 Esita Chattopadhyay
Esita Chattopadhyay Bidyut Roy
Bidyut Roy- Human Genetics Unit, Indian Statistical Institute, Kolkata, India
Mitochondria being the central organelle for metabolism and other cell signalling pathways have remained the topic of interest to tumour biologists. In spite of the wide acceptance of Warburg’s hypothesis, role of mitochondrial metabolism in cancer is still unclear. Uncontrolled growth and proliferation, hallmarks of tumour cells, are maintained when the cells adapt to metabolic reprogramming with the help of altered metabolism of mitochondria. This review has focussed on different aspects of mitochondrial metabolism and inter-related signalling pathways which have been found to be modified in cancer.
Introduction
Production of major amount of energy for the cell by oxidative phosphorylation is the most essential function of mitochondria. Other mitochondrial functions include apoptosis or programmed cell death, Ca2+ homoeostasis, etc. It has its own circular genome [mitochondrial DNA (mtDNA)], which codes for protein subunits for oxidative phosphorylation, tRNAs, and rRNAs. Some of the proteins involved in mitochondrial structure and functions are encoded by nuclear genome. Mutations in mtDNA are being studied and found to be causal for different mitochondrial diseases including cancer (1, 2). Implications of mitochondrial function in cancer seem to be well-debated question. Manifestation of cancer includes uncontrollable cell proliferation, inhibited cell death, angiogenesis, invasion into other tissues, etc. Proper functioning of mitochondria is required to maintain rapid growth and proliferation of cancer cells since tumour cell devoid of mitochondria grows very slowly (3–5). On the other hand, functional impairment of mitochondria is a common phenomenon in cancer cells since they undergo certain changes in metabolic pathways for their survival and maintenance (Figure 1). This review highlights metabolic reprogramming in cancer cell due to altered mitochondrial signalling.
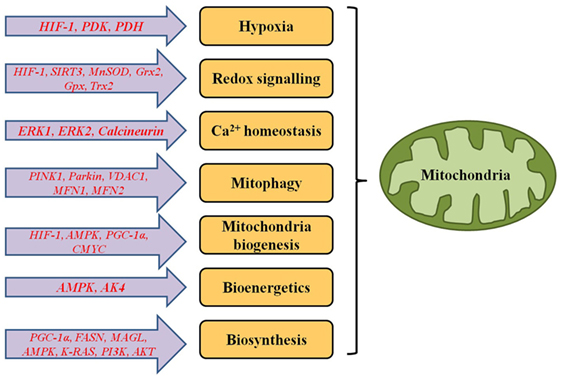
Figure 1. Genes responsible for functional alterations in metabolic and signalling pathways of mitochondria in cancer cells.
Hypoxia and Mitochondria
Hypoxia or oxygen deprivation is one of the key features of solid tumours and plays a significant role in different cellular functions including cell proliferation, survival, angiogenesis, metabolism, tumour evasion, and metastasis (6). It also regulates tumour cells to have reduced response to radiotherapy, resistance to chemotherapy and lower pH than normal cells (7–9). Proliferation rate of tumour cells is higher than the growth rate of new blood vessel formation, so newly generated cells are supplied with lower amount of oxygen. Depending on the aggressiveness of hypoxia, tumour cells either undergo apoptosis or adapt to the low oxygen environment and survive (10). The key coordinator of cellular mechanisms to adapt and survive in hypoxic condition is hypoxia-inducible factor 1 (HIF-1). It is a transcription factor which consists of two subunits: HIF-1α whose expression is regulated by abundance of oxygen and HIF-1β which is constitutively expressed. Limited oxygen availability induces expression of HIF-1 which regulates expression of several other genes (11) functionally involved in the pathways of angiogenesis, cell death/survival, metabolism, pH regulation, cell adhesion, extracellular matrix remodelling, cell migration, and metastasis (9).
In presence of low oxygen in cell, pyruvate is mostly converted to lactate instead of acetyl CoA (Figure 2) and HIF-1 induces expression of genes involved in the glycolytic pathway (such as glucose transporter, glycolytic enzymes, etc). HIF-1 increases expression of pyruvate dehydrogenase kinase 1, a subunit of PDK, which blocks function of pyruvate dehydrogenase (PDH) enzyme leading to increase production of lactate (12). Increased amount of lactate also induces HIF-1, which not only blocks acetyl-CoA metabolism in mitochondria, but also reduces mitochondrial biogenesis as well as oxygen consumption (13, 14).
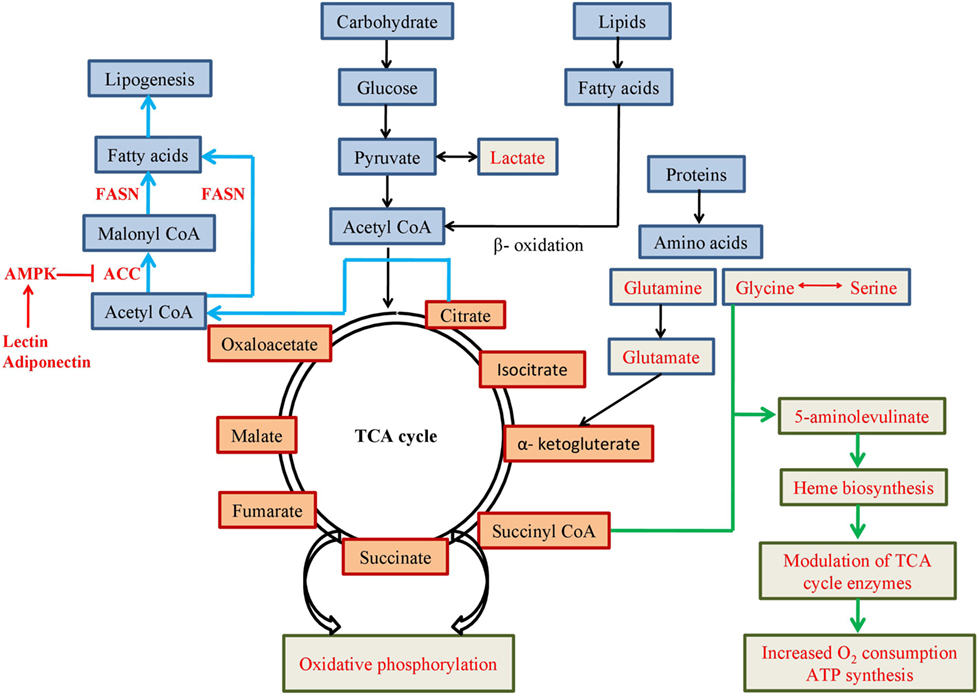
Figure 2. Schematic representation of metabolic pathways involving mitochondrial metabolites. Pathways or use of metabolites mentioned in red font are favoured in cancer cells for altered metabolism.
Although mitochondrial energy production is a more efficient method than that of glycolytic pathway, tumour cells perform glycolytic and other/altered metabolic pathways to generate energy. Mitochondrial electron transport chain (ETC) is involved in oxygen sensing since ETC consume most of the cellular oxygen. Certain inhibitors of ETC can block stabilisation of HIF-1α in hypoxic conditions, which signifies that functioning of ETC is required for hypoxia-mediated activities of HIF-1. During hypoxia, ETC complex III can release reactive oxygen species (ROS) into mitochondrial inter-membrane space and subsequently into the cytosol. Thus, mitochondrial ROS generation contributes to HIF-1α stabilisation under hypoxic condition of the cell (15).
