 Matthew Scott Schrier
Matthew Scott Schrier Malav Suchin Trivedi
Malav Suchin Trivedi Richard Carlton Deth
Richard Carlton Deth- Department of Pharmaceutical Sciences, College of Pharmacy, Nova Southeastern University, Fort Lauderdale, FL, USA
Glioblastoma is an exceptionally difficult cancer to treat. Cancer is universally marked by epigenetic changes, which play key roles in sustaining a malignant phenotype, in addition to disease progression and patient survival. Studies have shown strong links between the cellular redox state and epigenetics. Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a redox-sensitive transcription factor that upregulates endogenous antioxidant production, and is aberrantly expressed in many cancers, including glioblastoma. Methylation of DNA and histones provides a mode of epigenetic regulation, and cobalamin-dependent reactions link the redox state to methylation. Antagonists of dopamine receptor subtype 4 (D4 receptor) were recently shown to restrict glioblastoma stem cell growth by downregulating trophic signaling, resulting in inhibition of functional autophagy. In addition to stimulating glioblastoma stem cell growth, D4 receptors have the unique ability to catalyze cobalamin-dependent phospholipid methylation. Therefore, D4 receptors represent an important node in a molecular reflex pathway involving Nrf2 and cobalamin, operating in conjunction with redox status and methyl group donor availability. In this article, we describe the redox-related effects of Nrf2, cobalamin metabolism, and the D4 receptor on the regulation of the epigenetic state in glioblastoma.
Introduction
Gliomas are the most common primary central nervous system (CNS) tumors in adults, accounting for 78% of all primary malignant CNS tumors (1). Glial tumors are classified based on the type of glial cell giving rise to the tumor as oligodendrogliomas (oligodendrocytes), ependymomas (ependymal cells), and astrocytomas (astrocytes). Astrocytomas, which account for most cases of glioma (1), can be ranked as low-grade (WHO grade I–II) or high-grade (WHO grade III–IV), depending on their clinical behavior and genetic features (2).
High-grade glioma (WHO grade IV) is typically divided into four subtypes: classical, neural, proneural, and mesenchymal (3). Classical glioblastoma presents with chromosomal abnormalities, including epidermal growth factor receptor (EGFR) mutation and/or increased EGFR expression, but not protein 53 (p53) mutations (3). The neural subtype shares some similarities with the classical subtype, but frequently has p53 mutations and is positive for certain neuronal cell markers, such as neurofilament light peptide (NEFL) and gamma-aminobutyric acid receptor subunit α-1 (3). Proneural glioblastoma is associated with elevated plasma membrane levels of platelet-derived growth factor receptor and often have isocitrate dehydrogenase 1/2 (IDH1/2) mutations. The mesenchymal subtype uniquely displays activation of inflammatory mediators, especially NF-κB (3).
Surgical removal of primary glioblastoma is the preferred treatment option to prolong survival. While life expectancy can be prolonged by several months, the outcome is poor in the majority of patients who undergo resection because the tumor often continues to progress (4). At this time, there are two main non-surgical treatment protocols for glioblastoma. The first protocol comprises temozolomide, an orally administered DNA alkylating drug given in combination with radiation as first-line therapy (5). The second protocol comprises administration of the anti-angiogenic drug bevacizumab in patients with recurrent glioblastoma (6).
Despite treatment, the median survival time of patients with glioblastoma is between 9 and 12 months (1). A major cause of treatment failure is persistence of the epigenetic signatures, which define the malignant state (7). Epigenetic regulation provides dynamic control over gene expression. The methylation states of DNA and histones, which are influenced by intracellular S-adenosylmethionine (SAM) levels, are crucial determinants of this mode of regulation. DNA methylation at CpG sites promotes heterochromatin formation and gene silencing. Additionally, methylation of specific residues on the tails of histones, such as histone 3 lysine residue 9 (H3K9me) and histone 3 lysine residue 27 (H3K27me), can promote transcriptional silencing. However, other histone tail methylation marks, such as H3K4me, can lead to less dense nucleosome packing, thereby increasing promoter accessibility to transcription factors and favoring gene transcription.
Cancer is marked by genome-wide hypomethylation, chromosomal aberrations, and active transposable elements. Furthermore, cancer often presents with promoter hypomethylation or hypermethylation of genes expressing the components of major signaling pathways. The epigenetic landscape of glioblastoma includes differential expression patterns of long non-coding RNAs, such as increased HOTAIR (8). LncRNAs cooperate with proteins to modulate gene expression. Another typical epigenetic feature of glioblastomas is upregulation of EZH2 (9), the catalytic component of polycomb repressive complex 2, that methylates H3K27 (histone 3 lysine residue 27) (9), leading to condensation of nucleosomes. Paradoxically, several proteins that promote gene expression are also overexpressed, such as BRD2 (bromodomain-containing 2) and BRD4 (10). BRD2 and BRD4 are members of the bromodomain and extra-terminal (BET) family of proteins, which recognize acetylated lysine residues on histones and promote transcription of the histone-associated genes. Additionally, certain lysine-specific demethylases (KDMs) and 5-methylcytosine hydroxylation enzymes, called ten-eleven translocation (TET) enzymes, are inhibited by oncometabolites, such as 2-hydroxyglutarate (2-HG) and succinate (11).
