 Mev Dominguez-Valentin1*
Mev Dominguez-Valentin1* Patrik Wernhoff2
Patrik Wernhoff2 Andrea R. Cajal3
Andrea R. Cajal3 Pablo G. Kalfayan4
Pablo G. Kalfayan4 Tamara A. Piñero4
Tamara A. Piñero4 Maria L. Gonzalez4
Maria L. Gonzalez4 Alejandra Ferro4
Alejandra Ferro4 Ines Sammartino4
Ines Sammartino4 Natalia S. Causada Calo4
Natalia S. Causada Calo4 Carlos A. Vaccaro4
Carlos A. Vaccaro4
- 1Department of Tumor Biology, Institute for Cancer Research, Oslo University Hospital, Oslo, Norway
- 2Unit of Muscle Biology, Lund Transgenic Core Facility/Reproductive Immunology, Department of Experimental Medical Science, Lund University, Lund, Sweden
- 3Institute of Basic Sciences and Experimental Medicine (ICBME), Instituto Universitario Hospital Italiano, Buenos Aires, Argentina
- 4Programa de Cancer Hereditario (ProCanHe), Hospital Italiano de Buenos Aires, Buenos Aires, Argentina
Introduction
Heredity is a major risk factor for colorectal cancer (CRC). Identification of individuals and families at increased risk allows for targeted surveillance, which has been shown to reduce morbidity and mortality from CRC (1, 2).
Lynch syndrome is a multi-tumor syndrome with particularly high risks for colorectal, endometrial, and ovarian cancer (3–6). The syndrome is caused by germline DNA-mismatch repair (MMR) gene mutations with major contributions from MLH1 (MIM#120436) (42%), MSH2 (MIM#609309) (33%), MSH6 (MIM#600678) (18%), and PMS2 (MIM#600259) (8%). Only about one-third of the Lynch syndrome families fulfill the Amsterdam criteria (AC) (7–9). The cumulative incidence of any cancer at 70 years of age is 72% for MLH1 and MSH2 mutation carriers but lower in MSH6 (52%) and PMS2 (18%) mutation carriers. MSH6 and PMS2 carriers developed no cancers before 40 years of age (10).
Mutation screening in a relatively large proportion of South American families with suspected Lynch syndrome has recently identified 99 disease-predisposing mutations in MLH1 and MSH2, which mutation spectrum is predominated by MLH1 (60%) and MSH2 (40%). Among the reported mutations, genetic hot-spot regions, new and potential founder mutations have been described in the South American population (11, 12).
Several genome-wide association studies have identified single nucleotide polymorphisms (SNPs) in at least 15 independent loci associated with CRC risk (odds ratio ranging from 1.10 to 1.26 per risk allele) (13–15). Although there is no evidence that these SNPs associated with CRC in the general population are modifiers of the risk for MMR gene mutation carriers overall and therefore any evidence of proven clinical utility in Lynch syndrome (16).
The MLH1 Ile219Val (rs1799977) is a common germline alteration, located in exon 8 at the nucleotide 655. This polymorphism has been described in a high frequency of the South American Lynch syndrome population, but no modifier effect of CRC risk and MMR disease-predisposing mutation carriers was observed (17). However, it has been reported to confer a twofold-increased risk of CRC development in sporadic Mexican patients (18). Other conditions that have been associated with this polymorphism include childhood acute lymphoblastic leukemia, breast cancer, radiation-induced rectal or bladder toxicity, and ulcerative colitis (19–23). It is unknown whether the MLH1 Ile219Val polymorphism has an effect on cancer risk and in the MMR capacity in Argentinean families with suspected Lynch syndrome. Thus, we aim to determine its frequency, its correlation with disease-predisposing MMR gene mutations, and to delineate the clinical characteristics from these families.
Patients and Methods
Ethics Statement
The study was performed in compliance with the Helsinki Declaration and approved by the Ethics Committee of the Hospital Italiano de Buenos Aires in Argentina. Written informed consent was obtained from all participants during genetic counseling sessions.
