 Kaori Sasai
Kaori Sasai Warapen Treekitkarnmongkol
Warapen Treekitkarnmongkol Kazuharu Kai
Kazuharu Kai Hiroshi Katayama
Hiroshi Katayama Subrata Sen
Subrata Sen- 1Department of Molecular Oncology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan
- 2Department of Translational Molecular Pathology, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Aurora kinases play critical roles in regulating spindle assembly, chromosome segregation, and cytokinesis to ensure faithful segregation of chromosomes during mitotic cell division cycle. Molecular and cell biological studies have revealed that Aurora kinases, at physiological levels, orchestrate complex sequential cellular processes at distinct subcellular locations through functional interactions with its various substrates. Aberrant expression of Aurora kinases, on the other hand, cause defects in mitotic spindle assembly, checkpoint response activation, and chromosome segregation leading to chromosomal instability. Elevated expression of Aurora kinases correlating with chromosomal instability is frequently detected in human cancers. Recent genomic profiling of about 3000 human cancer tissue specimens to identify various oncogenic signatures in The Cancer Genome Atlas project has reported that recurrent amplification and overexpression of Aurora kinase-A characterize distinct subsets of human tumors across multiple cancer types. Besides the well-characterized canonical pathway interactions of Aurora kinases in regulating assembly of the mitotic apparatus and chromosome segregation, growing evidence also supports the notion that deregulated expression of Aurora kinases in non-canonical pathways drive transformation and genomic instability by antagonizing tumor suppressor and exacerbating oncogenic signaling through direct interactions with critical proteins. Aberrant expression of the Aurora kinases–p53 protein family signaling axes appears to be critical in the abrogation of p53 protein family mediated tumor suppressor pathways frequently deregulated during oncogenic transformation process. Recent findings reveal the existence of feedback regulatory loops in mRNA expression and protein stability of these protein families and their consequences on downstream effectors involved in diverse physiological functions, such as mitotic progression, checkpoint response pathways, as well as self-renewal and pluripotency in embryonic stem cells. While these investigations have focused on the functional consequences of Aurora kinase protein family interactions with wild-type p53 family proteins, those involving Aurora kinases and mutant p53 remain to be elucidated. This article presents a comprehensive review of studies on Aurora kinases–p53 protein family interactions along with a prospective view on the possible functional consequences of Aurora kinase–mutant p53 signaling pathways in tumor cells. Additionally, we also discuss therapeutic implications of these findings in Aurora kinases overexpressing subsets of human tumors.
Introduction
Gain-of-function alterations in the Aurora kinase protein family member, Aurora kinase-A (AURKA), due to amplification and/or overexpression of the gene- and loss-of-function changes in the TP53 tumor suppressor protein have been associated with multiple cellular phenotypes of similar nature, such as centrosome amplification, override of spindle assembly, and DNA damage checkpoint response, aneuploidy, and cellular transformation. Induction of such shared cellular phenotypes consequent to AURKA overexpression or functional inactivation of TP53 as well as reported localization of the two proteins at the centrosomes indicate that AURKA and TP53 (hereafter referred to as Aurora-A and p53) are involved in overlapping signaling pathways regulating the abovementioned cancer-associated aberrant cellular phenotypes through direct or indirect functional interactions (1–5). Evidence in support of this concept first became available following demonstration that p53 could suppress Aurora-A’s oncogenic potential through physiological interaction in transactivation-independent manner in mammalian cells (6). Similarly, Xenopus p53 was shown to inhibit Aurora-A kinase activity, indicating that the inhibitory role of p53 on Aurora-A kinase enzyme activity is conserved among vertebrates (7). Later studies have revealed that p53, besides inhibiting the kinase activity of Aurora-A through direct interaction, also regulates Aurora-A function in transactivation-dependent manner, as discussed below.
In addition to the findings mentioned above, a number of studies have identified Aurora kinases regulating p53 function through phosphorylation-mediated posttranslational modification of either p53 protein directly or a p53 interacting protein at multiple residues with each phosphorylation event having distinct functional consequence. Aurora-A phosphorylates p53 at serine 315, facilitating MDM2-mediated p53 ubiquitination and degradation (8), whereas phosphorylation of serine 215 inhibits p53 DNA-binding and transactivation function (9). These findings demonstrated that Aurora-A phosphorylation of p53 negatively regulates p53 tumor suppressor functions, resulting in abrogation of DNA damage checkpoint and induction of cell death responses in Aurora-A overexpressing cells. As a consequence, Aurora-A overexpressing cancer cells with wild-type p53 acquire cellular phenotypes associated with p53 loss-of-function mutant harboring cancer cells. A more recent finding of a novel Aurora-A phosphorylation residue, serine 106 of p53, was, however, reported to have an opposing effect on p53 stability compared with the destabilization effect of Aurora-A-mediated phosphorylation of p53 at serine 315. Phosphorylation of p53 serine 106 was shown to inhibit the interaction of p53 with MDM2 and prolong the half-life of p53 protein (10). Physiological significance of Aurora-A-mediated p53 phosphorylation at serine 106 in vivo and its functional implications in Aurora-A overexpressing tumor cells remain unknown. The possibility of enhanced p53 protein stability in Aurora-A overexpressing tumor cells appears intriguing since steady-state levels of Aurora-A and p53 proteins have been reported to be inversely correlated in most human tumors. Molecular characterization studies have shown that serine 215 phosphorylation is associated with loss of serine 33 phosphorylation of p53, mediated by p38 critical for p53 activation stabilization and induction of apoptosis, indicating that Aurora-A mediates cross-talk between N- and C-terminal posttranslational modifications of p53 (11, 12). In addition, Aurora-A also indirectly compromises p53 function by phosphorylating positive and negative regulators of p53, such as hnRNPK and MDM2 proteins, respectively. The RNA-binding protein, such as hnRNPK, is a p53 transcriptional cofactor that promotes gene expression in response to DNA damage and is also a target of MDM2 (13, 14). While Aurora-A-mediated hnRNPK phosphorylation at serine 379 disrupts its interaction with p53 and impairs DNA damage-induced gene expression, MDM2 phosphorylation at serine 166 enhances its protein stability and in turn destabilizes p53 (15–17). These findings demonstrate that Aurora-A is involved in regulating p53 downstream signaling negatively affecting growth arrest and apoptotic response pathways.
Aurora-B has also been shown to interact with and phosphorylate p53 at multiple residues in DNA-binding domain. Similar to the effect of Aurora-A phosphorylation on p53 activity and stability, Aurora-B phosphorylations of p53 at serine 269 and threonine 284 inhibit p53 transactivation activity, whereas phosphorylations at serine 183, threonine 211, and serine 215 accelerate the degradation of p53 through polyubiquitination-mediated proteasome pathway (18, 19). However, these studies have been performed with phosphor mutants of p53 under conditions of ectopic expression in cells and thus physiological relevance of identical in vivo phosphorylations have not been well validated. Further investigations of endogenous protein modifications are required to verify the role of Aurora-B-mediated p53 phosphorylations in vivo and to determine how Aurora-A and Aurora-B may be coordinately regulating p53 function through the cell cycle. It is worth noting that exogenously expressed p53 colocalizes with Aurora-B at centromeres during mitosis. This observation may be biologically significant since several spindle assembly checkpoint (SAC) kinases such as MPS1/TTK, BUB1, and BUBR1, localized at kinetochores, have been reported to functionally interact with p53 in activating spindle assembly and postmitotic checkpoint response pathways (20–23). In view of these findings and those demonstrating Aurora kinases regulating functions of p53 family proteins, it is likely that varying levels of Aurora kinases in tumor cells influence the extent of deregulations in checkpoint response pathway activation downstream of p53 family proteins in tumor cells. We discuss the role of Aurora kinases–p53 protein family signaling axis in SAC response pathway later in this review.