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is a transcription factor activated during oxidative stress that upregulates the expression of genes involved in antioxidant production. Glioblastoma and numerous other cancers are characterized by aberrant Nrf2 expression. Methylcobalamin (MeCbl) and 5′-deoxyadenosylcobalamin (AdoCbl)—the active forms of cobalamin (vitamin B12)—play central roles in regulating DNA and histone methylation by serving as cofactors for methionine synthase (MS) and methylmalonyl-CoA mutase (MCM), respectively. Their levels are sensitive to the cellular antioxidant status, making them responsive to Nrf2. The dopamine receptor subtype 4 (D4 receptor) is a G protein-coupled receptor (GPCR) with neurotransmission and metabolic roles, which include effects on the redox and methylation states. D4 receptors are expressed in glioblastoma and D4 receptor antagonists inhibit the growth of glioblastoma stem cells (12), although the precise relationship between Nrf2 and D4 receptors has not yet been established. This article is the first to explore the relationships between Nrf2, cobalamin, and D4 receptors in cancer, specifically in glioblastoma. Recognition of these relationships opens new opportunities for identifying novel drug targets.
The Role of Nrf2 in Cancer and Glioblastoma
Nuclear factor (erythroid-derived 2)-like 2 is a cap-n-collar basic region leucine zipper transcription factor. Under normal conditions, Nrf2 is sequestered through its Neh2 domain by a ubiquitin ligase substrate adaptor called Kelch-like ECH-associated protein 1 (Keap1) (13) (Figure 1). Upon interacting with electrophiles, such as reactive oxygen species (ROS), Keap1 releases Nrf2, which relocates to the nucleus, where it heterodimerizes with musculoaponeurotic fibrosarcoma proteins and binds to antioxidant response elements (AREs) with the core consensus sequence 5′-TGACnnnGC-3′ (14). In this way, Nrf2 regulates ≥1% of genes (15). ARE-regulated genes are typically involved in the production of antioxidants (e.g., glutathione) and detoxification enzymes (e.g., glutathione peroxidase 2).
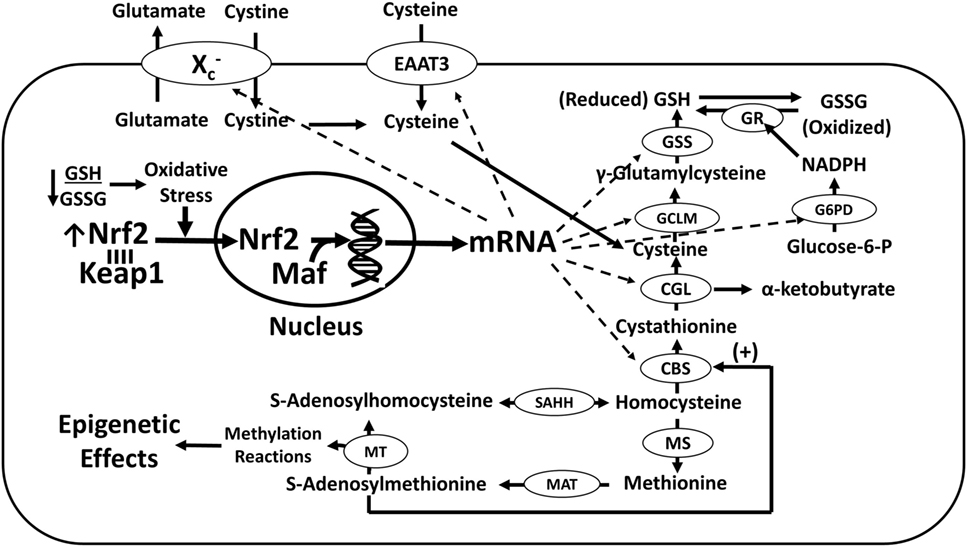
Figure 1. Nrf2-dependent transcription of genes promoting GSH synthesis. Oxidative stress induces the dissociation of Nrf2 from cytosolic Keap1, allowing its relocation to the nucleus where it augments the transcription of multiple genes directly or indirectly involved in GSH synthesis (dashed arrows), thereby increasing antioxidant capacity. Nrf2 activation of GSH synthesis negatively affects the methylation capacity by promoting transsulfuration of homocysteine to cysteine. Abbreviations: CBS, cystathionine-β-synthase; CGL, cystathionine-γ-lyase, EAAT3, excitatory amino acid transporter-3; GCLM, γ-glutamylcysteine ligase modulatory subunit; GR, glutathione reductase; GSH, glutathione; GSS, glutathione synthetase; GSSG, glutathione disulfide; G6PD, glucose-6-phosphate dehydrogenase; Keap1, kelch-like ECH-associated protein 1; Maf, musculoaponeurotic fibrosarcoma proteins; MAT, methionine adenosyltransferase; MS, methionine synthase; MT, methyltransferase; Nrf2, nuclear factor (erythroid-derived 2)-like 2; SAHH, S-adenosylhomocysteine hydrolase.
