 Juan L. Iovanna
Juan L. Iovanna- Centre de Recherche en Cancérologie de Marseille (CRCM), INSERM U1068, CNRS UMR 7258, Aix-Marseille Université et Institut Paoli-Calmettes, Parc Scientifique et Technologique de Luminy, Marseille, France
Pancreatitis is an inflammatory disease that both facilitates and accelerates the transformation of pancreatic cells upon activation of the KRAS oncogene. Autophagy is proposed to be one of the cellular mechanisms contributing to pancreatic carcinogenesis, especially during initial stages in which the KRAS oncogene appears to play a key role. Autophagy is also strongly induced during pancreatitis by the overexpression of VMP1. We recently developed a genetically engineered mouse model in which the VMP1 protein is induced simultaneously with the activation of the oncogene KrasG12D specifically in the pancreas, by the addition of doxycycline to a water drink. Using this sophisticated animal model, we can affirm that pancreatic autophagy, induced during pancreatitis by the overexpression of VMP1, promotes the development of precancerous lesions when induced by the mutated KRAS. In addition, the treatment of these mice with chloroquine, an inhibitor of autophagic flux, reverses the effects of VMP1 in pancreatic cancer induced by the KRAS oncogene. Overall, these results bear both mechanistic and biomedical relevance for further understanding and potentially targeting pathways that are critical for initiating pancreatic carcinogenesis, particularly if associated with pancreatitis.
Pancreatic Ductal Adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer death in the Western world, with prediction curves demonstrating it will become the second leading cause of death by cancer in 2030, just after lung cancer (1). Both the initiation and progression of this pathology result from the interaction of complex genetic events with multiple less characterized factors (2, 3). Genetic alterations that contribute to the pathogenesis of pancreatic adenocarcinoma have been widely studied and definitively determined. Among these alterations, oncogenic mutations in the KRAS gene have been frequently detected (more than 90% of cases), not only in the established disease but also in preneoplastic lesions known as pancreatic intraductal neoplasia (PanINs). Activation of the oncogene KRAS signals pancreatic cells to undergo acinar-to-ductal metaplasia, an essential step in the formation of premalignant lesions, which together with the inactivation of tumor suppressor genes, such as CDKN2A, TP53, and SMAD4, allow the progression of premalignant lesions to invasive cancer (4). As the activating mutation in the KRAS oncogene is almost systematically associated with PDAC, its role in cancer development has been the subject of numerous studies (5).
Autophagy has been proposed as a cellular process contributing to pancreatic carcinogenesis, particularly in the initial stages in which the KRAS oncogene is a key element (6–9). Indeed, activation of the pathway controlled by the KRAS oncogene generates a metabolic stress, characterized by a temporary deficit in energy, which must be compensated by an increase in metabolism, through activation of autophagy (6–10). Although this concept appears clear and simple, the role of autophagy in protumor or antitumor development is still debated in the context of PDAC, since multiple factors appear to modulate this process, such as regulatory pathways, the genomic status of transformed pancreatic cells, as well as the physiological and pathological contexts in which the process is enabled (11, 12).
Pancreatitis-Associated Autophagy Promotes the Protumoral Effect of the KRAS Oncogene
Pancreatitis, an inflammatory disease of the pancreas, enables and accelerates the transformation of pancreatic cells when the KRAS oncogene is activated (13). Exactly how pancreatitis promotes the development of PDAC is a fundamental question in the field of pancreatology, which has not yet been clearly answered. However, this has been partly answered by studies showing that the systematic activation of autophagy during pancreatitis, often for the protection of pancreatic cells, decreases disease progression and aids the recovery phase (14, 15). We have demonstrated that induction of autophagy in pancreatic acinar cells is accompanied by the overexpression of the VMP1 gene. VMP1 mRNA encodes a transmembrane protein that we cloned in 2002 due to its extraordinary pancreatic activation during the acute phase of pancreatitis (16). Overexpression of VMP1 triggers autophagy in numerous types of cells (16–19). Concerning its mechanistic activity, VMP1 is involved in the formation of the phagophore (18) following a direct interaction with beclin 1 (17), TP53INP2, a scaffold protein (20), and possibly its homolog, TP53INP1 (21). The main physiological role of autophagy during pancreatitis is the removal of damaged organelles to maintain cellular homeostasis and ensure improved survival of pancreatic cells (22). It is likely that the protective effect of autophagy during the acute phase of the disease is at least partly related to the sequestration of zymogen granules that contain digestive enzymes responsible for autodigestion during pancreatitis. This may have a dual effect on pancreatic cells: first, zymophagy (autophagy of zymogen granules) could reduce the availability of digestive enzymes, which when released into the pancreatic parenchyma destroys the pancreatic gland by necrosis; second, these organelles could meet the unique metabolic needs that accompany cell growth during the regeneration phase (23).