Patient Selection
The Hereditary cancer registry of the Hospital Italiano de Buenos Aires is a national Argentinean registry that contains information of all families identified with proven or suspected hereditary cancer. Through research collaborations, data from the registry is freely available. This prospective database contains clinical, molecular, and familial data, which is clinically relevant. In addition, the electronic medical records of these patients can be reviewed to retrieve further data.
We obtained molecular (genetic testing) and epidemiological data (family history) from 48 families that fulfilled AC (n = 33), Bethesda guidelines criteria (n = 8), and families suggestive of a dominant CRC inheritance syndrome (n = 7) (7, 8, 24) between 2009 and 2016. Family pedigree was constructed based on the information provided by the proband during one or more genetic counseling sessions.
Disease-Predisposing MMR Gene Mutations and Nomenclature
Germline mutation screening of MLH1, MSH2, MSH6, and PMS2 was performed by next generation sequencing platform using the personal genome machine® system (PGM, LT) according to the manufacturer’s instructions. Mutation nomenclature followed the Human Genome Variation Society (HGVS) guidelines.1 All identified mutations were compared to previously reported in MMR databases, maintained by the International Society for Gastrointestinal Hereditary Tumors2 and the French MMR network.3
Statistical Analysis
Age comparison between the groups was performed using Mann–Whitney U-test. In the tables, results were expressed, as mean age ± SD. Significance for expected frequencies was tested using Chi-squared test. The analyses were performed using Statview (SAS Institute Inc. 100 SAS Campus Drive Cary, NC 27513-2414, USA) and Chi-squared Calculator (Social Science Statistics4). For all tests, p < 0.05 was considered statistically significant.
Results
Overall, forty-eight hereditary CRC families were identified from the Hereditary cancer register of the Hospital Italiano de Buenos Aires. Of them, 19/48 (40%) of the families carried disease-predisposing mutations in the MMR genes. The mean age at first diagnosis in Lynch syndrome families was 42 ± 7.3 years (range 27–55). Mutations in MLH1 and MSH2 genes were identified in 17/33 (52%) of the families that fulfilled the AC. However, no disease-predisposing mutations in MSH6 and PMS2 genes were found in this series (Table 1). Mutational and clinical information from the Argentinean Lynch syndrome families is described in Table 1.
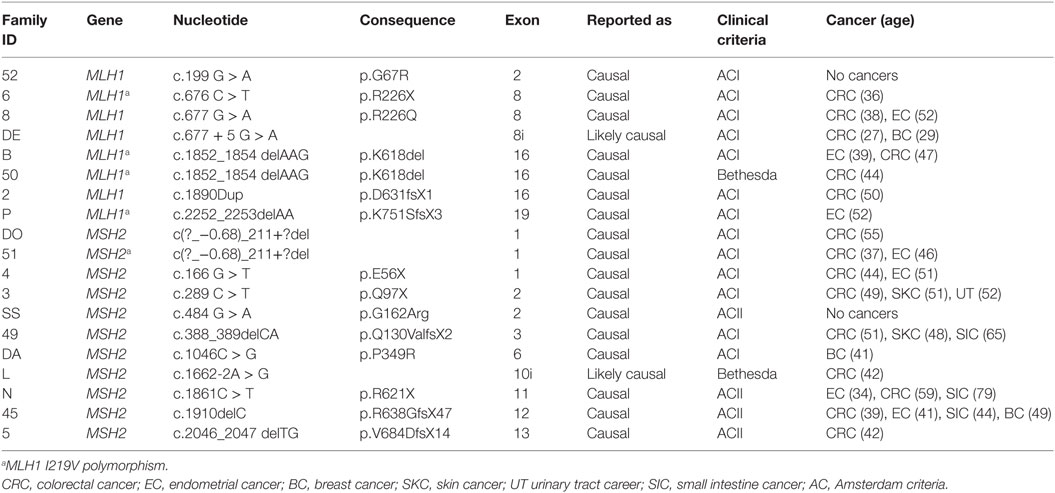
Table 1. Disease-predisposing MMR gene mutations in Argentinean Lynch syndrome families.