Mitochondrial Metabolism and Tumour Growth
Mitochondria coordinate anabolic as well as catabolic reactions combined with energy production to achieve the needs of cellular bioenergetics and biosynthesis. Acetyl CoA, the key ingredient of mitochondrial metabolism and energy production is generated from breakdown of glucose, amino acids, and fatty acids. Necessity of mitochondrial metabolism for tumour growth is a well-discussed but yet it is an inconclusive area of research. According to one hypothesis, rapid proliferation and growth of tumour cells require functional mitochondria as it is the major source of energy as well as supplier of metabolic building blocks for tumour cells. Anaplerotic pathways which maintain pools of metabolic intermediates for repeated usage of rapid growth and proliferation are well-adapted in cancer cells. TCA cycle intermediates are also utilised as carbon sources, such as production of α-ketoglutarate (α-KG) from glutamine by glutaminolysis, oxaloacetate production from pyruvate by pyruvate carboxylation, oxidation of branched chain amino acids, etc., to the anaplerotic activity of cancer cells (16–18).
Perturbation of cell signalling pathways, such as K-Ras, PI3K–Akt–mTROC1, Myc signalling, play significant roles in mitochondrial metabolism of cancer cells. K-Ras oncogene decouples glucose and glutamine metabolism. It exhibits enhanced glycolytic activity as well as increased usage of glutamine as a carbon source of TCA cycle (19). PI3K–Akt signalling pathway is found to be altered very frequently in different cancers. mTORC1, being one of the major targets of activated Akt, regulates growth factor signalling, energy state, and nutrient and oxygen availability in cancer cells (20–23). Oncogenic activation of Myc leads to activation of genes involved in glycolysis, glutamine metabolism, and mitochondrial biogenesis (24–27).
Mitochondrial TCA cycle enzymes, such as succinate dehydrogenase (SDH) and fumarate hydratase (FH), could function as mitochondrial tumour suppressors (28). Individuals with germline loss-of-function mutations in FH gene are predisposed to hereditary paraganglioma, pheochromocytoma, leiomyomatosis, and renal carcinoma (29, 30). Mutation-induced inactivation of SDH and FH results in accumulation of succinate and fumarate, respectively. These two metabolites leak out to cytosol to inhibit prolyl hydroxylase enzymes, which can promote cellular resistance to apoptotic signals or can activate pseudohypoxic response for triggering HIF-1-mediated glycolysis in cancer (31, 32). Elevated succinate and fumarate can also lead to consequent alteration of genome-wide histone and DNA methylation by inhibiting α-KG dependent dioxygenase (such as histone demethylases and TET family of 5-methylcytosine hydroxylases) activity (30, 33).
Bioenergetics and Altered Metabolism
Warburg’s hypothesis (1956) suggested cancer cells exhibit increased glycolysis and lactate production irrespective of the presence of oxygen (34). Loss of tumour suppressors, activation of oncogenes, upregulation of PI3K pathway, and altered expression of mitochondrial metabolic enzymes may also result in increased glycolysis (35, 36). Rate of glycolysis is increased in cancer cells but glycolytic products may enter mitochondria at different points of TCA cycle for energy production and growth, migration and metastasis of cancer cells (37). On the other hand, cancer cells with mitochondrial respiration defects rely on energy generated by glycolysis. Inhibiting glycolysis in these cancers results in depletion in ATP production and finally leads to apoptosis of the cells (35, 38). But in general, mitochondrial metabolism is necessary for cancer cell survival, proliferation, and growth. So, cancer cells adopt multiple mechanisms to maintain proper functioning of mitochondria. Moreover, in hypoxia and nutrient deficient conditions, mitochondria can modify its energy production as well as utilisation to adapt to the tumour microenvironment since mitochondrial ETC can produce ATP even at very low oxygen level (39, 40). In these conditions, cancer cells can adapt different mechanisms to maintain their ATP/ADP ratio and decrease their demand for ATP as well as cellular functions which are ATP-dependent. In lower energy state (higher ADP/ATP or AMP/ATP ratio), activation of AMP kinase (AMPK) is triggered by mitochondrial adenylate kinase (AK4) and activates catabolic pathways (such as fatty acid oxidation) to stimulate ATP production (41, 42).
Biosynthesis of Metabolites
Rapid cell division and growth of cancer cells require good supply of macromolecules. Various anabolic pathways utilise simpler and smaller nutrient molecules such as glucose, fatty acids, and amino acids to produce larger molecules or building blocks for the cells. Mitochondria which work as a central organelle of cellular metabolic pathways perform different anabolic reactions to generate intermediate products for macromolecules.
Biosynthetic pathways of fatty acids and amino acids were found to be upregulated in cancer cells indicating their importance as metabolites (43, 44). Except pyruvate from glycolysis, other metabolic substrates from amino acid and fatty acid metabolism are also transported to mitochondria for further metabolic activities. When pyruvate is mostly being used for lactate production, the metabolites from fatty acid and amino acid metabolism pathways play central role for providing metabolic substrates to mitochondria. In hypoxic condition, when acetyl CoA production from pyruvate is impaired, glutamine acts as a biosynthetic precursor of acetyl CoA for sustainability of the tumour (40).
Glucose
Glucose is the most widely used nutrient in the body. Uptake of fluorodeoxyglucose (Fl-18), a glucose analogue, measured by positron emission tomography in cancer cells, was found to be increased as the cancer progresses (45, 46). Recent evidences indicate, although lactate production is induced in cancer cells, glucose also produces pyruvate which enters into mitochondrial TCA cycle. Pyruvate is metabolized into acetyl CoA by mitochondrial PDH complex and it is then converted to citrate by citrate synthase. Citrate is either converted to isocitrate within TCA cycle or is transported to the cytosol to yield cytosolic acetyl CoA which is used as a substrate for lipogenesis and acetylation.
The final and rate limiting factor of glycolytic pathway is the M2 isoform of pyruvate kinase (PKM2). PKM2 dimer has low kinase activity and drives pyruvate to lactate formation, whereas PKM2 tetramer which has higher kinase activity promotes the pyruvate to enter mitochondria for ATP production via oxidative phosphorylation pathway (47). PKM2 is commonly found to be highly expressed in cancers and induces rate of glycolysis, cell proliferation, migration, and invasion (48–50). It functions as a coactivator of HIF-1 by enhancing the Warburg effect in cancer. PKM2 can also regulate cancer progression by activating mTOR or EGFR signalling pathway (47, 51, 52).
Amino Acids
Besides glucose, amino acids are also important substrates in mitochondrial biosynthesis of lipid and protein molecules. In absence of glucose, glutamine acts as major nutrient in cancer cells. Importance of glutamine in cellular metabolism is due to its ability to donate its carbon and nitrogen into different growth promoting pathways. Although, glutamine is a non-essential amino acid, during rapid growth and cell proliferation in tumour, it is required to be imported from outside source to meet the high demands of it. Glutamine catabolism occurs inside mitochondria by glutaminase to produce glutamate and ammonia. Glutamate functions as a precursor of cellular antioxidant glutathione and donates amino groups to synthesise non-essential amino acids like glycine, alanine, aspartate, and serine. Glutamate is also converted to α-KG, enters mitochondrial TCA cycle to provide carbon skeleton for macromolecules and contribute in ATP synthesis by oxidative phosphorylation (53). Requirement of glutamine can be variable in different tumours. In some tumours, glutamine plays as an essential amino acid, whereas some tumours seem to be independent of glutamine and utilise glucose-derived pyruvate as major source of nutrient (54).
Glycine and serine are two other amino acids which have been identified to have significant importance in cancer metabolism. They are inter-convertible and biosynthesis of both the amino acids comprises cytosolic as well as mitochondrial enzymatic pathways. Hyper-activation of serine and glycine biosynthesis can accelerate tumourigenesis (55). Activation of mitochondrial, but not cytosolic, enzymes for glycine biosynthesis, which is known to be significantly correlated with cancer cell proliferation, implicates importance of mitochondria in cell proliferation in cancer (56). Moreover, glycine and succinyl CoA might condense to form 5-aminolevulinate which is key precursor for haem biosynthesis in mitochondria (57). Iron modulates expression of four enzymes of TCA cycle, aconitase, citrate synthase, isocitrate dehydrogenase, and SDH. It also reduces glucose utilisation by increasing oxygen consumption and ATP synthesis in mitochondria. When iron is depleted, glycolysis and lactate formation are significantly increased to compensate for ATP production in mitochondria (58). Increased supply and generation of haem induces oxygen consumption and energy production in mitochondria for progression of cancer cells, such as lung cancer (59).
