 Haim Werner
Haim Werner Rive Sarfstein
Rive Sarfstein Derek LeRoith
Derek LeRoith Ilan Bruchim
Ilan Bruchim- 1Department of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel
- 2Yoran Institute for Human Genome Research, Tel Aviv University, Tel Aviv, Israel
- 3Diabetes and Metabolism Clinical Research Center, Rambam Health Care Center, Haifa, Israel
- 4Department of Obstetrics and Gynecology, Hillel Yaffe Medical Center, Hadera, Israel
Clinical, epidemiological, and experimental evidence indicate that the insulin-like growth factors (IGFs) are important mediators in the biochemical chain of events that lead from a phenotypically normal to a neoplastic cell. The IGF1 receptor (IGF1R), which mediates the biological actions of IGF1 and IGF2, exhibits potent pro-survival and antiapoptotic activities. The IGF1R is highly expressed in most types of cancer and is regarded as a promising therapeutic target in oncology. p53 is a transcription factor with tumor suppressor activity that is usually activated in response to DNA damage and other forms of cellular stress. On the basis of its protective activities, p53 is commonly regarded as the guardian of the genome. We provide evidence that the IGF signaling axis and p53 genome protection pathways are tightly interconnected. Wild-type, but not mutant, p53 suppresses IGF1R gene transcription, leading to abrogation of the IGF signaling network, with ensuing cell cycle arrest. Gain-of-function, or loss-of-function, mutations of p53 in tumor cells may disrupt its inhibitory activity, thus generating oncogenic molecules capable of transactivating the IGF1R gene. The interplay between the IGF1 and p53 pathways is also of major relevance in terms of metabolic regulation, including glucose transport and glycolysis. A better understanding of the complex physical and functional interactions between these important signaling pathways will have major basic and translational relevance.
The Insulin-Like Growth Factor Network: Ligands, Receptors, and Binding Proteins
The processes of growth, development, and cell death are tightly regulated by multiple cellular and secreted factors that, in a highly orchestrated fashion, control the stage- and tissue-specific expression of a wide array of genes. Disruption of this finely tuned genetic program may lead to a pathological phenotype, including tumor formation. Modern biological research is aimed at dissecting physiological and pathological processes at defined levels of regulation and tackling biological questions in a comprehensive and integrated manner.
The insulin-like growth factors (IGF1 and IGF2) are a family of mitogenic peptides with important roles in diverse aspects of body function (1). The mature circulating IGF1 and IGF2 contain, respectively, 70 and 67 amino acids and display a marked similarity to proinsulin. Both peptides contain A and B domains, analogous to the A and B chains of insulin. However, unlike insulin, IGF1 and IGF2 retain the C-peptide, which is absent in the mature insulin molecule. For more than 50 years, the IGFs have attracted enormous scientific and clinical interest. This broad attention emanates from the appreciation that IGFs mediate fundamental biological processes at multiple ontogenetic stages (e.g., embryonic, infancy, adolescence, adulthood) and at virtually every level of organization (e.g., cells, tissues, organs, organisms) (2–5).
Insulin-like growth factors were originally identified as liver-secreted hormones, primarily involved in mediating the endocrine actions of growth hormone (GH, somatotropin) (6). Appropriately, the term somatomedin was initially adopted. Subsequently, the IGFs were conclusively demonstrated to be synthesized by most extrahepatic organs, sites in which they exhibit autocrine and paracrine modes of action. As explained in the next section, IGF1 is a progression factor that is needed to advance throughout the various phases of the cell cycle. The biological actions of IGF1 and IGF2 are mediated via activation of the IGF1 receptor (IGF1R), a transmembrane heterotetramer whose cytoplasmic tyrosine kinase domain is linked to the ras–raf–MAPK and PI3K–PKB/Akt signal transduction cascades (7–9). The IGF1R exhibits marked structural and functional homology to the insulin receptor (INSR), a finding that reflects a common evolutionary origin (10). The divergent biological activities of INSR and IGF1R are described in the next section. Unlike insulin that circulates in a free form in serum, IGF1 and IGF2 are bound in the circulation and extracellular spaces to a family of IGF-binding proteins (IGFBP1–6). IGFBPs control the bioavailability of both IGFs by modulating their release, transport, and degradation (11). The various components of the IGF network are illustrated in Figure 1.
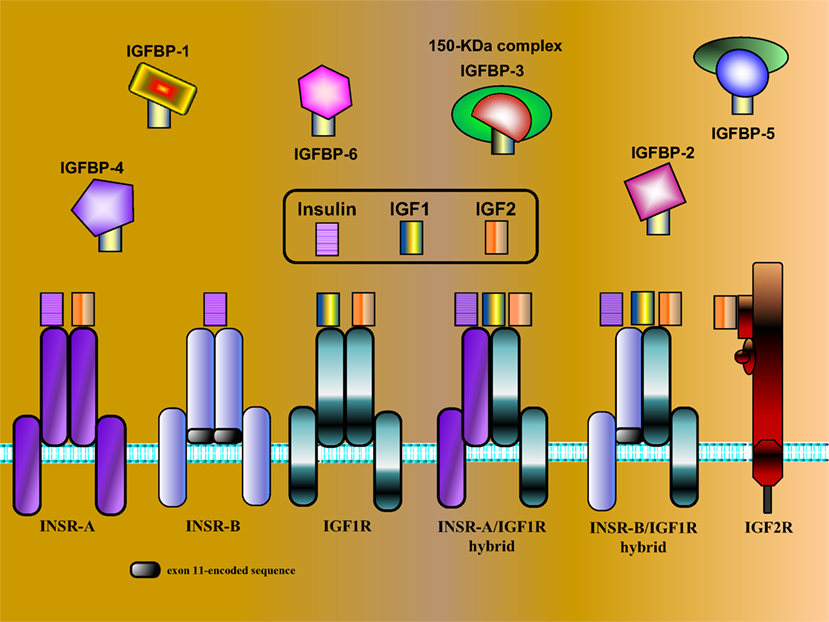
Figure 1. Schematic representation of IGF network components. The IGF system is comprised of three ligands (insulin, IGF1, and IGF2), three typical cell-surface receptors [insulin receptor (INSR), IGF1 receptor (IGF1R), and IGF2 receptor (IGF2R)], and at least six IGF-binding proteins (IGFBP1–6). The INSR has two isoforms, INSR-A and INSR-B, which differ in the absence or presence, respectively, of exon 11-encoded sequences. In addition to the typical receptors, naturally occurring hybrid receptors have been described in which an α/β INSR hemi-receptor is linked to an α/β IGF1R hemi-receptor. The IGF2R is a single-chain polypeptide composed of 15 repeat sequences and a short cytoplasmic domain. The IGF2R is homologous to the mannose 6-phosphate receptor and is involved in the recycling of lysosomal enzymes. IGFBP3 is the most abundant IGFBP in serum, and it is usually present as a ternary complex that includes the ligand and an acid-labile subunit (ALS). IGF bioavailability is also modulated by IGFBP proteases that cleave IGFBPs in a tissue-specific manner.
IGFs Play Key Roles in Homeostasis Regulation: Physiological and Pathological Aspects
The concept that INSR activation (mainly by insulin) leads primarily to metabolic activities while IGF1R activation (mainly by IGF1 or IGF2) leads to proliferative events was the prevalent dogma for more than 45 years (12, 13). These beliefs have been challenged in recent years due, in part, to the availability of transgenic and knockout animal models with organ-specific disruption or, alternatively, overexpression of IGF axis components. Whereas the general notion, in broad terms, is still regarded as correct, there is ample evidence in support of the view that INSR is also capable of mediating proliferative types of activities while, on the other hand, IGF1R is responsible for specific metabolic actions (14–16).
The structural and functional homology between INSR and IGF1R suggest that both molecules are derived from a common ancestral precursor that probably participated in food intake and regulation of cellular growth (17). A divergence of functions most likely occurred before the appearance of the first vertebrates (18). However, in view of their common evolutionary origins and semi-conserved architecture, there is a certain degree of cross talk between the insulin and IGF ligands and their receptors (19).
As mentioned above, IGF1 stimulates mitogenic responses and inhibits cell death in a wide variety of cell types (20). Quiescent cells in G0 can be induced to enter G1 by competence factors (e.g., PDGF, bFGF). Once the cell enters into G1, sub-physiological doses of IGF1 will allow the cell to evade arrest in G1 and to progress through the cell cycle (21), hence complying with the definition of progression factors (22). IGF1 exhibits a variety of cellular functions, including regulation of hormone synthesis and secretion, chemoattractant migration, immune cell recognition, and neuromodulation. Metabolic effects of IGF1 include, among others, elevation of glucose uptake and hypoglycemia, without lowering free fatty acid levels (1, 19). In addition, IGF1 improves renal function by increasing renal blood flow and glomerular filtration rate (23). The cardinal role of the IGF axis in growth and development was demonstrated by the severe growth deficits observed in mice in which components of the IGF system, each one individually and in combination, were disrupted by homologous recombination (24).