When we analyzed the genotypic and allelic frequencies of the MLH1 Ile219Val polymorphism, we found Val-carriers in 44% (21/48) of the Argentinean families with suspected of Lynch syndrome. Of them, 95% (20/21) were Ile/Val heterozygotes and 5% (1/21) were Val/Val homozygotes. The allelic frequency of Ile and Val were 0.77 and 0.23, respectively (Table 2).
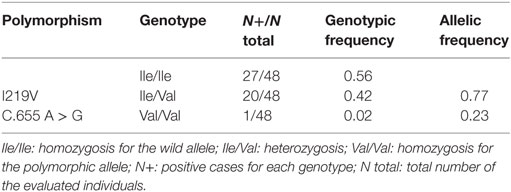
Table 2. Genotypic and allelic frequency of the MLH1 I219V polymorhism in Argentinean families with suspected Lynch syndrome.
Regarding the clinical characteristics, a slightly early mean age at CRC and endometrial cancer diagnoses was observed in the families without the MLH1 Ile219Val polymorphism in comparison to the families harboring the MLH1 polymorphism (45 and 44 years vs. 49 and 46 years, respectively) (p > 0.05). The presence of AC and Bethesda guidelines was not associated with the MLH1 Ile219Val polymorphism (p > 0.05). As expected, CRC and endometrial cancer were the most frequent tumors among the MLH1 Ile219Val carriers and non-carriers. While skin tumors were observed in both groups of families, breast and small intestine tumors were observed only in the non-MLH1 Ile219Val families.
Of the 21 families with the MLH1 Ile219Val polymorphism, 5 (24%) harbored disease-predisposing MMR gene mutations; MLH1 was the most affected gene occurring in 4/5 (80%) of the cases. In the 27 non-MLH1 Ile219Val polymorphism families, 14 (52%) harbored MLH1 or MSH2 disease predisposing mutations; MHS2 the most frequently affected gene occurring in 10 of them (71%) (available upon request). In the group of MLH1 Ile219Val polymorphism (n = 21) that displayed disease-predisposing MMR gene mutations (n = 5/21), the Val-allele was significantly associated with MLH1 and/or MSH2 mutations (p < 0.05).
In addition, we analyzed the clinical features of the 21 families harboring MLH1 Ile219Val polymorphism in the presence of disease-predisposing MMR gene mutations, MMR carrier’s families showed an early mean age at CRC diagnosis (41 ± 5.35) years, although these differences were not significant (p > 0.05) (available upon request).
In order to evaluate if the MLH1 Ile219Val polymorphism exerts any influence at the age of CRC onset, we analyzed the mean age at CRC diagnosis of families harboring or not the MLH1 Ile219Val polymorphism according to their MMR status (MLH1 or MSH2). The MLH1 Ile219Val polymorphism group (n = 21), which harbored disease-predisposing mutations in MLH1, or MSH2 showed a mean age at CRC onset of 42.3 and 37 years, respectively. In the non-MLH1 Ile219Val carriers, the mean age among those who displayed the mutations was 38.3 and 47.6 years, respectively. However, no statistically significant association was found between these groups (available upon request).
Discussion
In the Argentinean suspected Lynch syndrome families, the genetic polymorphism (Ile219Val) in the MLH1 gene was found in 44% of the population, which fits with the previous genetic studies that reported an incidence of 31–80% from different populations (17, 24–26). The allelic frequencies for Ile and Val (0.77 and 0.23) were similar to the reports from South American suspected Lynch syndrome families (0.7 and 0.3), German Lynch syndrome families (0.69 and 0.31), Swedish Lynch syndrome families (0.64 and 0.34), and Italian suspected Lynch syndrome families (0.33 for Val) (17, 24–27). This similarity may be explained by the influence of European ancestors in the South American population, particularly in Brazil, Uruguay, and Argentina (11). However, our allelic frequencies are slightly higher than the minor allele frequency (MAF) reported by the 1000 Genomes Project (0.87 and 0.13, respectively) although similar to Puerto Rico when analyzed into the American population (28).