Autophagy Induced by Overexpression of VMP1 Enhances Transformation of Pancreatic Cells
It is interesting to note that the expression of VMP1 is also transcriptionally activated by the mutated KRAS oncogene through a mechanism dependent on GLI3 and p300 (24). The KRAS oncogene possibly induces VMP1 expression to meet the increased energy needs of the cell during the transformation process. Expression of the VMP1 protein, and its triggered autophagy, is therefore induced and maintained by mutation of the KRAS oncogene, which is strongly reinforced during the course of pancreatitis. The most likely hypothesis is that autophagy induced by pancreatitis, and mediated by overexpression of VMP1, provides the energy required of cells harboring an activating mutation in the KRAS oncogene, therefore allowing their transformation. To test this hypothesis, we have recently developed an animal model wherein the genetically modified VMP1 protein is induced simultaneously with the activation of the oncogene KrasG12D specifically in the pancreas, by the addition of doxycycline to a water drink (25). This model was developed with the objective to first assess the effects of overexpressed VMP1 on initiation of pancreatic cancer, and second, to define the role of pharmacological inhibitors of autophagy in the development of pancreatic cancer. The results of these experiments in mice affirm our hypothesis that autophagy, induced by overexpressing VMP1 in the pancreas, significantly increases the protumor effect of the KRAS oncogene (Figure 1). In addition, we demonstrated that chloroquine, a classical inhibitor of autophagic flux (26), can reverse the effect of VMP1 overexpression on pancreatic cancer induced by the KRAS oncogene in a preclinical trial using our mouse model (25). Overall, these observations support the idea that pathways activated by pancreatitis, particularly those regulating autophagy, can promote pancreatic carcinogenesis. Finally, the results support the concept that inhibition of autophagy could be used to prevent the progression of pancreatic pre-tumoral lesions to pancreatic cancer.
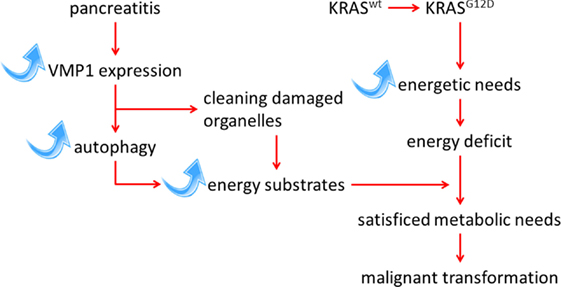
Figure 1. Schematic representation of the interaction between KRAS-mediated transformation in PDAC and autophagy induced by the pancreatitis-associated protein VMP1 pathways.
Mechanisms of Action of VMP1
In light of these clinically relevant results, it is important to review and discuss the identified functions of the VMP1 protein, which will consequently improve the interpretation of its role in pancreatic tumor progression. For example, it has been established that this protein is involved in the initiation of autophagy since cells engineered to be deficient in VMP1 have high levels of PtdIns3P and trigger autophagic signaling by the resulting aberrant endoplasmic reticulum, with subsequent recruitment of ATG18 and other autophagic proteins (19). In addition, although ULK1 and ATG5 are separated in the genetic hierarchy during autophagy, both proteins accumulate synchronously within punctate structures containing VMP1, followed by recruitment of ATG14, ZFYVE1, and WIPI1 (27). Moreover, VMP1 protein directly binds to the BH3 motif of beclin 1 to induce the formation of a complex with hVps34, a key phosphatidylinositol-3 kinase class III regulator of autophagy, on the site where autophagosomes are generated. Importantly, the interaction between beclin 1 and VMP1 proteins leads to the dissociation of the Bcl-2 protein with beclin 1, therefore increasing intracellular levels of beclin 1 available to induce autophagy (28). In addition, the presence of the VMP1 protein regulates the formation of autophagosomes by shortening the training time of the omegasome and therefore significantly accelerating autophagic flux (18). Finally, the production of cells inactivated for VMP1 protein in Dictyostelium revealed a massive accumulation of protein aggregates, both poly- and multi-ubiquitinated, containing the autophagic markers ATG8 counterparts and p62 but presenting strong defects in autophagy process. Altogether, these observations demonstrate that expression of the stress protein VMP1 is essential for unloading cells of these protein aggregates by autophagy (29) and recycling them to provide the energy substrate required by the cell under these stress conditions.