The IGF1R: A Potent Cell Survival Mediator
Clinical and experimental studies conducted since the early 1980s have established that nearly all human tumors display augmented IGF1R concentrations (25, 26). Elevated receptor levels are correlated with enhanced IGF1 and IGF2 binding and amplified IGF1R activation (phosphorylation). However, the levels of expression of the IGF1R gene as a determinant of IGF action and, in particular, the pathological significance of IGF1R overexpression, are still open questions (27–32). The archetypal features of the IGF1R include the following.
[1] potent antiapoptotic and cell survival capacities;
[2] critical roles in invasion, metastasis, and angiogenesis;
[3] contribution to malignant transformation (4, 5).
Experimental corroboration in favor of a critical role for the IGF1R in oncogenesis was provided by studies showing that fibroblasts derived from IGF1R knockout mice, with a few exceptions, do not undergo transformation when exposed to cellular or viral oncogenes (33, 34). However, it is important to emphasize that IGF1R, per se, is not oncogenic. In other words, the ligand-activated receptor is unable to induce cellular transformation. Furthermore, high IGF1R levels do not necessarily reflect the existence of a malignant phenotype. Thus, reduced circulating IGF1 values, such as those associated with congenital IGF1 deficiencies, usually lead to IGF1R upregulation. However, there is no evidence that these elevated IGF1R concentrations are correlated with a malignant phenotype (28). As mentioned above, IGF1 functions as a progression factor capable of “pushing” cells, including already transformed cells, through the cell cycle. Clinical and experimental data are consistent with the view that IGF1R expression and activation are fundamental prerequisites for acquisition of a malignant phenotype (10, 35). The converse scenario (i.e., that enhanced IGF1R gene expression in cancer is a consequence of the malignant phenotype) is, similarly, a biologically plausible theory that merits consideration. The molecular mechanisms responsible for the regulation of IGF1R gene expression are described in the next section.
Transcriptional and Epigenetic Regulation of the IGF1R Gene
Investigation of the IGF1R gene promoter is helping define the functional and physical foundations for transcriptional control of the gene. Comprehensive promoter analyses generated valuable information regarding cis-elements as well as trans-acting factors that are responsible for IGF1R gene expression under physiological and pathological conditions. Transcription rate of the IGF1R gene is primarily dependent on a number of stimulatory nuclear proteins, including zinc-finger protein Sp1 (36, 37), E2F1 (38), Krüppel-like factor-6 (KLF6) (39), high-mobility group AT-hook (HMGA1) (40), etc. Some of these transcription factors directly bind specific cis-elements, including arrays of GC boxes located in the proximal IGF1R promoter region (i.e., protein–DNA interactions), whereas other nuclear proteins interact with members of the basal transcription machinery (i.e., protein–protein interactions). Recent DNA affinity chromatography-based proteomic analyses using nuclear extracts of breast tumor cells along with biotin-labeled IGF1R promoter fragments, followed by mass spectroscopy, led to the identification of a large set of IGF1R promoter-binding transcription factors (41). These transcription factors fall into a number of functional categories:
[1] cytoskeleton-associated proteins;
[2] proteins involved in transcription and regulation of nucleic acid metabolism;
[3] proteins involved in nuclear stability, chromatin structure, cell cycle, and gene expression;
[4] proteins involved in DNA repair, breaking, replication, and cell death;
[5] proteins involved in RNA splicing and processing, and translation.
Hence, Bioinformatic analyses suggest that the IGF1R gene plays key roles in multiple, seemingly unrelated, cellular pathways.
The interplay between steroid hormones and the IGF1 axis is of major clinical relevance in specific types of cancer, in particular adult epithelial tumors with a strong endocrine background (42–44). In this context, nuclear receptors, such as the estrogen (ER) and androgen (AR) receptors, were shown to stimulate IGF1R gene expression via mechanisms that involve ER and AR binding to IGF1R promoter elements (45). On the other hand, mutant versions of AR were unable to enhance IGF1R promoter activity (46). Transcriptional regulation of the IGF1R gene by ER and AR is of major importance in the etiology of breast and prostate cancers, respectively (47, 48). In addition, the question whether DNA methylation is involved in epigenetic regulation of the IGF1R gene was recently investigated in a series of prostate cancer cell lines representing early or advanced (metastatic) stages of the disease. Results of methylation specific PCR, sodium bisulfite-direct DNA sequencing, and 5-Aza-2′-deoxycytidine experiments revealed that the IGF1R promoter is, most likely, not subject to DNA methylation at any stage of the disease (49).
Finally, a relevant question is whether genetic events might be linked to IGF1R overexpression in cancer. Most available clinical data support the notion that IGF1R gene mutations constitute a very rare event and no substantial evidence links IGF1R mutations and cancer. In fact, heterozygous IGF1R mutations were reported in cases of intrauterine and postnatal growth restriction, but not in association with cancer (28, 50). Similarly, IGF1R gene amplification has been reported only in a small number of breast cancer and melanoma cases, suggesting that this genetic event is not a common mechanism in malignancy.
p53: A Key Player in Genome Integrity Protection
p53 is a transcription factor with tumor suppressor activity that typically accumulates in the cell in response to DNA damage (51). In its hyperphosphorylated state, p53 is capable of arresting cell cycle progression at the G1 phase. Mutation of the p53 tumor suppressor gene is the most common event in human cancer (52, 53). The p53 pathway is activated in response to a wide spectrum of cellular stress signals. These insults include DNA damage and telomere shortening, hypoxia, low nucleoside triphosphate pool sizes, spindle damage, heat and cold shock, inflammation and nitric oxide production, and, finally, activation of oncogenes by mutations (54, 55). These various strains bear the potential to decrease the fidelity of cell cycle progression and DNA replication, hence leading to increased mutation rates (56). Accumulation of mutations constitutes an early event in cellular transformation and may, eventually, lead to the establishment of a cancerous phenotype. p53-mediated cell cycle arrest enables damaged DNA to be repaired before the replicative phase of the cell cycle (53, 57). Alternatively, p53 can elicit an apoptotic program. Based on these important protective roles, p53 is commonly regarded as the “guardian of the genome.” As described below, there is solid experimental and clinical data that suggest that p53 interacts with the IGF1 pathway at a number of levels, including (1) transcriptional regulation of IGF axis components, including the IGF1R gene; and (2) convergence of cytoplasmic and nuclear IGF1 and p53 signaling pathways. Finally, evidence assembled in recent years indicate that, in addition to its well-documented capacity to govern cell cycle progression, p53 activation also has a major impact on metabolic processes, including glucose transport (58) and obesity (59). This topic is described below.
Differential Regulation of IGF1R Gene Expression by Wild-Type and Mutant p53
The essential role of IGF1R in cell cycle progression and transformation led to the hypothesis that a potential mechanism by which the postmitotic, terminally differentiated cell kept out of the cell cycle may involve the constitutive inhibition of the IGF1R gene by wild-type forms of tumor suppressor genes (35). In accordance with this hypothesis, p53, the most frequently mutated tumor suppressor in human cancer, was identified as a bona fide negative regulator of the IGF1R gene. Cotransfection of a wild-type p53-encoding expression vector along with an IGF1R promoter luciferase reporter construct led to an ~90% suppression of IGF1R promoter activity (60, 61) (Figure 2). In contrast, tumor-derived mutant versions of p53-containing mutations at codons 143, 248, and 273 of the p53 molecule enhanced IGF1R promoter activity by 227, 319, and 406%, respectively.