Aurora-A involvement in regulating p73 function first became evident from a study in which Aurora-A inhibitor treatment or knockdown of Aurora-A in p53-deficient cells induced p73-mediated expression of apoptosis-related genes and also cell death (16). Further investigation revealed that Aurora-A directly interacts with and phosphorylates p73 at serine 235 in the DNA-binding domain, an equivalent site of serine 215 in p53, resulting in loss of its DNA-binding and transactivation activity. As a result, cells become resistant to DNA damage-induced cell death (24). Importantly, this study uncovered that Aurora-A phosphorylation of p73 leads to the formation of a large molecular complex that includes the chaperon protein Mortalin promoting translocation of the Mortalin–p73 complex into cytoplasm. Similar cytoplasmic distribution of Aurora-A phosphorylated p53 at serine 215 in a complex with Mortalin was observed as well. As a corollary to this finding, cytoplasmic distribution of p73 was found to correlate with Aurora-A expression levels in human primary pancreatic cancer tissues. Moreover, consistent with the earlier findings that p73 deficiency causes relaxation of the SAC reflected in the mislocalization of BUB1 and BUBR1 at kinetochores and reduced BUBR1 kinase activity (25–27), Aurora-A phosphorylation of p73 in a constitutive manner was found to facilitate accelerated mitotic progression and exit accompanied with relaxation of SAC due to premature dissociation of the MAD2–CDC20 complex in proliferating cells in vitro. SAC inactivation correlated with significant increase in multinucleated cells. These findings indicate that the mitotic checkpoint functions of p53 family proteins are regulated in a complex manner involving Aurora kinase-mediated posttranslational modifications during mitotic progression. It is currently unknown whether p73 reciprocally controls Aurora-A kinase function and if Aurora-B and Aurora-C also regulate p73 function.
Along with the discovery of crosstalk between Aurora kinases and p53 family proteins, there is growing evidence that these protein complexes directly or indirectly participate in various cellular processes and inappropriate activation of Aurora kinases can have dominant-negative effects on the phenotypes of normal cells involving pathways regulated by a variety of proteins functionally interacting with p53 protein family (Figure 1; Table 1). In the following sections, we summarize the current knowledge of Aurora kinases–p53 protein family signaling cascades relevant to the regulation of posttranslational modifications and stability of proteins, activity and integrity of centrosomes, checkpoint pathways in normal and aberrant mitosis, as well as protein–protein interactions and transcription and translation of genes involved in the development of pluripotent embryonic stem cells (ESC) and cancer stem cells (CSC), as outlined in the schematic overview diagram in Figure 2.
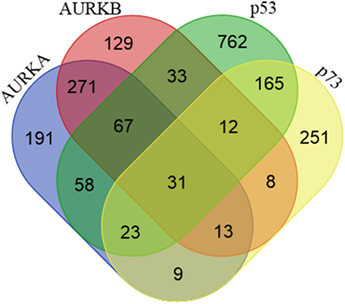
Figure 1. Proteins interacting with Aurora-A, Aurora-B, p53, and p73. Venn diagram showing the number of shared and unique proteins interacting with Aurora-A, Aurora-B, p53, and p73. Protein–protein interaction data were downloaded from the BioGRID (v3.4) and STRING (v9.1) databases.
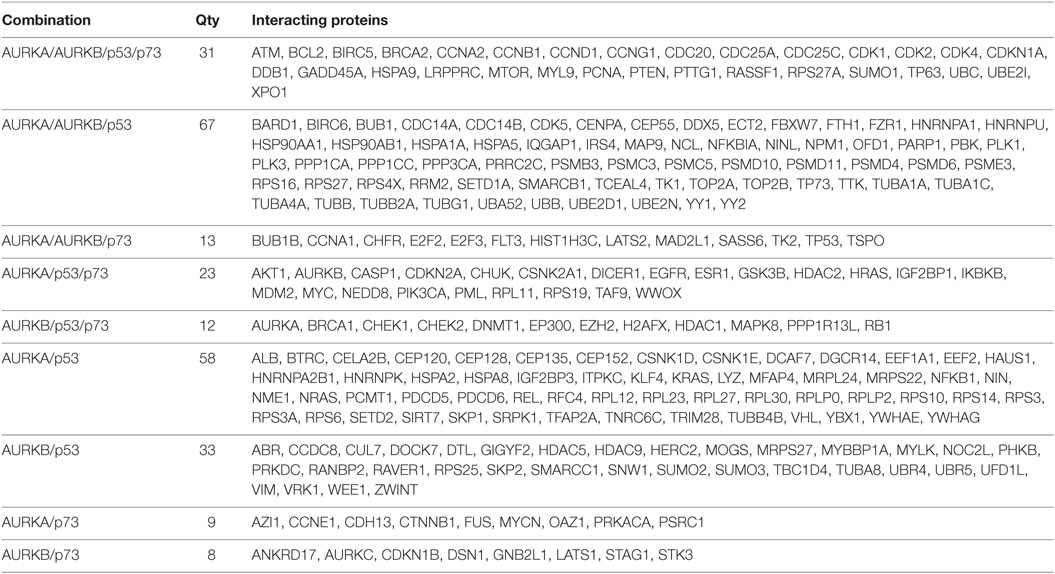
Table 1. List of proteins interacting with Aurora–p53 family protein complex represented in Venn diagram in Figure 1.
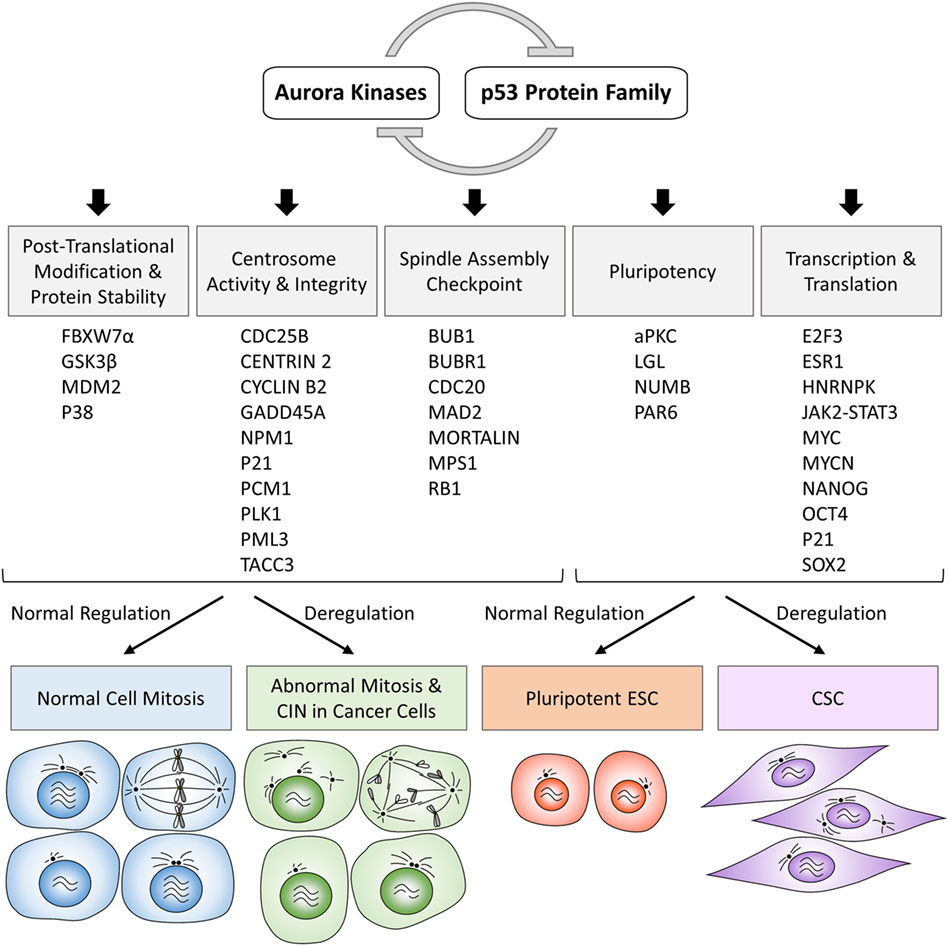
Figure 2. Schematic overview diagram showing phenotypic consequences of physiologically regulated interactions in normal cells and deregulated interactions in cancer cells involving Aurora kinases–p53 protein family. CIN, chromosomal instability; ESC, embryonic stem cells; CSC, cancer stem cells.
