 Markus Hartl
Markus Hartl- Institute of Biochemistry and Center of Molecular Biosciences (CMBI), University of Innsbruck, Innsbruck, Austria
MYC represents a transcription factor with oncogenic potential converting multiple cellular signals into a broad transcriptional response, thereby controlling the expression of numerous protein-coding and non-coding RNAs important for cell proliferation, metabolism, differentiation, and apoptosis. Constitutive activation of MYC leads to neoplastic cell transformation, and deregulated MYC alleles are frequently observed in many human cancer cell types. Multiple approaches have been performed to isolate genes differentially expressed in cells containing aberrantly activated MYC proteins leading to the identification of thousands of putative targets. Functional analyses of genes differentially expressed in MYC-transformed cells had revealed that so far more than 40 upregulated or downregulated MYC targets are actively involved in cell transformation or tumorigenesis. However, further systematic and selective approaches are required for determination of the known or yet unidentified targets responsible for processing the oncogenic MYC program. The search for critical targets in MYC-dependent tumor cells is exacerbated by the fact that during tumor development, cancer cells progressively evolve in a multistep process, thereby acquiring their characteristic features in an additive manner. Functional expression cloning, combinatorial gene expression, and appropriate in vivo tests could represent adequate tools for dissecting the complex scenario of MYC-specified cell transformation. In this context, the central goal is to identify a minimal set of targets that suffices to phenocopy oncogenic MYC. Recently developed genomic editing tools could be employed to confirm the requirement of crucial transformation-associated targets. Knowledge about essential MYC-regulated genes is beneficial to expedite the development of specific inhibitors to interfere with growth and viability of human tumor cells in which MYC is aberrantly activated. Approaches based on the principle of synthetic lethality using MYC-overexpressing cancer cells and chemical or RNAi libraries have been employed to search for novel anticancer drugs, also leading to the identification of several druggable targets. Targeting oncogenic MYC effector genes instead of MYC may lead to compounds with higher specificities and less side effects. This class of drugs could also display a wider pharmaceutical window because physiological functions of MYC, which are important for normal cell growth, proliferation, and differentiation would be less impaired.
MYC is an Endpoint of Multiple Signaling Pathways
Cancer cells are featured by deregulated activation and suppression of proto-oncogenes and tumor suppressor genes, respectively. Tumor cells evolve from a multistep process, resulting in sustained proliferation, inactivation of growth suppressors, immortalization, accelerated angiogenesis, metastasis, and resistance to programed cell death. In normal tissues, growth-promoting signals are carefully controlled leading to cellular homeostasis, whereas in cancer cells, these biological signals are deregulated. Signals are transmitted by growth factors, which bind to cell surface receptors containing intracellular tyrosine kinase domains. From here, the signal branches into multiple and complex signal transduction pathways to regulate cell cycle progression, cell growth, survival, and energy metabolism (1). Key players in these processes are encoded by genes, which are normally required to coordinate proper cell metabolism, proliferation, and differentiation. The functions of many of these genes had been elucidated after their identification as transforming principles in oncogenic retroviruses, which carry mutated versions in their genomes (2).
Some of the most intensively studied oncogenes encode transcription factors that are functionally located at the end of several signaling cascades, thereby integrating multiple cellular signals. Transcription factors regulate gene expression and similar to cytoplasmic key regulators, the deregulation of many transcription factors is associated with human oncogenesis. Transcription factors bind to the DNA control regions of target genes and activate or suppress their expression, which is important for cell proliferation and differentiation. In case of aberrant gene regulator activities caused by mutations, distinct target genes become abnormally activated or deactivated, which can ultimately lead to oncogenic transformation and malignant cell growth. MYC represents a prototypic transcription factor and a nuclear end point of several signaling pathways (3) (Figure 1). Hence, identification and characterization of transformation-relevant target genes acting downstream of MYC is a prerequisite to understand molecular mechanisms of tumor development in which this oncogenic transcription factor is involved.
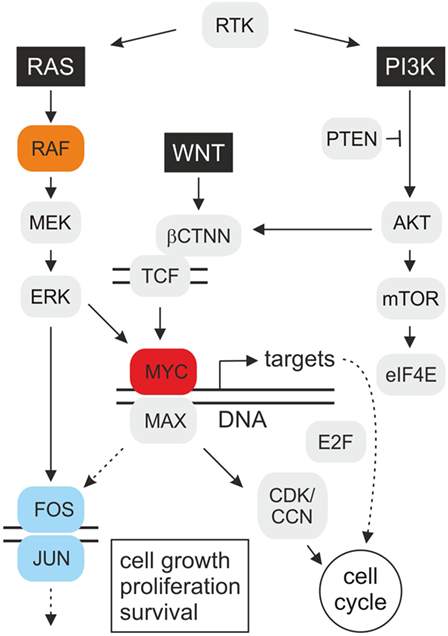
Figure 1. Schematic depiction of oncogenic MYC signal transduction. The highly simplified cartoon shows key pathways operating upstream and downstream of the MYC oncoprotein. Mitogenic signal transduction starts with stimulated receptor tyrosine kinases (RTK) transmitting signals via guanine nucleotide exchange factors onto the G protein RAS. RAS binds and activates the serine/threonine protein kinase RAF(Mil), which leads to consecutive phosphorylation of the mitogen-activated protein kinase kinase (MAPKK) MEK, the MAP kinase (MAPK) ERK, and of transcription factor complexes, such as MYC/MAX or JUN/FOS (AP-1), regulating the expression of numerous target genes. Based on the observed synergy between MYC and RAF(Mil), distinct MYC targets may enhance RAS/RAF-induced cell transformation via a positive feedback loop. On the other hand, MYC could also directly stimulate AP-1 by transcriptional activation of JUN or FOS encoding genes. The c-MYC gene is activated at the transcriptional level by the wingless/int-1 (WNT) signaling pathway, resulting in nuclear translocation of β-catenin (βCTNN) where it binds to T-cell factor (TCF). Several transforming MYC targets are involved in cell cycle regulation (Table 1), which encode inter alia E2F transcription factors, cyclins (CCN), and cyclin-dependent kinases (CDK), resulting in accelerated cell proliferation. Besides the extracellular signal-regulated kinase (RAS–ERK) pathway, phosphatidylinositol 3-kinase (PI3K)–mammalian target of rapamycin (mTOR) signaling is a central mechanism to control cell growth, proliferation, and survival in response to extracellular stimuli. The protein kinase AKT phosphorylates many survival factors, and mTOR-mediated signaling modulates ribosome biogenesis and translation of proteins, such as c-MYC and cyclin D, that promote cell growth and proliferation [adapted from Ref. (1, 4, 5)].