Mutations in isocitrate dehydrogenase enzymes IDH1 (cytosolic) and IDH2 (mitochondrial) are highly frequent in glioma and AML, though rare in other cancers. Recurrent mutations in Arg132 in IDH1 and Arg140 and Arg172 in IDH2 gene comprise 90% of the total mutations (60–63). Mutant IDH1 or IDH2 produces 2-hydroxyglutarate (2-HG), instead of α-KG (64, 65), which functions as an oncometabolite. Accumulation of 2-HG in cells results in epigenetic dysregulation followed by aberrant gene expression (62). IDH1 and IDH2 mutations are also responsible for hypermethylation, decreased differentiation, and increased stemness of cancer cells as well as HIF-1α-mediated angiogenesis and growth of tumour (66–68).
Fatty Acids
Lipogenesis is a common feature of different cancers. Although normal cells mostly depend on exogenous sources of lipid, cancer cells can execute de novo synthesis of fatty acids. It is one of the major components of cellular lipid which is needed for cell membrane synthesis, energy production, lipid modification of proteins, and signalling molecule production in highly proliferative cancer cells. In most of the cancers, cause of increased lipogenesis is due to overproduction of enzyme, fatty acid synthase (FASN). It performs the final catalytic step to convert acetyl CoA and malonyl CoA to fatty acids and functions as an oncogene by promoting cancer cell proliferation and growth. Suppression of FASN results in cell cycle arrest, reduction in cancer cell proliferation, and increase in apoptosis (69–74). Another enzyme monoacylglycerol lipase that controls monoacylglycerol levels in normal cells is highly expressed in aggressive cancers and regulates a fatty acid network. This network has lipids with oncogenic potential and increases pathogenicity by promoting invasion, survival, and growth of the tumour (44). Alternatively, low glucose content or food deprivation has been found to increase fatty acid oxidation in cancer (75). Fatty acids are considered major source of energy in healthy cells as they can provide twice ATP than carbohydrates. Fatty acid oxidation (also known as β-oxidation) promotes cell proliferation and inhibits apoptosis in tumour cells (76, 77). Other than ATP production, β-oxidation-derived NADPH is the key mediator of oxidative stress as well as the coenzyme for anabolic reactions (78). PPAR genes play essential roles in β-oxidation by modulating enzymes involved in the pathway (79). PPAR-gamma coactivator-1α (PGC-1α) is activated by low glucose content and activates SIRT1. Both SIRT1 and Sirtuin-3 (SIRT3) activate metabolic enzymes for β-oxidation of fatty acids into acetyl CoA (80, 81) in mitochondria. Acetyl-CoA carboxylase (ACC) converts acetyl CoA to malonyl CoA in fatty acid synthetic pathway and is suppressed by AMPK. Hormones, such as leptin and adiponectin, activate AMPK and suppress ACC to inhibit fatty acid synthesis and increase fatty acid oxidation (82, 83).
Acetate
Recent studies on cancer metabolism have identified that certain tumours use exogenous acetate as an alternative source for producing acetyl CoA (84–86). Even in a glucose abundant scenario, glucose and acetate are simultaneously used to produce TCA cycle intermediates. Tumours, especially those with hypoxic conditions can produce 50% of their acetyl CoA from acetate, while the remaining amount is produced by utilising glucose and glutamine as carbon sources (87). Disease progression is correlated to the activity of the nucleo-cytosolic acetyl-CoA synthetase (ACSS2) (88). ACSS2 uses acetate as a carbon source to generate acetyl CoA. Tumours which utilise acetate as a carbon source also show elevated activity of ACSS2. Mutations causing PTEN and BRAF inactivation driving AKT and ERK pathways have been found to play role in increased expression of ACSS2. Tumours devoid of this enzyme consume less amount of acetate, undergo cell death, and subsequently reach to a reduced tumour size (86).
Mevalonate–Isoprenoid Pathway
Mevalonate–isoprenoid pathway is used for cellular cholesterol biosynthesis and protein prenylation and hyper-activated in many cancers. It activates proteins of RAS signalling pathway by prenylation leading to cell transformation and malignancy (89, 90). Activation of this pathway shows chemoresistance in cancer cell line (91) and also induces cancer metastasis by epithelial mesenchymal transition, remodelling of cytoskeleton, cell motility and cell polarity (non-canonical Wnt/planar pathway) (92). Multiple factors can regulate mevalonate pathway. Mutant form of tumour suppressor protein P53 induces protein prenylation of mevalonate pathway, thus maintaining malignancy, three-dimensional growth of tumour, and invasive growth (90, 93, 94). Activation of ARID1A (a subunit of SWI/SNF chromatin remodelling complex) and IGF-1R/AKT/mTOR axis result in increased activity of mevalonate pathway in ovarian and colorectal cancer, respectively (95, 96). Kallikrein-related peptidase 5 (KLK5) protease can inhibit enzymes of mevalonate pathway in breast cancer (97). Drugs like statin, quinazoline, and simvastatin show anticancer effects by inhibiting mevalonate pathway, which indicates possibility of building new therapeutic strategies targetting this pathway (92, 98–103).
Mitophagy
Mitophagy or selective degradation of damaged mitochondria is a quality control mechanism for maintaining homoeostasis of functional mitochondria in cell (104). Key regulation of mitophagy is executed through PINK1–Parkin pathway. Under stress conditions like hypoxia and nutrient deficiency, Parkin (E3 ubiquitin ligase) is recruited by PINK1 for proteasomal degradation of target proteins (VDAC1, mitofusin 1, mitofusin 2) at mitochondrial outer membrane (105–108). Loss-of-function mutation, copy number variation, or deletion of PARK2 (encoding Parkin protein) resulted in retaining damaged mitochondria in different cancers and, thus, indicates tumour suppressing role of mitophagy. In solid tumours, loss of Perkin induces aerobic glycolysis, thus supporting Warburg’s hypothesis (109).
Redox Signalling
Reactive oxygen species are, mostly, by-products of electron transporting systems in mitochondria. Although ROS have some important roles in transcriptional activation, cell proliferation, and other signalling pathways but excess amount of ROS is responsible for damaging cellular DNA, lipids, and proteins (110). Antioxidant systems of the cell provide protection against excess amount of ROS. Excess redox signalling leads to carcinogenesis, tumour development and progression, cell migration, and angiogenesis. An increased quantity of ROS can activate hypoxia-mediated HIF-1α signalling pathway which might cause metabolic shift from oxidative phosphorylation to glycolysis by increasing expression of glycolytic enzymes. HIF-1α also reduces expression of tumour suppressor SIRT3 which functions in activating antioxidants in mitochondria of healthy cell. Loss of SIRT3 is reported in many cancers and it results in continuous steady state level of ROS and oxidative stress. Mitochondrial antioxidant systems which regulate ROS level are manganese superoxide dismutase (MnSOD or SOD2), mitochondrial glutaredoxin, glutathione peroxidise, and thioredoxin 2 (111–114). Antioxidants have dual role in redox signalling pathway. In physiological ROS signalling, they function as tumour suppressor by inhibiting ROS-induced cell proliferation and survival needed for cancer progression. By contrast, excess ROS in tumour microenvironment promotes apoptotic signals and, then, antioxidants suppress those signals and act as tumour promoters. But, generally, these antioxidant systems protect cells against oxidative stress and ROS-induced cell death.