Mechanism of Downregulation of Aurora Kinases by p53
In addition to direct inhibition of Aurora-A by p53 via protein–protein interaction, p53 has been shown to downregulate Aurora-A expression, kinase activity and stability through its binding to Aurora-A promoter or transactivation of its target genes including p21, Gadd45a, and Fbxw7α. Genome-wide chromatin occupancy of p53 analyzed by chromatin immunoprecipitation-seq (ChIP-seq) following activation with non-genotoxic molecules and genotoxic chemotherapeutic drugs revealed AURKA gene promoter as one of the novel p53 target sequences and that direct p53 binding to the promoter of AURKA gene repressed expression in MCF-7 and HCT-116 cells (28). This study also found that STAT3 binds to AURKA promoter and antagonizes p53-mediated repression of AURKA. Intriguingly, a recent study has shown that Aurora-A promotes STAT3 activity through regulating expression and phosphorylation levels of JAK2 in gastric and esophageal cancers (29), indicating the existence of negative feedback regulation of p53 function by Aurora-A–JAK2–STAT3 axis. These results suggest that the combination of Aurora-A and JAK2 inhibitors with p53 activators might be an effective therapeutic approach for the treatment of cancer. Both p21 and Gadd45a are transcriptionally activated by p53 upon DNA damage and play important roles in DNA repair and cell cycle checkpoint response. The E2F family transcription factor, E2F3 is known to be involved in the transactivation of Aurora-A gene expression during G2–M cell cycle progression (30). Induction of the cyclin-dependent kinase inhibitor, p21 leads to inhibition of Cdk kinase activity resulting in the maintenance of RB1 in hypo-phosphorylated state in a complex with E2F3, thereby impairing activation of Aurora-A gene expression, an indirect downstream effect of p53–p21 signaling axis. It is noteworthy that Aurora-B phosphorylates RB1 at serine 780, a known inhibitory phosphorylation site for Cdk4. Thus, deregulation of Aurora-B might lead to Aurora-A overexpression through direct downregulation of both p53 and RB1 functions. In fact, co-occurrence of increased gene expression of both Aurora-A and Aurora-B is observed in some human tumors. On the other hand, Gadd45a inhibits Aurora-A kinase activity via direct interaction to prevent cells from Aurora-A-induced centrosome amplification and aborted cytokinesis (31). These results indicate that cooperative inhibition of Aurora-A activity by p53 and Gadd45a is important for cells to maintain centrosome number and chromosomal/genomic stability.
Besides regulating Aurora kinase function through transcription-dependent and -independent mechanisms, p53 also downregulates Aurora-A activity by modulating its degradation pathway. Fbxw7α is a p53-dependent haploinsufficient tumor suppressor protein and a component of the SCF-like ubiquitin ligase complex that targets both Aurora-A and Aurora-B for proteasome degradation (32–34). Fbxw7α is frequently mutated or downregulated in tumors. Importantly, Fbxw7α cooperates with PTEN to regulate Aurora-A degradation via the PI3K/AKT/GSK3β pathway and Fbxw7α also preferentially degrades active Aurora-A (33, 35). It has been demonstrated that Aurora-A-mediated centrosome amplification and subsequent induction of aneuploidy is mediated in part through dysfunction of p53–Fbxw7α axis, commonly detected in human tumors and also in mouse models (33, 36). It is relevant in this context to mention that synthetic lethal screening of protein interacting with N-Myc in N-Myc amplified neuroblastoma has identified that Aurora-A stabilizes N-Myc by directing a K48 to K63/K11 switch in its ubiquitylation by Fbxw7α (37). Although this interaction was reported to be independent of Aurora-A kinase activity, recent finding have demonstrated that inhibitor of Aurora-A kinase activity can disrupt interaction between Aurora-A and Fbxw7α, leading to N-Myc destabilization and tumor regression in mouse model of N-Myc-driven neuroblastoma xenograft (38). Similarly, Aurora-B inhibitor treatment also showed profound growth inhibition and tumor regression in N-Myc-driven neuroblastoma, although the underlying mechanism of this finding remains unclear (39, 40).
Recent studies have identified an important role of microRNA functional networks in the control of gene expression and protein stability of Aurora-A and Myc involving the p53–Fbxw7α axis in neuroblastoma and other tumors. A well-characterized tumor suppressor micoRNA, let-7, regulated by p53 directly targets Aurora-A, c-Myc, N-Myc, and RAN-binding protein 2 (RANBP2). In normal cells, let-7-mediated suppression of c-Myc expression helps maintain basal low level expression of Aurora-A mRNA, while miR-25-targeted Fbxw7α regulates basal level protein expression (41–45). In p53-deficient and p53-mutant cells, these regulatory mechanisms are disrupted, and Aurora-A expression and stability are elevated. Functional genomic studies in N-Myc-amplified neuroblastoma have revealed that LIN28B RNA-binding protein promotes RAN level by directly binding to RAN mRNA and via RANBP2 by inhibiting let-7 expression, consequently facilitating Aurora-A activation and stabilization which in turn promote N-Myc stabilization (44). It was recently been reported that Aurora-A acts as a transactivating factor for hnRNPK, a known transcriptional cofactor of p53, to promote c-Myc expression and reciprocal c-Myc-mediated transactivation of Aurora-A gene in breast cancer stem-like cells (46). This finding on apparent absence of p53 inhibitory role in Aurora-A–c-Myc positive regulatory circuit is associated with frequent observation of centrosome amplification in N-Myc-amplified neuroblastoma cells compared to non-amplified neuroblastoma cells. Mechanistically, N-Myc directly transactivates MDM2 and Aurora-A stabilizes MDM2 by phosphorylating at Ser-166 both of which impair p53 function, resulting in centrosome amplification (17, 47, 48). Taken together, these data indicate that p53 controls Aurora-A function through multiple inhibitory signaling pathways and lack of p53 function results in deregulation of Aurora-A oncogenic signaling cascades which lead to profoundly aberrant phenotypes associated with tumor cells. Involvement of additional signaling pathways regulating centrosome activity and integrity mediated by Aurora-A–p53 interaction is discussed below.
Involvement of Aurora-A–p53 Signaling Pathway in Centrosome Activity and Integrity
A common phenotypic change in cells with gain of Aurora-A and loss of p53 function is manifested in the form of increased number of centrosomes. Multiple investigations have revealed that p53 controls centrosome duplication and separation in both transactivation activity-dependent and -independent manner (Figure 3). In transactivation activity-dependent mechanism, p21 expression plays a key role in synchronizing DNA replication and centrosome duplication by inhibiting Cdk2/Cyclin E activity which phosphorylates Nucleophosmin/NPM1 at centrosomes to promote its dissociation from the centrosomes to allow initiation of centrosome duplication (49). On the other hand, p53 downregulates PLK4 gene expression which is essential for centriole biogenesis through regulation of phosphorylations of centrosomal protein GCP6 and STIL (50–52).
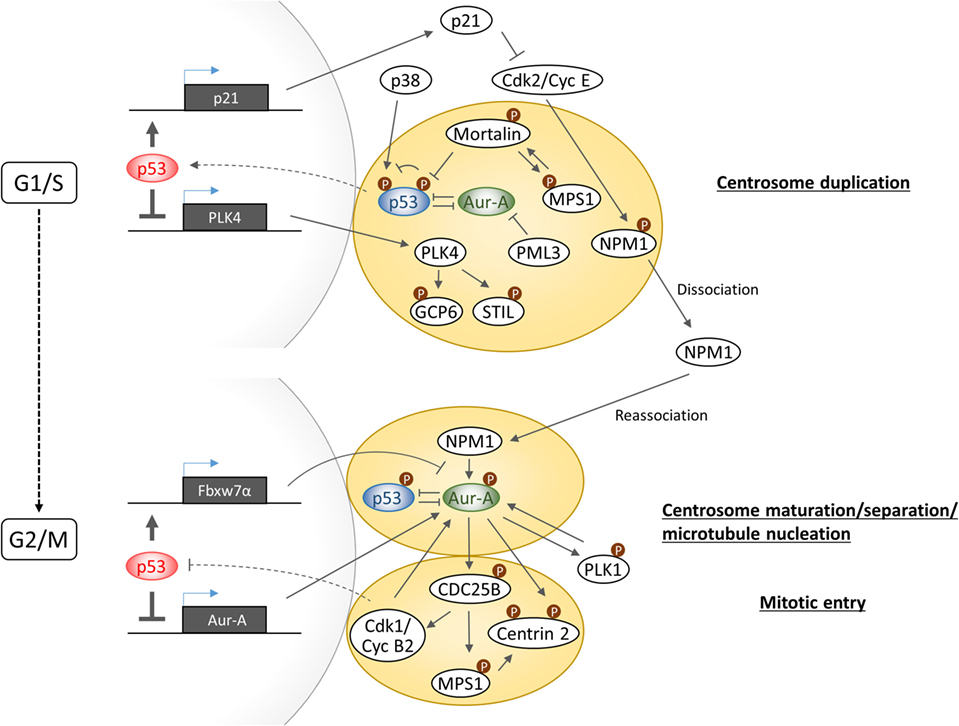
Figure 3. Schematic diagram illustrating the complexity of Aurora-A–p53-mediated signaling in centrosome biogenesis.