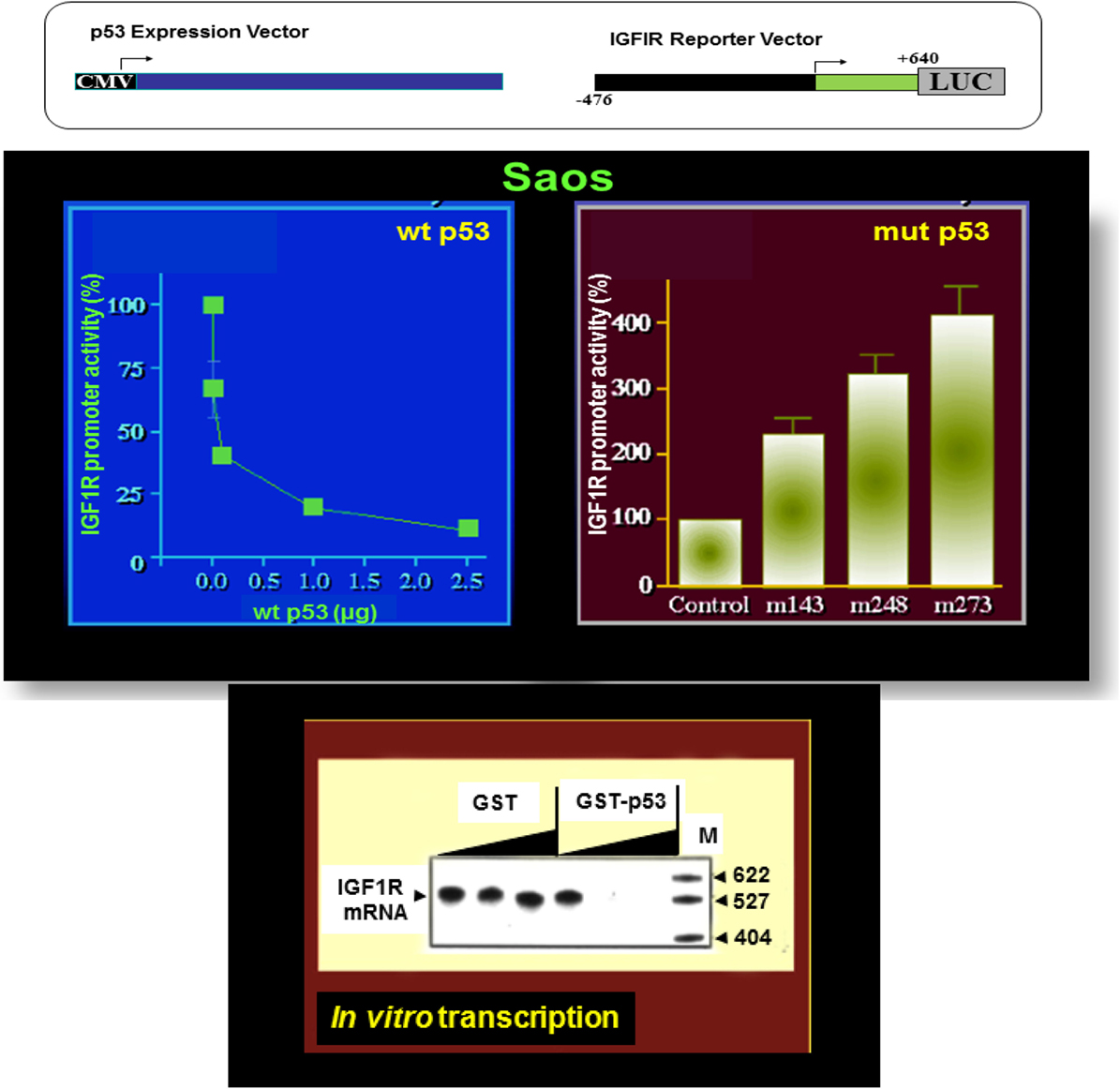
Figure 2. Differential regulation of IGF1R gene expression by wild-type and mutant p53. To evaluate the effect of p53 on IGF1R promoter activity, transient cotransfection experiments were performed in Saos-2 cells (an osteosarcoma cell line lacking endogenous p53) using a p53 expression vector along with an IGF1R promoter (nt −476 to +640) luciferase reporter construct (upper panel). Wild-type p53 suppressed IGF1R promoter activity in a dose-dependent manner, with maximal repression obtained with a dose of 2.5 μg DNA. Co-expression of tumor-derived mutant forms of p53 (bearing mutations at codons 143, 248, and 273) along with an IGF1R promoter led to transcriptional stimulation of the IGF1R gene. In vitro transcription assays using the purified wild-type p53 along with an IGF1R promoter template indicate that the effect of p53 was indeed mediated at the level of transcription.
Analyses aimed at defining the mechanistic basis of p53 regulation of IGF1R gene expression included, among others, transcription, mobility shift, and proteomic assays (41, 60, 61). Results of in vitro transcription assays using purified GST-p53 protein established that the tumor suppressor abolished transcription of an IGF1R gene template in a dose-dependent fashion (Figure 2). In addition, mobility shift assays indicate that p53 seems to exert its effects via protein–protein interactions with members of the basal transcription machinery, including the TATA-binding protein (TBP). The question whether p53 can directly bind IGF1R promoter DNA sequences is still under investigation. Proteomic analyses showed p53 binding to the IGF1R promoter, however, it is still unclear whether the tumor suppressor is part of a large multimeric protein complex or, alternatively, whether it binds IGF1R DNA in a sequence-specific manner (41). Combined data indicate that the mechanism of action of wild-type p53 involves transcriptional suppression of the IGF1R gene. Gain-of-function, or loss-of-function, mutations of p53 in tumor cells seem to disrupt its inhibitory activity, hence generating oncogenic molecules capable of transactivating the IGF1R gene. Because wild-type p53 is a potent inducer of apoptosis, we assume that the effect of p53 on apoptosis is mediated, at least in part, via suppression of the IGF1R promoter. Lack of IGF1R inhibition by mutant p53 molecules may help expand cancer cell populations that are otherwise destined to die (Figure 3).
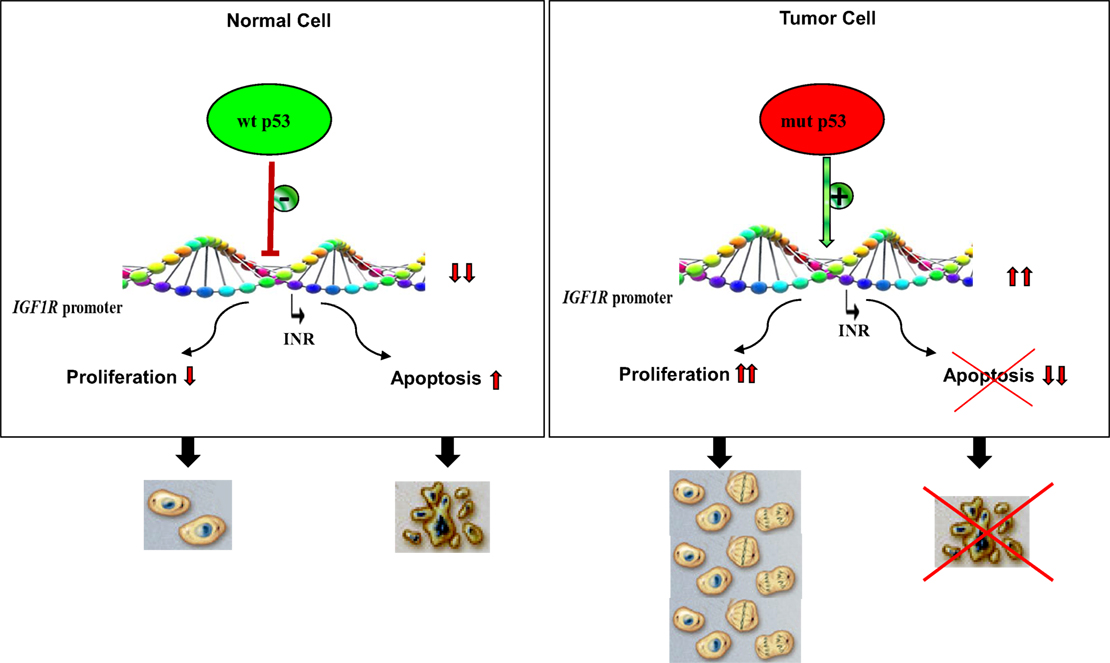
Figure 3. Impact of p53 status on cell proliferation and apoptosis. Under normal physiological conditions, activation of wild-type p53 following DNA damage, or other forms of cellular stress, may lead to stimulation or repression of various target genes. The IGF1R promoter has been identified as a bona fide target of p53. IGF1R transcription begins from a discrete promoter element termed initiator (INR). Transcriptional repression of IGF1R may lead to cell cycle arrest or, alternatively, increased apoptosis. p53 is the most frequently mutated gene in human cancer. Tumor cells containing mutant forms of p53 are typically expected to display large concentrations of IGF1R mRNA and cell-surface receptors. Increased IGF1 binding and IGF1R activation (phosphorylation) result in uncontrolled proliferation and/or abrogation of apoptosis, two critical traits of malignant cells.
Finally, wild-type p53 was also shown to inhibit transcription of the antiapoptotic IGF2 gene and to enhance transcription of the proapoptotic IGFBP3 (62, 63). Hence, tumor suppressor p53 governs the activity of the entire IGF network by modulating in a coordinated fashion the expression of ligands, receptors, and binding proteins. A schematic representation of the interactions between the IGF axis and p53 is presented in Figure 4.
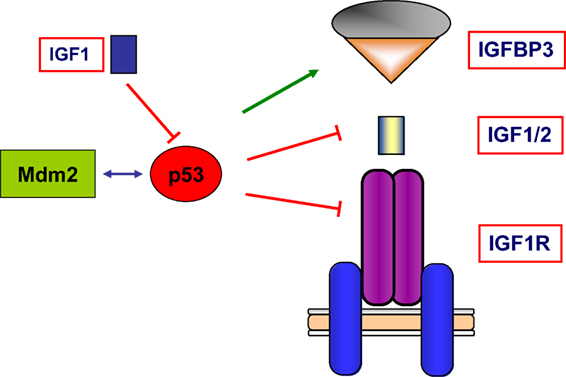
Figure 4. Schematic representation of the interplay between the IGF1 signaling axis and p53 genome protection pathway. p53 is activated in response to a broad spectrum of cellular insults and, in its hyperphosphorylated state, it can lead to cell cycle arrest and/or apoptosis. p53 was shown to inhibit transcription of the antiapoptotic genes IGF2 and IGF1R. On the other hand, p53 enhanced expression of the proapoptotic gene IGFBP3. Thus, p53 is capable of regulating the activity of the entire IGF signaling axis by controlling expression and activity of ligands, receptors, and IGFBPs in a coordinated fashion. The abundance and activity of p53 itself was shown to be regulated by IGF1, which induced p53 degradation in an Mdm2-dependent fashion.