Mitochondria Biogenesis
Biogenesis of mitochondria is required in cells with high energy demands. DNA double strand breaks, induced by anticancer drugs, activate ataxia telangiectasia mutated to activate α subunit of AMPK to increase mitochondrial biogenesis (115). Contradicting Warburg’s hypothesis, recent studies have proposed a two-compartmental metabolic system in cancer (116, 117). Cancer cell and surrounding stromal cells undergo metabolically symbiotic relationship, where the cancer cells have active mitochondria and increased mitochondrial biogenesis but the stromal cells contain dysfunctional mitochondria and take up glycolytic pathway (117). Ketone bodies are also major energy sources for mitochondria and they are synthesized in tumour stroma with the help of enzymes, such as HMGCS2, HMGCL, BDH1, and re-utilised in tumour cells (116). Severe oxidative stress leads to apoptotic cell death, whereas mild oxidative stress can increase mitochondrial biogenesis as well as mtDNA content in cancer cells. Key regulator of mitochondrial biogenesis is PGC-1α, which regulates expression of nuclear genes for respiratory chain function, transcription, and replication of mtDNA by activating transcription factors (NRF-1 and NRF-2), tumour suppressor genes (SIRT3), nuclear coded mitochondrial enzymes (POLRMT), and transcription factor (mtTFA) (25, 118–121). Activated PGC-1α in invasive cancer cells increases oxidative phosphorylation, oxygen consumption, and mitochondrial biogenesis and finally cell’s potential for distant metastasis. Activation of PGC-1α for mitochondrial biogenesis can occur through different signalling pathways (such as AMPK, NO–cGMP, cAMP–PKA–CREB, p38, and ERRα pathways) (122–124). Alternatively, HIF-1 negatively regulates mitochondrial biogenesis and oxygen consumption by inhibiting C-MYC via MXI-1 dependent and MXI-1 independent pathways in renal carcinoma (14). Translation of nuclear-encoded mitochondrial function-related genes, protein folding, and entry in mitochondrial sub-compartments are regulated by mTOR which is often found to be hyper-activated in cancer (125–128). Suppression of mTOR results in damage and loss of mitochondria in cancer (129). MYC, which has known oncogenic effects in various cancers, induces nuclear-encoded mitochondrial gene expression and mitochondrial biogenesis in cancer (14, 25, 130). Sustained expression of MYC can lead to increased production of ROS and subsequent genomic instability and mitochondrial dysfunction (131, 132).
Mitochondrial Fission and Fusion
Fission and fusion, which are the key components in mitochondrial dynamics, modulate mitochondrial morphology and subsequently regulate essential cellular mechanisms such as cell growth, cell division, and distribution of mitochondria during differentiation (133–135). Imbalance in expression of fission controlling protein dynamin-related protein 1 (Drp1) and fusion controlling protein Mfn1 (mitofusin 1) is observed in different cancers. Increased fission or mitochondrial fragmentation due to high expression and activity of Drp1 and decreased fusion due to loss of Mfn1 activity are often linked to cancer cell migration, invasiveness, and metastasis (136–140). Pathways mediated by p53, PINK1, and mitochondrial membrane proteins are also found to be involved in regulation of mitochondrial fission as well as chemosensitivity of cancer cells (141–144). Other than proteins, mitochondrial lipids (cardiolipin, phosphatidylethanolamine, phosphatidic acid, diacylglycerol) also play important role in controlling mitochondrial dynamics (140).
Calcium Homoeostasis
Calcium ion concentration is a key regulator of various signalling pathways of the cytosol and cellular organelles. Under physiological conditions, Ca2+ plays a beneficiary role by producing higher glycolytic and mitochondrial pathway enzymes (such as PDH, isocitrate dehydrogenase, α-KG dehydrogenase, ATP synthase, and α-glycerophosphate dehydrogenase), increasing oxidative phosphorylation activity and activating metabolite carriers (aspartate/glutamate carrier) of mitochondria. On the contrary, higher concentration of Ca2+ within mitochondria induces several negative effects on mitochondrial function which finally leads to apoptosis (145, 146). Mitochondrial stress (such as mtDNA depletion) in cancer cells results in increased cytosolic Ca2+, activation of calcium dependent MAPK (ERK1 and ERK2) and calcineurin, increased anti-apoptotic proteins, and loss of pro-apoptotic proteins (147). Mitochondria-associated membrane (MAM) structure which is the interacting interface between ER and mitochondrial outer membrane, functions as the gateway of Ca2+ release from ER to mitochondria. Oncoproteins and tumour suppressor proteins residing on MAM control apoptosis via Ca2+ homoeostasis. Ca2+ release from ER as well as uptake by mitochondria are inhibited by several oncogenes like AKT, Bcl2, and K-Ras to trigger anti-apoptotic signalling in cancer cells (148–150). Functional loss of ER protein PERK and mitochondrial calcium channel (MCU) are also known to have anti-apoptotic effects in cancer (151–153).
Cell Death
Cell death is a physiological regulator for development, tissue homoeostasis, stress, and also functions as tumour suppressor. Besides apoptosis, mitochondria are also found to be involved in other cell death mechanisms such as autophagy, necrosis, and necroptosis (programmed necrosis) (154–156). Proteins known as inhibitors of apoptosis (IAPs) are overexpressed in cancer and inhibit caspases or procaspases (primarily caspase-3 and caspase-7) to suppress apoptosis. Cancer cells with activated IAPs become highly resistant to radiation or chemotherapy (157). Anti-apoptotic proteins of BCL2 family are overexpressed in cancer and inhibit the pro-apoptotic proteins to initiate the process of cell death. Thus, BCL2 proteins are targetted by BCL2-inhibitors in cancer therapy to promote apoptosis (158, 159). Tumour suppressor, P53, plays an important role in promoting cell death. It is activated in ROS-dependent pathway and inhibits oncogenes via JNK-mediated signalling pathway leading to apoptosis in cancer (160, 161). Inhibited cell growth and increased apoptosis in cancer by P53 activation are also regulated by miRNA or SIRT2 dependent pathways (162, 163).
Mitochondrial fission- and fusion-related proteins, Drp1 and mitofusin (Mfn1 and Mfn2), are found to be involved in cell death (164). Drp1 induces mitochondrial fragmentation and apoptosis in a BAX/BAK-mediated pathway. Overexpression of Drp1 increases ROS production, release of cytochrome c, and PARP cleavage (165, 166). Being phosphorylated by ERK, Mfn1 modulates apoptosis and fusion. Mutant Mfn1 binds to BAK more strongly inducing BAK activation and cell death (167). Mfn2 promotes anti-proliferative and pro-apoptotic effects via PI3K–AKT signalling pathway and lower expression of Mfn2 is associated with poor survival in cancer (168, 169).
Mutations in Mitochondrial Genome
Somatic mutations in mitochondrial genome (mtDNA) are common and frequently reported in different types of cancer (170–177). Functional consequences of these mutations are not well understood. These mutations are mostly point mutations, small insertion–deletions, or large scale deletions distributed in protein coding genes (177, 178). These mutations are thought to arise due to poor DNA repair mechanism and direct exposure to ROS, although oxidative stress is not always considered as a major contributor to somatic mutations (179). Mutations in coding genes might cause functional imbalance in respiratory chain. Mutant respiratory chain proteins promote elevation of ROS, tumour size, and glycolysis via HIF-1-mediated pathway in head and neck and prostate cancer (180, 181).
Conclusion
Mitochondria are essential organelles for energy production but play important roles in carcinogenesis, cancer progression, and metastasis helping altered energy metabolism in cancer cells. Mitochondrial metabolism is also connected with other mitochondrial pathways such as redox signalling, Ca2+ signalling, mitophagy, and mitochondrial biogenesis. These pathways cross talk and seem to play important roles in cancer. Targeting mitochondrial pathways individually or in combination might be considered as future cancer therapy. Recently, cancer researchers are focussing on the metabolic reprogramming of cancer cells to use altered metabolites/oncometabolites for therapeutic approach.
Author Contributions
EC wrote the draft of the manuscript and BR revised and finished it.
Conflict of Interest Statement
Authors declare that the manuscript was written in absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
Acknowledgments
Indian Statistical Institute provided fellowship to EC.
Funding
No funding was needed for the manuscript.
Abbreviations
mtDNA, mitochondrial DNA; ETC, electron transport chain; ROS, reactive oxygen species.