In transactivation activity-independent mechanism, centrosomal localization of p53 appears to be critical for negatively regulating centrosome biogenesis and its dissociation from centrosome appears to be sufficient to initiate centrosome duplication. p38–p53 axis was reported to play a central role in inhibition of G1–S cell cycle progression in response to loss of centrosome integrity. Centrosome perturbation caused by depletion of centrosomal proteins such as PCM1, centrobin, and TACC3 promotes the recruitment of both p38 and p53 to centrosomes and facilitate p53 phosphorylation by p38 at serine 33, which in turn transduces the inhibitory signal for cell cycle arrest by inducing p21 expression (53–55). However, the precise function of phosphorylated p53 on centrosome and the molecular mechanism of signal transduction from impaired centrosomes to the nucleus remain unknown. Regarding the mechanism of p53 dissociation from centrosome, a study has revealed that Mortalin through binding to p53 facilitates dissociation of p53 from centrosomes, which in turn results in release of the p53-mediated suppression of centrosome duplication (56). Interestingly, centrosome localization of Mortalin depends on the presence of centrosomal MPS1 kinase which is implicated in the regulation of centrosome duplication and mitotic spindle checkpoint response (57). MPS1 phosphorylates Mortalin, which in turn hyperactivates MPS1 kinase in a feed-forward regulatory manner. Importantly, Mortalin phosphorylation-activated MPS1 can drive centrosome overduplication. Although MPS1 phosphorylation of p53 positively regulates postmitotic checkpoint response (20), the precise role of MPS1 in the regulation of p53 function at the centrosome remains uncertain. Interestingly, the promyelocytic leukemia gene 3 (PML3) was shown to physically interact with Aurora-A and inhibit its kinase activity, while loss of PML3 shown to increase Aurora-A kinase activity and reduced protein stability of p53 along with decreased p21 expression, leading to activation of Cdk2/Cyclin E activity (58). Therefore, since there is no direct evidence supporting a role of centrosome localized Aurora-A in centrosome duplication, it would be imperative to further investigate whether or not increased p53–Mortalin interaction mediated by Aurora-A promotes p53 dissociation from centrosome and accompanying reduction of serine 33 phosphorylation is a cause of centrosome amplification induced in Aurora-A overexpressing cells.
At G1–S transition phase, Nucleophosmin/NPM1 is dissociated from unduplicated centrosome and at G2 phase is again recruited to duplicated centrosome to activate Aurora-A through phosphorylation of serine 89 (59). Activated Aurora-A cooperates with PLK1 to produce the onset signal for entry into mitosis as well as centrosome maturation. Since PLK1 has been shown to induce p53 degradation through phosphorylation of Topors (60), Aurora-A–PLK1 functional interaction, therefore, could interfere with p53 function on the centrosome at G2/M phase. NPM1-activated Aurora-A has also shown to induce phosphorylation of Centrin 2 at serine170 for stabilization of the protein (61). Phosphorylation of CDC25B at serine 353, which in turn stabilizes MPS1, also leads to stabilization of Centrin 2 through phosphorylation (62, 63). These findings indicate that Aurora-A and MPS1 cooperatively regulates Centrin 2 stability to induce centrosome maturation and separation. Activation of CDC25B is also pivotal for activation of Cdk1/Cyclin B, and a recent study has revealed that Cyclin B2 antagonizes p53 inhibitory activity against Aurora-A to control proper timing of centrosome separation at the onset of mitosis (64). Taken together, Aurora-A signaling branches off from CDC25B toward MPS1 for control of Centrin 2 stabilization regulating centrosome activity and toward Cdk1/CyclinB for positive feedback toward activation of Aurora-A in part by preventing p53 inhibitory action on Aurora-A.
The studies mentioned above clearly present evidence in support of a critical role for p53 signaling in regulating centrosome biogenesis and activity through cell cycle. In view of Aurora-A expression levels correlating with centrosome number and activity as well as known Aurora-A functional interactions with p53, Mortalin, PLK1, CDC25B, and, possibly MPS1, it will be interesting to investigate how the entire signaling axis involving these proteins maintains centrosomal homeostasis in proliferating cells.
Aurora-A–p73 Interaction in Spindle Assembly Checkpoint
A number of studies have shown the association of deregulated Aurora-A expression and activity with SAC override in cells irrespective of the p53 functional status in cells. Therefore, it is currently unclear whether or not p53 is involved in Aurora-A-mediated signal for SAC override. Accumulating evidence consistently suggest that p53 also functions in mitotic cell death and postmitotic checkpoint activated following aberrant mitosis and/or spindle damage through interaction with and phosphorylation by SAC proteins rather than being involved in the activation of SAC (20, 65–68). On the contrary, the role of Aurora-A–p73 interaction in SAC is relatively better defined. In vitro studies have shown roles of p73 in G2–M transition, mitotic exit, and mitotic cell death (69–72), while analysis of transgenic mouse lacking transactivation competent p73 (TAp73) revealed frequent occurrence of aberrant spindle structure associated with aneuploidy, chromosome instability, and mitotic slippage with spindle poisons (26). Further biochemical studies have also shown interaction of TAp73 with SAC proteins BUB1, BUB3, and BUBR1, and this interaction is crucial for BUB1 and BUBR1 localization at kinetochores and BUBR1 kinase activity (26, 27). These results suggest that TAp73 is directly involved in regulating SAC pathway to maintain chromosome stability. More recent study has demonstrated that TAp73 interacts with the inhibitory mitotic checkpoint complex of MAD2 and CDC20, preventing activation of the E3 ubiquitin ligase APC/C, and that Aurora-A phosphorylation of TAp73 at serine 235 causes dissociation of the MAD2–CDC20 complex, facilitating mitotic exit (24), suggesting that Aurora-A–TAp73 interaction is essential for a critical step in the SAC inactivation pathway (Figure 4). Unlike its effect on MAD2–CDC20 interaction and p73 depletion induced mislocalization of BUBR1 from the kinetochore, phosphorylation of p73 does not affect interaction of BUBR1 with CDC20 and its kinetochore localization, indicating that p73 participates in distinct pathway to control SAC activation. Although serine 235 phosphorylation of p73 enhances its interaction with Mortalin as described above, a more detailed investigation on the role of Aurora-A–Mortalin signaling axis in mitotic progression and SAC is warranted.
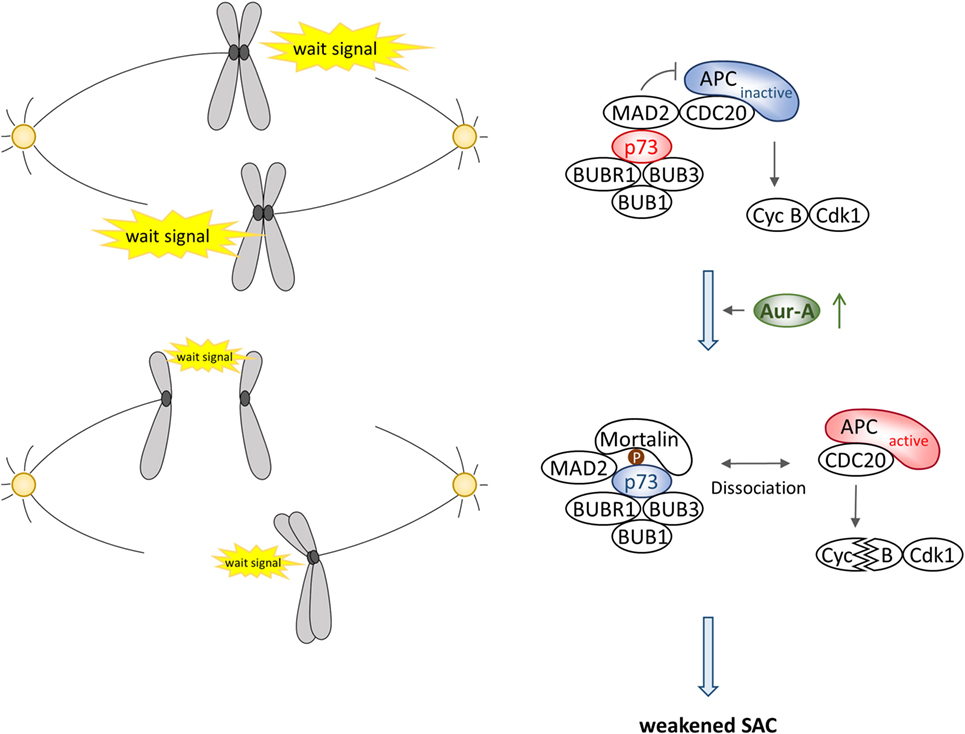
Figure 4. Schematic illustration of Aurora-A–p73 interaction in spindle assembly checkpoint.
