 Pratikkumar Harsukhbhai Vekaria
Pratikkumar Harsukhbhai Vekaria Trisha Home1
Trisha Home1 Rekha Rao
Rekha Rao- 1Division of Hematologic Malignancies and Cellular Therapeutics, Kansas University Medical Center, Kansas City, KS, USA
- 2The University of Kansas Cancer Center, University of Kansas, Kansas City, KS, USA
- 3Specialized Chemistry Center, University of Kansas, Lawrence, KS, USA
Cancer cells are addicted to numerous non-oncogenic traits that enable them to thrive. Proteotoxic stress is one such non-oncogenic trait that is experienced by all tumor cells owing to increased genomic abnormalities and the resulting synthesis and accumulation of non-stoichiometric amounts of cellular proteins. This imbalance in the amounts of proteins ultimately culminates in proteotoxic stress. p97, or valosin-containing protein (VCP), is an ATPase whose function is essential to restore protein homeostasis in the cells. Working in concert with the ubiquitin proteasome system, p97 promotes the retrotranslocation from cellular organelles and/or degradation of misfolded proteins. Consequently, p97 inhibition has emerged as a novel therapeutic target in cancer cells, especially those that have a highly secretory phenotype. This review summarizes our current understanding of the function of p97 in maintaining protein homeostasis and its inhibition with small molecule inhibitors as an emerging strategy to target cancer cells.
Introduction
Cell division control protein 48 (CDC48), or p97, is a member of the type II AAA+ protein family of ATPases that participates in a diverse array of cellular activities (1–3). AAA+ proteins are evolutionary conserved class of proteins found in archaea, bacteria, viruses, and all eukaryotes. They typically assemble into hexameric complexes that utilize the energy of ATP-hydrolysis to facilitate macromolecular remodeling (4). Consequently, AAA+ proteins are involved in numerous processes including protein folding, DNA recombination, repair or replication, metal chelation, and proteasome-associated activities (2). p97 is an abundantly expressed cellular protein involved in the disassembly of protein complexes particularly, in proteasomal protein degradation, chromatin remodeling, autophagosome maturation, immune signaling, endoplasmic reticulum (ER) membrane fusion, and assembly of Golgi membranes (5). This review highlights the role of p97 in protein homeostasis and describes the consequences of its chemical or genetic inhibition in cancer cells.
p97 comprises two AAA domains called D1 and D2, as well as an N-terminal domain that is involved in substrate and/or adaptor molecule recognition (6–8). The C-terminal tail of p97 is fairly unstructured and usually terminates with a hydrophobic residue, a tyrosine, and variable amino acid residues. The C-terminus tail also binds numerous adaptors, thus increasing the repertoire of p97 interacting proteins (7). p97 assembles into a homohexameric, barrel-like structure, in which the D1 and D2 domains are stacked in a head-to-tail manner. The axial channel resulting from the hexameric organization of p97 is used to translocate proteins and/or nucleic acid substrates and aid in their turnover or remodeling (9–11). The cryo-EM structure of p97 reveals the upward displacement of the N-terminal domain upon ATPγS binding to D1 and D2 domains. These structural changes are essential for 97 activity and are blocked by the p97 inhibitor UPCDC3024 (Figure 1) (11). The D1 domain is important for mediating the assembly of p97 hexamers and has very low ATPase activity (4, 12). The D2 domain contributes to the major share of ATPase activity in p97 (13). However, a functional D1 domain and cooperativity between both D1 and D2 domains is necessary for optimal p97 function and cell growth (13–15). The linker region between the D1 and D2 domains has also been reported to be important for ATPase activity and asymmetric assembly of p97 (16). p97 interacts with its ubiquitinated substrates through a diverse array of ubiquitin adapters that contain the ubiquitin-binding domain (UBD), or p97-binding (UBX), or UBX-like (UBX-L) domain. It also employs numerous cofactors that aid in its cellular functions (17) (Table 1).