It is also important to discuss the broader role that autophagy plays in the development of PDAC as it is so complex and varied. Indeed, it was previously demonstrated that autophagy participates in the transition from mitosis to senescence (30), and certain molecules can induce both autophagy and senescence, such as kinase ULK3 (30). Senescence is known to be an important anticancer pathway set up in response to the oncogenic activation of mutated KRAS. In this context, senescence enhanced by activation of autophagy might partially inhibit the oncogenic effect of the KRAS oncogene rather than increase it. Furthermore, activation of autophagy in certain tissues, either dependent or independent of VMP1 overexpression, can act as an antiapoptotic factor, according to the biological circumstances (31, 32). Finally, as mentioned earlier, the oncogenic activation of KRAS induces a strong metabolic stress to cells due to their exceptional energy requirements that can be partially counterbalanced with the contribution of energy sources through the activation of autophagy. Autophagy can therefore play important roles in either promoting or, on the contrary, antagonizing the development of PDAC, depending on the activated intracellular pathways by cells harboring KRAS mutations. This possibly explains the contrasting results reported in the literature on the role of autophagy in cancer. Another important note is that a large majority of these studies were performed in vitro, therefore the cellular environment has not, or only partially, been taken into account, possibly causing a bias in data interpretation. Regarding the pancreatic autophagy induced by VMP1 overexpression in mice, we have established that the development of pancreatic precancerous lesions is associated with a significant reduction of apoptosis with a concomitant increase in cell proliferation (25). In other words, autophagy is clearly a pro-tumor cellular event, at least in this context.
Importantly, autophagy has been considered an important mediator of the resistance to radiotherapy and chemotherapy, at least with particular anticancer drugs and for certain cancers (33, 34), although this point still remains controversial. Nevertheless, the fact that cancer treatments systematically induce autophagy has now been clearly established (35). However, the mechanism by which autophagy is involved in resistance to cancer treatments seem to be initiated by the removal of damaged intracellular organelles to improve cell viability. Furthermore, autophagy has also been reported as a mediator of cell death induced by chemotherapy in several cancers (36). Although the mechanism by which autophagy induces cell death is not yet clearly established, it appears to be mediated by the activation of caspase 3 (36). Therefore, in line with such knowledge, co-treatment with chloroquine appears to enhance the effect of many anticancer drugs in vitro as well as in some preclinical models (37–41), although a clinical study has yet to confirm its benefit as a co-treatment.
Conclusion and Perspectives
In conclusion, many aspects concerning the role of autophagy during PDAC development are still not clearly defined. However, we can confirm that pancreatic autophagy induced during pancreatitis through the overexpression of VMP1, a protein associated with pancreatitis, promotes PanINs when activated by the KRAS oncogene. In addition, inhibition of autophagic flux by chloroquine almost completely abolishes the KRAS pro-tumor effect in the pancreas. Overall, these results bear both mechanistic and biomedical relevance for further understanding and potentially targeting those pathways critical for initiating pancreatic carcinogenesis, particularly if associated with pancreatitis. In the near future, it will be necessary to take into account not only the role of autophagy activation in transformed cells but also in the stromal non-transformed cells. Recently, it was clearly evidenced that the activation of autophagy in cancer-activated fibroblast (CAF cells) is an essential mechanism to produce and secrete non-essential amino acids into the microenvironment, which serves as a major source of energy for transformed cells (42). This may be the starting point of a novel time in which the autophagy may be considered as the fuel source for other cells. All in all, these facts are revealing a more complex scenario than suspected and therefore are opening news ways for treating diseases in which autophagy seems to be strongly involved, such as PDAC. An interesting observation to be noted was recently pointed out by Guo and colleagues who demonstrated that the loss of VMP1 expression in colorectal cancer is associated with a poor prognosis and aggressiveness of the cancer cells. In addition, in vitro assays revealed that colon cancer-derived cells in which VMP1 was knocked down gained significant aggressive properties in regards to proliferation and invasion. Remarkably, in vivo studies revealed a higher number of formed nodules in mice after intraperitoneal injection of VMP1 knocked down cells (43). Another recent work reports that approximately 10% of esophageal adenocarcinomas present a RPS6KB1–VMP1 gene fusion as a recurrent event. Notably, esophageal adenocarcinoma cases harboring RPS6KB1–VMP1 fusions exhibited significantly poorer overall survival as compared to fusion-negative cases. Mechanistically, the RPS6KB1–VMP1 fusion protein promotes cell growth in vitro, but it is ineffective in triggering autophagy (44). Altogether, these studies suggest that the role of VMP1, and perhaps autophagy, in cancer development and progression is organ or context dependent.