Expression level of transactivation-defective ΔNp73 is known to be elevated in many tumors and ectopic expression of transactivation-defective ΔNp73 has been implicated in abnormal mitotic progression accompanied with multipolar spindle and cytokinesis failure resulting in multinucleated cells. However, ΔNp73 neither affects SAC activation in the presence of spindle poison nor is it known to interact with BUBR1 (26, 73), indicating that expression of ΔNp73 helps bypass SAC. Intriguingly, Aurora-A also interacts with and phosphorylates ΔNp73 with similar efficacy as that of TAp73 but its phosphorylation site is different from TAp73 that remains to be mapped (24). Thus, characterization of physiological role of Aurora-A phosphorylated ΔNp73 could provide evidence of a novel signaling pathway affecting SAC.
Aurora-A–p53 Signaling in Pluripotent Cells
Aurora-A has been reported to suppresses p53 function via phosphorylation of cell-fate determinant protein NUMB. While NUMB interacts with and helps stabilize and activate the tumor suppressor protein p53 (74, 75), Aurora-A initiates a phosphorylation cascade of aPKC–PAR6–Lgl cell polarity complex that ultimately leads to NUMB phosphorylation during mitosis to commit to asymmetric cell division (76–78). A recent study has revealed that phosphorylation of NUMB by Aurora–aPKC cascade disrupts its binding to p53 and promotes MDM2-mediated p53 degradation in cancer initiating cells of liver cancer (79). Thus, Aurora-A also antagonizes p53 activity indirectly through aPKC activation, resulting in maintenance of pluripotent state of cells and possibly promoting tumorigenesis. It would be interesting to examine if Aurora-A phosphorylation of p53 and NUMB synergistically affect disruption of their bindings.
A number of studies on cancer stem-like cells have revealed strong association of Aurora-A expression with gene expression of core stemness markers, such as Myc, Sox2, and Oct4. Additionally, Aurora-A–p53 functional interaction in the regulation of self-renewal and differentiation of mouse embryonic stem cells (mESC) and somatic cell reprograming has also been investigated (80, 81). Loss-of-function screening for protein kinases and phosphatases essential in mESC development and subsequent functional studies revealed strong correlation between elevated expression of Aurora-A and the undifferentiated state of mESC. Furthermore, loss of Aurora-A, but not loss of Aurora-A, mitotic substrates compromised self-renewal and triggered differentiation of mESC, indicating that non-canonical function of Aurora-A, unrelated to its role in mitosis, is possibly involved in regulating self-renewal potential of mESC (82). This observation also showed inverse correlation with p53 activity in mESC and attributed this finding to Aurora-A-mediated inactivation of p53 function. The study also revealed that Aurora-A-mediated serine 215 phosphorylation rather than serine 315 phosphorylation is more critical in antagonizing p53-induced mESC differentiation and p53-mediated suppression of induced Pluripotent Stem Cells (iPSC) reprograming via activation of gene expression program associated with pluripotency. Phosphorylation of serine 315, on the other hand, was shown to cause partial impairments of both mESC differentiation and suppression of iPSC reprograming correlating with lower expression of pluripotency markers. The varying degree of downstream effects of the two Aurora-A-mediated p53 phosphorylated residues possibly represents stronger inhibition of p53 function following serine 215 phosphorylation resulting in complete loss of its transactivation activity and cytoplasmic sequestration reflecting the naturally observed localization of endogenous p53 in mESC (83). The study concluded that Aurora-A controls pluripotency through inhibition of p53 target gene expression required for ectodermal and mesodermal differentiation. The observation regarding serine 315 phosphorylation showing less pronounced phenotype in this study appeared conflicting to an earlier report showing elevated serine 315 phosphorylation during mESC differentiation and knockin of serine 315 phosphor-deficient mutant impairing mESC differentiation. Importantly, serine 315 phosphorylation was also reported to enable the recruitment of the corepressor mSin3a to the NANOG promotor, resulting in complete suppression of NANOG transcription and primitive endodermal differentiation (84–86). Serine 315 phosphorylation is known to be mediated not only by Aurora-A but also by Cdk/cyclin complex. In view of the observed loss of serine 33 phosphorylation in serine 215 phosphorylated p53, it is plausible that serine 215 phosphorylation might inhibit serine 315 phosphorylation by Cdk1 or Aurora-A. Alternatively, Aurora-A phosphorylation of the two p53 residues may be playing non-overlapping physiological roles in Aurora-A-mediated cellular processes.
In contrast to the requirement of Aurora-A in maintenance of pluripotency and induction of iPSC state mentioned above, a study reported that loss of Aurora-A function is essential for somatic cell reprograming (87). In this study, authors reported that loss of Aurora-A function following small-molecule inhibitor treatment or siRNA knockdown enhanced efficacy of iPSC generation with cells reaching a fully reprogramed state. The iPSC generated by this approach possessed ability to differentiate into different lineages in vitro and in vivo. Moreover, p53 depletion could further enhance the effect of loss of Aurora-A function. The underlying reasons for these contradictory findings are not known at this time and need to be investigated.
Genetically Engineered Aurora-A–p53 Targeted Mouse Models
Comprehensive genomic analyses have identified Aurora-A as a low penetrance tumor-susceptibility gene and elevated expression was reported to play an essential pathological in tumor development correlating with prognosis and resistance to therapy (80, 88–91). Several transgenic mouse models have been developed to gain direct evidence of Aurora-A tumorigenic potential and associated phenotypic alternations in vivo, which have yielded somewhat conflicting and distinct results (92–94). While Wap-Cre mouse model system in which Aurora-A was constitutively overexpressed under CAG-CAT promoter in mammary gland after one cycle of pregnancy developed hyperplasia in p53 wild-type background and precancerous atypical ductal hyperplasia in p53–null background (92, 94), MMTV promoter-driven mouse model was reported to develop mammary tumors in both p53 wild-type and heterozygous background after four to five cycles of pregnancy (93). Notably, centrosome amplification and chromosome instability were detected in all mouse models, suggesting that Aurora-A overexpression affects p53 function in the maintenance of centrosome homeostasis and chromosomal stability in vivo. Consistent with in vitro studies, activation of AKT signaling pathway leading to Cyclin D overexpression was seen in the tumors developed in MMTV–Aurora-A mice. We have recently reported a mammary gland targeted Aurora-A mouse model in a p53 wild-type background in which Aurora-A expression is driven by ovine β-lactoglobulin promoter led to the development of mammary tumors after four to five of pregnancy cycles (95). In addition to genomic instability, reduced expression of p53 protein and activation of AKT signal pathway was detected in tumors similar to MMTV–Aurora-A mouse model, again suggesting that elevated levels of Aurora-A can be oncogenic with inhibitory effects on p53-mediated tumor suppressor signaling pathways. It is relevant to mention, in this context, that an inducible gene switch mouse model overexpressing Aurora-A in skin epidermis exposed to tumor promoter 12-O-tetradecanoylphorbol 13-acetate (TPA) and the mutagen 7,12-dimethylbenz(a)anthracene, developed by us earlier, revealed malignant progression of skin tumors with centrosome amplification, abnormal spindle formation, and genomic instability (96). Expression of p53 protein was lost, and amplification of MDM2 gene was concurrently found in these tumors. Taken together, Aurora-A overexpressing mouse models of organ-specific tumors have revealed loss of p53 expression recapitulating naturally occurring Aurora-A and p53 expression changes seen in human tumors. Further in-depth studies to elucidate the role of Aurora-A–p53 signaling cascades relevant to human tumor development utilizing Aurora-A overexpressing mouse models are warranted.