Origin of MYC and Biological Functions
MYC has been originally identified as the transforming determinant (v-myc) of avian acute leukemia virus MC29 in chicken (myelocytomatosis virus 29) (6). MYC was also isolated from the avian leukemia- and carcinoma-inducing MH2 virus, which carries in addition the v-mil(RAF) allele encoding a serine/threonine protein kinase (7). The presence of two oncogenes significantly increases the oncogenicity of MH2, which is due to cooperative effects between the v-Myc and v-Mil(RAF) proteins (8, 9).
The v-myc allele is derived from the cellular c-myc proto-oncogene by retroviral transduction (10, 11). C-MYC encodes the c-MYC protein, a transcription factor with oncogenic potential representing the central hub of a network controlling the expression of at least 15% of all human genes, and regulating fundamental cellular processes, such as growth, proliferation, differentiation, metabolism, pluripotency, and apoptosis (3, 12). Transcriptional deregulation of human c-MYC caused by chromosomal translocation was first observed in Burkitt’s lymphoma (13).
Besides retroviral insertion or transduction of human c-MYC leading to the development of lymphomas and carcinomas, amplification of MYC alleles has been observed in colon carcinoma, neuroblastoma, and lung cancer leading to the discovery of the N-MYC and L-MYC paralogs (11). Constitutive activation of MYC is required for oncogenesis and occurs in many human tumor cell lines indicating that deregulated expression of this oncoprotein may contribute to cancer formation. In fact, besides the K-RAS and B-RAF oncoproteins, c-MYC represents a major driver in human tumorigenesis (11, 14). Ectopic expression of c-MYC suffices to induce metastasis in a murine non-small-cell lung cancer (NSCLC) model featuring the most lethal human cancer due to its high metastasis rate. Likewise, in prostate and pancreatic cancer, c-MYC is upregulated upon constitutive stimulation of the RAS and WNT pathways (15–19) (Figure 1). Immortalization and transformation of human epithelial cells occur after overexpressing c-MYC and simultaneously inactivating cyclin-dependent kinase inhibitor 2A (CDKN2A), leading to specific gene expression changes (20, 21). Today, it is known that deregulation of MYC genes is a frequent event in animal and human tumorigenesis taking place in more than 50% of all human cancers (3, 22). MYC proteins therefore belong to those crucial master switches in most human cancers, from which many of them are associated with a poor clinical outcome (12, 23).
Principal Biochemical Functions of MYC
MYC is a bHLHZip protein encompassing protein dimerization domains (helix–loop–helix, leucine zipper) and a DNA contact surface (basic region) that forms heterodimers with the MAX protein and binds typically to specific DNA sequence elements termed E-boxes (5′-CACGTG-3′) (10, 11). MYC and MAX homologs with conserved basic functions were found in primitive metazoans (24, 25) and premetazoans (26), suggesting that principal functions of the MYC master regulator arose very early in the evolution of multicellular animals.
MYC is regulated at transcriptional and translational levels and stabilized by post-translational modifications, such as RAS-dependent phosphorylation (27). In fact, it has been shown that RAS/ERK and PI3K/AKT signaling cascades significantly increase the half-life of MYC, which is normally subjected to rapid ubiquitin-mediated protein degradation (28, 29) (Figure 1). Although MYC is also involved in DNA replication and cell cycle checkpoint processes (30), its major function is transcriptional regulation (11, 12). MYC binds to multiple coactivators representing components of histone acetyltransferase complexes, to ubiquitin ligases, or to other transcription factors, thereby inducing transcriptional activation or repression (10, 11).
Amplification of Gene Expression by MYC
Previous global analyses, using techniques such as serial analysis of gene expression, DNA microarrays, chromatin immunoprecipitation coupled with high through-put sequencing (ChIP-Seq), promoter scanning, or proteomics, have led to the identification of thousands of genes controlled by the MYC/MAX network, which are involved in fundamental cellular processes, including growth, proliferation, metabolism, differentiation, and apoptosis (31–37). Many of the MYC-activated genes are broadly related to processes of nucleotide synthesis, cell growth, and metabolism, including protein synthesis, ribosomal biogenesis, glycolysis, mitochondrial function, and cell cycle progression (11, 12, 38). In addition, several cell cycle-related genes whose protein products initiate DNA replication are transcriptional MYC targets, which could explain why deregulated DNA synthesis, chromosomal abnormalities, and genomic instability frequently occurs in human tumor cells containing activated MYC (39).
Deregulated MYC target genes have been identified in numerous human tumors (11, 40), but so far it has been difficult to ascribe the oncogenic properties of MYC to a defined set of target genes. In fact, results from recent studies indicate that MYC acts as a general amplifier of gene expression (41–43). According to this theory, the promoters of all actively transcribed genes are occupied and activated by c-MYC in tumor cells expressing high levels of this transcription factor, leading to non-linear amplification of existing transcriptional activities (41, 42, 44–46). The observed differential expression of multiple genes in cells containing aberrantly activated MYC is therefore due to individually enhanced gene expression occurring at varying levels. The amplifier model also explains how ectopic c-MYC increases the efficiencies of other transcription factor programs (46), e.g., during generation of pluripotent stem cells from fibroblasts. This re-programing of cells is achieved by overexpressing the transcription factors OCT4, SOX2, and KLF4 (47). On the other hand, gene repression in cells transformed by MYC is caused by MYC interaction with specific transcription factors or indirectly by increasing the expression of repressive transcriptional and chromatin components.