Nuclear factor (erythroid-derived 2)-like 2 acts as a tumor suppressor or an oncoprotein (16), depending upon the nature of the cell expressing it. In normal physiologic conditions, Nrf2 activation is brief and its transcriptional effects can restore redox homeostasis (13). The tight regulation of Nrf2 reduces the risk of cellular transformation from a benign state to a malignant state, reflecting its status as a tumor suppressor in typical cells. This tumor-suppressing effect is attributed to the ability of Nrf2 to decrease ROS levels and to restrict DNA damage by promoting the expression of antioxidant and detoxification systems (13). Although Nrf2 generally suppresses cellular transformation, it may actually enhance this process in some situations. This appears to be the case in BEAS-2B lung bronchial epithelial cells chronically exposed to the metal cadmium (17). In these cells, Nrf2 binds to AREs on the antiapoptotic genes Bcl-2 and Bcl-xL and to an ARE at p62, which codes a ubiquitin-binding protein (17). Phosphorylated p62 maintains constitutive Nrf2 activity in transformed cancer cells, such as transformed BEAS-2B cells, by binding to Keap1 and blocking its suppressive interaction with Nrf2 (17, 18). Consequently, the transcription of ARE-containing genes is increased and loses responsiveness to intracellular redox status in malignant cells. In this setting, Nrf2 promotes tumor cell proliferation by acting as an oncoprotein and increasing tumor resistance to chemotherapy and radiation (19).
The expression of Nrf2-regulated genes is essential for the survival of many cancer types because high levels of ROS are produced during cell growth, implying that cancer cells are burdened with higher levels of ROS than non-neoplastic cells (20). Higher ROS levels can impair enzyme function, and several enzymes involved in epigenetic regulation are especially sensitive to ROS, including enzymes discussed in this article: MS, MCM, histone deacetylases (HDACs) 2 and 4 (HDAC2/4) (21, 22), and propionyl-CoA carboxylase (23).
Mitochondrial respiration is a major source of ROS production and increased antioxidant capacity secondary to Nrf2 activation increases mitochondrial aerobic activity, while Nrf2 interaction with other transcriptional factors increases mitochondrial biogenesis. Indeed, Nrf2 and Keap1 are tethered to the outer mitochondrial membrane phosphatase phosphoglycerate mutase family member PGAM5, representing a third site of distribution (beyond cytoplasmic and nuclear) (24). PGAM5 degradation is in turn regulated by a Keap1-dependent ubiquitin ligase complex (25), indicating that Nrf2/Keap1 and mitochondrial activities are coordinated.
Nuclear factor (erythroid-derived 2)-like 2 is overexpressed in glioblastoma (26) and elevated expression of genes under the transcriptional influence of Nrf2 occurs in 13.7 and 32.7% of anaplastic gliomas and glioblastomas, respectively (27). There is a strong connection between ROS (and other radicals) and the epigenetic mechanisms that regulate gene expression. Because Nrf2 is a central regulator of the intracellular balance between antioxidants and free radicals, its overexpression may account for much of the epigenetic divergence of glioblastoma from its precursor cells.
Nrf2, Antioxidants, and Cobalamins
Cobalamin is a corrin ring-containing structure with a central cobalt atom. Different cobalamin species have a variable upper axial ligand bound to the cobalt, enabling cobalamin to act as a cofactor for two enzymes in humans. MeCbl is utilized by MS, while AdoCbl is necessary for the activity of MCM. Cobalt contributes to the propensity of cobalamin to be a highly potent reducing agent, and thus an easily oxidized molecule.
For cobalamin to be converted into its active forms, the cobalt atom must be in its (II) transition state (28). This is achieved enzymatically after the cellular uptake of cobalamin in a complex with transcobalamin, a cobalamin-carrier protein (29). The complex is initially imported into cells via the transcobalamin receptor (30). As cobalamin accumulates in tumors (31), both transcobalamin and transcobalamin receptor have been explored as potential tumor biomarkers and their expression is elevated in numerous canine and feline cancers (32). Cobalamin exits lysosomes and interacts with a cytoplasmic enzyme, methylmalonic aciduria, and homocystinuria type C protein (MMACHC), that removes the upper axial ligand and reduces the cobalt atom to its (II) oxidized state (29). To accomplish this, MMACHC uses NADPH to remove cyano groups (from cyanocobalamin) and reduced glutathione (GSH) to remove methyl and adenosyl groups (33).