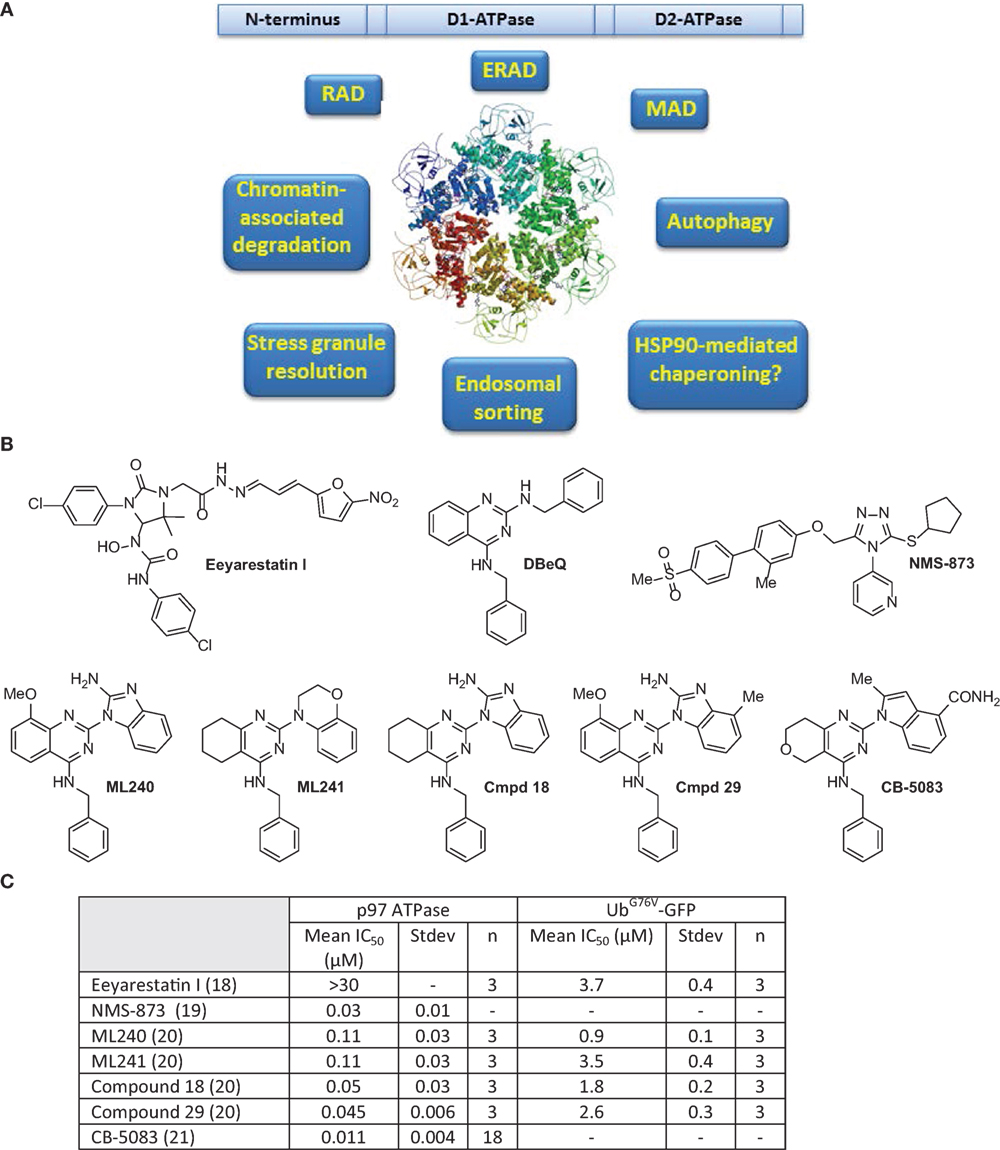
Figure 1. (A) Domain structure, ribbon diagram of p97 hexameric structure (11), and functions of p97 are summarized. (B) Select inhibitors of p97 ATPase. Chemical structures for Eeyarestatin I, DBeQ, NMS-873, ML240, ML241, compound 18, compound 29, and CB-5083. (C) Comparison of IC50 values for shown compounds for p97 in biochemical and cell-based assays as reported in Ref. (18–21).
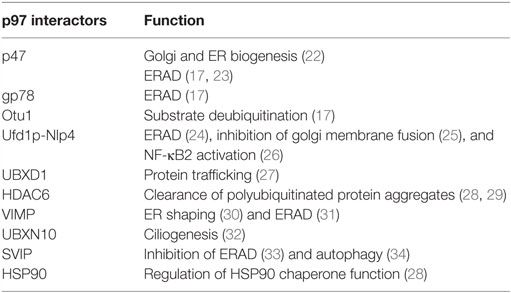
Table 1. A partial list p97 interacting proteins and their associated functions.
p97 and Its Role in Cellular Adaptations to Proteotoxic Stress
Tumors are “addicted” to oncogenic mutations such as gene amplification, loss of tumor suppressor function, or gain-of-function mutations. However, it has become increasingly clear that several non-oncogenic traits or “stress phenotypes” enable them to thrive in the hostile tumor microenvironment (35). Examples of the stress phenotype include replicative/DNA damage stress, proteotoxic stress, oxidative stress, and metabolic stress (36). Due to altered genomic copy numbers and the resultant synthesis of non-stoichiometric amounts of cellular proteins, tumors are experience heightened proteotoxic stress. The proteotoxic stress phenotype of tumors, in turn, makes them highly dependent on heat shock protein 90 (HSP90) chaperone function to promote protein folding and maturation (35, 37, 38). The following section highlights key aspects of p97-dependent cellular adaptions to proteotoxic stress. Other functions of p97, such as its chromatin-associated functions, etc., are beyond the scope of this review and are covered in many excellent publications (39, 40).
HSP90-Dependent Chaperoning of Misfolded Proteins
p97 is a key player in the clearance of misfolded proteins in concert with the HSPs and the stress-inducible transcription factor, heat shock factor 1 (HSF1) (41). HSF1-induced HSPs (HSP90 and its co-chaperones (HSP70, HSP40, and HSP27) participate in the refolding and maturation of proteins. Misfolded proteins that cannot be refolded are polyubiquitinated and degraded by the ubiquitin proteasome system (41–43). p97 exists in a “repressive” complex with HSP90, histone deacetylase 6 (HDAC6), and HSF1. Accumulation of misfolded proteins in the cell and the ensuing proteotoxic stress disrupt the repressive complex. This, in turn, results in the activation of HSF1 by a process that involves its phosphorylation, trimerization, and nuclear translocation (28). Nuclear HSF1 activates the transcription of HSPs that promote protein refolding. The HDAC6 released from the complex binds to misfolded, polyubiquitinated proteins and facilitates their shuttling into peri-nuclear structures called aggresomes. p97 has been demonstrated to be involved in the HDAC6-dependent fusion of aggresomes with an autophagolysosomes – a vesicular body that promotes the clearance of ubiquitinated proteins and/or damaged organelles (2, 44, 45). By virtue of its segregase activity, p97 dissociates HDAC6-polyubiquitin complexes and determines the extent of HDAC6-dependent shuttling (and accumulation) or clearance of polyubiquitinated proteins by proteasomes or autophagosomes (29, 46). p97 is also required for the reformation of the HSP90-HDAC6-HSF1 complex, which occurs only when HDAC6 is not bound to polyubiquitinated proteins (47). The dissociation of HDAC6 from the repressive complex could potentially affect the acetylation status of HSP90, which negatively regulates HSP9 chaperone function (48, 49). The impact p97 inhibitors on HSP90 chaperone function has, however, not been studied.