MYC Targets with Oncogenic or Transformation-Suppressive Activities
The conversion of a normal into a tumorigenic cell could be caused by the products of multiple transformation-associated MYC target genes, from which more than 40 have been identified so far (Table 1). Some of these genes exhibit transforming activity upon ectopic expression, suggesting that they contribute to MYC-induced oncogenesis. Furthermore, there is evidence that MYC enhances the effects of other oncogenic gene regulators, such as E2F (48) or AP-1 (9) (Figure 1). In addition to the implication of MYC/MAX heterodimers in transcriptional activation, MYC has been also associated with transcriptional repression, thereby in many cases not binding directly to E-boxes but instead involving other transcription factors such as MIZ-1 or SP1 (3, 12, 46, 49, 50). Most of the genes repressed by MYC are involved in cell cycle arrest, cell adhesion, and cell-to-cell communication (11).
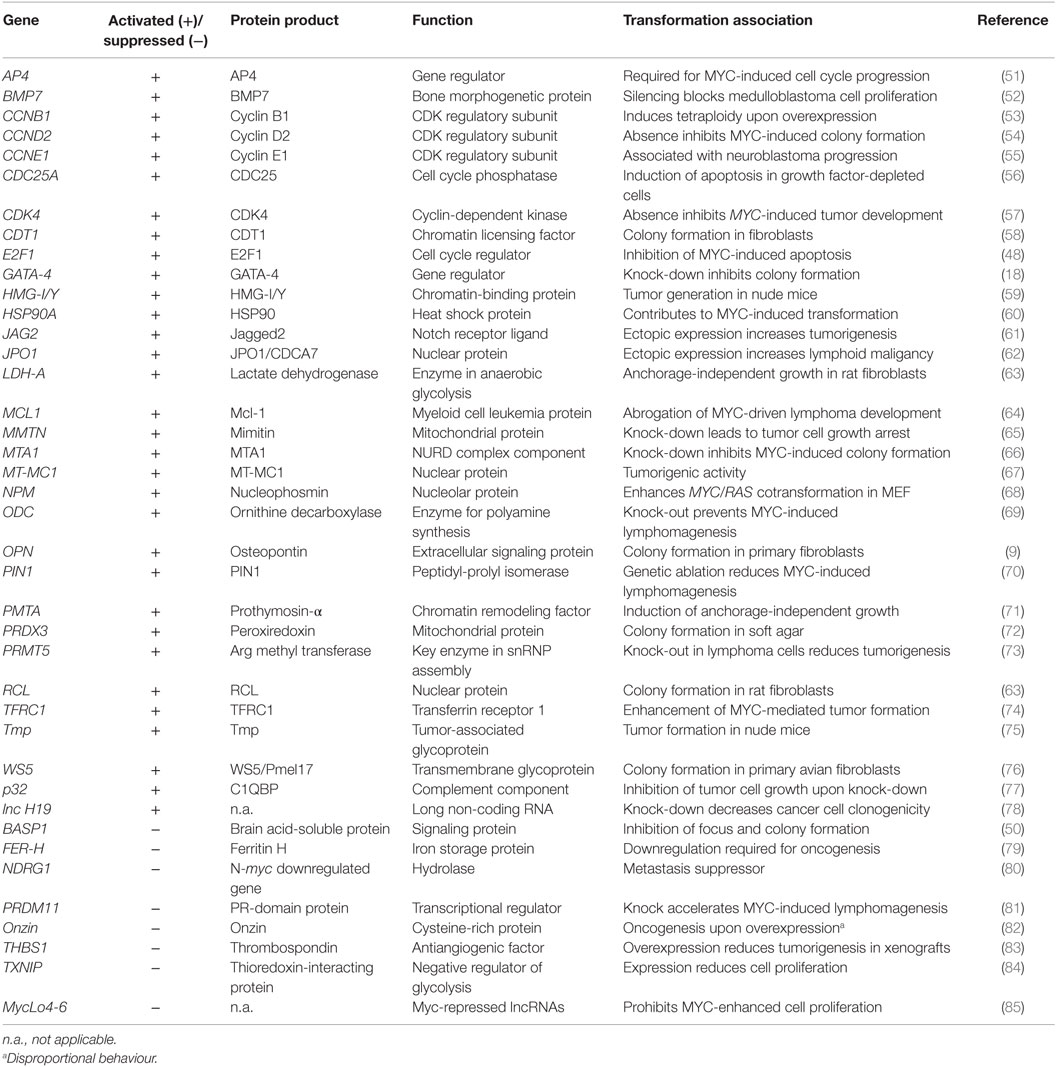
Table 1. Activated and suppressed MYC target genes associated with cell transformation.
Besides regulating the expression of protein-encoding genes, MYC also controls the expression of distinct long non-coding ribonucleic acids (lncRNA) (78, 85–87) and of multiple small non-coding regulatory microRNAs (miRNA) (3, 88–91). Some of the miRNAs have oncogenic properties such as the miR-17–92 cluster (oncomir-1) or have tumor suppressor functions (16, 85, 88, 92) (Table 1). miRNAs inhibit protein translation or lead to degradation of their target messenger RNAs (mRNA) and have been implicated in cancer by inactivating distinct mRNAs encoding oncogenes or tumor suppressors (93).