References
1. Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet (2005) 6(5):389–402. doi:10.1038/nrg1606
2. Falk MJ, Sondheimer N. Mitochondrial genetic diseases. Curr Opin Pediatr (2010) 22(6):711–6. doi:10.1097/MOP.0b013e3283402e21
3. Holmuhamedov E, Lewis L, Bienengraeber M, Holmuhamedova M, Jahangir A, Terzic A. Suppression of human tumor cell proliferation through mitochondrial targeting. FASEB J (2002) 16(9):1010–6. doi:10.1096/fj.01-0996com
4. Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature (2011) 481(7381):385–8. doi:10.1038/nature10642
5. Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab (2015) 21(1):81–94. doi:10.1016/j.cmet.2014.12.003
6. Ruan K, Song G, Ouyang G. Role of hypoxia in the hallmarks of human cancer. J Cell Biochem (2009) 107(6):1053–62. doi:10.1002/jcb.22214
7. Brahimi-Horn MC, Chiche J, Pouyssegur J. Hypoxia and cancer. J Mol Med (Berl) (2007) 85(12):1301–7. doi:10.1007/s00109-007-0281-3
8. Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med (2010) 14(4):771–94. doi:10.1111/j.1582-4934.2009.00994.x
9. Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev (2010) 20(1):51–6. doi:10.1016/j.gde.2009.10.009
10. Greijer AE, van der Wall E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol (2004) 57(10):1009–14. doi:10.1136/jcp.2003.015032
11. Mole DR, Blancher C, Copley RR, Pollard PJ, Gleadle JM, Ragoussis J, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem (2009) 284(25):16767–75. doi:10.1074/jbc.M901790200
12. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab (2006) 3(3):177–85. doi:10.1016/j.cmet.2006.02.002
13. Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab (2006) 3(3):187–97. doi:10.1016/j.cmet.2006.01.012
14. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell (2007) 11(5):407–20. doi:10.1016/j.ccr.2007.04.001
15. Klimova T, Chandel NS. Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ (2008) 15(4):660–6. doi:10.1038/sj.cdd.4402307
16. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab (2008) 7(1):11–20. doi:10.1016/j.cmet.2007.10.002
17. Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature (2011) 476(7360):346–50. doi:10.1038/nature10350
18. Sellers K, Fox MP, Bousamra M II, Slone SP, Higashi RM, Miller DM, et al. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest (2015) 125(2):687–98. doi:10.1172/JCI72873
19. Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol (2011) 7:523. doi:10.1038/msb.2011.56
20. Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis (2004) 9(6):667–76. doi:10.1023/B:APPT.0000045801.15585.dd
21. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov (2005) 4(12):988–1004. doi:10.1038/nrd1902
22. Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol (2006) 18(1):77–82. doi:10.1097/01.cco.0000198021.99347.b9
23. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer (2009) 9(8):550–62. doi:10.1038/nrc2664
24. Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem (2000) 275(29):21797–800. doi:10.1074/jbc.C000023200
25. Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, et al. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol Cell Biol (2005) 25(14):6225–34. doi:10.1128/MCB.25.14.6225-6234.2005
26. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature (2009) 458(7239):762–5. doi:10.1038/nature07823
28. Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer (2005) 5(11):857–66. doi:10.1038/nrc1737
29. Kuwada M, Chihara Y, Lou Y, Torimoto K, Kagebayashi Y, Tamura K, et al. Novel missense mutation in the FH gene in familial renal cell cancer patients lacking cutaneous leiomyomas. BMC Res Notes (2014) 7:203. doi:10.1186/1756-0500-7-203
30. Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget (2015) 6(36):38777–88. doi:10.18632/oncotarget.6091
31. Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell (2005) 8(2):143–53. doi:10.1016/j.ccr.2005.06.017
32. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene (2006) 25(34):4675–82. doi:10.1038/sj.onc.1209594
33. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev (2012) 26(12):1326–38. doi:10.1101/gad.191056.112
34. Warburg O. On the origin of cancer cells. Science (1956) 123(3191):309–14. doi:10.1126/science.123.3191.309
35. Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene (2006) 25(34):4633–46. doi:10.1038/sj.onc.1209597
36. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science (2010) 330(6009):1340–4. doi:10.1126/science.1193494
37. Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle (2010) 9(17):3506–14. doi:10.4161/cc.9.17.12731
38. Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res (2005) 65(2):613–21.
39. Chandel NS, Budinger GR, Choe SH, Schumacker PT. Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J Biol Chem (1997) 272(30):18808–16. doi:10.1074/jbc.272.30.18808
40. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv (2016) 2(5):e1600200. doi:10.1126/sciadv.1600200
41. Lanning NJ, Looyenga BD, Kauffman AL, Niemi NM, Sudderth J, DeBerardinis RJ, et al. A mitochondrial RNAi screen defines cellular bioenergetic determinants and identifies an adenylate kinase as a key regulator of ATP levels. Cell Rep (2014) 7(3):907–17. doi:10.1016/j.celrep.2014.03.065
42. Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol (2016) 26(3):190–201. doi:10.1016/j.tcb.2015.10.013
43. Denkert C, Budczies J, Weichert W, Wohlgemuth G, Scholz M, Kind T, et al. Metabolite profiling of human colon carcinoma – deregulation of TCA cycle and amino acid turnover. Mol Cancer (2008) 7:72. doi:10.1186/1476-4598-7-72
44. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell (2010) 140(1):49–61. doi:10.1016/j.cell.2009.11.027
45. Som P, Atkins HL, Bandoypadhyay D, Fowler JS, MacGregor RR, Matsui K, et al. A fluorinated glucose analog, 2-fluoro-2-deoxy-d-glucose (F-18): nontoxic tracer for rapid tumor detection. J Nucl Med (1980) 21(7):670–5.
46. Duhaylongsod FG, Lowe VJ, Patz EF Jr, Vaughn AL, Coleman RE, Wolfe WG. Lung tumor growth correlates with glucose metabolism measured by fluoride-18 fluorodeoxyglucose positron emission tomography. Ann Thorac Surg (1995) 60(5):1348–52. doi:10.1016/0003-4975(95)00754-9
47. Wong N, De Melo J, Tang D. PKM2, a central point of regulation in cancer metabolism. Int J Cell Biol (2013) 2013:242513. doi:10.1155/2013/242513
48. Luo W, Semenza GL. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab (2012) 23(11):560–6. doi:10.1016/j.tem.2012.06.010
49. Keller KE, Doctor ZM, Dwyer ZW, Lee YS. SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol Cell (2014) 53(5):700–9. doi:10.1016/j.molcel.2014.02.015
50. Tang R, Yang C, Ma X, Wang Y, Luo D, Huang C, et al. miR-let-7a inhibits cell proliferation, migration, and invasion by down-regulating PKM2 in gastric cancer. Oncotarget (2016) 7(5):5972–84. doi:10.18632/oncotarget.6821
51. Hsu MC, Hung WC, Yamaguchi H, Lim SO, Liao HW, Tsai CH, et al. Extracellular PKM2 induces cancer proliferation by activating the EGFR signaling pathway. Am J Cancer Res (2016) 6(3):628–38.