Unfolded Protein Response and ER-associated Degradation
Nearly thirty percent of mammalian proteins pass through the ER where they are folded, glycosylated, and assembled before they reach their final destination within or outside the cell (50). A series of glycosylation/deglucosylation steps as well as the unique oxidative environment in the ER promote the proper folding of membrane-resident and/or secretory proteins (51). Perturbations in ER function due to intrinsic and extrinsic signals, such as hypoxia, nutrient deprivation, deregulation of Ca homeostasis, enhanced rates of protein synthesis, etc., lead to the accumulation of misfolded proteins in the ER (52). These perturbations result in ER stress – a stress phenotype that is prevalent in both normal and malignant secretory cells (53, 54).
Unfolded protein response (UPR) is a cellular adaption that ensues following induction of ER stress. UPR activates three important signaling pathways: the inositol-requiring enzyme 1α (IRE-1α), PRKR-like ER kinase (PERK) kinases, and as the activating transcription factor 6 (ATF6) (reviewed in detail elsewhere) to restore proteostasis (55). Activation of UPR induces the phosphorylation of the transmembrane kinase IRE-1α, which catalyzes the splicing of X-box-binding protein (1) (XBP1) which, in turn, induces the transcription of HSP40 family members ERdj4 and the ER degradation enhancer, mannosidase alpha-like 1 (EDEM1), a protein required for ERAD (discussed below) (56). Activation of PERK phosphorylation induces the phosphorylation of eukaryotic transcription factor 2α (eIF2α), which halts overall protein translation but induces the translation of activating transcription 4 (ATF4) and other selected mRNAs. ATF4 also induces the pro-apoptotic transcription factor CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), which upregulates numerous apoptotic targets genes including BIM, death receptor 5 (DR5), and ER oxidase 1α (ERO1α) (57, 58). ATF6α is also a transmembrane ER protein, which is proteolytically processed in response to ER stress (59). ATF6α enters the nucleus to activate the transcription of chaperones glucose regulated proteins 78 and 94 (GRP78 and GRP94) as well as XBP1. Overwhelming amounts of misfolded proteins or prolonged ER stress activate the PERK pathway to upregulate CHOP as well as ATF4 transcription factors. These events promote ERO1α and protein disulfide isomerase-dependent production of reactive oxygen species (ROS) through protein disulfide oxidation, leading to oxidative stress and apoptosis (60).
Endoplasmic reticulum-associated degradation (ERAD) is a process by which misfolded proteins that cannot be refolded in the ER are retrotranslocated to the cytosol and degraded by the proteasomes (61–64). p97 uses its energy of ATP-hydrolysis to structurally remodel and extract ERAD substrates through the ER membrane into the cytosol. p97 works in association with a complex of ER membrane protein channels (also called dislocons) including Sec61 and Sel1L and adaptors, ubiquitin fusion degradation 1 (Ufd1)-nuclear pore localization 4 (Npl4) (24, 65, 66). Owing to the fact that misfolded proteins are “tagged” with mono or polyubiquitin before they can be degraded by the proteasome, another important component of the ERAD machinery is the ubiquitin ligase complex (65). Both luminal and membrane-associated misfolded ERAD substrates are ubiquitinated by E3 ubiquitin ligases (see below) (66, 67). The process of substrate dislocation and proteasomal degradation is tightly coupled in order to prevent the toxic aggregation of hydrophobic misfolded proteins in the cytosol. Non-glycosylated proteins that cannot be folded in the ER are also subjected to proteasomal degradation by ERAD through mechanisms that utilize GRP78 and SEL1L (68, 69). The last step in ERAD is the deubiquitination of ERAD substrates before they can begin their translocation into the proteasomes. p97 is, therefore, also found to be associated with several deubiquitinating enzymes (DUBs), including ataxin3, TOD1, Otu1, and VCIP135 (70).
Ubiquitination and p97
p97 interacts either directly (e.g., Gp78 and HRD1) or through adaptors (e.g., UBXD7/Ubx5) to E3 ubiquitin ligases such as cullin RING ligases (CRL) to facilitate the ubiquitination of numerous ERAD substrates (67, 71, 72). CRLs comprise multi-protein complexes that include the cullin scaffold, a RING-domain protein, and a cullin-specific adaptor that recruits both ERAD and non-ERAD substrates for ubiquitination. CRLs make up more than 240 different ligases that enable p97 to ubiquitinate numerous substrates including hypoxia-inducible factor 1 (HIF1) α, RNA polymerase II large subunit (Rpb1), and Aurora B (72, 73). Additionally, p97 is involved in the polyubiquitin chain assembly of its substrates in concert with the E4 ubiquitin ligase, Ufd2 (74).
Ribosome-Associated Degradation
mRNA with no stop codons also called non-stop mRNA or those with a polybasic tract, such as polylysine, which occurs during the translation of a poly(A) tail of a non-stop mRNA, are subjected to ribosome stalling and degradation of the nascent peptide (75). Accumulation of such aberrant peptides could potentially lead to reduced translation-competent ribosomes or produce aggregation-prone polypeptides. Two independent studies identified p97 as a component of the complex that is involved in the ubiquitination and proteasomal degradation of aberrant tRNA-linked nascent peptides from the ribosomes (76, 77). Additional components of this complex include the E3 ubiquitin ligase Listerin (Ltn1), translation-associated element (Tae2), and the ribosome quality control 1 (Rqc1) (76, 77).