Regulation of IGF1R Gene Expression by p53 Homologs
Studies have identified a family of proteins that are structurally and functionally related to p53. Members of this family, specifically p63 and p73, retain the basic features of the p53 protein, including an acidic, N-terminal transactivation domain, a central DNA-binding domain, and a C-terminal oligomerization domain (64–66). In addition, p63 and p73 exhibit some of the biological properties of p53, including the ability to recognize and bind p53 target sequences, transactivate p53-responsive genes, and induce apoptosis. However, unlike p53, the genetic structures of p63 and p73 are extremely complex, leading to the synthesis of several isoforms. To investigate whether novel members of the p53 family share the paradigm of p53 suppression of the IGF1R gene, the regulation of IGF1R by p63 and p73 was evaluated in colon cancer cells. Results of coexpression studies using the various p63 and p73 isoforms demonstrate that p63/p73 proteins suppressed IGF1R promoter activity in a dose-dependent manner, suggesting an anti-oncogenic role for p53 homologs (67). On the other hand, mutant proteins were impaired in their ability to inhibit the IGF1R gene.
Although the multiplicity of p63/p73 isoforms precludes any generalization regarding their roles in cancer biology, it is evident that this family of p53 homologs is involved in acquisition and maintenance of a malignant phenotype (64, 65). As depicted above for p53, negative regulation of IGF1R by p63/p73 leads to diminished IGF binding, a characteristic feature of terminally differentiated cells. On the other hand, disruption of p53/p63/p73 pathways in cancer cells may result in impaired suppression of IGF1R transcription, with enhanced binding and cell-surface receptor activation by endocrine or locally produced IGF1 and/or IGF2.
Convergence of Tumor Suppressor p53 and IGF1 Signaling Pathways
Early studies suggested a potential convergence of the p53 and IGF1 signaling pathways (68). Binding of IGF1 to the IGF1R results in the recruitment and activation of PI3K to the plasma membrane receptor and activation of the Akt protein kinase. Akt has several antiapoptotic substrates, such as BAD and Mdm2 (69). Akt is translocated to the cell nucleus where it phosphorylates forkhead transcription factors, leading to antiapoptotic signaling and cell growth (70). PI3K activity is counteracted by PTEN, a lipid phosphatase. PTEN functions as a tumor suppressor and, similar to p53, is frequently mutated in breast cancer as well as other sporadic and familial malignancies (56). p53 regulates PTEN expression, while PTEN and inhibitors of Akt signaling upregulate p53 expression (71). As described below, the functional and physical connections between the IGF1 signaling pathways and tumor suppressor p53 take place at multiple levels of biological regulation (e.g., transcription, translation, stability, etc.) and occur in several cellular compartments (e.g., nucleus, cytoplasm).
The ubiquitin ligase Mdm2 is of primary importance in regulation of p53 activity (72), and IGF1 was shown to induce p53 degradation in an Mdm2-dependent manner (73). Girnita et al. have shown that Mdm2 physically associates with IGF1R and causes IGF1R ubiquitination and degradation (74). Mdm2 serves as a ligase in ubiquitination of the IGF1R and thereby causes its degradation by the proteasome system. Consequently, by sequestering Mdm2 in the cell nuclei, the level of p53 may indirectly influence the expression of IGF1R. This function of Mdm2 and p53 constitutes a potential mechanism for the regulation of IGF1R and cell growth. Various other mechanisms and signaling pathways have been suggested to participate in the convergent tasks of p53 and the IGF system and are thus regarded as potential therapeutic targets (75). This multifocal signal modulation therapy includes, in addition to the IGF1 axis and the Mdm2–p53 loop, additional targets, including the epidermal growth factor receptor, mammalian target of rapamycin, AR and ER, NFkB, etc.
An intact p53 signaling pathway is a critical prerequisite for many of the biological actions of IGF1. KLF6, a zinc-finger of the Krüppel-like family, was identified as a transcription factor involved in the regulation of genes associated with response to injury (76, 77). In addition, KLF6 plays a tumor suppressor role in colon and prostate cancer (78). IGF1 was shown to stimulate KLF6 gene transcription in cells with normal, but not disrupted, p53 (79). These results identify the KLF6 gene as a downstream target for IGF1 action and suggest that the mechanism of action of IGF1 requires an intact p53 pathway. An additional example of the critical need for p53 in IGF1 action is provided by folic acid, a member of the vitamin B family. Folic acid exhibits chemopreventive activity, and part of the antiproliferative action of this micronutrient can be attributed to its ability to inhibit IGF1R gene expression in a p53-dependent manner (80). The protective effect of folic acid was abrogated in cells lacking p53.
Of major interest, a recent study identified the GH gene as a direct transcriptional target of p53. Pituitary adenomas secreting GH are always benign and exhibit DNA damage and a senescent phenotype. The laboratory of Shlomo Melmed tested the effect of nutlin-induced p53-mediated senescence in pituitary cells (81). The authors showed that DNA damage induced by nutlin triggers the p53 senescent pathway, with subsequent induction of intracellular pituitary GH. In contrast, GH was not induced in p53-null cells. p53 was capable of binding specific GH promoter motifs and enhanced GH gene transcription and secretion in senescent pituitary adenoma cells. In summary, intracrine GH acts in pituitary cells as an apoptosis switch for p53-mediated senescence, likely protecting the pituitary adenoma from progression to malignancy. Given the key role of GH in regulation of IGF1 biosynthesis and secretion, the identification of the GH gene as a novel target for p53 action is consistent with the concept that the tumor suppressor constitutes an important systemic regulator of the entire GH–IGF1 endocrine axis.
IGF1, IGF1R, and DNA Damage
Evidence accumulated in recent years revealed a strong link between the IGF1R gene and radiosensitivity, defined as cell killing after exposure to ionizing radiation. IGF1R overexpression in fibroblasts conferred radioresistance and, conversely, addition of antisense oligomers against IGF1R mRNA reversed the radioresistant phenotype (82). Immunohistochemical analysis of primary breast tumors indicated that high levels of IGF1R correlated with ipsilateral tumor recurrence following lumpectomy and radiation therapy. Of interest, the only growth factor receptor that provided protection from ultraviolet-induced apoptosis in keratinocytes was the IGF1R (83). It is conceivable that the activated IGF1R may, on one hand, function as a survival factor for irradiated cells and, on the other hand, induce a postmitotic state that prevents passage of damaged DNA to daughter cells.
The ATM (ataxia-telangiectasia mutated) gene encodes a 350-kDa protein whose mutation in ataxia-telangiectasia, a genetic neurological disorder, leads to progressive neuronal degeneration, premature aging, immunological abnormalities, and an increased risk of cancer (84). The central role of the ATM protein in signaling DNA damage is now well established. Ionizing, but not ultraviolet, radiation enhances ATM kinase activity and phosphorylates a series of target proteins, including p53 and BRCA1, which, as described here, are involved in cell cycle control and repair of DNA damage (85). The potential role of the IGF1R gene as a target in an ATM-dependent pathway involved in regulating the radiation response was inferred from studies demonstrating that IGF1R levels were reduced in cells carrying mutations in the ATM gene (86).
Complementation of mutant cells with the ATM cDNA resulted in increased IGF1R promoter activity and elevated IGF1R levels (87). Furthermore, forced expression of the IGF1R in ataxia-telangiectasia cells conferred increased radioresistance. Hence, data indicate that the IGF1R gene is a novel downstream target in an ATM-mediated DNA damage response pathway. The fact that p53 has been characterized as a target for ATM action suggests that the regulation of IGF1R by ATM is, most probably, a p53-dependent process. Deregulated expression of the IGF1R gene after ionizing radiation is linked to genomic instability and increased cancer rates.
Finally, the laboratory of Valentine Macaulay has shown that downregulation of IGF1R in melanoma cells was associated with enhanced radiosensitivity (88). The authors also showed that IGF1R depletion delays repair of radiation-induced DNA double-strand breaks (DSB) and demonstrated that IGF1R affects DSB repair by modulating both major DSB repair pathways, i.e., non-homologous end-joining and homologous recombination repair (20, 89, 90).
Regulation of the IGF Axis by Genome Protection Genes: A Common Theme in Cell Biology
The identification of the IGF1R gene as a downstream target for members of the p53 family of genome protection genes led us to postulate the hypothesis that the expression and activity of the IGF1 axis, in general, and the IGF1R in particular, is governed by multiple families of negative regulators, i.e., tumor suppressor genes (35, 44). The rationale for this hypothesis lies in the fact that the IGF1R is usually overexpressed in tumors displaying loss-of function mutations of tumor suppressor genes (7–9, 26). In other words, our model proposes that the IGF1R gene constitutes a common downstream target for multiple tumor suppressors. While tumor suppressors might differ in their organ-specific expression, mechanisms of activation, type of tumors involved, and other parameters, they share the IGF1R pathway as a shared response path.