The potential for Nrf2 to regulate cobalamin status is related to its ability to transcriptionally activate enzymes involved in regulating the intracellular concentrations of the metabolic reducing agents (i.e., GSH and NADPH), which are necessary to process and protect cobalamins. Indeed, Nrf2 may be a key factor that increases the cobalamin content of glioblastoma cells in response to activation of the phosphoinositide 3-kinase (PI3K) or extracellular signal-regulated kinase (ERK) pathway induced by growth factor receptor signaling, irrespective of whether these pathways are stimulated by their respective ligands or transactivated by D4 receptors (34). Of note, some mutations may result in hyperactivity of these transduction pathways (Figure 2). In SH-SY5Y neuroblastoma cells, stimulation of the PI3K pathway by the growth factor neuregulin-1 was associated with increased intracellular concentrations of cobalamin owing to activation of the cysteine transporter excitatory amino acid transporter-3 (EAAT3) (35). EAAT3 translocation to the plasma membrane in response to PI3K activation is associated with an increase in GSH, and Nrf2 is a known transcriptional activator of EAAT3 (36). PI3K and ERK are known to promote the activity of Nrf2 in glioblastoma (37), which may increase cobalamin levels by upregulating NADPH and GSH.
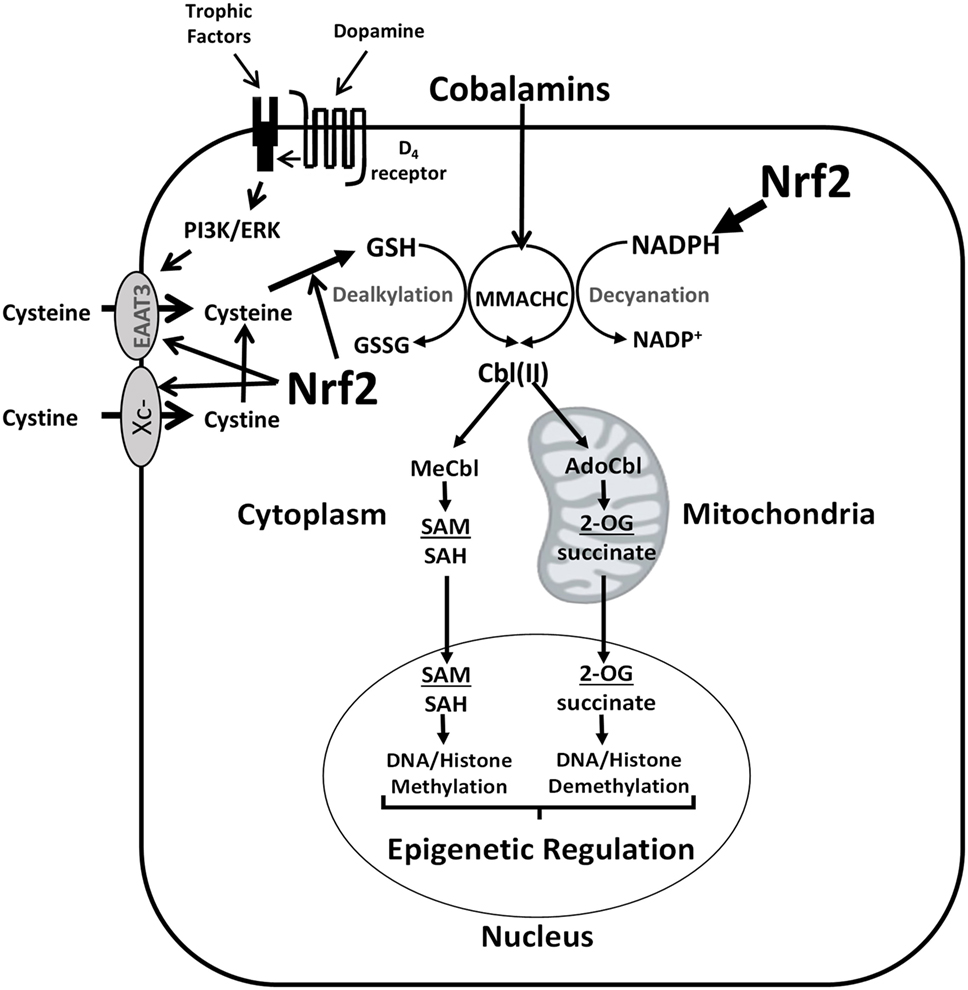
Figure 2. Nrf2 regulation of cobalamin metabolism. Nrf2 upregulates Xc− and EAAT3 transcription, increasing cysteine availability for GSH synthesis. Nrf2 also upregulates the formation of GSH and NADPH, promoting MMACHC-dependent conversion of cobalamin to its active forms (MeCbl and AdoCbl). Mutations in PI3K- and ERK-linked trophic receptors (e.g., epidermal growth factor receptor and platelet-derived growth factor receptor beta), can increase their activities, augmenting cystine/cysteine uptake and increasing the levels of active cobalamin species, with epigenetic consequences. Transactivation of PDGFR by the D4 receptor also promotes PI3K/ERK signaling and cystine/cysteine uptake. Abbreviations: 2-OG, 2-oxoglutarate; AdoCbl, adenosylcobalamin; Cbl, cobalamin; EAAT3, excitatory amino acid transporter-3; GSH, glutathione; GSSG, glutathione disulfide; MeCbl, methylcobalamin; MMACHC, methylmalonic aciduria and homocystinuria type C protein; Nrf2, nuclear factor (erythroid-derived 2)-like 2; PI3K, phosphoinositide 3-kinase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Excitatory amino acid transporter-3 levels are increased by neuregulin-1 in C6 glioma cells (38), a cell line that serves as a useful model for the study of glioblastoma. The cystine/glutamate antiporter, also known as system Xc−, provides an important source of cysteine for GSH synthesis, as cystine is reduced to two cysteine molecules in the cytoplasm. Nrf2 induces transcription of xCT (SLC7A11), a component of Xc−, along with other GSH synthesis and redox-related genes (Table 1).