Mitochondria-Associated Degradation
The process of extracting misfolded peptides from mitochondrial outer membranes to facilitate their proteasomal disposal (MAD) also involves p97 (78). MAD employs the p97 cofactors, Ufd1-Npl4, for the translocation of damaged mitochondrial proteins. p97 also participates in the elimination damaged mitochondria by mitophagy, a process that is dependent on the E3 ubiquitin ligase Parkin and autophagolysosomal function (79). Recent studies in yeast have identified that Ufd3 or Doa1 and Npl4 both serve as substrate recruitment cofactors for p97. Doa1 is a member of the WD40 family of proteins that functions specifically in the degradation of mitochondrial substrates. Deletion of Doa1 does not affect the degradation of non-mitochondrial substrates nor does it sensitize cells to ER stress-inducing agents. The absence of Doa1, however, inhibits growth of cells in response to increased oxidative stress (80). An analogous role of the human homolog of Doa1, phospholipase A2 activating protein, in MAD or mitophagy has not been reported.
p97 Inhibitors: From Hit to Lead
It is clear from the above description that p97 is involved in the clearance of misfolded proteins by affecting numerous protein homeostatic mechanisms. Other than the processes mentioned above, p97 is involved in resolution of stress granules (SGs), endosomal sorting, and chromatin remodeling, among other functions. SGs are cytoplasmic aggregates formed due to impaired translation initiation of mRNAs in complex with ribosomal subunits (77, 81, 82). Owing to its pivotal role in HSP90 chaperone function, ERAD, MAD, RAD, and autophagy, it is conceivable that p97 inhibition would induce the accumulation of misfolded proteins or toxic protein aggregates in the cell. Consequently, p97 inhibitors selectively target cancer cells because of their heightened sensitivity to agents that disturb protein homeostasis. Eeyrestatins (EerI and II) were the earliest identified p97 inhibitors that inhibited ERAD, albeit with low potency (83, 84). Several chemically distinct classes of p97 inhibitors have been identified since then (85–88). The compounds, described below, have led to the identification of a molecule (CB-5083) with drug-like properties, which is currently being tested in the clinic (21).
Given that the ATPase activity of p97 is essential for its segregase activity, high-throughput assays designed to measure the ATPase activity of recombinant p97 were utilized to screen compound libraries to identify p97 inhibitors (89). N2,N4-dibenzylquinazoline-2,4-diamine (DBeQ) was identified as a reversible, ATP-competitive p97 inhibitor that showed IC50 value of <10 μM in the ATPase activity assay (89). The inhibitory activity of DBeQ was further confirmed in cellular screens by monitoring the degradation of a p97-dependent substrate UbG67V-GFP. DBeQ was also 50-fold less potent in inhibiting other unrelated ATPase activities, such as those of N-ethylmaleimide-sensitive factor (NSF) and the ATP-dependent chymotryptic activity of the 26S proteasome. Mechanistically, DBeQ upregulated CHOP and resulted in cell death suggesting that DBeQ-dependent inhibition of p97 resulted in a lethal ER stress response. DBeQ also induced autophagy, as evidenced by the enhanced lipidation of LC3 (microtubule associated protein light chain) or LC3-B (89, 90).
To develop more potent p97 inhibitors, two promising hits, DBeQ and N-benzyl-2-(2-fluorophenyl) quinazolin-4-amine, from the high-throughput screen were optimized through structure–activity relationship (SAR) studies (20). These studies resulted in the identification of two compounds ML240 and ML241 bearing different substitutions at the N2 position on distinct quinazoline core scaffolds (Figure 1) (20). Both ML240 and ML241 were ATP-competitive p97 inhibitors and possessed similar in vitro ATPase activity, with ML240 being modestly more potent than ML241 in the stabilizing UbG67V-GFP. ML241 showed greater induction accumulation of polyubiquitinated proteins compared to ML240. However, ML240 induced more apoptosis and efficient poly(ADP-ribose) polymerase cleavage compared ML241. These findings suggest that there are mechanistic differences in the modes of action of ML240 and ML241. Among the two compounds, only ML240 induced the accumulation of LC3-B (a marker of early autophagosome formation or impaired autophagic maturation) and thus behaved similar to DBeQ (20, 90). ML240 also outperformed ML241 in its antiproliferative effects as assessed in the NCI-60 cancer cell line panel.
NMS-873, unlike the other p97 inhibitors described above, is an allosteric p97 inhibitor that induces the accumulation of polyubiquitinated proteins, leading to ER stress and inhibition of autophagosome maturation (19). NMS-873 is thought to disrupt the ATPase activity of p97 by binding to the linker domain between the D1 and D2 ATPase domains of p97, leading to the stabilization of D2-ADP bound p97. Consequently, NMS-873 showed reduced sensitivity to limited trypsin digestion, which is associated to its higher stability and potency (19). Both the ATP-competitive (DBeQ, ML240, and ML241) inhibitors as well as the allosteric inhibitor NMS-873 have differential effects on the D1 and D2 ATPase activity of p97. In vitro ATPase activity assays revealed that NMS-873 and DBeQ inhibit ATPase activity from both D1 and D2 domains. ML240 and ML241, however, seem to selectively inhibit the D2 domain of p97 (15). The inhibitory activity of ML240 and ML241 is also dramatically decreased by the presence of the p97 cofactor p47 (49- to 37-fold) compared to DBeQ and NMS-873 (2- to 6-fold) (15, 91). In contrast, cofactors p37 and Npl4-Ufd1 did not change the potency of ML240 and ML241 (92). These findings suggest that it is possible to develop p97 inhibitors that exhibit complex-specific inhibitory activities, which could be used to inhibit specific functions of p97.