The breast and ovarian cancer susceptibility gene (BRCA1) is a transcription factor with well-defined roles in DNA damage repair, cell growth, and apoptosis (91, 92). Mutations in the BRCA1 gene are detected in a large proportion of families with inherited breast and/or ovarian cancer (93, 94). BRCA1 mutation carriers have up to 87% estimated cumulative risk of developing breast cancer by age 70. Consistent with its tumor suppressor role, forced expression of BRCA1 in breast cancer cells led to a marked reduction in endogenous IGF1R levels and promoter activity (95–97). In contrast, a mutant BRCA1 gene encoding a truncated version of the molecule (del185AG, a mutation with a high incidence among Ashkenazi Jews) had no effect on IGF1R expression. These results are consistent with the above postulated hypothesis, which proposes that the IGF1R gene is a downstream target for BRCA1 (and other tumor suppressors) action. Activation of BRCA1 in response to DNA damage, oxidative stress, or other cellular insults, may lead to a reduction in IGF1R levels and IGF action (98). Mobility shift assays performed with the full-length, in vitro-translated, BRCA1 failed to reveal binding of the protein to IGF1R promoter sequences. However, BRCA1 was shown to bind zinc-finger protein Sp1, a potent IGF1R gene transactivator, hence preventing it from binding to the IGF1R promoter.
A manifestation of the mechanistic interplay between BRCA1/BRCA2 and the IGF axis is seen in the clinics. Immunohistochemical analysis of IGF1R levels in breast tumor specimens derived from BRCA1/BRCA2 mutation carriers, compared to matched sporadic breast cancer patients, revealed higher IGF1R levels in tumors of BRCA mutation carriers (99). Furthermore, evidence in support of a complex interplay between the IGF1 axis and tumor suppressor BRCA1 was provided by studies showing that IGF1 increases BRCA1 gene expression and enhances BRCA1 promoter activity (100). Inhibitory control of IGF1R gene expression by BRCA1 may constitute a protection mechanism that prevents from normal breast cells from engaging in mitogenic activity. Lack of IGF1R inhibition by mutant BRCA1 may lead to enhanced IGF1R levels, an important prerequisite for malignant transformation (101).
Additional examples of tumor suppressor genes whose mechanisms of action involve transcriptional suppression of the IGF1R gene are the von-Hippel–Lindau (VHL) protein, a gene with important roles in the etiology of renal cancer (102), and WT1, a zinc-finger tumor suppressor with key roles in Wilm’s tumor, or nephroblastoma (103, 104). Of cardinal importance, the ability of WT1 to suppress IGF1R transcription is strictly dependent on the cellular status of p53. Thus, WT1 exerts its inhibitory role only in cells expressing a wild-type p53 gene, whereas it is unable to repress the IGF1R gene in cells with a mutant p53.
In summary, the IGF1R gene constitutes a common target for multiple oncogenes and antioncogenes. Inhibition of IGF1R expression and activation by negative growth regulators is expected to keep IGF1R levels below a certain threshold. We assume that low IGF1R levels are, for the most part, incompatible with the execution of mitogenic activities (Figure 3). p53 displays both direct and indirect roles in regulation of the IGF1R gene by virtue of its capacity to modulate the transcriptional activities of multiple transcription factors.
Can p53 Status Predict Responsiveness to IGF1R-Directed Targeted Therapies?
Given its strong pro-survival activity along with its universal expression in cancer cells, the IGF1R emerged in recent years as a promising therapeutic target in oncology (105–107). Unfortunately, results of phase I/II clinical trials have shown variable responses to IGF1R-directed therapies. The reasons for the failure to translate solid experimental and preclinical data into the clinics are complex and not fully explored. However, it has been suggested that most clinical trials using IGF1R inhibitors were conducted on unselected patients, and this fact had a negative impact on trials outcome. Therefore, identification of biomarkers that can predict response to targeted therapy is a major goal in current cancer treatment (108, 109).
While in certain cancers, including Ewing’s and rhabdomyosarcoma, tumor IGF1R levels were correlated with responsiveness to IGF1R-directed therapies, most clinical data seem to indicate that IGF1R expression levels, per se, do not predict sensitivity to IGF1R inhibition (110). Among a number of potential scenarios, it has been suggested that the specific molecular context and the complexity of the IGF1R–INSR hybrid receptors formation may explain part of the contradictory results. Likewise, circulating ligand and/or IGFBP levels are not always associated with the degree of anti-IGF1R effectiveness. Regarding the impact of IGF1R subcellular distribution on tumor phenotype and responsiveness to IGF1R-directed therapies, Aleksic et al. (111) reported the presence of nuclear IGF1R in primary renal cancer cells, formalin-fixed tumors, preinvasive lesions of the breast, and rapidly proliferating non-malignant tissues, and they also demonstrated that nuclear IGF1R was associated with poor prognosis in renal cancer. Asmane et al. (112) performed an immunohistochemical analysis of nuclear IGF1R in patients with unresectable or metastatic soft tissue sarcomas, Ewing’s sarcoma, and osteosarcoma treated with anti-IGF1R. In contrast to the previous study, exclusive intranuclear IGF1R presence was correlated with a better progression-free survival. Further studies are expected to shed light on this emerging aspect of IGF1R biology.
The impact of p53 mutational status on selective IGF1R-targeted therapies is of major translational relevance. A recent study has shown that picropodophyllin, an IGF1R inhibitor of the cyclolignan family, prevented the growth of wild-type, but not mutant, p53-expressing colorectal carcinoma cell lines (113). Likewise, cixutumumab, an IGF1R monoclonal antibody, inhibited proliferation of a uterine papillary serous carcinoma cell line expressing a wild-type p53 gene but had no effect on uterine cells containing a mutant p53 (114). In conclusion, the potential role of p53 as a biomarker for IGF1R-directed therapies in human cancers must be confirmed by large cell-based and patients’ analyses.
Finally, a number of obstacles must be resolved in order to translate the success of preclinical studies into the clinical setting. These difficulties are primarily due to the considerable similarity between the mature forms of IGF1R and INSR. The homology between the tyrosine kinase domains of both receptors reaches 84%, and the downstream pathways elicited by IGF1R and INSR are almost identical (14). The possible effect of IGF1R-targeted therapy on INSR action is of major concern. Impaired insulin signaling in classical insulin target organs (e.g., muscle, adipose tissue, etc.) may lead to metabolic complications, including development of insulin resistance. On the other hand and given the well-documented involvement of INSR in various types of cancer, in particular breast tumors, experts in the field advise the combined targeting of both IGF1R and INSR.
IGF1 and p53 Collaborate in Regulation of Metabolism
In addition to the well-established interplay between the IGF1 and p53 signaling pathways in the context of cell survival, proliferation, and cancer, as described above, evidence is mounting in support of novel homeostatic and metabolic activities of p53. Since its discovery in the mid-1950s, the insulin-like activities of IGF1 have been particularly well described. IGF1 has direct effects on fuel metabolism and, when given acutely, it enhances glucose uptake by muscle and abrogates hepatic glucose production (1). p53 has an important role in promoting oxidative phosphorylation (115). In addition, p53 has been shown to contribute to mitochondrial mass, a parameter of energetic expenditure (116), and cells expressing the tumor suppressor were shown to derive a larger portion of their ATP from oxidative phosphorylation, in comparison to cells devoid of p53 (117). Further evidence of p53 involvement in energy balance arises from the observation that, in addition to its nuclear localization, p53 is also present in mitochondria (118). It has been suggested that the mitochondrial presence of p53 in resting cells attests to the involvement of the tumor suppressor in normal mitochondrial activity.
p53 has an important role in the regulation of glycolysis. Most experimental evidence seems to indicate that, in agreement with its tumor suppressor role, p53 is capable of lowering glycolysis (119, 120). This activity can be regarded as an attempt by p53 to counter the acquisition of aerobic glycolysis usually associated with cancer cells (115). However, this intuitive rationalization is obscured by the finding that enhancement of glycolysis has also been associated, under certain circumstances, with p53 action. Of major interest, p53 has been identified as an important regulator of glucose transport, and wild-type p53 was shown to repress transcription of both GLUT1 and GLUT4 promoters in transfection assays (32). The inhibitory effect of wild-type p53 was abolished when cells were transfected with tumor-derived mutant versions of p53, harboring mutations in codons 143, 248, or 273. Evidence in support of a direct effect of p53 was provided by results of mobility shift assays showing binding of p53 to GLUT4 promoter sequences. These results suggest that the ability of p53 to prevent tumor initiation is, in part, mediated by its ability to inhibit glucose uptake and cell energy supply. In contrast, mutant p53s have an impaired capacity to repress GLUT1 and GLUT4 promoters. Of interest, p53 was also shown to promote the expression of gluconeogenesis-related genes (e.g., G6PC, PCK2, etc.) and to enhance hepatic glucose production (121). By facilitating glucose export, p53 may prevent it from being shunted to proliferative pathways, such as glycolysis.
In the context of lipid metabolism, both IGF1 and p53 have been linked to sterol regulatory element binding protein (SREBP) activity. SREBPs are transcription factors that bind to sterol regulatory element DNA sequences, leading to enhanced synthesis of enzymes involved in sterol biosynthesis. SREBPs are required for cholesterol and fatty acid biosynthesis. p53 was shown to be induced in adipocytes of ob/ob mice, leading to inhibition of SREBP-1 and lipogenic genes (122). Luciferase assays revealed that p53 expression suppressed the promoter activity of the SREBP-1c gene. Hence, p53 activation may constitute a negative feedback loop with important roles in prevention of fat accumulation in adipocytes. On the other hand, IGF1 was shown to stimulate SREBP expression and lipogenesis via activation of the PI3K pathway (123). Taken together, the interplay between the IGF1 and p53 pathways may have major implications in lipid metabolism and, probably, obesity.