Author Contributions
The author confirms being the sole contributor of this work and approved it for publication.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was supported by La Ligue Contre le Cancer, INCa, Canceropôle PACA, DGOS (labellisation SIRIC) and INSERM.
References
1. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res (2014) 74(11):2913–21. doi:10.1158/0008-5472.CAN-14-0155
2. Iovanna JL, Marks DL, Fernandez-Zapico ME, Urrutia R. Mechanistic insights into self-reinforcing processes driving abnormal histogenesis during the development of pancreatic cancer. Am J Pathol (2013) 182(4):1078–86. doi:10.1016/j.ajpath.2012.12.004
3. Lomberk GA, Iovanna J, Urrutia R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics (2016) 8(6):831–42. doi:10.2217/epi-2015-0016
4. Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev (2006) 20(10):1218–49. doi:10.1101/gad.1415606
5. Tsuchida N, Murugan AK, Grieco M. Kirsten Ras* oncogene: significance of its discovery in human cancer research. Oncotarget (2016). doi:10.18632/oncotarget.8773
6. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev (2011) 25(5):460–70. doi:10.1101/gad.2016311
7. Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem (2011) 286(15):12924–32. doi:10.1074/jbc.M110.138958
8. Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell (2011) 22(2):165–78. doi:10.1091/mbc.E10-06-0500
9. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev (2011) 25(7):717–29. doi:10.1101/gad.2016111
10. Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn’t kill you makes you stronger. Cold Spring Harb Symp Quant Biol (2011) 76:389–96. doi:10.1101/sqb.2012.76.011015
11. Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature (2013) 504(7479):296–300. doi:10.1038/nature12865
12. Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov (2014) 4(8):905–13. doi:10.1158/2159-8290.CD-14-0362
13. Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell (2007) 11(3):291–302. doi:10.1016/j.ccr.2007.01.012
14. Gukovsky I, Gukovskaya AS. Impaired autophagy underlies key pathological responses of acute pancreatitis. Autophagy (2010) 6(3):428–9. doi:10.4161/auto.6.3.11530
15. Diakopoulos KN, Lesina M, Wormann S, Song L, Aichler M, Schild L, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology (2015) 148(3):626–638e617. doi:10.1053/j.gastro.2014.12.003
16. Dusetti NJ, Jiang Y, Vaccaro MI, Tomasini R, Azizi Samir A, Calvo EL, et al. Cloning and expression of the rat vacuole membrane protein 1 (VMP1), a new gene activated in pancreas with acute pancreatitis, which promotes vacuole formation. Biochem Biophys Res Commun (2002) 290(2):641–9. doi:10.1006/bbrc.2001.6244
17. Ropolo A, Grasso D, Pardo R, Sacchetti ML, Archange C, Lo Re A, et al. The pancreatitis-induced vacuole membrane protein 1 triggers autophagy in mammalian cells. J Biol Chem (2007) 282(51):37124–33. doi:10.1074/jbc.M706956200
18. Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell (2010) 141(6):1042–55. doi:10.1016/j.cell.2010.04.034
19. Calvo-Garrido J, King JS, Munoz-Braceras S, Escalante R. Vmp1 regulates PtdIns3P signaling during autophagosome formation in Dictyostelium discoideum. Traffic (2014) 15(11):1235–46. doi:10.1111/tra.12210
20. Nowak J, Archange C, Tardivel-Lacombe J, Pontarotti P, Pebusque MJ, Vaccaro MI, et al. The TP53INP2 protein is required for autophagy in mammalian cells. Mol Biol Cell (2009) 20(3):870–81. doi:10.1091/mbc.E08-07-0671
21. Seillier M, Peuget S, Gayet O, Gauthier C, N’Guessan P, Monte M, et al. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ (2012) 19(9):1525–35. doi:10.1038/cdd.2012.30
22. Antonucci L, Fagman JB, Kim JY, Todoric J, Gukovsky I, Mackey M, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A (2015) 112(45):E6166–74. doi:10.1073/pnas.1519384112
23. Grasso D, Ropolo A, Lo Re A, Boggio V, Molejon MI, Iovanna JL, et al. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J Biol Chem (2011) 286(10):8308–24. doi:10.1074/jbc.M110.197301