While the compounds described, so far, in this review have excellent ATPase-inhibitory activities, they lack “drug-like” properties, thus making them unsuitable for in vivo studies. Utilizing DBeQ, ML240, and ML241 and cogeners such as Compound 18 and Compound 29 as the starting point, CB-5083 (1-[4-(benzylamino)-5H,7H,8H-pyrano[4,3-d]pyrimidin-2-yl]-2-methyl-1H-indole-4-carboxamide) was developed as a D2 domain selective, first-in-class p97 inhibitor with an IC50 of 11 nM (21). An in-depth analysis of the pathways affected by CB-5083 revealed that it affected the expression of mediators of the UPR (93). Significantly altered genes in response to CB-5083 treatment include CHOP, DR5, HSPA5, HERPUD1, SEL1L, SYVN1, and EDEM1. CB-5083 showed promising antitumor responses in colorectal, lung cancer, and plasmacytoma tumor xenografts as well as patient-derived xenograft models of colorectal cancer (93). The study also determined that resistance to CB-5083 was dictated by both mRNA and protein levels of p97 as well as possibly other cell-specific factors such as expression of EDEM1 and autocrine motility factor (AMFR). Additionally, activation of the mitogen-activated protein kinase (MAPK) pathway and phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) correlated with the activity of CB-5083. Owing to its promising pre-clinical activity, CB-5083 is being tested in the clinic against relapsed/refractory multiple myeloma and advanced solid tumors (NCT02243917 and NCT02223598).
Potential Pitfalls and Advantages of p97 Inhibition
Activation of the IRE1α-XBP1 pathway in response to misfolded proteins in the ER induces ER chaperones that promote efficient protein refolding and promotes ERAD. However, XBP1 and PERK activation has also been reported to activate an epithelial–mesenchymal transition (EMT) phenotype in cancer cells (94–96). XBP1-induced EMT is associated with the upregulation of transcription factor Snail in breast cancer (96). Similar findings have also been reported in colorectal carcinoma (CRC) where the IRE1α-XBP1 pathway promotes proliferation and invasion of CRC cells (95). Consistent with these findings, siRNA-knockdown of p97 or treatment with Eeyarestatin I was reported to induce an EMT-like phenotype in lung adenocarcinoma cells (97). These studies indicate that p97 inhibition in cancer cells need to be considered with caution.
While studies have shown that the induction of ER stress in cancers promotes EMT and invasion, this can be considered as their vulnerability. This is because ER stress activation in cancers can potentially sensitize them to agents that accentuate ER stress (94). p97 inhibitors (EerI and DBeQ) have been reported to induce synergistic cell death in combination with the proteasome inhibitor bortezomib in mantle cell lymphoma and multiple myeloma (84, 98). Genetic knockdown of p97 inhibits cancer cell viability and synergizes with a wide variety of agents that induce DNA damage, growth inhibition, and cellular stress in vitro (19). Interestingly, no evidence of EMT-like phenotype has been reported for CB-5083 in in vivo models. Future studies to disrupt specific adaptor/cofactor-p97 associations will likely lead to the identification of novel p97 inhibitors with enhanced specificity and/or superior anticancer activity.
Author Contributions
RR, SW, and FS conceptualized and prepared the mini review. PV and TH helped in manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
RR is a recipient of P30 CA168524 award from NCI.
References
1. Frohlich KU, Fries HW, Rudiger M, Erdmann R, Botstein D, Mecke D. Yeast cell cycle protein CDC48p shows full-length homology to the mammalian protein VCP and is a member of a protein family involved in secretion, peroxisome formation, and gene expression. J Cell Biol (1991) 114(3):443–53. doi:10.1083/jcb.114.3.443
2. Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol (2012) 14(2):117–23. doi:10.1038/ncb2407ncb2407
3. Chapman E, Maksim N, de la Cruz F, La Clair JJ. Inhibitors of the AAA+ chaperone p97. Molecules (2015) 20(2):3027–49. doi:10.3390/molecules20023027
4. Zhang X, Shaw A, Bates PA, Newman RH, Gowen B, Orlova E, et al. Structure of the AAA ATPase p97. Mol Cell (2000) 6(6):1473–84. doi:10.1016/S1097-2765(00)00143-X
5. Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci (2014) 127(Pt 18):3877–83. doi:10.1242/jcs.093831jcs.093831
6. Huyton T, Pye VE, Briggs LC, Flynn TC, Beuron F, Kondo H, et al. The crystal structure of murine p97/VCP at 3.6A. J Struct Biol (2003) 144(3):337–48. doi:10.1016/j.jsb.2003.10.007
7. DeLaBarre B, Brunger AT. Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Biol (2003) 10(10):856–63. doi:10.1038/nsb972
8. Dreveny I, Kondo H, Uchiyama K, Shaw A, Zhang X, Freemont PS. Structural basis of the interaction between the AAA ATPase p97/VCP and its adaptor protein p47. EMBO J (2004) 23(5):1030–9. doi:10.1038/sj.emboj.7600139
9. Pye VE, Dreveny I, Briggs LC, Sands C, Beuron F, Zhang X, et al. Going through the motions: the ATPase cycle of p97. J Struct Biol (2006) 156(1):12–28. doi:10.1016/j.jsb.2006.03.003