52. Li N, Feng L, Liu H, Wang J, Kasembeli M, Tran MK, et al. PARP inhibition suppresses growth of EGFR-mutant cancers by targeting nuclear PKM2. Cell Rep (2016) 15(4):843–56. doi:10.1016/j.celrep.2016.03.070
53. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest (2013) 123(9):3678–84. doi:10.1172/JCI69600
54. Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, et al. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A (2011) 108(21):8674–9. doi:10.1073/pnas.1016627108
55. Amelio I, Cutruzzola F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci (2014) 39(4):191–8. doi:10.1016/j.tibs.2014.02.004
56. Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, et al. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science (2012) 336(6084):1040–4. doi:10.1126/science.1218595
57. Ferreira GC, Gong J. 5-Aminolevulinate synthase and the first step of heme biosynthesis. J Bioenerg Biomembr (1995) 27(2):151–9. doi:10.1007/BF02110030
58. Oexle H, Gnaiger E, Weiss G. Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta (1999) 1413(3):99–107. doi:10.1016/S0005-2728(99)00088-2
59. Hooda J, Cadinu D, Alam MM, Shah A, Cao TM, Sullivan LA, et al. Enhanced heme function and mitochondrial respiration promote the progression of lung cancer cells. PLoS One (2013) 8(5):e63402. doi:10.1371/journal.pone.0063402
60. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (2008) 321(5897):1807–12. doi:10.1126/science.1164382
61. Dang L, Jin S, Su SM. IDH mutations in glioma and acute myeloid leukemia. Trends Mol Med (2010) 16(9):387–97. doi:10.1016/j.molmed.2010.07.002
62. Prensner JR, Chinnaiyan AM. Metabolism unhinged: IDH mutations in cancer. Nat Med (2011) 17(3):291–3. doi:10.1038/nm0311-291
63. Ghiam AF, Cairns RA, Thoms J, Dal Pra A, Ahmed O, Meng A, et al. IDH mutation status in prostate cancer. Oncogene (2012) 31(33):3826. doi:10.1038/onc.2011.546
64. Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med (2010) 207(2):339–44. doi:10.1084/jem.20092506
65. Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell (2010) 17(3):225–34. doi:10.1016/j.ccr.2010.01.020
66. Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science (2009) 324(5924):261–5. doi:10.1126/science.1170944
67. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18(6):553–67. doi:10.1016/j.ccr.2010.11.015
68. Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science (2013) 340(6132):626–30. doi:10.1126/science.1236062
69. Pizer ES, Wood FD, Heine HS, Romantsev FE, Pasternack GR, Kuhajda FP. Inhibition of fatty acid synthesis delays disease progression in a xenograft model of ovarian cancer. Cancer Res (1996) 56(6):1189–93.
70. Baron A, Migita T, Tang D, Loda M. Fatty acid synthase: a metabolic oncogene in prostate cancer? J Cell Biochem (2004) 91(1):47–53. doi:10.1002/jcb.10708
71. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer (2007) 7(10):763–77. doi:10.1038/nrc2222
72. Mashima T, Seimiya H, Tsuruo T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer (2009) 100(9):1369–72. doi:10.1038/sj.bjc.6605007
73. Migita T, Ruiz S, Fornari A, Fiorentino M, Priolo C, Zadra G, et al. Fatty acid synthase: a metabolic enzyme and candidate oncogene in prostate cancer. J Natl Cancer Inst (2009) 101(7):519–32. doi:10.1093/jnci/djp030
74. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab (2013) 18(2):153–61. doi:10.1016/j.cmet.2013.05.017
75. Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis (2006) 9(3):230–4. doi:10.1038/sj.pcan.4500879
76. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer (2013) 13(4):227–32. doi:10.1038/nrc3483
77. Harris FT, Rahman SM, Hassanein M, Qian J, Hoeksema MD, Chen H, et al. Acyl-coenzyme A-binding protein regulates beta-oxidation required for growth and survival of non-small cell lung cancer. Cancer Prev Res (Phila) (2014) 7(7):748–57. doi:10.1158/1940-6207.CAPR-14-0057
78. Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome – a key determinant of cancer cell biology. Nat Rev Cancer (2012) 12(11):741–52. doi:10.1038/nrc3340
79. Fan W, Evans R. PPARs and ERRs: molecular mediators of mitochondrial metabolism. Curr Opin Cell Biol (2015) 33:49–54. doi:10.1016/j.ceb.2014.11.002
80. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J (2007) 26(7):1913–23. doi:10.1038/sj.emboj.7601633
81. Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature (2010) 464(7285):121–5. doi:10.1038/nature08778
82. Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature (2002) 415(6869):339–43. doi:10.1038/415339a
83. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med (2002) 8(11):1288–95. doi:10.1038/nm788
84. Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, et al. Acetate dependence of tumors. Cell (2014) 159(7):1591–602. doi:10.1016/j.cell.2014.11.020
85. Lyssiotis CA, Cantley LC. Acetate fuels the cancer engine. Cell (2014) 159(7):1492–4. doi:10.1016/j.cell.2014.12.009
86. Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell (2014) 159(7):1603–14. doi:10.1016/j.cell.2014.11.025
87. Kamphorst JJ, Chung MK, Fan J, Rabinowitz JD. Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab (2014) 2:23. doi:10.1186/2049-3002-2-23140
88. Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, et al. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell (2015) 27(1):57–71. doi:10.1016/j.ccell.2014.12.002
89. Thurnher M, Gruenbacher G, Nussbaumer O. Regulation of mevalonate metabolism in cancer and immune cells. Biochim Biophys Acta (2013) 1831(6):1009–15. doi:10.1016/j.bbalip.2013.03.003
90. Gruenbacher G, Thurnher M. Mevalonate metabolism in cancer. Cancer Lett (2015) 356(2 Pt A):192–6. doi:10.1016/j.canlet.2014.01.013
91. Greife A, Tukova J, Steinhoff C, Scott SD, Schulz WA, Hatina J. Establishment and characterization of a bladder cancer cell line with enhanced doxorubicin resistance by mevalonate pathway activation. Tumour Biol (2015) 36(5):3293–300. doi:10.1007/s13277-014-2959-9
92. Likus W, Siemianowicz K, Bienk K, Pakula M, Pathak H, Dutta C, et al. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist Updat (2016) 25:13–25. doi:10.1016/j.drup.2016.02.001
93. Freed-Pastor W, Prives C. Targeting mutant p53 through the mevalonate pathway. Nat Cell Biol (2016) 18(11):1122–4. doi:10.1038/ncb3435
94. Greenaway JB, Virtanen C, Osz K, Revay T, Hardy D, Shepherd T, et al. Ovarian tumour growth is characterized by mevalonate pathway gene signature in an orthotopic, syngeneic model of epithelial ovarian cancer. Oncotarget (2016) 7(30):47343–65. doi:10.18632/oncotarget.10121
95. Sharon C, Baranwal S, Patel NJ, Rodriguez-Agudo D, Pandak WM, Majumdar AP, et al. Inhibition of insulin-like growth factor receptor/AKT/mammalian target of rapamycin axis targets colorectal cancer stem cells by attenuating mevalonate-isoprenoid pathway in vitro and in vivo. Oncotarget (2015) 6(17):15332–47. doi:10.18632/oncotarget.3684
96. Goldman AR, Bitler BG, Schug Z, Conejo-Garcia JR, Zhang R, Speicher DW. The primary effect on the proteome of ARID1A-mutated ovarian clear cell carcinoma is downregulation of the mevalonate pathway at the post-transcriptional level. Mol Cell Proteomics (2016) 15(11):3348–60. doi:10.1074/mcp.M116.062539
97. Pampalakis G, Obasuyi O, Papadodima O, Chatziioannou A, Zoumpourlis V, Sotiropoulou G. The KLK5 protease suppresses breast cancer by repressing the mevalonate pathway. Oncotarget (2014) 5(9):2390–403. doi:10.18632/oncotarget.1235
98. Jiang P, Mukthavaram R, Chao Y, Nomura N, Bharati IS, Fogal V, et al. In vitro and in vivo anticancer effects of mevalonate pathway modulation on human cancer cells. Br J Cancer (2014) 111(8):1562–71. doi:10.1038/bjc.2014.431
99. Warita K, Warita T, Beckwitt CH, Schurdak ME, Vazquez A, Wells A, et al. Statin-induced mevalonate pathway inhibition attenuates the growth of mesenchymal-like cancer cells that lack functional E-cadherin mediated cell cohesion. Sci Rep (2014) 4:7593. doi:10.1038/srep07593
100. Yeganeh B, Wiechec E, Ande SR, Sharma P, Moghadam AR, Post M, et al. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol Ther (2014) 143(1):87–110. doi:10.1016/j.pharmthera.2014.02.007
101. Karlic H, Thaler R, Gerner C, Grunt T, Proestling K, Haider F, et al. Inhibition of the mevalonate pathway affects epigenetic regulation in cancer cells. Cancer Genet (2015) 208(5):241–52. doi:10.1016/j.cancergen.2015.03.008
102. Lin Z, Bishop KS, Sutherland H, Marlow G, Murray P, Denny WA, et al. A quinazoline-based HDAC inhibitor affects gene expression pathways involved in cholesterol biosynthesis and mevalonate in prostate cancer cells. Mol Biosyst (2016) 12(3):839–49. doi:10.1039/c5mb00554j
103. Wang T, Seah S, Loh X, Chan CW, Hartman M, Goh BC, et al. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget (2016) 7(3):2532–44. doi:10.18632/oncotarget.6304
104. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12(1):9–14. doi:10.1038/nrm3028
105. Kim Y, Park J, Kim S, Song S, Kwon SK, Lee SH, et al. PINK1 controls mitochondrial localization of Parkin through direct phosphorylation. Biochem Biophys Res Commun (2008) 377(3):975–80. doi:10.1016/j.bbrc.2008.10.104
106. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol (2010) 12(2):119–31. doi:10.1038/ncb2012
107. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol (2010) 8(1):e1000298. doi:10.1371/journal.pbio.1000298
108. Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature (2014) 510(7503):162–6. doi:10.1038/nature13392
109. Bernardini JP, Lazarou M, Dewson G. Parkin and mitophagy in cancer. Oncogene (2016) 36:1315–27. doi:10.1038/onc.2016.302
110. Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol (2000) 279(6):L1005–28.