In the current study, no association was observed between the MLH1 Ile219Val polymorphism and cancer risk. This is consistent with several functional analyses indicating that the variant has binding properties to PMS2 and DNA repair efficiency similar to the wild type (29, 30). A recent CRC meta-analysis including 8068 cases and 6568 controls from Australia, Czech Republic, Spain, Germany, and Sweden with frequencies of 0.678 and 0.671, respectively, found not associations of CRC risk and MLH1 Ile219Val polymorphism (31). However, Kim et al. suggested that the homozygosity for the 219V variant was correlated with a significantly reduced MLH1 expression among sporadic CRC cases (32). The difference might be a reflection of environmental impact on gene distribution, ethnic background or small-sized family cohort (33–35).
We found that the absence of the MLH1 Ile219Val polymorphism was associated with an early mean age at CRC cancer diagnosis, and the development of breast tumors was the main clinical features. This is in line with the Brazilian hereditary non-polyposis colorectal cancer (HNPCC) familial study that reported that breast cancer followed by endometrial and uterine cervix cancer are the most frequent extracolonic tumors found in women (36). Although, this is a small increase in breast cancer risk, it might point out interesting biological connections.
Regarding the association of the MLH1 Ile219Val polymorphism and disease-predisposing MMR gene mutations, we described a lower frequency (24%) of disease-predisposing MMR gene mutations compared to the reported by the South American Lynch syndrome families (33%) (17). In families harboring the MLH1 Ile219Val polymorphism and disease-predisposing MMR mutations, MLH1 was the most affected gene. The seemingly unique contribution than the 57 (MLH1) and 43% (MSH2) reported by the South American population could reflect random variation, population structure, size sample and genetic heterogeneity (17). In this series, we found a statistically significant link between the presence of the MLH1 Ile219Val polymorphism and disease-predisposing MMR gene mutations carriers, which suggest that these genetic discriminators may be relevant for molecular diagnostics in this population. However, our study has a limited sample size and the results need further validation. Studies with larger sample size are needed to further evaluate the role of this polymorphism.
In summary, our findings point to a high frequency of the MLH1 Ile219Val polymorphism in the Argentinean families with suspected Lynch syndrome and its modifier effect with the disease-predisposing MLH1/MSH2 genes mutations. Our data may provide important clues to contribute to molecular diagnostics, improved risk stratification, and targeted therapeutic strategies in hereditary CRC.
Author Contributions
MD-V, PW, AC, PK, AF, MG, IS, NC, TP, and CV participated in the acquisition of data, or analysis, interpretation of data and have been involved in drafting the manuscript. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
1. Mecklin JP, Jarvinen HJ. Surveillance in Lynch syndrome. Fam Cancer (2005) 4(3):267–71. doi: 10.1007/s10689-005-1475-x
2. Balmana J, Castells A, Cervantes A, Group EGW. Familial colorectal cancer risk: ESMO Clinical Practice Guidelines. Ann Oncol (2010) 21(Suppl 5):v78–81. doi:10.1093/annonc/mdq161
3. Stoffel E, Mukherjee B, Raymond VM, Tayob N, Kastrinos F, Sparr J, et al. Calculation of risk of colorectal and endometrial cancer among patients with Lynch syndrome. Gastroenterology (2009) 137(5):1621–7. doi:10.1053/j.gastro.2009.07.039
4. Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer (1999) 81(2):214–8. doi:10.1002/(SICI)1097-0215(19990412)81:2<214::AID-IJC8>3.0.CO;2-L
5. Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA (2011) 305(22):2304–10. doi:10.1001/jama.2011.743
6. Win AK, MacInnis RJ, Hopper JL, Jenkins MA. Risk prediction models for colorectal cancer: a review. Cancer Epidemiol Biomarkers Prev (2012) 21(3):398–410. doi:10.1158/1055-9965.EPI-11-0771
7. Lynch HT, Lynch PM. Molecular screening for the Lynch syndrome – better than family history? N Engl J Med (2005) 352(18):1920–2. doi:10.1056/NEJMe058058
8. Vasen HFA, Moslein G, Alonso A, Bernstein I, Bertario L, Blanco I, et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet (2007) 44(6):353–62. doi:10.1136/jmg.2007.048991