Approaches to Identify Critical Targets Executing MYC-Induced Cell Transformation
Transformation-associated targets of MYC either display intrinsic transforming activities or inhibit oncogenesis depending whether they are activated or repressed (Table 1). Assuming that MYC transformation is mainly based on transcriptional deregulation, only the combined effects of multiple activated or suppressed targets may suffice to induce a “MYC-like” transformed phenotype. However, just systematically analyzing known transformation-relevant MYC targets is not constructive because the list in Table 1 is not exhaustive, and even more important, many of these targets have been isolated from different cellular systems under in vitro cell culture conditions. This may not reflect the real situation in vivo in which oxygen, nutrients, or growth factors are limited. Furthermore, the hypothesis in earlier reductionists’ approaches assuming that a tumor consists of a homogenous collection of cancer cells and its biology is accessible by elucidating all cell autonomous properties and is not valid any more. In human carcinogenesis, diverse cell types from cancer stem cells give rise to intra-tumor heterogeneity, thus further increasing the genetic complexity and representing a major cause of cancer recurrence (1, 94, 95). Cancer cells progressively evolve from normal cells in a multistep process, thereby acquiring distinct characteristic features in an additive manner. Thus, a succession of clonal expansions occurs also involving epigenetic mechanisms such as methylation or histone modification. In particular, the transition to invasion and metastasis encompasses several discrete steps. With regard to this complex scenario, certain in vitro environmental pertubations have to be reconsidered and better adapted to the in vivo situation, for instance, by using isogenic cell lines, which differ only in single allelic mutations (95, 96). More unbiased approaches based on oncogenic functions are required to identify a putative magic target gene set which suffices to phenocopy MYC transformation, supposed that such Holy Grail exists at all. The following approaches are suggested to dissect the complexity of the oncogenic MYC transcriptional program:
Functional Expression Cloning
Isolation of novel coding and non-coding MYC targets with strong oncogenic activities could be done by cDNA expression cloning using MYC-dependent tumor cells as a source for RNA isolation. The application of retroviral cDNA expression libraries has already successfully led to the isolation of transforming genes from human tumor cells (97–100). Thereby, the selection for distinct genes is based exclusively on function, in this case the capacity to transform cells. Appropriate gene-transfer tools are retroviral vectors, allowing the efficient introduction of complex cDNA libraries (99, 101) and appropriate screening procedures.
Combinatorial Gene Expression
Due to the pleiotropic MYC effect leading to the development of multiple different tumor forms, one could postulate that simultaneous perturbation of multiple targets suffices to convert a normal cell into a cancer cell displaying a MYC-transformed phenotype. Due to the capacity of MYC to enhance existing transcriptional programs (see above), the identification of transcription factors, which are involved in executing the oncogenic MYC program, should be straightforward. Critical MYC targets can then be overexpressed and inactivated depending on whether they are activated and suppressed in MYC-transformed cells, respectively. To simultaneously overexpress multiple genes or interfering RNAs in single cells, several established techniques exist. They are based on different principles such as co-transfection of multiple plasmids, usage of bicistronic vectors containing an internal ribosomal-binding site, infection with retroviruses containing different envelope subtypes, or self-processing peptides (47, 102).
Analysis of Targets by Permanent Gene Inactivation
The functionality of critical target genes can be tested by genomic inactivation and the usage of appropriate in vivo tumor model systems. To analyze if expression of a distinct target is required for maintenance of cell transformation, its inactivation should be performed in MYC-dependent tumor cells. Otherwise, to test if a target is required for the initiation of MYC-induced cell transformation, the relevant gene has to be disrupted in normal cells prior to MYC transduction. An appropriate tool for genomic inactivation is the recently developed clustered regularly interspaced short palindromic repeats (CRISPR) system (103, 104). Precise genome editing is achieved by creating specific double-stranded breaks, which allow the generation of homozygous knock-out or knock-in genotypes. Specific MYC target gene inactivation could lead to inhibition of the tumorigenic phenotype, cell cycle arrest, or apoptosis. Suitable in vivo techniques to quantify gene inactivation effects on tumor growth and angiogenesis are, e.g., the generation of mouse xenoplants, and the chicken chorioallanthoic membrane assay. Inhibition of tumorigenesis caused by inactivation of MYC distinct targets would indicate essential functions of the tested genes.
MYC Targets as Templates for Inhibitor Design
Because of its pivotal role in cancer, MYC has become an obvious target in the treatment of human cancer cells. Several approaches to interfere with MYC gene transcription, MYC protein function, or with the functions of distinct targets have been pursued to inhibit MYC-dependent pathogenesis.
Intracellular signal transduction pathways regulating MYC expression and protein stability have been targeted by using chemical inhibitors, which are in the trial phase or already applied in the clinic. Thereby, key proteins of the two main signaling cascades responsible for cell survival, differentiation, proliferation, metabolism, and motility were inhibited: the RAS–extracellular signal-regulated kinase (ERK) and the phosphatidylinositol 3-kinase (PI3K) pathways (4, 105, 106) (Figure 1).
Direct inhibition of MYC functions has been achieved by using different strategies. c-MYC transcription has been targeted by inhibiting the chromatin acetyl-lysine recognition domain (bromodomain) of a MYC-specific coactivator. This led to suppression of c-MYC transcription followed by genome-wide downregulation of MYC-dependent target genes (107). C-MYC transcription has been blocked by the miRNA miR-494 leading to inhibition of proliferation, invasion, and chemoresistance in pancreatic cancer (108). Furthermore, a dominant negative mutant of the MYC dimerization domain termed Omomyc is effective against glioma thereby inhibiting cell proliferation and increasing apoptosis (109). Perturbation of MYC/MAX interaction by synthetic α-helix mimetics or by the homeobox protein Hhex led to impaired DNA binding suppressed transcriptional activation and inhibition of cell growth and tumorigenesis (110, 111). Efficient interference with MYC functions has been also achieved by using novel pyridine inhibitors leading to specific inhibition of MYC/MAX dimerization, transcriptional regulation, and oncogenesis (112, 113). These novel compounds reveal a unique inhibitory potential even at nanomolar concentrations combined with the specific inhibition of MYC-driven tumor growth in vivo (112).
However, under normal physiological conditions MYC is required for many cell physiological processes and for homeostasis. A complete block of the MYC protein by binding to efficient inhibitors may result into undesired side effects or into drug resistance after prolonged application. Approaches based on the principle of synthetic lethality using MYC-overexpressing cancer cells have lead to the identification of targets, which may be susceptible towards appropriate drugs. Synthetic lethality is defined by cell death induced by mutation or inhibition of two different genes, whereas the dysfunction of one gene has no effect on cell viability. This principle can be exploited to screen for anticancer drugs by mimicking the effect of the second genetic mutation using chemical or inhibiting-RNA libraries (114). For instance, pharmacological inhibition of the eukaryotic translation factor eIF4F is synthetic lethal in an Eμ-MYC lymphoma model (115). Likewise, selective death of MYC-dependent human breast cancer cells was achieved by siRNA-mediated inhibition of cyclin-dependent kinase 1 (CDK1) (116). Another example is the identification of a DNA repair protein kinase (PRKDC) as a synthetic lethal target in MYC-overexpressing lung cancer cells, which was identified in RNAi library screen (117). Therefore, attacking oncogenic MYC effectors may increase the specificity of MYC-dependent tumor treatment and enlarge the arsenal of available drugs.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
The work was supported by the Austrian Science Foundation grant P18148.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi:10.1016/j.cell.2011.02.013
2. Vogt PK. Retroviral oncogenes: a historical primer. Nat Rev Cancer (2012) 12:639–48. doi:10.1038/nrc3320
4. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci (2011) 36:320–8. doi:10.1016/j.tibs.2011.03.006
5. Hartl M, Bister K. Oncogenes. 2nd ed. In: Maloy S, Hughes K, editors. Brenner’s Encyclopedia of Genetics. (Vol. 5), San Diego: Academic Press (2013). p. 164–6.