Concluding Remarks
Overexpression of the IGF1R gene constitutes a common trait of many, if not most, tumors. The expression of the IGF1R gene is determined, to a large extent, at the transcriptional level, and the IGF1R promoter has been identified as a molecular target to a family of stimulatory transcription factors as well as nuclear proteins with tumor suppressor activity. Tumor suppressor p53 is the most frequently mutated molecule in human cancer. p53 is capable of arresting cell cycle progression at the G1 phase, thus enabling damaged DNA to be repaired before the replicative phase of the cell cycle. Alternatively, p53 can elicit an apoptotic program. In the present review article, we provided evidence that the mechanisms of action of wild-type p53 and p53 homologs (e.g., p63, p73) involve transcriptional suppression of the IGF1R gene. Gain-of-function or loss-of-function mutations of p53 disrupt its inhibitory activity and generate potentially oncogenic molecules capable of transactivating the IGF1R gene. While the interplay between the IGF1 signaling axis and the p53 genome protection pathways has been primarily investigated in the context of cancer cells, new evidence links these converging networks to the regulation of metabolism, including glucose transport, glycolysis, mitochondrial biology, and energy generation. Dissection of these complex regulatory loops will have a major impact on our comprehension of basic physiopathological processes as well as on our ability to personalize cancer therapies.
Author Contributions
IB and HW: substantial contributions to the conception and design of the work and interpretation of data for the work; drafting the work and revising it critically for important intellectual content; final approval of the version to be published; and agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. RS and DL: substantial contributions to the design of the work and interpretation of data; revising it critically for important intellectual content; final approval of the version to be published; and agreement to be accountable for all aspects of the work.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Work in the laboratory of HW is supported by grants from the Israel Science Foundation, US-Israel Binational Science Foundation, and the Dotan Research Center in Hemato-Oncology (Tel Aviv University). IB wishes to thank the Israel Cancer Research Fund (Montreal, Canada) for their generous support. HW is the incumbent of the Lady Davis Chair in Biochemistry.
References
1. LeRoith D, Yakar S. Mechanisms of disease: metabolic effects of growth hormone and insulin-like growth factor-1. Nat Clin Pract Endocrinol Metab (2007) 3:302–10. doi: 10.1038/ncpendmet0427
2. Benarroch EE. Insulin-like growth factors in the brain and their potential clinical implications. Neurology (2012) 79:2148–53. doi:10.1212/WNL.0b013e3182752eef
3. Wit JM, Oostdijk W, Losekoot M. Spectrum of insulin-like growth factor deficiency. Endocr Dev (2012) 23:30–41. doi:10.1159/000341739
4. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8:915–28. doi:10.1038/nrc2536
5. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev (2007) 28:20–47. doi:10.1210/er.2006-0001
6. Salmon WD, Daughaday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med (1957) 49:825–36.
7. Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer (2003) 107:873–7. doi:10.1002/ijc.11487
8. Werner H, Bruchim I. The insulin-like growth factor-I receptor as an oncogene. Arch Physiol Biochem (2009) 115:58–71. doi:10.1080/13813450902783106
9. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer (2012) 12:159–69. doi:10.1038/nrc3215
10. Bentov I, Werner H. IGF, IGF receptor and overgrowth syndromes. Pediatr Endocrinol Rev (2004) 1:352–60.
11. Bach LA, Headey SJ, Norton RS. IGF-binding proteins – the pieces are falling into place. Trends Endocrinol Metab (2005) 16:228–34. doi:10.1016/j.tem.2005.05.005
12. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev (2001) 22:818–35. doi:10.1210/edrv.22.6.0452
13. LeRoith D, Bondy C, Yakar S, Liu JL, Butler A. The somatomedin hypothesis: 2001. Endocr Rev (2001) 22:53–74. doi:10.1210/edrv.22.1.0419
14. Belfiore A, Malaguarnera R. The insulin receptor and cancer. Endocr Relat Cancer (2011) 18:R125–47. doi:10.1530/ERC-11-0074
15. Yakar S, LeRoith D, Brodt P. The role of the growth hormone/insulin-like growth factor axis in tumor growth and progression: lessons from animal models. Cytokine Growth Factor Rev (2005) 16:407–20. doi:10.1016/j.cytogfr.2005.01.010
16. Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (2014) 5:10. doi:10.3389/fendo.2014.00010
17. Werner H, Weinstein D, Bentov I. Similarities and differences between insulin and IGF-I: structures, receptors, and signaling pathways. Arch Physiol Biochem (2008) 114:17–22. doi:10.1080/13813450801900694
18. Bentov I, Werner H. Insulin-like growth factor-I. In: Kastin A, editor. Handbook of Biologically Active Peptides. San Diego: Elsevier Press (2006). p. 1385–92.
19. Yakar S, Sun H, Zhao H, Pennisi P, Toyoshima Y, Setser J, et al. Metabolic effects of IGF-I deficiency: lessons from mouse models. Pediatr Endocrinol Rev (2005) 3:11–9.
20. Chitnis MM, Yuen JSP, Protheroe AS, Pollak M, Macaulay VM. The type I insulin-like growth factor-I receptor pathway. Clin Cancer Res (2008) 14:6364–70. doi:10.1158/1078-0432.CCR-07-4879
21. Lu K, Campisi J. Ras proteins are essential and selective for the action of insulin-like growth factor 1 late in the G1 phase of the cell cycle in BALB/c murine fibroblasts. Proc Natl Acad Sci U S A (1992) 89:3889–93. doi:10.1073/pnas.89.9.3889
22. Moschos SJ, Mantzoros CS. The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology (2002) 63:317–32. doi:10.1159/000066230
23. Guler HP, Eckardt KU, Zapf J, Bauer C, Froesch ER. Insulin-like growth factor-I increases glomerular filtration rate and renal plasma flow in man. Acta Endocrinol (Copenh) (1989) 121:101–6.
24. Baker J, Liu J-P, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell (1993) 75:73–82. doi:10.1016/S0092-8674(05)80085-6
25. Baserga R. The IGF-I receptor in cancer research. Exp Cell Res (1999) 253:1–6. doi:10.1006/excr.1999.4667
26. Werner H. Tumor suppressors govern insulin-like growth factor signaling pathways: implications in metabolism and cancer. Oncogene (2012) 31:2703–14. doi:10.1038/onc.2011.447
27. Werner H, LeRoith D. The role of the insulin-like growth factor system in human cancer. Adv Cancer Res (1996) 68:183–223. doi:10.1016/S0065-230X(08)60354-1
28. Werner H. The pathophysiological significance of IGF-I receptor overexpression: new insights. Pediatr Endocrinol Rev (2009) 7:2–5.
29. Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Akiyama M, et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell (2004) 5:221–30. doi:10.1016/S1535-6108(04)00050-9
30. LeRoith D, Helman LJ. The new kid on the block(ade) of the IGF-1 receptor. Cancer Cell (2004) 5:403. doi:10.1016/S1535-6108(04)00093-5
31. Turney BW, Turner GDH, Brewster SF, Macaulay VM. Serial analysis of resected prostate cancer suggests up-regulation of type 1 IGF receptor with disease progression. BJU Int (2011) 107:1489–99. doi:10.1111/j.1464-410X.2010.09556.x
32. Liao Y, Abel U, Grobholz R, Hermani A, Trojan L, Angel P, et al. Up-regulation of insulin-like growth factor axis components in human primary prostate cancer correlates with tumor grade. Hum Pathol (2005) 36:1186–96. doi:10.1016/j.humpath.2005.07.023
33. Sell C, Rubini M, Rubin R, Liu J-P, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A (1993) 90:11217–21. doi:10.1073/pnas.90.23.11217
34. Baserga R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene (2000) 19:5574–81. doi:10.1038/sj.onc.1203854
35. Werner H, Maor S. The insulin-like growth factor-I receptor gene: a downstream target for oncogene and tumor suppressor action. Trends Endocrinol Metab (2006) 17:236–42. doi:10.1016/j.tem.2006.06.007
36. Beitner-Johnson D, Werner H, Roberts CT Jr, LeRoith D. Regulation of insulin-like growth factor I receptor gene expression by Sp1: physical and functional interactions of Sp1 at GC boxes and at a CT element. Mol Endocrinol (1995) 9:1147–56. doi:10.1210/me.9.9.1147
37. Werner H, Bach MA, Stannard B, Roberts CT Jr, LeRoith D. Structural and functional analysis of the insulin-like growth factor I receptor gene promoter. Mol Endocrinol (1992) 6:1545–58. doi:10.1210/mend.6.10.1448110
38. Schayek H, Bentov I, Rotem I, Pasmanik-Chor M, Ginsberg D, Plymate SR, et al. Transcription factor E2F1 is a potent transactivator of the insulin-like growth factor-I receptor gene. Growth Horm IGF Res (2010) 20:68–72. doi:10.1016/j.ghir.2009.08.001
39. Rubinstein M, Idelman G, Plymate SR, Narla G, Friedman SL, Werner H. Transcriptional activation of the IGF-I receptor gene by the Kruppel-like factor-6 (KLF6) tumor suppressor protein: potential interactions between KLF6 and p53. Endocrinology (2004) 145:3769–77. doi:10.1210/en.2004-0173
40. Aiello A, Pandini G, Sarfstein R, Werner H, Manfioletti G, Vigneri R, et al. HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur J Cancer (2010) 46:1919–26. doi:10.1016/j.ejca.2010.02.050
41. Sarfstein R, Belfiore A, Werner H. Identification of insulin-like growth factor-I receptor gene promoter-binding proteins in estrogen receptor (ER)-positive and ER-depleted breast cancer cells. Cancers (Basel) (2010) 2:233–61. doi:10.3390/cancers2031642
42. Lee AV, Hilsenbeck SG, Yee D. IGF system components as prognostic markers in breast cancer. Breast Cancer Res Treat (1998) 47:295–302. doi:10.1023/A:1005915420341
43. Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res (2005) 65:1849–57. doi:10.1158/0008-5472.CAN-04-1837
44. Oesterreich S, Zhang P, Guler RL, Sun X, Curran EM, Welshons WV, et al. Re-expression of estrogen receptor a in estrogen receptor a-negative MCF-7 cells restores both estrogen and insulin-like growth factor-mediated signaling and growth. Cancer Res (2001) 61:5771–7.