9. Plazzer JP, Sijmons RH, Woods MO, Peltomaki P, Thompson B, Den Dunnen JT, et al. The InSiGHT database: utilizing 100 years of insights into Lynch syndrome. Fam Cancer (2013) 12(2):175–80. doi:10.1007/s10689-013-9616-0
10. Moller P, Seppala T, Bernstein I, Holinski-Feder E, Sala P, Evans DG, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut (2015) 1–9. doi:10.1136/gutjnl-2015-309675
11. Dominguez-Valentin M, Nilbert M, Wernhoff P, Lopez-Kostner F, Vaccaro C, Sarroca C, et al. Mutation spectrum in South American Lynch syndrome families. Hered Cancer Clin Pract (2013) 11:18. doi:10.1186/1897-4287-11-18
12. Vaccaro CA, Sarroca C, Rossi B, Lopez-Kostner F, Dominguez M, Calo NC, et al. Lynch syndrome in South America: past, present and future. Fam Cancer (2016) 15(3):437–45. doi:10.1007/s10689-016-9903-7
13. Tenesa A, Dunlop MG. New insights into the aetiology of colorectal cancer from genome-wide association studies. Nat Rev Genet (2009) 10(6):353–8. doi:10.1038/nrg2574
14. von Holst S, Picelli S, Edler D, Lenander C, Dalen J, Hjern F, et al. Association studies on 11 published colorectal cancer risk loci. Br J Cancer (2010) 103(4):575–80. doi:10.1038/sj.bjc.6605774
15. Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet (2010) 42(11):973–U89. doi:10.1038/ng.670
16. Win AK, Jenkins MA. Is the reported modifying effect of 8q23.3 and 11q23.1 on colorectal cancer risk for MLH1 mutation carriers valid? Int J Cancer (2013) 133(7):1762–3. doi:10.1002/ijc.28179
17. Valentin MD, Da Silva FC, Santos EMM, Da Silva SD, Ferreira FD, Aguiar S, et al. Evaluation of MLH1 I219V polymorphism in unrelated South American individuals suspected of having Lynch syndrome. Anticancer Res (2012) 32(10):4347–51.
18. Muniz-Mendoza R, Ayala-Madrigal ML, Partida-Perez M, Peregrina-Sandoval J, Leal-Ugarte E, Macias-Gomez N, et al. MLH1 and XRCC1 polymorphisms in Mexican patients with colorectal cancer. Genet Mol Res (2012) 11(3):2315–20. doi:10.4238/2012.June.27.6
19. Mathonnet G, Krajinovic M, Labuda D, Sinnett D. Role of DNA mismatch repair genetic polymorphisms in the risk of childhood acute lymphoblastic leukaemia. Br J Haematol (2003) 123(1):45–8. doi:10.1046/j.1365-2141.2003.04551.x
20. Listgarten J, Damaraju S, Poulin B, Cook L, Dufour J, Driga A, et al. Predictive models for breast cancer susceptibility from multiple single nucleotide polymorphisms. Clin Cancer Res (2004) 10(8):2725–37. doi:10.1158/1078-0432.CCR-1115-03
21. Damaraju S, Murray D, Dufour J, Carandang D, Myrehaug S, Fallone G, et al. Association of DNA repair and steroid metabolism gene polymorphisms with clinical late toxicity in patients treated with conformal radiotherapy for prostate cancer. Clin Cancer Res (2006) 12(8):2545–54. doi:10.1158/1078-0432.CCR-05-2703
22. Bagnoli S, Putignano AL, Melean G, Baglioni S, Sestini R, Milla M, et al. Susceptibility to refractory ulcerative colitis is associated with polymorphism in the hMLH1 mismatch repair gene. Inflamm Bowel Dis (2004) 10(6):705–8. doi:10.1097/00054725-200411000-00001
23. Plotz G, Raedle J, Spina A, Welsch C, Stallmach A, Zeuzem S, et al. Evaluation of the MLH1 I219V alteration in DNA mismatch repair activity and ulcerative colitis. Inflamm Bowel Dis (2008) 14(5):605–11. doi:10.1002/ibd.20358
24. RodriguezBigas MA, Boland CR, Hamilton SR, Henson DE, Jass JR, Khan PM, et al. A National Cancer Institute workshop on hereditary nonpolyposis colorectal cancer syndrome: meeting highlights and Bethesda guidelines. J Natl Cancer Inst (1997) 89(23):1758–62. doi:10.1093/jnci/89.23.1758
25. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst (2004) 96(4):261–8. doi:10.1093/jnci/djh034
26. Trojan J, Zeuzem S, Randolph A, Hemmerle C, Brieger A, Raedle J, et al. Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology (2002) 122(1):211–9. doi:10.1053/gast.2002.30296
27. Hudler P, Voulk K, Liovic M, Repse S, Juvan R, Komel R. Mutations in the hMLH1 gene in Slovenian patients with gastric carcinoma. Clin Genet (2004) 65:405. doi:10.1111/j.0009-9163.2004.0234.x 2004;66(1):81
28. Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. An integrated map of genetic variation from 1,092 human genomes. Nature (2012) 491(7422):56–65. doi:10.1038/nature11632
29. Kondo E, Suzuki H, Horii A, Fukushige S. A yeast two-hybrid assay provides a simple way to evaluate the vast majority of hMLH1 germ-line mutations. Cancer Res (2003) 63(12):3302–8.