6. Bister K, Hayman MJ, Vogt PK. Defectiveness of avian myelocytomatosis virus MC29: isolation of long-term nonproducer cultures and analysis of virus-specific polypeptide synthesis. Virology (1977) 82:431–48. doi:10.1016/0042-6822(77)90017-4
7. Jansen HW, Lurz R, Bister K, Bonner TI, Mark GE, Rapp UR. Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature (1984) 307:281–4. doi:10.1038/307281a0
8. Bister K, Jansen HW. Oncogenes in retroviruses and cells: biochemistry and molecular genetics. Adv Cancer Res (1986) 47:99–188. doi:10.1016/S0065-230X(08)60199-2
9. Hartl M, Karagiannidis AI, Bister K. Cooperative cell transformation by Myc/Mil(Raf) involves induction of AP-1 and activation of genes implicated in cell motility and metastasis. Oncogene (2006) 25:4043–55. doi:10.1038/sj.onc.1209441
10. Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol (2005) 6:635–45. doi:10.1038/nrm1703
11. Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med (2014) 4:a014357. doi:10.1101/cshperspect.a014357
13. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A (1982) 79:7824–7. doi:10.1073/pnas.79.24.7824
14. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer (2014) 14:455–67. doi:10.1038/nrc3760
15. Marampon F, Ciccarelli C, Zani BM. Down-regulation of c-MYC following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol Cancer (2006) 5:31. doi:10.1186/1476-4598-5-31
16. Coppola V, De Maria R, Bonci D. microRNAs and prostate cancer. Endocr Relat Cancer (2010) 17:F1–17. doi:10.1677/ERC-09-0172
17. Wang J, Kobayashi T, Floc’h N, Kinkade CW, Aytes A, Dankort D, et al. B-Raf activation cooperates with PTEN loss to drive c-MYC expression in advanced prostate cancer. Cancer Res (2012) 72:4765–76. doi:10.1158/0008-5472.CAN-12-0820
18. Castro IC, Breiling A, Luetkenhaus K, Ceteci F, Hausmann S, Kress S, et al. MYC-induced epigenetic activation of GATA4 in lung adenocarcinoma. Mol Cancer Res (2013) 11:161–72. doi:10.1158/1541-7786.MCR-12-0414-T
19. Hessmann E, Schneider G, Ellenrieder V, Siveke JT. MYC in pancreatic cancer: novel insights and their translation into therapeutic strategies. Oncogene (2016) 35:1609–18. doi:10.1038/onc.2015.216
20. Garbe JC, Vrba L, Sputova K, Fuchs L, Novak P, Brothman AR, et al. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle (2014) 13:3423–35. doi:10.4161/15384101.2014.954456
21. Vrba L, Garbe JC, Stampfer MR, Futscher BW. A lincRNA connected to cell mortality and epigenetically-silenced in most common human cancers. Epigenetics (2015) 10:1074–83. doi:10.1080/15592294.2015.1106673
22. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene (1999) 18:3004–16. doi:10.1038/sj.onc.1202746
23. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature (2010) 463:899–905. doi:10.1038/nature08822
24. Hartl M, Mitterstiller AM, Valovka T, Breuker K, Hobmayer B, Bister K. Stem cell-specific activation of an ancestral myc protooncogene with conserved basic functions in the early metazoan Hydra. Proc Natl Acad Sci U S A (2010) 107:4051–6. doi:10.1073/pnas.0911060107
25. Hartl M, Glasauer S, Valovka T, Breuker K, Hobmayer B, Bister K. Hydra myc2, a unique pre-bilaterian member of the myc gene family, is activated in cell proliferation and gametogenesis. Biol Open (2014) 3:397–407. doi:10.1242/bio.20147005
26. Young SL, Diolaiti D, Conacci-Sorrell M, Ruiz-Trillo I, Eisenman RN, King N. Premetazoan ancestry of the Myc-Max network. Mol Biol Evol (2011) 28:2961–71. doi:10.1093/molbev/msr132
27. Jackstadt R, Menssen A, Hermeking H. Genome-wide analysis of c-MYC-regulated mRNAs and miRNAs, and c-MYC DNA binding by next-generation sequencing. Methods Mol Biol (2013) 1012:145–85. doi:10.1007/978-1-62703-429-6_11
28. Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell (1999) 3:169–79. doi:10.1016/S1097-2765(00)80308-1
29. Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, et al. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res (2012) 72:2622–33. doi:10.1158/0008-5472.CAN-11-3605
30. Herold S, Herkert B, Eilers M. Facilitating replication under stress: an oncogenic function of MYC? Nat Rev Cancer (2009) 9:441–4. doi:10.1038/nrc2640
31. Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci U S A (2002) 99:6274–9. doi:10.1073/pnas.082005599
32. Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-MYC in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A (2003) 100:8164–9. doi:10.1073/pnas.1332764100
33. Marinkovic D, Marinkovic T, Kokai E, Barth T, Möller P, Wirth T. Identification of novel Myc target genes with a potential role in lymphomagenesis. Nucleic Acids Res (2004) 32:5368–78. doi:10.1093/nar/gkh877
34. Kim J, Lee JH, Iyer VR. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One (2008) 3:e1798. doi:10.1371/journal.pone.0001798
35. Seitz V, Butzhammer P, Hirsch B, Hecht J, Gütgemann I, Ehlers A, et al. Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One (2011) 6:e26837. doi:10.1371/journal.pone.0026837