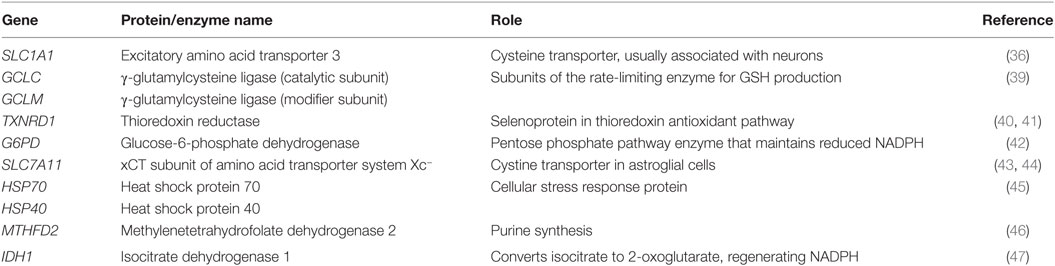
Table 1. Genes and corresponding proteins regulated by Nrf2.
Epidermal growth factor receptor activation in C6 glioma cells upregulates xCT in association with increased GSH levels (48). In mice, EGFR-overexpressing gliomas are associated with greater invasiveness, and the xCT inhibitor sulfasalazine suppressed tumor growth and invasiveness. Taken together, these findings suggest that increased EGFR signaling may increase xCT and GSH synthesis via increased Nrf2 expression, contributing to the aggressive properties of glioblastoma.
Nrf2, SAM, and Glioblastoma
Many biological processes are regulated by methylation reactions, including DNA and histone methylation. These reactions are governed by the availability of the universal methyl donor SAM, and methyltransferases are inhibited by the demethylated end-product S-adenosylhomocysteine (SAH). The SAM to SAH ratio is affected by the kinetics of the cobalamin- and folate-dependent enzyme MS, which converts homocysteine to methionine.
While normal cells can adapt to a lack of methionine, many cancer cell lines require methionine, as demonstrated by their inability to survive just on methionine provided by MS activity (i.e., methionine derived from homocysteine) (49). While the cause of this condition varies, it may reflect a limitation in cobalamin or folate availability. By contrast, methionine-independent cancer cells can maintain sufficient levels of MS activity and methionine synthesis to sustain their rapid growth rates.
By augmenting GSH synthesis, Nrf2 activity can influence methionine dependence and the cell’s methylation state in multiple ways. As illustrated in Figure 3, left panel, the normal adaptive response to oxidative stress involves a transient increase in Nrf2 activity together with a decrease in the ratio of reduced GSH to oxidized glutathione disulfide (GSSG) (i.e., [GSH]2/[GSSG]). Increased transcription of ARE-regulated genes serves to augment antioxidant capacity, restoring redox status and allowing Nrf2 activity to return to its basal level. However, methylation activity decreases during periods of elevated oxidative stress, affecting DNA and histone methylation, with the potential for persistent epigenetic effects.
