D, Sapoznik S, Arditi-Duvdevany A, et al. The cytoskeletal network controls c-Jun translation in a UTR-dependent manner. Oncogene (2006) 25(5):665–76. doi:10.1038/sj.onc.1209114
115. Spangler B, Vardimon L, Bosserhoff AK, Kuphal S. Post-transcriptional regulation controlled by E-cadherin is important for c-Jun activity in melanoma. Pigment Cell Melanoma Res (2011) 24(1):148–64. doi:10.1111/j.1755-148X.2010.00787.x
116. Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem (2003) 253(1–2):269–85. doi:10.1023/A:1026028303196
117. Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J (2002) 21(15):4104–13. doi:10.1093/emboj/cdf389
118. Lopez-Bergami P, Huang C, Goydos JS, Yip D, Bar-Eli M, Herlyn M, et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell (2007) 11(5):447–60. doi:10.1016/j.ccr.2007.03.009
119. Vleugel MM, Greijer AE, Bos R, van der Wall E, van Diest PJ. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum Pathol (2006) 37(6):668–74. doi:10.1016/j.humpath.2006.01.022
120. Knirsh R, Ben-Dror I, Spangler B, Matthews GD, Kuphal S, Bosserhoff AK, et al. Loss of E-cadherin-mediated cell-cell contacts activates a novel mechanism for up-regulation of the proto-oncogene c-Jun. Mol Biol Cell (2009) 20(7):2121–9. doi:10.1091/mbc.E08-12-1196
121. Oren A, Herschkovitz A, Ben-Dror I, Holdengreber V, Ben-Shaul Y, Seger R, et al. The cytoskeletal network controls c-Jun expression and glucocorticoid receptor transcriptional activity in an antagonistic and cell-type-specific manner. Mol Cell Biol (1999) 19(3):1742–50. doi:10.1128/MCB.19.3.1742
122. Lamb RF, Hennigan RF, Turnbull K, Katsanakis KD, MacKenzie ED, Birnie GD, et al. AP-1-mediated invasion requires increased expression of the hyaluronan receptor CD44. Mol Cell Biol (1997) 17(2):963–76. doi:10.1128/MCB.17.2.963
Keywords: IRES, cap-independent, tumorigenesis, p53, Apaf-1, c-Jun, apoptosis, translation
Citation: Walters B and Thompson SR (2016) Cap-Independent Translational Control of Carcinogenesis. Front. Oncol. 6:128. doi: 10.3389/fonc.2016.00128
Received: 14 January 2016; Accepted: 10 May 2016;
Published: 25 May 2016
Edited by:
Francois X. Claret, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Min Hee Kang, Texas Tech University Health Sciences Center, USAShiu-Wan Chan, The University of Manchester, UK
Copyright: © 2016 Walters and Thompson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sunnie R. Thompson, c3VubmllQHVhYi5lZHU=