30. Raevaara TE, Korhonen MK, Lohi H, Hampel H, Lynch E, Lonnqvist KE, et al. Functional significance and clinical phenotype of nontruncating mismatch repair variants of MLH1. Gastroenterology (2005) 129(2):537–49. doi:10.1053/j.gastro.2005.06.005
31. Picelli S, Lorenzo Bermejo J, Chang-Claude J, Hoffmeister M, Fernandez-Rozadilla C, Carracedo A, et al. Meta-analysis of mismatch repair polymorphisms within the cogent consortium for colorectal cancer susceptibility. PLoS One (2013) 8(9):e72091. doi:10.1371/journal.pone.0072091
32. Kim JC, Roh SA, Koo KH, Ka IH, Kim HC, Yu CS, et al. Genotyping possible polymorphic variants of human mismatch repair genes in healthy Korean individuals and sporadic colorectal cancer patients. Fam Cancer (2004) 3(2):129–37. doi:10.1023/B:FAME.0000039919.66461.8f
33. Hirschhorn JN, Altshuler D. Editorial: once and again – issues surrounding replication in genetic association studies. J Clin Endocrinol Metab (2002) 87(10):4438–41. doi:10.1210/jc.2002-021329
34. Woo YL, Cheah PL, Shahruddin SI, Omar SZ, Arends M. The immunohistochemistry signature of mismatch repair (MMR) proteins in a multiethnic Asian cohort with endometrial carcinoma. Int J Gynecol Pathol (2014) 33(6):554–9. doi:10.1097/PGP.0000000000000099
35. Panigrahi GB, Slean MM, Simard JP, Pearson CE. Human mismatch repair protein hMutL alpha is required to repair short slipped-DNAs of trinucleotide repeats. J Biol Chem (2012) 287(50):41844–50. doi:10.1074/jbc.M112.420398
Keywords: Ile219Val, Lynch syndrome, MLH1, MSH2, mutation
Citation: Dominguez-Valentin M, Wernhoff P, Cajal AR, Kalfayan PG, Piñero TA, Gonzalez ML, Ferro A, Sammartino I, Causada Calo NS and Vaccaro CA (2016) MLH1 Ile219Val Polymorphism in Argentinean Families with Suspected Lynch Syndrome. Front. Oncol. 6:189. doi: 10.3389/fonc.2016.00189
Received: 23 March 2016; Accepted: 08 August 2016;
Published: 24 August 2016
Edited by:
Rupert Langer, University of Bern, SwitzerlandReviewed by:
Gisela Keller, Technische Universität München, GermanyElgin Ozkan, Ankara University Medical School, Turkey
Angel Lanas, University of Zaragoza, Spain
Copyright: © 2016 Dominguez-Valentin, Wernhoff, Cajal, Kalfayan, Piñero, Gonzalez, Ferro, Sammartino, Causada Calo and Vaccaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mev Dominguez-Valentin, bWV2X2R2QHlhaG9vLmNvbQ==