36. Yap CS, Peterson AL, Castellani G, Sedivy JM, Neretti N. Kinetic profiling of the c-MYC transcriptome and bioinformatic analysis of repressed gene promoters. Cell Cycle (2011) 10:2184–96. doi:10.4161/cc.10.13.16249
37. Perna D, Fagà G, Verrecchia A, Gorski MM, Barozzi I, Narang V, et al. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene (2012) 31:1695–709. doi:10.1038/onc.2011.359
38. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov (2015) 5:1024–39. doi:10.1158/2159-8290.CD-15-0507
39. Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature (2007) 448:445–51. doi:10.1038/nature05953
40. Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One (2011) 6:e26057. doi:10.1371/journal.pone.0026057
41. Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell (2012) 151:56–67. doi:10.1016/j.cell.2012.08.026
42. Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, et al. Revisiting global gene expression analysis. Cell (2012) 151:476–82. doi:10.1016/j.cell.2012.10.012
43. Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, et al. c-MYC is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell (2012) 151:68–79. doi:10.1016/j.cell.2012.08.033
44. Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature (2014) 511:488–92. doi:10.1038/nature13537
45. Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature (2014) 511:483–7. doi:10.1038/nature13473
46. Wolf E, Lin CY, Eilers M, Levens DL. Taming of the beast: shaping Myc-dependent amplification. Trends Cell Biol (2015) 25:241–8. doi:10.1016/j.tcb.2014.10.006
47. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell (2006) 126:663–76. doi:10.1016/j.cell.2006.07.024
48. Andrechek ER. HER2/Neu tumorigenesis and metastasis is regulated by E2F activator transcription factors. Oncogene (2015) 34:217–25. doi:10.1038/onc.2013.540
49. Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, et al. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci U S A (2001) 98:4510–5. doi:10.1073/pnas.081074898
50. Hartl M, Nist A, Khan MI, Valovka T, Bister K. Inhibition of Myc-induced cell transformation by brain acid-soluble protein 1 (BASP1). Proc Natl Acad Sci U S A (2009) 106:5604–9. doi:10.1073/pnas.0812101106
51. Jackstadt R, Hermeking H. AP4 is required for mitogen- and c-MYC-induced cell cycle progression. Oncotarget (2014) 5:7316–27. doi:10.18632/oncotarget.2348
52. Fiaschetti G, Castelletti D, Zoller S, Schramm A, Schroeder C, Nagaishi M, et al. Bone morphogenetic protein-7 is a MYC target with prosurvival functions in childhood medulloblastoma. Oncogene (2011) 30:2823–35. doi:10.1038/onc.2011.10
53. Yin XY, Grove L, Datta NS, Katula K, Long MW, Prochownik EV. Inverse regulation of cyclin B1 by c-MYC and p53 and induction of tetraploidy by cyclin B1 overexpression. Cancer Res (2001) 61:6487–93.
54. Kozar K, Ciemerych MA, Rebel VI, Shigematsu H, Zagozdzon A, Sicinska E, et al. Mouse development and cell proliferation in the absence of D-cyclins. Cell (2004) 118:477–91. doi:10.1016/j.cell.2004.07.025
55. Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature (1997) 387:422–6. doi:10.1038/387422a0
56. Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature (1996) 382:511–7. doi:10.1038/382511a0
57. Miliani de Marval PL, Macias E, Rounbehler R, Sicinski P, Kiyokawa H, Johnson DG, et al. Lack of cyclin-dependent kinase 4 inhibits c-myc tumorigenic activities in epithelial tissues. Mol Cell Biol (2004) 24:7538–47. doi:10.1128/MCB.24.17.7538-7547.2004
58. Valovka T, Schönfeld M, Raffeiner P, Breuker K, Dunzendorfer-Matt T, Hartl M, et al. Transcriptional control of DNA replication licensing by Myc. Sci Rep (2013) 3:3444. doi:10.1038/srep03444
59. Wood LJ, Mukherjee M, Dolde CE, Xu Y, Maher JF, Bunton TE, et al. HMG-I/Y, a new c-MYC target gene and potential oncogene. Mol Cell Biol (2000) 20:5490–502. doi:10.1128/MCB.20.15.5490-5502.2000
60. Teng SC, Chen YY, Su YN, Chou PC, Chiang YC, Tseng SF, et al. Direct activation of HSP90A transcription by c-MYC contributes to c-MYC-induced transformation. J Biol Chem (2004) 279:14649–55. doi:10.1074/jbc.M308842200
61. Yustein JT, Liu YC, Gao P, Jie C, Le A, Vuica-Ross M, et al. Induction of ectopic Myc target gene JAG2 augments hypoxic growth and tumorigenesis in a human B-cell model. Proc Natl Acad Sci U S A (2010) 107:3534–9. doi:10.1073/pnas.0901230107
62. Osthus RC, Karim B, Prescott JE, Smith BD, McDevitt M, Huso DL, et al. The Myc target gene JPO1/CDCA7 is frequently overexpressed in human tumors and has limited transforming activity in vivo. Cancer Res (2005) 65:5620–7. doi:10.1158/0008-5472.CAN-05-0536
63. Lewis BC, Prescott JE, Campbell SE, Shim H, Orlowski RZ, Dang CV. Tumor induction by the c-MYC target genes rcl and lactate dehydrogenase A. Cancer Res (2000) 60:6178–83.