10. DeLaBarre B, Brunger AT. Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol (2005) 347(2):437–52. doi:10.1016/j.jmb.2005.01.060
11. Banerjee S, Bartesaghi A, Merk A, Rao P, Bulfer SL, Yan Y, et al. 2.3 Å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science (2016) 351(6275):871–5. doi:10.1126/science.aad7974
12. Wang Q, Song C, Li CC. Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem Biophys Res Commun (2003) 300(2):253–60. doi:10.1016/S0006-291X(02)02840-1
13. Song C, Wang Q, Li CC. ATPase activity of p97-valosin-containing protein (VCP). D2 mediates the major enzyme activity, and D1 contributes to the heat-induced activity. J Biol Chem (2003) 278(6):3648–55. doi:10.1074/jbc.M208422200
14. Briggs LC, Baldwin GS, Miyata N, Kondo H, Zhang X, Freemont PS. Analysis of nucleotide binding to P97 reveals the properties of a tandem AAA hexameric ATPase. J Biol Chem (2008) 283(20):13745–52. doi:10.1074/jbc.M709632200
15. Chou TF, Bulfer SL, Weihl CC, Li K, Lis LG, Walters MA, et al. Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J Mol Biol (2014) 426(15):2886–99. doi:10.1016/j.jmb.2014.05.022
16. Tang WK, Xia D. Role of the D1-D2 linker of human VCP/p97 in the asymmetry and ATPase activity of the D1-domain. Sci Rep (2016) 6:20037. doi:10.1038/srep20037
17. Chapman E, Fry AN, Kang M. The complexities of p97 function in health and disease. Mol Biosyst (2011) 7(3):700–10. doi:10.1039/c0mb00176g
18. Chou TF, Deshaies RJ. Development of p97 AAA ATPase inhibitors. Autophagy (2011) 7(9):1091–2. doi:10.4161/auto.7.9.16489
19. Magnaghi P, D’Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol (2013) 9(9):548–56. doi:10.1038/nchembio.1313
20. Chou TF, Li K, Frankowski KJ, Schoenen FJ, Deshaies RJ. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem (2013) 8(2):297–312. doi:10.1002/cmdc.201200520
21. Zhou HJ, Wang J, Yao B, Wong S, Djakovic S, Kumar B, et al. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083). J Med Chem (2015) 58(24):9480–97. doi:10.1021/acs.jmedchem.5b01346
22. Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, et al. p47 is a cofactor for p97-mediated membrane fusion. Nature (1997) 388(6637):75–8. doi:10.1038/40411
23. Woodman PG. p97, a protein coping with multiple identities. J Cell Sci (2003) 116(Pt 21):4283–90. doi:10.1242/jcs.00817
24. Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature (2001) 414(6864):652–6. doi:10.1038/414652a
25. Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J (2000) 19(10):2181–92. doi:10.1093/emboj/19.10.2181
26. Zhang Z, Wang Y, Li C, Shi Z, Hao Q, Wang W, et al. The transitional endoplasmic reticulum ATPase p97 regulates the alternative nuclear factor NF-kappaB signaling via partial degradation of the NF-kappaB subunit p100. J Biol Chem (2015) 290(32):19558–68. doi:10.1074/jbc.M114.630061
27. Haines DS, Lee JE, Beauparlant SL, Kyle DB, den Besten W, Sweredoski MJ, et al. Protein interaction profiling of the p97 adaptor UBXD1 points to a role for the complex in modulating ERGIC-53 trafficking. Mol Cell Proteomics (2012) 11(6):M111016444. doi:10.1074/mcp.M111.016444
28. Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev (2007) 21(17):2172–81. doi:10.1101/gad.436407
29. Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, et al. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J (2006) 25(14):3357–66. doi:10.1038/sj.emboj.7601210
30. Noda C, Kimura H, Arasaki K, Matsushita M, Yamamoto A, Wakana Y, et al. Valosin-containing protein-interacting membrane protein (VIMP) links the endoplasmic reticulum with microtubules in concert with cytoskeleton-linking membrane protein (CLIMP)-63. J Biol Chem (2014) 289(35):24304–13. doi:10.1074/jbc.M114.571372
31. Wang Q, Li L, Ye Y. Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J Cell Biol (2006) 174(7):963–71. doi:10.1083/jcb.200605100
32. Raman M, Sergeev M, Garnaas M, Lydeard JR, Huttlin EL, Goessling W, et al. Systematic proteomics of the VCP-UBXD adaptor network identifies a role for UBXN10 in regulating ciliogenesis. Nat Cell Biol (2015) 17(10):1356–69. doi:10.1038/ncb3238
33. Ballar P, Zhong Y, Nagahama M, Tagaya M, Shen Y, Fang S. Identification of SVIP as an endogenous inhibitor of endoplasmic reticulum-associated degradation. J Biol Chem (2007) 282(47):33908–14. doi:10.1074/jbc.M704446200
34. Wang Y, Ballar P, Zhong Y, Zhang X, Liu C, Zhang YJ, et al. SVIP induces localization of p97/VCP to the plasma and lysosomal membranes and regulates autophagy. PLoS One (2011) 6(8):e24478. doi:10.1371/journal.pone.0024478
35. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell (2009) 136(5):823–37. doi:10.1016/j.cell.2009.02.024
36. Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell (2007) 130(6):986–8. doi:10.1016/j.cell.2007.09.007
37. Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature (2003) 425(6956):407–10. doi:10.1038/nature01913
38. Donnelly N, Storchova Z. Aneuploidy and proteotoxic stress in cancer. Mol Cell Oncol (2015) 2(2):e976491. doi:10.4161/23723556.2014.976491
39. Franz A, Ackermann L, Hoppe T. Ring of change: CDC48/p97 drives protein dynamics at chromatin. Front Genet (2016) 7:73. doi:10.3389/fgene.2016.00073