Conclusion
Functional interactions between Aurora kinases and p53 family proteins coordinately regulate diverse cellular pathways by modulating activity and subcellular localization of each other and their downstream effector proteins. Deregulations of these interactions in cells undergoing tumorigenic transformation have significant functional consequences on induction of chromosome instability, development of different tumor-associated phenotypes including resistance to therapy. In addition to Aurora kinase functional interactions with wild-type p53 and p73, there is evidence of Aurora-A interacting with and phosphorylating mutant p53 protein. Physiological function of Aurora-A–mutant p53 interactions have not been elucidated yet. Mutant p53 and transactivation-deficient mutant of p73 also phenocopy some of the Aurora-A overexpression-induced phenotypes. It would be interesting to investigate the functional consequences of Aurora-A phosphorylation of mutant p53 family members in the p53 signaling cascades and their significance in the development of tumorigenic phenotypes.
Author Contributions
All authors contributed to writing the review and preparing figures.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This study was supported by National Cancer Institute grants (R01CA089716, NCI/EDRN UO1CA111302 to SS); University Cancer Foundation, MD Anderson (to SS); JSPS KAKENHI (JP25893137 to HK); and Grant-in-Aid for Research Activity Start-up from Okayama University (to KS and HK).
References
1. Fukasawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science (1996) 271(5256):1744–7. doi:10.1126/science.271.5256.1744
2. Bischoff FZ, Yim SO, Pathak S, Grant G, Siciliano MJ, Giovanella BC, et al. Spontaneous abnormalities in normal fibroblasts from patients with Li-Fraumeni cancer syndrome: aneuploidy and immortalization. Cancer Res (1990) 50(24):7979–84.
3. Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet (1998) 20(2):189–93. doi:10.1038/2496
4. Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, et al. A homologue of Drosophila Aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J (1998) 17(11):3052–65. doi:10.1093/emboj/17.11.3052
5. Marumoto T, Hirota T, Morisaki T, Kunitoku N, Zhang D, Ichikawa Y, et al. Roles of Aurora-A kinase in mitotic entry and G2 checkpoint in mammalian cells. Genes Cells (2002) 7(11):1173–82. doi:10.1046/j.1365-2443.2002.00592.x
6. Chen SS, Chang PC, Cheng YW, Tang FM, Lin YS. Suppression of the STK15 oncogenic activity requires a transactivation-independent p53 function. EMBO J (2002) 21(17):4491–9. doi:10.1093/emboj/cdf409
7. Eyers PA, Maller JL. Regulation of Xenopus Aurora A activation by TPX2. J Biol Chem (2004) 279(10):9008–15. doi:10.1074/jbc.M312424200
8. Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, et al. Phosphorylation by Aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet (2004) 36(1):55–62. doi:10.1038/ng1279
9. Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem (2004) 279(50):52175–82. doi:10.1074/jbc.M406802200
10. Hsueh KW, Fu SL, Chang CB, Chang YL, Lin CH. A novel Aurora-A-mediated phosphorylation of p53 inhibits its interaction with MDM2. Biochim Biophys Acta (2013) 1834(2):508–15. doi:10.1016/j.bbapap.2012.11.005
11. Warnock LJ, Raines SA, Milner J. Aurora A mediates cross-talk between N- and C-terminal post-translational modifications of p53. Cancer Biol Ther (2011) 12(12):1059–68. doi:10.4161/cbt.12.12.18141
12. Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW, Appella E, et al. Phosphorylation of human p53 by p38 kinase coordinates N-terminal phosphorylation and apoptosis in response to UV radiation. EMBO J (1999) 18(23):6845–54. doi:10.1093/emboj/18.23.6845
13. Moumen A, Masterson P, O’Connor MJ, Jackson SP. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell (2005) 123(6):1065–78. doi:10.1016/j.cell.2005.09.032
14. Enge M, Bao W, Hedstrom E, Jackson SP, Moumen A, Selivanova G. MDM2-dependent downregulation of p21 and hnRNP K provides a switch between apoptosis and growth arrest induced by pharmacologically activated p53. Cancer Cell (2009) 15(3):171–83. doi:10.1016/j.ccr.2009.01.019
15. Hsueh KW, Fu SL, Huang CY, Lin CH. Aurora-A phosphorylates hnRNPK and disrupts its interaction with p53. FEBS Lett (2011) 585(17):2671–5. doi:10.1016/j.febslet.2011.07.031
16. Dar AA, Zaika A, Piazuelo MB, Correa P, Koyama T, Belkhiri A, et al. Frequent overexpression of Aurora kinase A in upper gastrointestinal adenocarcinomas correlates with potent antiapoptotic functions. Cancer (2008) 112(8):1688–98. doi:10.1002/cncr.23371
17. Sehdev V, Katsha A, Arras J, Peng D, Soutto M, Ecsedy J, et al. HDM2 regulation by AURKA promotes cell survival in gastric cancer. Clin Cancer Res (2014) 20(1):76–86. doi:10.1158/1078-0432.CCR-13-1187
18. Wu L, Ma CA, Zhao Y, Jain A. Aurora B interacts with NIR-p53, leading to p53 phosphorylation in its DNA-binding domain and subsequent functional suppression. J Biol Chem (2011) 286(3):2236–44. doi:10.1074/jbc.M110.174755
19. Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proc Natl Acad Sci U S A (2012) 109(24):E1513–22. doi:10.1073/pnas.1110287109
20. Huang YF, Chang MD, Shieh SY. TTK/hMps1 mediates the p53-dependent postmitotic checkpoint by phosphorylating p53 at Thr18. Mol Cell Biol (2009) 29(11):2935–44. doi:10.1128/MCB.01837-08
21. Ha GH, Baek KH, Kim HS, Jeong SJ, Kim CM, McKeon F, et al. p53 activation in response to mitotic spindle damage requires signaling via BubR1-mediated phosphorylation. Cancer Res (2007) 67(15):7155–64. doi:10.1158/0008-5472.CAN-06-3392
22. Wang YC, Tsai YS, Huang JL, Lee KW, Kuo CC, Wang CS, et al. Arecoline arrests cells at prometaphase by deregulating mitotic spindle assembly and spindle assembly checkpoint: implication for carcinogenesis. Oral Oncol (2010) 46(4):255–62. doi:10.1016/j.oraloncology.2010.01.003
23. Ha GH, Breuer EK. Mitotic kinases and p53 signaling. Biochem Res Int (2012) 2012:195903. doi:10.1155/2012/195903
24. Katayama H, Wang J, Treekitkarnmongkol W, Kawai H, Sasai K, Zhang H, et al. Aurora kinase-A inactivates DNA damage-induced apoptosis and spindle assembly checkpoint response functions of p73. Cancer Cell (2012) 21(2):196–211. doi:10.1016/j.ccr.2011.12.025
25. Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev (2008) 22(19):2677–91. doi:10.1101/gad.1695308
26. Tomasini R, Tsuchihara K, Tsuda C, Lau SK, Wilhelm M, Ruffini A, et al. TAp73 regulates the spindle assembly checkpoint by modulating BubR1 activity. Proc Natl Acad Sci U S A (2009) 106(3):797–802. doi:10.1073/pnas.0812096106
27. Vernole P, Neale MH, Barcaroli D, Munarriz E, Knight RA, Tomasini R, et al. TAp73alpha binds the kinetochore proteins Bub1 and Bub3 resulting in polyploidy. Cell Cycle (2009) 8(3):421–9. doi:10.4161/cc.8.3.7623
28. Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ (2012) 19(12):1992–2002. doi:10.1038/cdd.2012.89
29. Katsha A, Arras J, Soutto M, Belkhiri A, El-Rifai W. AURKA regulates JAK2-STAT3 activity in human gastric and esophageal cancers. Mol Oncol (2014) 8(8):1419–28. doi:10.1016/j.molonc.2014.05.012