64. Grabow S, Delbridge AR, Aubrey BJ, Vandenberg CJ, Strasser A. Loss of a single Mcl-1 allele inhibits MYC-driven lymphomagenesis by sensitizing pro-B cells to apoptosis. Cell Rep (2016) 14:2337–47. doi:10.1016/j.celrep.2016.02.039
65. Tsuneoka M, Teye K, Arima N, Soejima M, Otera H, Ohashi K, et al. A novel Myc-target gene, mimitin, that is involved in cell proliferation of esophageal squamous cell carcinoma. J Biol Chem (2005) 280:19977–85. doi:10.1074/jbc.M501231200
66. Zhang XY, DeSalle LM, Patel JH, Capobianco AJ, Yu D, Thomas-Tikhonenko A, et al. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc Natl Acad Sci U S A (2005) 102:13968–73. doi:10.1073/pnas.0502330102
67. Rogulski KR, Cohen DE, Corcoran DL, Benos PV, Prochownik EV. Deregulation of common genes by c-MYC and its direct target, MT-MC1. Proc Natl Acad Sci U S A (2005) 102:18968–73. doi:10.1073/pnas.0507902102
68. Li Z, Boone D, Hann SR. Nucleophosmin interacts directly with c-MYC and controls c-MYC-induced hyperproliferation and transformation. Proc Natl Acad Sci U S A (2008) 105:18794–9. doi:10.1073/pnas.0806879105
69. Nilsson JA, Keller UB, Baudino TA, Yang C, Norton S, Old JA, et al. Targeting ornithine decarboxylase in Myc-induced lymphomagenesis prevents tumor formation. Cancer Cell (2005) 7:433–44. doi:10.1016/j.ccr.2005.03.036
70. D’Artista L, Bisso A, Piontini A, Doni M, Verrecchia A, Kress TR, et al. Pin1 is required for sustained B cell proliferation upon oncogenic activation of Myc. Oncotarget (2016). doi:10.18632/oncotarget.7846
71. Orre RS, Cotter MA II, Subramanian C, Robertson ES. Prothymosin alpha functions as a cellular oncoprotein by inducing transformation of rodent fibroblasts in vitro. J Biol Chem (2001) 276:1794–9. doi:10.1074/jbc.M008560200
72. Wonsey DR, Zeller KI, Dang CV. The c-MYC target gene PRDX3 is required for mitochondrial homeostasis and neoplastic transformation. Proc Natl Acad Sci U S A (2002) 99:6649–54. doi:10.1073/pnas.102523299
73. Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature (2015) 523:96–100. doi:10.1038/nature14351
74. O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, et al. Activation of transferrin receptor 1 by c-MYC enhances cellular proliferation and tumorigenesis. Mol Cell Biol (2006) 26:2373–86. doi:10.1128/MCB.26.6.2373-2386.2006
75. Ben-Porath I, Yanuka O, Benvenisty N. The tmp gene, encoding a membrane protein, is a c-MYC target with a tumorigenic activity. Mol Cell Biol (1999) 19:3529–39. doi:10.1128/MCB.19.5.3529
76. Reiter F, Hartl M, Karagiannidis AI, Bister K. WS5, a direct target of oncogenic transcription factor Myc, is related to human melanoma glycoprotein genes and has oncogenic potential. Oncogene (2007) 26:1769–79. doi:10.1038/sj.onc.1209975
77. Fogal V, Babic I, Chao Y, Pastorino S, Mukthavaram R, Jiang P, et al. Mitochondrial p32 is upregulated in Myc expressing brain cancers and mediates glutamine addiction. Oncotarget (2015) 6:1157–70. doi:10.18632/oncotarget.2708
78. Barsyte-Lovejoy D, Lau SK, Boutros PC, Khosravi F, Jurisica I, Andrulis IL, et al. The c-MYC oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res (2006) 66:5330–7. doi:10.1158/0008-5472.CAN-06-0037
79. Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science (1999) 283:676–9. doi:10.1126/science.283.5402.676
80. Li J, Kretzner L. The growth-inhibitory Ndrg1 gene is a Myc negative target in human neuroblastomas and other cell types with overexpressed N- or c-myc. Mol Cell Biochem (2003) 250:91–105. doi:10.1023/A:1024918328162
81. Fog CK, Asmar F, Côme C, Jensen KT, Johansen JV, Kheir TB, et al. Loss of PRDM11 promotes MYC-driven lymphomagenesis. Blood (2015) 125:1272–81. doi:10.1182/blood-2014-03-560805
82. Rogulski K, Li Y, Rothermund K, Pu L, Watkins S, Yi F, et al. Onzin, a c-MYC-repressed target, promotes survival and transformation by modulating the Akt-Mdm2-p53 pathway. Oncogene (2005) 24:7524–41. doi:10.1038/sj.onc.1208897
83. Watnick RS, Rodriguez RK, Wang S, Blois AL, Rangarajan A, Ince T, et al. Thrombospondin-1 repression is mediated via distinct mechanisms in fibroblasts and epithelial cells. Oncogene (2015) 34:2823–35. doi:10.1038/onc.2014.228
84. Shen L, O’Shea JM, Kaadige MR, Cunha S, Wilde BR, Cohen AL, et al. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc Natl Acad Sci U S A (2015) 112:5425–30. doi:10.1073/pnas.1501555112
85. Kim T, Cui R, Jeon YJ, Fadda P, Alder H, Croce CM. MYC-repressed long noncoding RNAs antagonize MYC-induced cell proliferation and cell cycle progression. Oncotarget (2015) 6:18780–9. doi:10.18632/oncotarget.3909
86. Kim T, Cui R, Jeon YJ, Lee JH, Lee JH, Sim H, et al. Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc Natl Acad Sci U S A (2014) 111:4173–8. doi:10.1073/pnas.1400350111
87. Hart JR, Roberts TC, Weinberg MS, Morris KV, Vogt PK. MYC regulates the non-coding transcriptome. Oncotarget (2014) 5:12543–54. doi:10.18632/oncotarget.3033
88. Frenzel A, Lovén J, Henriksson MA. Targeting MYC-regulated miRNAs to combat cancer. Genes Cancer (2010) 1:660–7. doi:10.1177/1947601910377488
89. Bui TV, Mendell JT. Myc: maestro of microRNAs. Genes Cancer (2010) 1:568–75. doi:10.1177/1947601910377491
90. Song R, Sponer N, He L. Methods to quantify microRNAs in the Myc gene network for posttranscriptional gene repression. Methods Mol Biol (2013) 1012:135–44. doi:10.1007/978-1-62703-429-6_10
91. Tao J, Zhao X, Tao J. c-MYC-miRNA circuitry: a central regulator of aggressive B-cell malignancies. Cell Cycle (2014) 13:191–8. doi:10.4161/cc.27646
92. Fontana L, Fiori ME, Albini S, Cifaldi L, Giovinazzi S, Forloni M, et al. Antagomir-17-5p abolishes the growth of therapy-resistant neuroblastoma through p21 and BIM. PLoS One (2008) 3:e2236. doi:10.1371/journal.pone.0002236
93. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet (2009) 10:704–14. doi:10.1038/nrg2634
94. Fisher R, Pusztai L, Swanton C. Cancer heterogeneity: implications for targeted therapeutics. Br J Cancer (2013) 108:479–85. doi:10.1038/bjc.2012.581
95. Vermeersch KA, Wang L, McDonald JF, Styczynski MP. Distinct metabolic responses of an ovarian cancer stem cell line. BMC Syst Biol (2014) 8:134. doi:10.1186/s12918-014-0134-y
96. Hart JR, Zhang Y, Liao L, Ueno L, Du L, Jonkers M, et al. The butterfly effect in cancer: a single base mutation can remodel the cell. Proc Natl Acad Sci U S A (2015) 112:1131–6. doi:10.1073/pnas.1424012112
97. Foster KW, Ren S, Louro ID, Lobo-Ruppert SM, McKie-Bell P, Grizzle W, et al. Oncogene expression cloning by retroviral transduction of adenovirus E1A-immortalized rat kidney RK3E cells: transformation of a host with epithelial features by c-MYC and the zinc finger protein GKLF1. Cell Growth Differ (1999) 10:423–34.