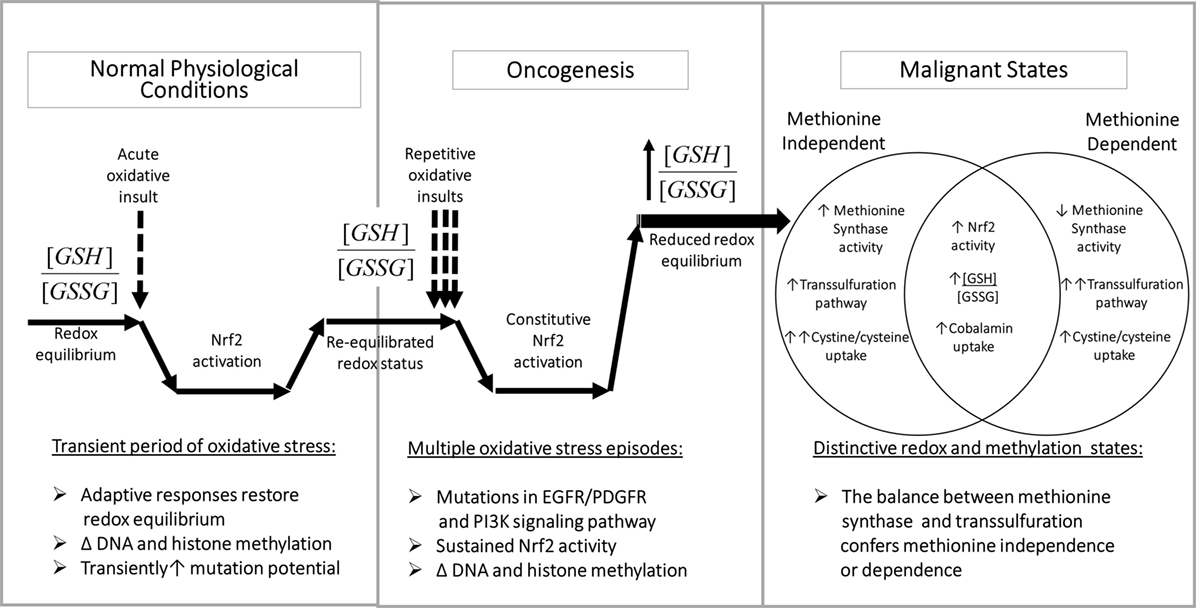
Figure 3. Redox-dependent epigenetic regulation and oncogenesis. Left panel: transient Nrf2 activation is an important component of the adaptive metabolic responses to oxidative stressors, accompanied by epigenetic changes, which promote homeostatic restoration of redox equilibrium. Middle panel: multiple oxidative challenges increase the potential for DNA mutations, including mutations of growth factor signaling pathway components, which can lead to sustained Nrf2 activation, resulting in abnormal redox and epigenetic status. Right panel: methionine-independent and methionine-dependent states can alternatively emerge from oncogenesis, depending upon the level of MS activity, which is influenced by the ratio of cystine/cysteine uptake vs. transsulfuration. Abbreviations: GSH, glutathione; GSSG, glutathione disulfide; MS, methionine synthase; Nrf2, nuclear factor (erythroid-derived 2)-like 2.
The potential for DNA mutations increases during transient oxidative stress (due to unquenched oxidizing species) and/or increased prevalence of euchromatin (non-compacted DNA). Cancer-inducing mutations in pathways controlling Nrf2 expression (e.g., PI3K) during repetitive or unresolved periods of oxidative stress can lead to constitutive Nrf2 activation (Figure 3, middle panel). Unlike in normal physiologic conditions, mutation-induced Nrf2 overexpression is not transient and can lead to an abnormally high antioxidant capacity, which promotes cell division and produces an abnormal pattern of DNA and histone methylation.
As illustrated in Figure 3 (right panel), sustained Nrf2 hyperactivity induced by mutations can promote methionine-independent or methionine-dependent phenotypes, depending on the level of MS activity. Under conditions where the increase in cystine/cysteine uptake is dominant, MS-mediated methylation of homocysteine is maintained, resulting in methionine independence. However, when the increase in transsulfuration of homocysteine toward GSH production is more dominant, MS activity is insufficient, resulting in methionine dependence. Thus, the balance between cystine/cysteine uptake and homocysteine transsulfuration is a major determinant of methionine dependence.
Increased cystine/cysteine uptake capacity secondary to Nrf2 activation may have implications on the tumor microenvironment. Increased expression of Xc− and EAAT3 enhances tumor uptake of antioxidant resources at the expense of other cells in the microenvironment. Glioblastoma cells may competitively impinge on EAAT3-dependent cysteine uptake by neuronal cells, with potential functional implications for neurons.
Nuclear factor (erythroid-derived 2)-like 2 can also affect MS activity by influencing cobalamin synthesis and cellular redox status. As described above, Nrf2 can promote cobalamin assimilation through its ability to increase GSH and NADPH level, and the resultant increase in cobalamin availability decreases methionine dependency by sustaining MS activity (50). In addition, a Nrf2-induced decrease in oxidative stress status decreases the frequency of cobalamin inactivation and promotes MS activity. When the cobalt atom of the cobalamin cofactor of MS is in its (I) oxidation state, it removes and transiently holds a methyl group from 5-methyltetrahydrofolate. The methyl group is subsequently transferred to homocysteine, forming methionine. However, cobalamin (I) is readily oxidized, depending upon the prevailing redox environment, temporarily halting enzyme activity, until it is reactivated by NADPH-dependent MS reductase. Thus, hyperactivity of Nrf2 decreases the rate of cobalamin oxidation by lowering oxidative stress and ROS levels, and facilitates MS reactivation by promoting higher levels of NADPH.
The availability of methionine is proportionally reflected by intracellular SAM concentrations (51). Therefore, the involvement of Nrf2 in maintaining a methionine-independent state may dictate how glioblastoma cells allocate SAM toward different reactions. SAM-dependent methylation reactions in the nucleus control gene expression through multiple mechanisms. Thus, the availability of SAM, as modulated by Nrf2, may affect the survival and proliferation of glioblastoma cells by influencing genome-wide gene transcription.