30. He L, Yang H, Ma Y, Pledger WJ, Cress WD, Cheng JQ. Identification of Aurora-A as a direct target of E2F3 during G2/M cell cycle progression. J Biol Chem (2008) 283(45):31012–20. doi:10.1074/jbc.M803547200
31. Shao S, Wang Y, Jin S, Song Y, Wang X, Fan W, et al. Gadd45a interacts with Aurora-A and inhibits its kinase activity. J Biol Chem (2006) 281(39):28943–50. doi:10.1074/jbc.M600235200
32. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, et al. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature (2004) 432(7018):775–9. doi:10.1038/nature03155
33. Wu CC, Yang TY, Yu CT, Phan L, Ivan C, Sood AK, et al. p53 negatively regulates Aurora A via both transcriptional and posttranslational regulation. Cell Cycle (2012) 11(18):3433–42. doi:10.4161/cc.21732
34. Teng CL, Hsieh YC, Phan L, Shin J, Gully C, Velazquez-Torres G, et al. FBXW7 is involved in Aurora B degradation. Cell Cycle (2012) 11(21):4059–68. doi:10.4161/cc.22381
35. Kwon YW, Kim IJ, Wu D, Lu J, Stock WA Jr, Liu Y, et al. Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Mol Cancer Res (2012) 10(6):834–44. doi:10.1158/1541-7786.MCR-12-0025
36. Mao JH, Wu D, Perez-Losada J, Jiang T, Li Q, Neve RM, et al. Crosstalk between Aurora-A and p53: frequent deletion or downregulation of Aurora-A in tumors from p53 null mice. Cancer Cell (2007) 11(2):161–73. doi:10.1016/j.ccr.2006.11.025
37. Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell (2009) 15(1):67–78. doi:10.1016/j.ccr.2008.12.005
38. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of Aurora-A induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer Cell (2013) 24(1):75–89. doi:10.1016/j.ccr.2013.05.005
39. Morozova O, Vojvodic M, Grinshtein N, Hansford LM, Blakely KM, Maslova A, et al. System-level analysis of neuroblastoma tumor-initiating cells implicates AURKB as a novel drug target for neuroblastoma. Clin Cancer Res (2010) 16(18):4572–82. doi:10.1158/1078-0432.CCR-10-0627
40. Bogen D, Wei JS, Azorsa DO, Ormanoglu P, Buehler E, Guha R, et al. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget (2015) 6(34):35247–62. doi:10.18632/oncotarget.6208
41. den Hollander J, Rimpi S, Doherty JR, Rudelius M, Buck A, Hoellein A, et al. Aurora kinases A and B are up-regulated by Myc and are essential for maintenance of the malignant state. Blood (2010) 116(9):1498–505. doi:10.1182/blood-2009-11-251074
42. Chou CH, Yang NK, Liu TY, Tai SK, Hsu DS, Chen YW, et al. Chromosome instability modulated by BMI1-AURKA signaling drives progression in head and neck cancer. Cancer Res (2013) 73(2):953–66. doi:10.1158/0008-5472.CAN-12-2397
43. Li Z, Sun Y, Chen X, Squires J, Nowroozizadeh B, Liang C, et al. p53 mutation directs AURKA overexpression via miR-25 and FBXW7 in prostatic small cell neuroendocrine carcinoma. Mol Cancer Res (2015) 13(3):584–91. doi:10.1158/1541-7786.MCR-14-0277-T
44. Schnepp RW, Khurana P, Attiyeh EF, Raman P, Chodosh SE, Oldridge DA, et al. A LIN28B-RAN-AURKA signaling network promotes neuroblastoma tumorigenesis. Cancer Cell (2015) 28(5):599–609. doi:10.1016/j.ccell.2015.09.012
45. Suh SS, Yoo JY, Nuovo GJ, Jeon YJ, Kim S, Lee TJ, et al. MicroRNAs/TP53 feedback circuitry in glioblastoma multiforme. Proc Natl Acad Sci U S A (2012) 109(14):5316–21. doi:10.1073/pnas.1202465109
46. Zheng F, Yue C, Li G, He B, Cheng W, Wang X, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun (2016) 7:10180. doi:10.1038/ncomms10180
47. Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A, et al. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci U S A (2005) 102(3):731–6. doi:10.1073/pnas.0405495102
48. Slack AD, Chen Z, Ludwig AD, Hicks J, Shohet JM. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res (2007) 67(6):2448–55. doi:10.1158/0008-5472.CAN-06-1661
49. Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell (2000) 103(1):127–40. doi:10.1016/S0092-8674(00)00093-3
50. Bahtz R, Seidler J, Arnold M, Haselmann-Weiss U, Antony C, Lehmann WD, et al. GCP6 is a substrate of Plk4 and required for centriole duplication. J Cell Sci (2012) 125(Pt 2):486–96. doi:10.1242/jcs.093930
51. Nakamura T, Saito H, Takekawa M. SAPK pathways and p53 cooperatively regulate PLK4 activity and centrosome integrity under stress. Nat Commun (2013) 4:1775. doi:10.1038/ncomms2752
52. Fischer M, Quaas M, Wintsche A, Müller GA, Engeland K. Polo-like kinase 4 transcription is activated via CRE and NRF1 elements, repressed by DREAM through CDE/CHR sites and deregulated by HPV E7 protein. Nucleic Acids Res (2014) 42(1):163–80. doi:10.1093/nar/gkt849
53. Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat Cell Biol (2007) 9(2):160–70. doi:10.1038/ncb1529
54. Song L, Dai T, Xiong H, Lin C, Lin H, Shi T, et al. Inhibition of centriole duplication by centrobin depletion leads to p38-p53 mediated cell-cycle arrest. Cell Signal (2010) 22(5):857–64. doi:10.1016/j.cellsig.2010.01.009
55. Suhail TV, Singh P, Manna TK. Suppression of centrosome protein TACC3 induces G1 arrest and cell death through activation of p38-p53-p21 stress signaling pathway. Eur J Cell Biol (2015) 94(2):90–100. doi:10.1016/j.ejcb.2014.12.001
56. Ma Z, Izumi H, Kanai M, Kabuyama Y, Ahn NG, Fukasawa K. Mortalin controls centrosome duplication via modulating centrosomal localization of p53. Oncogene (2006) 25(39):5377–90. doi:10.1038/sj.onc.1209543
57. Kanai M, Ma Z, Izumi H, Kim SH, Mattison CP, Winey M, et al. Physical and functional interaction between mortalin and Mps1 kinase. Genes Cells (2007) 12(6):797–810. doi:10.1111/j.1365-2443.2007.01091.x
58. Xu ZX, Zou WX, Lin P, Chang KS. A role for PML3 in centrosome duplication and genome stability. Mol Cell (2005) 17(5):721–32. doi:10.1016/j.molcel.2005.02.014
59. Reboutier D, Troadec MB, Cremet JY, Fukasawa K, Prigent C. Nucleophosmin/B23 activates Aurora A at the centrosome through phosphorylation of serine 89. J Cell Biol (2012) 197(1):19–26. doi:10.1083/jcb.201107134
60. Yang X, Li H, Zhou Z, Wang WH, Deng A, Andrisani O, et al. Plk1-mediated phosphorylation of Topors regulates p53 stability. J Biol Chem (2009) 284(28):18588–92. doi:10.1074/jbc.C109.001560
61. Lukasiewicz KB, Greenwood TM, Negron VC, Bruzek AK, Salisbury JL, Lingle WL. Control of centrin stability by Aurora A. PLoS One (2011) 6(6):e21291. doi:10.1371/journal.pone.0021291
62. Yang CH, Kasbek C, Majumder S, Yusof AM, Fisk HA. Mps1 phosphorylation sites regulate the function of centrin 2 in centriole assembly. Mol Biol Cell (2010) 21(24):4361–72. doi:10.1091/mbc.E10-04-0298
63. Boutros R, Mondesert O, Lorenzo C, Astuti P, McArthur G, Chircop M, et al. CDC25B overexpression stabilises centrin 2 and promotes the formation of excess centriolar foci. PLoS One (2013) 8(7):e67822. doi:10.1371/journal.pone.0067822
64. Nam HJ, van Deursen JM. Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol (2014) 16(6):538–49. doi:10.1038/ncb2952
65. Andreassen PR, Lohez OD, Lacroix FB, Margolis RL. Tetraploid state induces p53-dependent arrest of nontransformed mammalian cells in G1. Mol Biol Cell (2001) 12(5):1315–28. doi:10.1091/mbc.12.5.1315