98. Felts KA, Chen K, Zaharee K, Sundar L, Limjoco J, Miller A, et al. Functional cloning using pFB retroviral cDNA expression libraries. Mol Biotechnol (2002) 22:25–32. doi:10.1385/MB:22:1:025
99. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature (2007) 448:561–6. doi:10.1038/nature05945
100. Choi YL, Soda M, Ueno T, Hamada T, Haruta H, Yamoto A, et al. Oncogenic MAP2K1 mutations in human epithelial tumors. Carcinogenesis (2012) 33:956–61. doi:10.1093/carcin/bgs099
101. Loftus SK, Larson DM, Watkins-Chow D, Church DM, Pavan WJ. Generation of RCAS vectors useful for functional genomic analyses. DNA Res (2001) 8:216–21. doi:10.1093/dnares/8.5.221
102. Gao SY, Jack MM, O’Neill C. Towards optimising the production of and expression from polycistronic vectors in embryonic stem cells. PLoS One (2012) 7:e48668. doi:10.1371/journal.pone.0048668
103. Charpentier E, Marraffini LA. Harnessing CRISPR-Cas9 immunity for genetic engineering. Curr Opin Microbiol (2014) 19C:114–9. doi:10.1016/j.mib.2014.07.001
104. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science (2014) 346:6213. doi:10.1126/science.1258096
105. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget (2011) 2:135–64. doi:10.18632/oncotarget.240
106. Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem (2016) 109:314–41. doi:10.1016/j.ejmech.2016.01.012
107. Baratta MG, Schinzel AC, Zwang Y, Bandopadhayay P, Bowman-Colin C, Kutt J, et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc Natl Acad Sci U S A (2015) 112:232–7. doi:10.1073/pnas.1422165112
108. Liu Y, Li X, Zhu S, Zhang JG, Yang M, Qin Q, et al. Ectopic expression of miR-494 inhibited the proliferation, invasion and chemoresistance of pancreatic cancer by regulating SIRT1 and c-Myc. Gene Ther (2015) 22:729–38. doi:10.1038/gt.2015.39
109. Annibali D, Whitfield JR, Favuzzi E, Jauset T, Serrano E, Cuartas I, et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat Commun (2014) 5:4632. doi:10.1038/ncomms5632
110. Jung KY, Wang H, Teriete P, Yap JL, Chen L, Lanning ME, et al. Perturbation of the c-MYC-Max protein-protein interaction via synthetic α-helix mimetics. J Med Chem (2015) 58:3002–24. doi:10.1021/jm501440q
111. Marfil V, Blazquez M, Serrano F, Castell JV, Bort R. Growth-promoting and tumourigenic activity of c-MYC is suppressed by Hhex. Oncogene (2015) 34:3011–22. doi:10.1038/onc.2014.240
112. Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, et al. Inhibitor of MYC identified in a Kröhnke pyridine library. Proc Natl Acad Sci U S A (2014) 111:12556–61. doi:10.1073/pnas.1319488111
113. Raffeiner P, Röck R, Schraffl A, Hartl M, Hart JR, Janda KD, et al. In vivo quantification and perturbation of Myc-Max interactions and the impact on oncogenic potential. Oncotarget (2014) 5:8869–78. doi:10.18632/oncotarget.2588
114. Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov (2011) 10:351–64. doi:10.1038/nrd3374
115. Lin CJ, Nasr Z, Premsrirut PK, Porco JA Jr, Hippo Y, Lowe SW, et al. Targeting synthetic lethal interactions between Myc and the eIF4F complex impedes tumorigenesis. Cell Rep (2012) 1:325–33. doi:10.1016/j.celrep.2012.02.010
116. Kang J, Sergio CM, Sutherland RL, Musgrove EA. Targeting cyclin-dependent kinase 1 (CDK1) but not CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast cancer cells. BMC Cancer (2014) 14:32. doi:10.1186/1471-2407-14-32
Keywords: signal transduction, transcription, genetic, oncogenes, tumor suppressor, carcinogenesis
Citation: Hartl M (2016) The Quest for Targets Executing MYC-Dependent Cell Transformation. Front. Oncol. 6:132. doi: 10.3389/fonc.2016.00132
Received: 07 April 2016; Accepted: 20 May 2016;
Published: 02 June 2016
Edited by:
Francois X. Claret, The University of Texas MD Anderson Cancer Center, USAReviewed by:
Alexandre Arcaro, University of Bern, SwitzerlandMartha Ruskin Stampfer, University of California, USA
Giovanni Blandino, Regina Elena National Cancer Institute, Italy
Copyright: © 2016 Hartl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Markus Hartl, bWFya3VzLmhhcnRsQHVpYmsuYWMuYXQ=