40. Dantuma NP, Hoppe T. Growing sphere of influence: Cdc48/p97 orchestrates ubiquitin-dependent extraction from chromatin. Trends Cell Biol (2012) 22(9):483–91. doi:10.1016/j.tcb.2012.06.003
41. Home T, Jensen RA, Rao R. Heat shock factor 1 in protein homeostasis and oncogenic signal integration. Cancer Res (2015) 75(6):907–12. doi:10.1158/0008-5472.CAN-14-2905
42. Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, et al. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell (2014) 158(2):434–48. doi:10.1016/j.cell.2014.05.039
43. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer (2005) 5(10):761–72. doi:10.1038/nrc1716
44. Yao TP. The role of ubiquitin in autophagy-dependent protein aggregate processing. Genes Cancer (2010) 1(7):779–86. doi:10.1177/1947601910383277
45. Lee JY, Yao TP. Quality control autophagy: a joint effort of ubiquitin, protein deacetylase and actin cytoskeleton. Autophagy (2010) 6(4):555–7. doi:10.4161/auto.6.4.11812
46. Dargemont C, Ossareh-Nazari B. Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim Biophys Acta (2012) 1823(1):138–44. doi:10.1016/j.bbamcr.2011.07.011
47. Pernet L, Faure V, Gilquin B, Dufour-Guerin S, Khochbin S, Vourc’h C. HDAC6-ubiquitin interaction controls the duration of HSF1 activation after heat shock. Mol Biol Cell (2014) 25(25):4187–94. doi:10.1091/mbc.E14-06-1032
48. Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem (2005) 280(29):26729–34. doi:10.1074/jbc.C500186200
49. Rao R, Fiskus W, Yang Y, Lee P, Joshi R, Fernandez P, et al. HDAC6 inhibition enhances 17-AAG – mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood (2008) 112(5):1886–93. doi:10.1182/blood-2008-03-143644
50. Lodish HF, Kong N. The secretory pathway is normal in dithiothreitol-treated cells, but disulfide-bonded proteins are reduced and reversibly retained in the endoplasmic reticulum. J Biol Chem (1993) 268(27):20598–605.
51. Benham AM. Protein secretion and the endoplasmic reticulum. Cold Spring Harb Perspect Biol (2012) 4(8):a012872. doi:10.1101/cshperspect.a012872
52. Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol (2007) 18(6):716–31. doi:10.1016/j.semcdb.2007.09.003
53. Moore KA, Hollien J. The unfolded protein response in secretory cell function. Annu Rev Genet (2012) 46:165–83. doi:10.1146/annurev-genet-110711-155644
54. Clarke HJ, Chambers JE, Liniker E, Marciniak SJ. Endoplasmic reticulum stress in malignancy. Cancer Cell (2014) 25(5):563–73. doi:10.1016/j.ccr.2014.03.015
55. Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature (2016) 529(7586):326–35. doi:10.1038/nature17041
56. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol (2003) 23(21):7448–59. doi:10.1128/MCB.23.21.7448-7459.2003
57. Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell (2007) 129(7):1337–49. doi:10.1016/j.cell.2007.04.027
58. Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem (2004) 279(44):45495–502. doi:10.1074/jbc.M406933200
59. Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell (2000) 6(6):1355–64. doi:10.1016/S1097-2765(00)00133-7
60. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol (2013) 15(5):481–90. doi:10.1038/ncb2738
61. Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol (2008) 9(12):944–57. doi:10.1038/nrm2546
62. Avci D, Lemberg MK. Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol (2015) 25(10):611–22. doi:10.1016/j.tcb.2015.07.003
63. Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem (2004) 73:1019–49. doi:10.1146/annurev.biochem.73.011303.073752
64. Olzmann JA, Kopito RR, Christianson JC. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol (2013) 5(9):1–16. doi:10.1101/cshperspect.a013185
65. Lemus L, Goder V. Regulation of endoplasmic reticulum-associated protein degradation (ERAD) by ubiquitin. Cells (2014) 3(3):824–47. doi:10.3390/cells3030824
66. Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci U S A (2005) 102(40):14132–8. doi:10.1073/pnas.0505006102
67. Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang SAAA. ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplasmic reticulum-associated degradation. J Biol Chem (2004) 279(44):45676–84. doi:10.1074/jbc.M409034200
68. Shenkman M, Groisman B, Ron E, Avezov E, Hendershot LM, Lederkremer GZ. A shared endoplasmic reticulum-associated degradation pathway involving the EDEM1 protein for glycosylated and nonglycosylated proteins. J Biol Chem (2013) 288(4):2167–78. doi:10.1074/jbc.M112.438275
69. Ushioda R, Hoseki J, Nagata K. Glycosylation-independent ERAD pathway serves as a backup system under ER stress. Mol Biol Cell (2013) 24(20):3155–63. doi:10.1091/mbc.E13-03-0138
70. Tsai YC, Weissman AM. Ubiquitylation in ERAD: reversing to go forward? PLoS Biol (2011) 9(3):e1001038. doi:10.1371/journal.pbio.1001038
71. Kikkert M, Doolman R, Dai M, Avner R, Hassink G, van Voorden S, et al. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J Biol Chem (2004) 279(5):3525–34. doi:10.1074/jbc.M307453200
72. Alexandru G, Graumann J, Smith GT, Kolawa NJ, Fang R, Deshaies RJ. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell (2008) 134(5):804–16. doi:10.1016/j.cell.2008.06.048