24. Lo Re AE, Fernandez-Barrena MG, Almada LL, Mills LD, Elsawa SF, Lund G, et al. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J Biol Chem (2012) 287(30):25325–34. doi:10.1074/jbc.M112.370809
25. Loncle C, Molejon MI, Lac S, Tellechea JI, Lomberk G, Gramatica L, et al. The pancreatitis-associated protein VMP1, a key regulator of inducible autophagy, promotes Kras(G12D)-mediated pancreatic cancer initiation. Cell Death Dis (2016) 7:e2295. doi:10.1038/cddis.2016.202
26. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy (2016) 12(1):1–222. doi:10.1080/15548627.2015.1100356
27. Koyama-Honda I, Itakura E, Fujiwara TK, Mizushima N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy (2013) 9(10):1491–9. doi:10.4161/auto.25529
28. Molejon MI, Ropolo A, Re AL, Boggio V, Vaccaro MI. The VMP1-beclin 1 interaction regulates autophagy induction. Sci Rep (2013) 3:1055. doi:10.1038/srep01055
29. Calvo-Garrido J, Escalante R. Autophagy dysfunction and ubiquitin-positive protein aggregates in Dictyostelium cells lacking Vmp1. Autophagy (2010) 6(1):100–9. doi:10.4161/auto.6.1.10697
30. Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev (2009) 23(7):798–803. doi:10.1101/gad.519709
31. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell (2005) 122(6):927–39. doi:10.1016/j.cell.2005.07.002
32. Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol (2014) 15(2):81–94. doi:10.1038/nrm3735
33. Ding L, Ma G, Liu Y, Jia Y, Liu X. Autophagy blockage enhances radiosensitivity of osteosarcoma MG-63 cells in vitro. Clin Lab (2015) 61(10):1365–72. doi:10.7754/Clin.Lab.2015.150308
34. Yang MC, Wang HC, Hou YC, Tung HL, Chiu TJ, Shan YS. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol Cancer (2015) 14(1):179. doi:10.1186/s12943-015-0449-3
35. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer (2005) 5(9):726–34. doi:10.1038/nrc1692
36. Salazar M, Carracedo A, Salanueva IJ, Hernandez-Tiedra S, Lorente M, Egia A, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest (2009) 119(5):1359–72. doi:10.1172/JCI37948
37. Bellodi C, Lidonnici MR, Hamilton A, Helgason GV, Soliera AR, Ronchetti M, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest (2009) 119(5):1109–23. doi:10.1172/JCI35660
38. Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle (2010) 9(17):3515–33. doi:10.4161/cc.9.17.12928
39. Saleem A, Dvorzhinski D, Santanam U, Mathew R, Bray K, Stein M, et al. Effect of dual inhibition of apoptosis and autophagy in prostate cancer. Prostate (2012) 72(12):1374–81. doi:10.1002/pros.22487
40. Sasaki K, Tsuno NH, Sunami E, Kawai K, Hongo K, Hiyoshi M, et al. Resistance of colon cancer to 5-fluorouracil may be overcome by combination with chloroquine, an in vivo study. Anticancer Drugs (2012) 23(7):675–82. doi:10.1097/CAD.0b013e328353f8c7
41. Lefort S, Joffre C, Kieffer Y, Givel AM, Bourachot B, Zago G, et al. Inhibition of autophagy as a new means of improving chemotherapy efficiency in high-LC3B triple-negative breast cancers. Autophagy (2014) 10(12):2122–42. doi:10.4161/15548627.2014.981788
42. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature (2016) 536(7617):479–83. doi:10.1038/nature19084
43. Guo XZ, Ye XL, Xiao WZ, Wei XN, You QH, Che XH, et al. Downregulation of VMP1 confers aggressive properties to colorectal cancer. Oncol Rep (2015) 34(5):2557–66. doi:10.3892/or.2015.4240
Keywords: VMP1, pancreatic cancer, pancreatitis, autophagy, chloroquine, KRAS
Citation: Iovanna JL (2016) Autophagy Induced during Pancreatitis Promotes KRAS-Dependent Transformation in the Pancreas. Front. Oncol. 6:226. doi: 10.3389/fonc.2016.00226
Received: 30 August 2016; Accepted: 10 October 2016;
Published: 26 October 2016
Edited by:
Patrizia Agostinis, University of Leuven, BelgiumReviewed by:
Alessandro Rimessi, University of Ferrara, ItalyFrancisco X. Real, Spanish National Cancer Research Centre, Spain
Jun Yan, Nanjing University, China
Copyright: © 2016 Iovanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan L. Iovanna, anVhbi5pb3Zhbm5hQGluc2VybS5mcg==