66. Margolis RL, Lohez OD, Andreassen PR. G1 tetraploidy checkpoint and the suppression of tumorigenesis. J Cell Biochem (2003) 88(4):673–83. doi:10.1002/jcb.10411
67. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature (2005) 437(7061):1043–7. doi:10.1038/nature04217
68. Gao F, Ponte JF, Levy M, Papageorgis P, Cook NM, Ozturk S, et al. hBub1 negatively regulates p53 mediated early cell death upon mitotic checkpoint activation. Cancer Biol Ther (2009) 8(7):548–56. doi:10.4161/cbt.8.7.7929
69. Fulco M, Costanzo A, Merlo P, Mangiacasale R, Strano S, Blandino G, et al. p73 is regulated by phosphorylation at the G2/M transition. J Biol Chem (2003) 278(49):49196–202. doi:10.1074/jbc.M304921200
70. Merlo P, Fulco M, Costanzo A, Mangiacasale R, Strano S, Blandino G, et al. A role of p73 in mitotic exit. J Biol Chem (2005) 280(34):30354–60. doi:10.1074/jbc.M500635200
71. Niikura Y, Dixit A, Scott R, Perkins G, Kitagawa K. BUB1 mediation of caspase-independent mitotic death determines cell fate. J Cell Biol (2007) 178(2):283–96. doi:10.1083/jcb.200702134
72. Toh WH, Nam SY, Sabapathy K. An essential role for p73 in regulating mitotic cell death. Cell Death Differ (2010) 17(5):787–800. doi:10.1038/cdd.2009.181
73. Marrazzo E, Marchini S, Tavecchio M, Alberio T, Previdi S, Erba E, et al. The expression of the DeltaNp73beta isoform of p73 leads to tetraploidy. Eur J Cancer (2009) 45(3):443–53. doi:10.1016/j.ejca.2008.09.024
74. Colaluca IN, Tosoni D, Nuciforo P, Senic-Matuglia F, Galimberti V, Viale G, et al. NUMB controls p53 tumour suppressor activity. Nature (2008) 451(7174):76–80. doi:10.1038/nature06412
75. Carter S, Vousden KH. A role for Numb in p53 stabilization. Genome Biol (2008) 9(5):221. doi:10.1186/gb-2008-9-5-221
76. Wirtz-Peitz F, Nishimura T, Knoblich JA. Linking cell cycle to asymmetric division: Aurora-A phosphorylates the Par complex to regulate Numb localization. Cell (2008) 135(1):161–73. doi:10.1016/j.cell.2008.07.049
77. Bell GP, Fletcher GC, Brain R, Thompson BJ. Aurora kinases phosphorylate Lgl to induce mitotic spindle orientation in Drosophila epithelia. Curr Biol (2015) 25(1):61–8. doi:10.1016/j.cub.2014.10.052
78. Carvalho CA, Moreira S, Ventura G, Sunkel CE, Morais-de-Sa E. Aurora A triggers Lgl cortical release during symmetric division to control planar spindle orientation. Curr Biol (2015) 25(1):53–60. doi:10.1016/j.cub.2014.10.053
79. Siddique HR, Feldman DE, Chen CL, Punj V, Tokumitsu H, Machida K. NUMB phosphorylation destabilizes p53 and promotes self-renewal of tumor-initiating cells by a NANOG-dependent mechanism in liver cancer. Hepatology (2015) 62(5):1466–79. doi:10.1002/hep.27987
80. D’Assoro AB, Liu T, Quatraro C, Amato A, Opyrchal M, Leontovich A, et al. The mitotic kinase Aurora – a promotes distant metastases by inducing epithelial-to-mesenchymal transition in ERalpha(+) breast cancer cells. Oncogene (2014) 33(5):599–610. doi:10.1038/onc.2012.628
81. Zheng FM, Long ZJ, Hou ZJ, Luo Y, Xu LZ, Xia JL, et al. A novel small molecule Aurora kinase inhibitor attenuates breast tumor-initiating cells and overcomes drug resistance. Mol Cancer Ther (2014) 13(8):1991–2003. doi:10.1158/1535-7163.MCT-13-1029
82. Lee DF, Su J, Ang YS, Carvajal-Vergara X, Mulero-Navarro S, Pereira CF, et al. Regulation of embryonic and induced pluripotency by Aurora kinase-p53 signaling. Cell Stem Cell (2012) 11(2):179–94. doi:10.1016/j.stem.2012.05.020
83. Han MK, Song EK, Guo Y, Ou X, Mantel C, Broxmeyer HE. SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell (2008) 2(3):241–51. doi:10.1016/j.stem.2008.01.002
84. Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, et al. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell (2003) 113(5):631–42. doi:10.1016/S0092-8674(03)00393-3
85. Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol (2005) 7(2):165–71. doi:10.1038/ncb1211
86. Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, et al. Dissecting self-renewal in stem cells with RNA interference. Nature (2006) 442(7102):533–8. doi:10.1038/nature04915
87. Li Z, Rana TM. A kinase inhibitor screen identifies small-molecule enhancers of reprogramming and iPS cell generation. Nat Commun (2012) 3:1085. doi:10.1038/ncomms2059
88. Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci U S A (2003) 100(10):5974–9. doi:10.1073/pnas.0931261100
89. Ewart-Toland A, Briassouli P, de Koning JP, Mao JH, Yuan J, Chan F, et al. Identification of Stk6/STK15 as a candidate low-penetrance tumor-susceptibility gene in mouse and human. Nat Genet (2003) 34(4):403–12. doi:10.1038/ng1220
90. Weier HU, Mao JH. Meta-analysis of Aurora kinase A (AURKA) expression data reveals a significant correlation between increased AURKA expression and distant metastases in human ER-positive breast cancers. J Data Mining Genomics Proteomics (2013) 4(1):127. doi:10.4172/2153-0602.1000127
91. Opyrchal M, Salisbury JL, Zhang S, McCubrey J, Hawse J, Goetz MP, et al. Aurora-A mitotic kinase induces endocrine resistance through down-regulation of ERalpha expression in initially ERalpha+ breast cancer cells. PLoS One (2014) 9(5):e96995. doi:10.1371/journal.pone.0096995
92. Zhang D, Hirota T, Marumoto T, Shimizu M, Kunitoku N, Sasayama T, et al. Cre-loxP-controlled periodic Aurora-A overexpression induces mitotic abnormalities and hyperplasia in mammary glands of mouse models. Oncogene (2004) 23(54):8720–30. doi:10.1038/sj.onc.1208153
93. Wang X, Zhou YX, Qiao W, Tominaga Y, Ouchi M, Ouchi T, et al. Overexpression of Aurora kinase A in mouse mammary epithelium induces genetic instability preceding mammary tumor formation. Oncogene (2006) 25(54):7148–58. doi:10.1038/sj.onc.1209707
94. Zhang D, Shimizu T, Araki N, Hirota T, Yoshie M, Ogawa K, et al. Aurora A overexpression induces cellular senescence in mammary gland hyperplastic tumors developed in p53-deficient mice. Oncogene (2008) 27(31):4305–14. doi:10.1038/onc.2008.76
95. Treekitkarnmongkol W, Katayama H, Kai K, Sasai K, Jones JC, Wang J, et al. Aurora kinase-A overexpression in mouse mammary epithelium induces mammary adenocarcinomas harboring genetic alterations shared with human breast cancer. Carcinogenesis (2016). doi:10.1093/carcin/bgw097
Keywords: Aurora kinases, p53 tumor suppressor protein family, chromosome instability, centrosome amplification, pluripotency, tumorigenesis
Citation: Sasai K, Treekitkarnmongkol W, Kai K, Katayama H and Sen S (2016) Functional Significance of Aurora Kinases–p53 Protein Family Interactions in Cancer. Front. Oncol. 6:247. doi: 10.3389/fonc.2016.00247
Received: 01 September 2016; Accepted: 07 November 2016;
Published: 25 November 2016
Edited by:
Claude Prigent, Centre national de la recherche scientifique, FranceReviewed by:
Robert Friis, University of Bern, SwitzerlandEdward Prochownik, University of Pittsburgh, USA
Copyright: © 2016 Sasai, Treekitkarnmongkol, Kai, Katayama and Sen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroshi Katayama, aGthdGF5YW1hQGNjLm9rYXlhbWEtdS5hYy5qcA==;
Subrata Sen, c3NlbkBtZGFuZGVyc29uLm9yZw==