111. Esposito LA, Kokoszka JE, Waymire KG, Cottrell B, MacGregor GR, Wallace DC. Mitochondrial oxidative stress in mice lacking the glutathione peroxidase-1 gene. Free Radic Biol Med (2000) 28(5):754–66. doi:10.1016/S0891-5849(00)00161-1
112. Gladyshev VN, Liu A, Novoselov SV, Krysan K, Sun QA, Kryukov VM, et al. Identification and characterization of a new mammalian glutaredoxin (thioltransferase), Grx2. J Biol Chem (2001) 276(32):30374–80. doi:10.1074/jbc.M100020200
113. Patenaude A, Ven Murthy MR, Mirault ME. Mitochondrial thioredoxin system: effects of TrxR2 overexpression on redox balance, cell growth, and apoptosis. J Biol Chem (2004) 279(26):27302–14. doi:10.1074/jbc.M402496200M402496200
114. Li H, Kantoff PW, Giovannucci E, Leitzmann MF, Gaziano JM, Stampfer MJ, et al. Manganese superoxide dismutase polymorphism, prediagnostic antioxidant status, and risk of clinical significant prostate cancer. Cancer Res (2005) 65(6):2498–504. doi:10.1158/0008-5472.CAN-04-3535
115. Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS One (2008) 3(4):e2009. doi:10.1371/journal.pone.0002009
116. Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, Howell A, Lisanti MP, Sotgia F. Ketone bodies and two-compartment tumor metabolism: stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle (2012) 11(21):3956–63. doi:10.4161/cc.22136
117. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, et al. Mitochondria “fuel” breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle (2012) 11(23):4390–401. doi:10.4161/cc.22777
118. Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, et al. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One (2010) 5(7):e11707. doi:10.1371/journal.pone.0011707
119. Lee CH, Wu SB, Hong CH, Liao WT, Wu CY, Chen GS, et al. Aberrant cell proliferation by enhanced mitochondrial biogenesis via mtTFA in arsenical skin cancers. Am J Pathol (2011) 178(5):2066–76. doi:10.1016/j.ajpath.2011.01.056
120. Salem AF, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle (2012) 11(22):4174–80. doi:10.4161/cc.22376
121. LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol (2014) 16(10):992–1003. doi:10.1038/ncb3039
122. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science (2003) 299(5608):896–9. doi:10.1126/science.1079368
123. Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, et al. The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A (2004) 101(17):6472–7. doi:10.1073/pnas.0308686101
124. Favre C, Zhdanov A, Leahy M, Papkovsky D, O’Connor R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene (2010) 29(27):3964–76. doi:10.1038/onc.2010.146
125. Morita M, Gravel SP, Chenard V, Sikstrom K, Zheng L, Alain T, et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab (2013) 18(5):698–711. doi:10.1016/j.cmet.2013.10.001
126. Albert V, Hall MN. mTOR signaling in cellular and organismal energetics. Curr Opin Cell Biol (2015) 33:55–66. doi:10.1016/j.ceb.2014.12.001
127. Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J, et al. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle (2015) 14(4):473–80. doi:10.4161/15384101.2014.991572
128. Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J (2017) 284(2):183–95. doi:10.1111/febs.13820
129. Khan MI, Mohammad A, Patil G, Naqvi SA, Chauhan LK, Ahmad I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials (2012) 33(5):1477–88. doi:10.1016/j.biomaterials.2011.10.080
130. Kim J, Lee JH, Iyer VR. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS One (2008) 3(3):e1798. doi:10.1371/journal.pone.0001798
131. Dang CV, Li F, Lee LA. Could MYC induction of mitochondrial biogenesis be linked to ROS production and genomic instability? Cell Cycle (2005) 4(11):1465–6. doi:10.4161/cc.4.11.2121
132. Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res (2006) 66(18):8927–30. doi:10.1158/0008-5472.CAN-06-1501
133. Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol (2010) 11(12):872–84. doi:10.1038/nrm3013
134. Mitra K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. Bioessays (2013) 35(11):955–64. doi:10.1002/bies.201300011
135. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol (2013) 5(6):a011072. doi:10.1101/cshperspect.a011072
136. Rehman J, Zhang HJ, Toth PT, Zhang Y, Marsboom G, Hong Z, et al. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J (2012) 26(5):2175–86. doi:10.1096/fj.11-196543
137. Zhao J, Zhang J, Yu M, Xie Y, Huang Y, Wolff DW, et al. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene (2013) 32(40):4814–24. doi:10.1038/onc.2012.494
138. Tailor D, Hahm ER, Kale RK, Singh SV, Singh RP. Sodium butyrate induces DRP1-mediated mitochondrial fusion and apoptosis in human colorectal cancer cells. Mitochondrion (2014) 16:55–64. doi:10.1016/j.mito.2013.10.004
139. Ferreira-da-Silva A, Valacca C, Rios E, Populo H, Soares P, Sobrinho-Simoes M, et al. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS One (2015) 10(3):e0122308. doi:10.1371/journal.pone.0122308
140. Frohman MA. Role of mitochondrial lipids in guiding fission and fusion. J Mol Med (Berl) (2015) 93(3):263–9. doi:10.1007/s00109-014-1237-z
141. Kong B, Wang Q, Fung E, Xue K, Tsang BK. p53 is required for cisplatin-induced processing of the mitochondrial fusion protein l-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J Biol Chem (2014) 289(39):27134–45. doi:10.1074/jbc.M114.594812
142. Fan S, Liu B, Sun L, Lv XB, Lin Z, Chen W, et al. Mitochondrial fission determines cisplatin sensitivity in tongue squamous cell carcinoma through the BRCA1-miR-593-5p-MFF axis. Oncotarget (2015) 6(17):14885–904. doi:10.18632/oncotarget.3659
143. O’Flanagan CH, Morais VA, Wurst W, De Strooper B, O’Neill C. The Parkinson’s gene PINK1 regulates cell cycle progression and promotes cancer-associated phenotypes. Oncogene (2015) 34(11):1363–74. doi:10.1038/onc.2014.81
144. Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo X, et al. Increased mitochondrial fission promotes autophagy and hepatocellular carcinoma cell survival through the ROS-modulated coordinated regulation of the NFKB and TP53 pathways. Autophagy (2016) 12(6):999–1014. doi:10.1080/15548627.2016.1166318
145. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol (2004) 287(4):C817–33. doi:10.1152/ajpcell.00139.2004
146. Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer (2007) 7(7):519–30. doi:10.1038/nrc2171
147. Amuthan G, Biswas G, Ananadatheerthavarada HK, Vijayasarathy C, Shephard HM, Avadhani NG. Mitochondrial stress-induced calcium signaling, phenotypic changes and invasive behavior in human lung carcinoma A549 cells. Oncogene (2002) 21(51):7839–49. doi:10.1038/sj.onc.1205983
148. Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta (2013) 1833(1):213–24. doi:10.1016/j.bbamcr.2012.04.013
149. Marchi S, Giorgi C, Oparka M, Duszynski J, Wieckowski MR, Pinton P. Oncogenic and oncosuppressive signal transduction at mitochondria-associated endoplasmic reticulum membranes. Mol Cell Oncol (2014) 1(2):e956469. doi:10.4161/23723548.2014.956469956469
150. Yoon MJ, Lee AR, Jeong SA, Kim YS, Kim JY, Kwon YJ, et al. Release of Ca2+ from the endoplasmic reticulum and its subsequent influx into mitochondria trigger celastrol-induced paraptosis in cancer cells. Oncotarget (2014) 5(16):6816–31. doi:10.18632/oncotarget.2256
151. Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell (2012) 151(3):630–44. doi:10.1016/j.cell.2012.10.011
152. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ (2012) 19(11):1880–91. doi:10.1038/cdd.2012.74
153. Marchi S, Pinton P. Mitochondrial calcium uniporter, miRNA and cancer: live and let die. Commun Integr Biol (2013) 6(3):e23818. doi:10.4161/cib.238182013CIB0014
154. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene (2004) 23(16):2891–906. doi:10.1038/sj.onc.1207521
155. Zong WX, Thompson CB. Necrotic death as a cell fate. Genes Dev (2006) 20(1):1–15. doi:10.1101/gad.1376506
156. Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell (2008) 135(7):1161–3. doi:10.1016/j.cell.2008.12.004
157. Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis (2007) 12(9):1543–68. doi:10.1007/s10495-007-0087-3
158. Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res (2009) 15(4):1126–32. doi:10.1158/1078-0432.CCR-08-0144
159. Zhang H, Li Y, Huang Q, Ren X, Hu H, Sheng H, et al. miR-148a promotes apoptosis by targeting Bcl-2 in colorectal cancer. Cell Death Differ (2011) 18(11):1702–10. doi:10.1038/cdd.2011.28
160. Ma Y, Ha CS, Hwang SW, Lee HJ, Kim GC, Lee KW, et al. Non-thermal atmospheric pressure plasma preferentially induces apoptosis in p53-mutated cancer cells by activating ROS stress-response pathways. PLoS One (2014) 9(4):e91947. doi:10.1371/journal.pone.0091947PONE-D-13-39326
161. Shi Y, Nikulenkov F, Zawacka-Pankau J, Li H, Gabdoulline R, Xu J, et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ (2014) 21(4):612–23. doi:10.1038/cdd.2013.186
162. Zhang J, Sun Q, Zhang Z, Ge S, Han ZG, Chen WT. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene (2013) 32(1):61–9. doi:10.1038/onc.2012.28
163. Hoffmann G, Breitenbucher F, Schuler M, Ehrenhofer-Murray AE. A novel sirtuin 2 (SIRT2) inhibitor with p53-dependent pro-apoptotic activity in non-small cell lung cancer. J Biol Chem (2014) 289(8):5208–16. doi:10.1074/jbc.M113.487736
164. Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion (2010) 10(6):640–8. doi:10.1016/j.mito.2010.08.005
165. Jia Y, Zhou L, Tian C, Shi Y, Wang C, Tong Z. Dynamin-related protein 1 is involved in micheliolide-induced breast cancer cell death. Onco Targets Ther (2015) 8:3371–81. doi:10.2147/OTT.S91805
166. Wang P, Liu B, Zhao J, Pang Q, Agrawal SG, Jia L, et al. Dynamin-related protein Drp1 is required for Bax translocation to mitochondria in response to irradiation-induced apoptosis. Oncotarget (2015) 6(26):22598–612. doi:10.18632/oncotarget.4200
167. Pyakurel A, Savoia C, Hess D, Scorrano L. Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell (2015) 58(2):244–54. doi:10.1016/j.molcel.2015.02.021
168. Ma LI, Chang Y, Yu L, He W, Liu Y. Pro-apoptotic and anti-proliferative effects of mitofusin-2 via PI3K/Akt signaling in breast cancer cells. Oncol Lett (2015) 10(6):3816–22. doi:10.3892/ol.2015.3748OL-0-0-3748
169. Wu Y, Zhou D, Xu X, Zhao X, Huang P, Zhou X, et al. Clinical significance of mitofusin-2 and its signaling pathways in hepatocellular carcinoma. World J Surg Oncol (2016) 14(1):179. doi:10.1186/s12957-016-0922-5
170. Alonso A, Martin P, Albarran C, Aquilera B, Garcia O, Guzman A, et al. Detection of somatic mutations in the mitochondrial DNA control region of colorectal and gastric tumors by heteroduplex and single-strand conformation analysis. Electrophoresis (1997) 18(5):682–5. doi:10.1002/elps.1150180504
171. Liu VW, Shi HH, Cheung AN, Chiu PM, Leung TW, Nagley P, et al. High incidence of somatic mitochondrial DNA mutations in human ovarian carcinomas. Cancer Res (2001) 61(16):5998–6001.
172. Parrella P, Xiao Y, Fliss M, Sanchez-Cespedes M, Mazzarelli P, Rinaldi M, et al. Detection of mitochondrial DNA mutations in primary breast cancer and fine-needle aspirates. Cancer Res (2001) 61(20):7623–6.
173. Tan DJ, Bai RK, Wong LJ. Comprehensive scanning of somatic mitochondrial DNA mutations in breast cancer. Cancer Res (2002) 62(4):972–6.
174. Lee HC, Li SH, Lin JC, Wu CC, Yeh DC, Wei YH. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat Res (2004) 547(1–2):71–8. doi:10.1016/j.mrfmmm.2003.12.011
175. He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, et al. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature (2010) 464(7288):610–4. doi:10.1038/nature08802
176. Liu SA, Jiang RS, Wang WY, Lin JC. Somatic mutations in the D-loop of mitochondrial DNA in head and neck squamous cell carcinoma. Head Neck (2015) 37(6):878–83. doi:10.1002/hed.23680
177. Chattopadhyay E, De Sarkar N, Singh R, Ray A, Roy R, Paul RR, et al. Genome-wide mitochondrial DNA sequence variations and lower expression of OXPHOS genes predict mitochondrial dysfunction in oral cancer tissue. Tumour Biol (2016) 37(9):11861–71. doi:10.1007/s13277-016-5026-x
178. Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet (2012) 13(12):878–90. doi:10.1038/nrg3275
179. Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, et al. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet (2014) 10(2):e1003974. doi:10.1371/journal.pgen.1003974
180. Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A (2005) 102(3):719–24. doi:10.1073/pnas.0408894102
Keywords: mitochondria, cancer, metabolism, bioenergetics, hypoxia
Citation: Chattopadhyay E and Roy B (2017) Altered Mitochondrial Signalling and Metabolism in Cancer. Front. Oncol. 7:43. doi: 10.3389/fonc.2017.00043
Received: 17 January 2017; Accepted: 03 March 2017;
Published: 20 March 2017
Edited by:
Nadège Bellance University of Bordeaux, FranceReviewed by:
Giovanni Sorrentino, Laboratorio Nazionale CIB, ItalyJuan Antonio Rosado Dionisio, University of Extremadura, Spain
Copyright: © 2017 Chattopadhyay and Roy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bidyut Roy, YnJveUBpc2ljYWwuYWMuaW4=