In addition to its role in DNA and histone methylation, SAM is used for the synthesis of polyamines, which are involved in cell growth, transcription, translation, and numerous other processes (52). Indicative of the importance of polyamines in cancer, a polyamine analog was reported to induce apoptosis in glioblastoma cells by exhausting the cellular polyamine content (53). Polyamines are recycled back into the one-carbon cycle as methylthiooxobutyrate to re-form methionine and subsequently SAM. SAM may also influence many processes involved in the progression of glioblastoma, including cell signaling, cell cycle progression, protein translation, mRNA splicing, and possibly the health of telomeres (54).
Finally, SAM may influence glioblastoma by acting as a positive allosteric modulator of proteins involved in maintaining redox homeostasis, including cystathionine-β-synthase (CBS) and Nrf2. CBS is one of two Nrf2-regulated enzymes in the transsulfuration pathway. This pathway directs homocysteine away from the one-carbon cycle toward cysteine synthesis, accompanied by α-ketobutyrate formation (Figure 1). Notably, Nrf2 is post-translationally methylated at an arginine residue (R437) in its Neh1 domain by the arginine methyltransferase PRMT1 (55). R437 methylation of Nrf2 increases its DNA-binding ability and increases recruitment of transcriptional coactivator p300/CBP (CREB-binding protein) to an ARE in the heme oxygenase-1 promoter region. However, expression of both Nrf2 and its suppressor keap1 are also subject to epigenetic regulation by promoter methylation in several cancers types (56). Thus, the relationships between Nrf2, SAM, and glioblastoma are highly complex.
Nrf2 and Oncometabolites in Glioblastoma
Oncometabolites are metabolic intermediates that promote tumor growth and other malignant processes. These oncogenic metabolites are either produced excessively or are inefficiently metabolized in cancer and consequentially accumulate in the cytosol (57). Well-known examples of oncometabolites include succinate and fumarate. Mutations in succinate dehydrogenase (SDH) and fumarate hydratase prevent the passage of their respective substrates, succinate and fumarate, through the Krebs cycle, resulting in their accumulation. Mutations in these proteins have been linked to malignant pheochromocytoma, paraganglioma, and other forms of cancer (58–60). When mutated in glioblastoma, IDH1/2 yield another oncometabolite, 2-hydroxyglutarate (2-HG), from 2-oxoglutarate (2-OG) (also known as α-ketoglutarate) (61). While mutations in IDH1/2 are clinically relevant (62), only IDH1 is transcriptionally regulated by Nrf2.
Because Nrf2 enhances cobalamin processing, and thus AdoCbl levels, it can connect the transsulfuration pathway to the Krebs cycle. AdoCbl promotes the formation of succinate. As a cofactor for the mitochondrial enzyme MCM, AdoCbl is necessary for the catabolism of certain fats and amino acids in a pathway involved in the anaplerotic incorporation of succinate into the Krebs cycle as succinyl-CoA, which is then converted to succinate.
The amino acid methionine is catabolized through this anaplerotic pathway. The passage of methionine through the one-carbon cycle produces homocysteine, and any homocysteine that is not recycled back to methionine can enter the transsulfuration pathway and be converted to cystathionine. Cystathionine is then converted to cysteine and α-ketobutyrate. While cysteine is directed toward GSH production, α-ketobutyrate is converted to propionyl-CoA, which is subsequently converted to d-methylmalonyl-CoA by the biotin-dependent enzyme propionyl-CoA carboxylase. d-methylmalonyl-CoA is converted to l-methylmalonyl-CoA by epimerase, and MCM (with its AdoCbl cofactor) converts l-methylmalonyl-CoA to succinyl-CoA.
The product of this pathway, succinyl-CoA, enters the Krebs cycle and is converted to succinate by succinyl-CoA synthetase. Succinate is subsequently converted to fumarate by SDH, or complex II of the mitochondrial electron transport chain. Fumarate and succinate can act as retrograde signaling messengers, relaying information from the mitochondria through their influence over epigenetics and cell signaling. Both fumarate (63) and succinate (64) inhibit prolyl hydroxylases from ubiquitinating hypoxia-inducible factor-1α (HIF-1α) and allow its buildup in normoxic conditions. However, only fumarate binds to cysteine residues on keap1, increasing the amount of Nrf2 available to promote gene transcription (65). SDH subunit B was downregulated and HIF-1α was expressed in glioblastoma stem cells in normoxic conditions (66). However, certain glioblastoma-associated mutations promote oxidative phosphorylation rather than glycolysis (67). Therefore, the amount of succinate and fumarate in glioblastoma cells may vary according to how closely they resemble a stem cell phenotype and the presence of specific mutations in energy producing pathways, in addition to AdoCbl levels; factors that may be influenced by Nrf2.
Nuclear factor (erythroid-derived 2)-like 2 can increase the level of 2-HG, another retrograde signaling molecule in glioblastoma. 2-HG is produced by mutant forms of IDH1/2. Normally, IDH1/2 promote the conversion of isocitrate to 2-OG, while reducing NADP+ to NADPH. However, mutated forms of IDH1/2 convert 2-OG to 2-HG. Furthermore, these mutant forms oxidize NADPH to NADP+. Ordinarily, the expression of IDH2 (the mitochondrial isoform) is not directly induced by Nrf2. However, the activity of mutated IDH2 may be indirectly promoted by Nrf2 through the increased concentration of NADPH. Around 80% of low-grade gliomas and 5–10% of glioblastomas have IDH1/2 mutations (3) and display increased methylation in the gene promoters compared with other glioma subtypes.