73. Meyer H. p97 complexes as signal integration hubs. BMC Biol (2012) 10:48. doi:10.1186/1741-7007-10-48
74. Richly H, Rape M, Braun S, Rumpf S, Hoege C, Jentsch S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell (2005) 120(1):73–84. doi:10.1016/j.cell.2004.11.013
75. Lykke-Andersen J, Bennett EJ. Protecting the proteome: eukaryotic cotranslational quality control pathways. J Cell Biol (2014) 204(4):467–76. doi:10.1083/jcb.201311103
76. Defenouillere Q, Yao Y, Mouaikel J, Namane A, Galopier A, Decourty L, et al. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc Natl Acad Sci U S A (2013) 110(13):5046–51. doi:10.1073/pnas.1221724110
77. Verma R, Oania RS, Kolawa NJ, Deshaies RJ. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. Elife (2013) 2:e00308. doi:10.7554/eLife.00308
78. Taylor EB, Rutter J. Mitochondrial quality control by the ubiquitin-proteasome system. Biochem Soc Trans (2011) 39(5):1509–13. doi:10.1042/BST0391509
79. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol (2008) 183(5):795–803. doi:10.1083/jcb.200809125
80. Wu X, Li L, Jiang H. Doa1 targets ubiquitinated substrates for mitochondria-associated degradation. J Cell Biol (2016) 213(1):49–63. doi:10.1083/jcb.201510098
81. Ju JS, Weihl CC. p97/VCP at the intersection of the autophagy and the ubiquitin proteasome system. Autophagy (2010) 6(2):283–5. doi:10.4161/auto.6.2.11063
82. Buchan JR, Kolaitis RM, Taylor JP, Parker R. Eukaryotic stress granules are cleared by autophagy and Cdc48/VCP function. Cell (2013) 153(7):1461–74. doi:10.1016/j.cell.2013.05.037
83. Wang Q, Li L, Ye Y. Inhibition of p97-dependent protein degradation by Eeyarestatin I. J Biol Chem (2008) 283(12):7445–54. doi:10.1074/jbc.M708347200
84. Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc Natl Acad Sci U S A (2009) 106(7):2200–5. doi:10.1073/pnas.0807611106
85. Tao S, Tillotson J, Wijeratne EM, Xu YM, Kang M, Wu T, et al. Withaferin A analogs that target the AAA+ chaperone p97. ACS Chem Biol (2015) 10(8):1916–24. doi:10.1021/acschembio.5b00367
86. Alverez C, Arkin MR, Bulfer SL, Colombo R, Kovaliov M, LaPorte MG, et al. Structure-activity study of bioisosteric trifluoromethyl and pentafluorosulfanyl indole inhibitors of the AAA ATPase p97. ACS Med Chem Lett (2015) 6(12):1225–30. doi:10.1021/acsmedchemlett.5b00364
87. Alverez C, Bulfer SL, Chakrasali R, Chimenti MS, Deshaies RJ, Green N, et al. Allosteric indole amide inhibitors of p97: identification of a novel probe of the ubiquitin pathway. ACS Med Chem Lett (2016) 7(2):182–7. doi:10.1021/acsmedchemlett.5b00396
88. Wijeratne EM, Gunaherath GM, Chapla VM, Tillotson J, de la Cruz F, Kang M, et al. Oxaspirol B with p97 inhibitory activity and other oxaspirols from lecythophora sp. FL1375 and FL1031, endolichenic fungi inhabiting Parmotrema tinctorum and Cladonia evansii. J Nat Prod (2016) 79(2):340–52. doi:10.1021/acs.jnatprod.5b00986
89. Chou TF, Brown SJ, Minond D, Nordin BE, Li K, Jones AC, et al. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A (2011) 108(12):4834–9. doi:10.1073/pnas.1015312108
90. Tasdemir E, Galluzzi L, Maiuri MC, Criollo A, Vitale I, Hangen E, et al. Methods for assessing autophagy and autophagic cell death. Methods Mol Biol (2008) 445:29–76. doi:10.1007/978-1-59745-157-4_3
91. Fang CJ, Gui L, Zhang X, Moen DR, Li K, Frankowski KJ, et al. Evaluating p97 inhibitor analogues for their domain selectivity and potency against the p97-p47 complex. ChemMedChem (2015) 10(1):52–6. doi:10.1002/cmdc.201402420
92. Gui L, Zhang X, Li K, Frankowski KJ, Li S, Wong DE, et al. Evaluating p97 inhibitor analogues for potency against p97-p37 and p97-Npl4-Ufd1 complexes. ChemMedChem (2016) 11(9):953–7. doi:10.1002/cmdc.201600214
93. Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, et al. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell (2015) 28(5):653–65. doi:10.1016/j.ccell.2015.10.002
94. Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov (2014) 4(6):702–15. doi:10.1158/2159-8290.CD-13-0945
95. Jin C, Jin Z, Chen NZ, Lu M, Liu CB, Hu WL, et al. Activation of IRE1alpha-XBP1 pathway induces cell proliferation and invasion in colorectal carcinoma. Biochem Biophys Res Commun (2016) 470(1):75–81. doi:10.1016/j.bbrc.2015.12.119
96. Li H, Chen X, Gao Y, Wu J, Zeng F, Song F. XBP1 induces snail expression to promote epithelial- to-mesenchymal transition and invasion of breast cancer cells. Cell Signal (2015) 27(1):82–9. doi:10.1016/j.cellsig.2014.09.018
97. Shah PP, Beverly LJ. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget (2015) 6(19):17725–37. doi:10.18632/oncotarget.3918
Keywords: ERAD, cancer, p97 inhibitors, proteotoxic stress, VCP, CDC48, ER stress
Citation: Vekaria PH, Home T, Weir S, Schoenen FJ and Rao R (2016) Targeting p97 to Disrupt Protein Homeostasis in Cancer. Front. Oncol. 6:181. doi: 10.3389/fonc.2016.00181
Received: 03 May 2016; Accepted: 22 July 2016;
Published: 03 August 2016
Edited by:
Matiullah Khan, Asian Institute of Medicine, Science & Technology, MalaysiaCopyright: © 2016 Vekaria, Home, Weir, Schoenen and Rao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rekha Rao, cnJhb21hbmVwYWxsaUBrdW1jLmVkdQ==