45. Maor S, Mayer D, Yarden RI, Lee AV, Sarfstein R, Werner H, et al. Estrogen receptor regulates insulin-like growth factor-I receptor gene expression in breast tumor cells: involvement of transcription factor Sp1. J Endocrinol (2006) 191:605–12. doi:10.1677/joe.1.07016
46. Schayek H, Seti H, Greenberg NM, Werner H, Plymate SR. Differential regulation of IGF1-R gene transcription by wild type and mutant androgen receptor in prostate cancer cells. Mol Cell Endocrinol (2010) 323:239–45. doi:10.1016/j.mce.2010.04.017
47. Schnarr B, Strunz K, Ohsam J, Benner A, Wacker J, Mayer D. Down-regulation of insulin-like growth factor-I receptor and insulin receptor substrate-1 expression in advanced human breast cancer. Int J Cancer (2000) 89:506–13. doi:10.1002/1097-0215(20001120)89:6<506::AID-IJC7>3.0.CO;2-F
48. Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, et al. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol (1999) 13:787–96. doi:10.1210/mend.13.5.0274
49. Schayek H, Bentov I, Sun S, Plymate SR, Werner H. Progression to metastatic stage in a cellular model of prostate cancer is associated with methylation of the androgen receptor gene and transcriptional suppression of the insulin-like growth factor-I receptor gene. Exp Cell Res (2010) 316:1479–88. doi:10.1016/j.yexcr.2010.03.007
50. Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med (2003) 349:2211–22. doi:10.1056/NEJMoa010107
52. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer (2014) 14:359–70. doi:10.1038/nrc3711
53. Harris CC, Hollstein M. Clinical implications of the p53 tumor suppressor gene. N Engl J Med (1993) 329:1318–27. doi:10.1056/NEJM199310283291807
54. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell (1997) 88:323–31. doi:10.1016/S0092-8674(00)81871-1
55. Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol (2015) 16:393–405. doi:10.1038/nrm4007
56. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med (2004) 10:789–99. doi:10.1038/nm1087
57. Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science (1991) 252:1708–11. doi:10.1126/science.2047879
58. Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res (2004) 64:2627–33. doi:10.1158/0008-5472.CAN-03-0846
59. Molchadsky A, Ezra O, Amendola PG, Krantz D, Kogan-Sakin I, Buganim Y, et al. p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ (2013) 20:774–83. doi:10.1038/cdd.2013.9
60. Werner H, Karnieli E, Rauscher FJ III, LeRoith D. Wild type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc Natl Acad Sci U S A (1996) 93:8318–23. doi:10.1073/pnas.93.16.8318
61. Ohlsson C, Kley N, Werner H, LeRoith D. p53 regulates IGF-I receptor expression and IGF-I induced tyrosine phosphorylation in an osteosarcoma cell line: interaction between p53 and Sp1. Endocrinology (1998) 139:1101–7. doi:10.1210/endo.139.3.5832
62. Zhang L, Kashanchi F, Zhan Q, Zhan S, Brady JN, Fornace AJ, et al. Regulation of insulin-like growth factor II P3 promoter by p53: a potential mechanism for tumorigenesis. Cancer Res (1996) 56:1367–73.
63. Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR, et al. Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature (1995) 377:1367–73. doi:10.1038/377646a0
64. Irwin MS, Kaelin WG. p53 family update: p73 and p63 develop their own identities. Cell Growth Differ (2001) 12:337–49.
65. Yang A, McKeon F. P63 and p73: p53 mimics, menaces and more. Nat Rev Mol Cell Biol (2000) 1:199–207. doi:10.1038/35042066
66. Jost CA, Marin MC, Kaelin WG Jr. p73 is a simian p53-related protein that can induce apoptosis. Nature (1997) 389:191–4. doi:10.1038/38298
67. Nahor I, Abramovitch S, Engeland K, Werner H. The p53-family members p63 and p73 inhibit insulin-like growth factor-I receptor gene expression in colon cancer cells. Growth Horm IGF Res (2005) 15:388–96. doi:10.1016/j.ghir.2005.07.005
68. Levine AJ, Feng Z, Mak TW, You H, Jin S. Coordination and communication between the p53 and IGF-1-AKT-TOR signal transduction pathways. Genes Dev (2006) 20:267–75. doi:10.1101/gad.1363206
69. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated Mdm2 phosphorylation. Nat Cell Biol (2001) 3:973–82. doi:10.1038/ncb1101-973
70. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell (1999) 96:857–68. doi:10.1016/S0092-8674(00)80595-4
71. Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell (2001) 8:317–25. doi:10.1016/S1097-2765(01)00323-9
72. Lakin ND, Jackson SP. Regulation of p53 in response to DNA damage. Oncogene (1999) 18:7644–55. doi:10.1038/sj.onc.1203015
73. Heron-Milhavet L, LeRoith D. Insulin-like growth factor I induces MDM2-dependent degradation of p53 via the p38 MAPK pathway in response to DNA damage. J Biol Chem (2002) 277:15600–6. doi:10.1074/jbc.M111142200
74. Girnita L, Girnita A, Larsson O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor-I receptor. Proc Natl Acad Sci U S A (2003) 100:8247–52. doi:10.1073/pnas.1431613100
75. McCarty MF. Targeting multiple signaling pathways as a strategy for managing prostate cancer: multifocal signal modulation therapy. Integr Cancer Ther (2004) 3:349–80. doi:10.1177/1534735404270757
76. Kojima S, Hayashi S, Shimokado K, Suzuki Y, Shimada J, Crippa MP, et al. Transcriptional activation of urokinase by the Kruppel-like factor Zf9/COPEB activates latent TGF-beta1 in vascular endothelial cells. Blood (2000) 95:1309–16.
77. Narla G, Friedman SL, Martignetti JA. Kruppel cripples prostate cancer: KLF6 progress and prospects. Am J Pathol (2003) 162:1047–52. doi:10.1016/S0002-9440(10)63901-6
78. Narla G, Heath KE, Reeves HL, Li D, Giono LE, Kimmelman AC, et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science (2001) 294:2563–6. doi:10.1126/science.1066326
79. Bentov I, Narla G, Schayek H, Akita K, Plymate SR, LeRoith D, et al. Insulin-like growth factor-I regulates Kruppel-like factor-6 gene expression in a p53-dependent manner. Endocrinology (2008) 149:1890–7. doi:10.1210/en.2007-0844
80. Attias Z, Werner H, Vaisman N. Folic acid and its metabolites modulate IGF-I receptor gene expression in colon cancer cells in a p53-dependent manner. Endocr Relat Cancer (2006) 13:571–81. doi:10.1677/erc.1.01156
81. Chesnokova V, Zhou C, Ben-Shlomo A, Zonis S, Tani Y, Ren S, et al. Growth hormone is a cellular senescence target in pituitary and nonpituitary cells. Proc Natl Acad Sci U S A (2013) 110:E3331–9. doi:10.1073/pnas.1310589110
82. Turner BC, Haffty BG, Narayanan L, Yuan J, Havre PA, Gumbs AA, et al. Insulin-like growth factor-I receptor overexpression mediates cellular radioresistance and local breast cancer recurrence after lumpectomy and radiation. Cancer Res (1997) 57:3079–83.
83. Kuhn C, Hurwitz SA, Kumar MG, Cotton J, Spandau DF. Activation of the insulin-like growth factor-I receptor promotes the survival of human keratinocytes following ultraviolet B irradiation. Int J Cancer (1999) 80:431–8. doi:10.1002/(SICI)1097-0215(19990129)80:3<431::AID-IJC16>3.3.CO;2-X
84. Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet (2001) 27:247–54. doi:10.1038/85798
85. Pandita TK. A multifaceted role for ATM in genome maintenance. Expert Rev Mol Med (2003) 2003:1–21.
86. Peretz S, Jensen R, Baserga R, Glazer PM. ATM-dependent expression of the insulin-like growth factor-I receptor in a pathway regulating radiation response. Proc Natl Acad Sci U S A (2001) 98:1676–81. doi:10.1073/pnas.98.4.1676
87. Shahrabani-Gargir L, Pandita TK, Werner H. Ataxia-telangiectasia mutated gene controls IGF-IR gene expression in a DNA damage response pathway via mechanisms involving zinc-finger transcription factors Sp1 and WT1. Endocrinology (2004) 145:5679–87. doi:10.1210/en.2004-0613
88. Macaulay VM, Salisbury AJ, Bohula EA, Playford MP, Smorodinsky NI, Shiloh Y. Down regulation of the type 1 insulin-like growth factor receptor in mouse melanoma cells is associated with enhanced radiosensitivity and impaired activation of Atm kinase. Oncogene (2001) 20:4029–40. doi:10.1038/sj.onc.1204565
89. Turney BW, Kerr M, Chitnis MM, Lodhia K, Wang Y, Riedemann J, et al. Depletion of the type 1 IGF receptor delays repair of radiation-induced DNA double strand breaks. Radiother Oncol (2012) 103:402–9. doi:10.1016/j.radonc.2012.03.009
90. Chitnis MM, Lodhia KA, Aleksic T, Gao S, Protheroe AS, Macaulay VM. IGF-1R inhibition enhances radiosensitivity and delays double-strand break repair by both non-homologous end-joining and homologous recombination. Oncogene (2014) 33:5262–73. doi:10.1038/onc.2013.460
91. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science (1994) 266:66–71. doi:10.1126/science.7545954
92. Holt JT, Thompson ME, Szabo C, Robinson-Benion C, Arteaga CL, King MC, et al. Growth retardation and tumour inhibition by BRCA1. Nat Genet (1996) 12:298–301. doi:10.1038/ng0396-298
93. Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science (1994) 266:120–2. doi:10.1126/science.7939630
94. Wang Q, Zhang H, Fishel R, Greene MI. BRCA1 and cell signaling. Oncogene (2000) 19:6152–8. doi:10.1038/sj.onc.1203974
95. Maor SB, Abramovitch S, Erdos MR, Brody LC, Werner H. BRCA1 suppresses insulin-like growth factor-I receptor promoter activity: potential interaction between BRCA1 and Sp1. Mol Genet Metab (2000) 69:130–6. doi:10.1006/mgme.1999.2958
96. Abramovitch S, Glaser T, Ouchi T, Werner H. BRCA1-Sp1 interactions in transcriptional regulation of the IGF-IR gene. FEBS Lett (2003) 541:149–54. doi:10.1016/S0014-5793(03)00315-6
97. Abramovitch S, Werner H. Functional and physical interactions between BRCA1 and p53 in transcriptional regulation of the IGF-IR gene. Horm Metab Res (2003) 35:758–62. doi:10.1055/s-2004-814154
98. Werner H, Bruchim I. IGF-1 and BRCA1 signalling pathways in familial cancer. Lancet Oncol (2012) 13:e537–44. doi:10.1016/S1470-2045(12)70362-5
99. Maor S, Yosepovich A, Papa MZ, Yarden RI, Mayer D, Friedman E, et al. Elevated insulin-like growth factor-I receptor (IGF-IR) levels in primary breast tumors associated with BRCA1 mutations. Cancer Lett (2007) 257:236–43. doi:10.1016/j.canlet.2007.07.019
100. Maor S, Papa MZ, Yarden RI, Friedman E, Lerenthal Y, Lee SW, et al. Insulin-like growth factor-I controls BRCA1 gene expression through activation of transcription factor Sp1. Horm Metab Res (2007) 39:179–85. doi:10.1055/s-2007-970415
101. Voskuil DW, Bosma A, Vrieling A, Rookus MA, van’t Veer LJ. Insulin-like growth factor system mRNA quantities in normal and tumor breast tissue of women with sporadic and familial breast cancer risk. Breast Cancer Res Treat (2004) 84:225–33. doi:10.1023/B:BREA.0000019954.59130.d3
102. Yuen JSP, Cockman ME, Sullivan M, Protheroe A, Turner GDH, Roberts IS, et al. The VHL tumor suppressor inhibits expression of the IGF1R and its loss induces IGF1R upregulation in human clear cell renal carcinoma. Oncogene (2007) 26:6499–508. doi:10.1038/sj.onc.1210474
103. Werner H, Shen-Orr Z, Rauscher FJ III, Morris JF, Roberts CT Jr, LeRoith D. Inhibition of cellular proliferation by the Wilms’ tumor suppressor WT1 is associated with suppression of insulin-like growth factor I receptor gene expression. Mol Cell Biol (1995) 15:3516–22. doi:10.1128/MCB.15.7.3516
104. Idelman G, Glaser T, Roberts CT Jr, Werner H. WT1-p53 interactions in IGF-I receptor gene regulation. J Biol Chem (2003) 278:3474–82. doi:10.1074/jbc.M211606200
105. Scotlandi K, Picci P. Targeting insulin-like growth factor 1 receptor in sarcomas. Curr Opin Oncol (2008) 20:419–27. doi:10.1097/CCO.0b013e328302edab
106. Yuen JS, Macaulay VM. Targeting the type 1 insulin-like growth factor receptor as a treatment for cancer. Expert Opin Ther Targets (2008) 12:589–603. doi:10.1517/14728222.12.5.589
107. Bruchim I, Attias Z, Werner H. Targeting the IGF1 axis in cancer proliferation. Expert Opin Ther Targets (2009) 13:1179–92. doi:10.1517/14728220903201702
108. Baserga R. The decline and fall of the IGF-I receptor. J Cell Physiol (2013) 28:675–9. doi:10.1002/jcp.24217
109. Gualberto A, Pollak M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene (2009) 28:3009–21. doi:10.1038/onc.2009.172
110. King H, Aleksic T, Haluska P, Macaulay VM. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat Rev (2014) 40:1096–105. doi:10.1016/j.ctrv.2014.07.004
111. Aleksic T, Chitnis MM, Perestenko OV, Gao S, Thomas PH, Turner GD, et al. Type 1 insulin-like growth factor receptor translocates to the nucleus of human tumor cells. Cancer Res (2010) 70:6412–9. doi:10.1158/0008-5472.CAN-10-0052
112. Asmane I, Watkin E, Alberti L, Duc A, Marec-Berard P, Ray-Coquard I, et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: a predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur J Cancer (2012) 48:3027–35. doi:10.1016/j.ejca.2012.05.009
113. Wang Q, Wei F, Lv G, Li C, Liu T, Hadjipanayis CG, et al. The association of TP53 mutations with the resistance of colorectal carcinoma to the insulin-like growth factor-1 receptor inhibitor picropodophyllin. BMC Cancer (2013) 13:521. doi:10.1186/1471-2407-13-521
114. Attias-Geva Z, Bentov I, Kidron D, Amichay K, Sarfstein R, Fishman A, et al. p53 Regulates insulin-like growth factor-I receptor gene expression in uterine serous carcinoma and predicts responsiveness to an insulin-like growth factor-I receptor-directed targeted therapy. Eur J Cancer (2012) 48:1570–80. doi:10.1016/j.ejca.2011.09.014
115. Gottlieb E, Vousden KH. p53 regulation of metabolic pathways. Cold Spring Harb Perspect Biol (2010) 2:a001040. doi:10.1101/cshperspect.a001040
116. Kulawiec M, Ayyasamy V, Singh KK. p53 regulates mtDNA copy number and mitocheckpoint pathway. J Carcinog (2009) 8:8. doi:10.4103/1477-3163.50893
117. Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr (2007) 39:243–6. doi:10.1007/s10863-007-9083-0
118. Ferecatu I, Bergeaud M, Rodríguez-Enfedaque A, Le Floch N, Oliver L, Rincheval V, et al. Mitochondrial localization of the low level p53 protein in proliferative cells. Biochem Biophys Res Commun (2009) 387:772–7. doi:10.1016/j.bbrc.2009.07.111
119. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell (2006) 126:107–20. doi:10.1016/j.cell.2006.05.036
120. Li H, Jogl G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator). J Biol Chem (2009) 284:1748–54. doi:10.1074/jbc.M807821200
121. Goldstein I, Yizhak K, Madar S, Goldfinger N, Ruppin E, Rotter V. p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer Metab (2013) 1:9–15. doi:10.1186/2049-3002-1-9
122. Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, et al. p53 activation in adipocytes of obese mice. J Biol Chem (2003) 278:25395–400. doi:10.1074/jbc.M302364200
Keywords: insulin-like growth factor 1, IGF1 receptor, tumor suppressors, p53, BRCA1, transcription regulation, genome protection
Citation: Werner H, Sarfstein R, LeRoith D and Bruchim I (2016) Insulin-like Growth Factor 1 Signaling Axis Meets p53 Genome Protection Pathways. Front. Oncol. 6:159. doi: 10.3389/fonc.2016.00159
Received: 03 April 2016; Accepted: 10 June 2016;
Published: 23 June 2016
Edited by:
Wen Zhou, University of Miami Miller School of Medicine, USAReviewed by:
David R. Croucher, Garvan Institute of Medical Research, AustraliaGiovanni Sorrentino, Laboratorio Nazionale CIB (LNCIB), Italy
Copyright: © 2016 Werner, Sarfstein, LeRoith and Bruchim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haim Werner, aHdlcm5lckBwb3N0LnRhdS5hYy5pbA==