Epigenetic Effects of 2-HG, Succinate, and Fumarate
Epigenetic control of gene expression is mediated by numerous regulatory factors and represents an overlaying program that governs the manner in which the genetic code is read. Specifically, epigenetic alterations determine the probability of gene expression, the timing of expression, and which isoforms of a specific gene are produced. One of the most important epigenetic events is the chemical modification of amino acid residues in histone tails by ubiquitin, acetyl, phosphoryl, and methyl groups. Histone tail methylation marks are laid down by histone methyltransferases. These marks include H3K4me (which leads to chromatin opening and is positively associated with gene expression), as well as H3K9me and H3K27me (which lead to chromatin condensation and are associated with inactivation of gene expression).
Histone lysine demethylases (KDMs) remove methyl groups from lysine residues on histone tails. There are two classes of KDMs: 2-OG-dependent dioxygenases (2-OGDOs), which possess Jumonji C domains, and flavin-containing amino oxidases. 2-OGDOs can oxidize numerous substrates, which are not limited to methylated lysine and cytosine residues (68). 2-OGDO enzymes convert 2-OG to succinate and CO2. 2-OGDO activity is downregulated in a negative feedback loop by succinate, fumarate, and by 2-HG (68).
KDM2-7 are inhibited by the same, whereas KDM1a belongs to the flavin-containing amino oxidase subclass and is not inhibited by these oncometabolites. KDM1a regulates the transcription of embryonic transcription factors by altering Myc expression (69). The role of KDM1a in removing histone tail methylation marks may become more prominent following oncometabolite-mediated inhibition of KDM2-7. Although KDM2-7 are sensitive to succinate, fumarate, and 2-HG, KDM1a activity is regulated by flavin adenine dinucleotide. The reaction mediated by SDH results in the conversion of succinate to fumarate, in which flavin adenine dinucleotide is used as a cofactor to accept two hydrogen atoms and two electrons. Therefore, SDH may act as a junction that regulates both KDM1a and KDM2-7. However, more studies are needed to elucidate the dynamics between SDH, KDM1a, KDM2-7, and histone tail marks in glioblastoma.
Other members of the 2-OGDO family include the TET enzymes. TET enzymes catalyze the first step of cytosine nucleotide demethylation by converting 5-methylcytosine to 5-hydroxymethylcytosine (5hmeC) (70). This step is essential for demethylating CpG sites to allow gene reactivation because methyl-binding proteins cannot block access to transcriptional machinery following demethylation.
Like KDM2-7, TET1-3 are inhibited by succinate, fumarate, and 2-HG. The levels of 5hmeC are low in many cancers, including glioblastoma. A deficit in 5hmeC may indicate a significant reduction in TET enzyme activity that might arise from gene deletions or mutations (71). However, genetic sequencing does not support the hypothesis that mutations are a major causal factor of 5hmC depletion in colorectal, breast, and prostate cancer (72). The expression of mutant IDH1 in the subventricular zone of mice was associated with decreased 5hmeC genomic content and increased 5-methylcytosine levels, mimicking gliomagenesis (73). Therefore, it is plausible that oncometabolite-mediated inhibition of TET activity contributes to the reduced 5hmeC expression observed in these cancers and in glioblastoma.
Redox and Transcriptionally Induced Epigenetic Effects of Nrf2
Nuclear factor (erythroid-derived 2)-like 2 can exert epigenetic effects via multiple mechanisms. As described in the Section “The Role of Nrf2 in Cancer and Glioblastoma,” Nrf2 influences the transcription of ARE-containing genes to increase the levels of antioxidants and 2-HG (in IDH1 mutants). This increase in antioxidant levels protects cobalamin from oxidation, sustaining the one-carbon cycle. Antioxidants also protect the enzyme propionyl-CoA carboxylase, which is upstream of MCM in an anaplerotic pathway that leads to the formation of succinate. The metabolic information conveyed by the intracellular concentrations of SAM and the oncometabolites succinate and 2-HG is integrated at the nucleus and determines global DNA and histone methylation and demethylation.
While Nrf2 indirectly regulates epigenetics through chemical messengers, such as succinate and 2-HG, it can also regulate epigenetics in a more direct manner. Many epigenome-modifying proteins are sensitive to the intracellular redox balance, the prime examples being HDACs 2 and 4 (21, 74). HDACs remove acetyl groups from histone tail residues and non-histone proteins, thereby decreasing gene transcription. HDAC4 expression is downregulated in glioblastoma compared with other gliomas and normal brain tissue, although there is considerable variation in its expression levels between resected tumors from glioblastoma patients and other gliomas (75). The protective effect of Nrf2 on HDAC4 was reported to precede HDAC4-mediated downregulation of the microRNAs miR-206 and miR-1 in lung and prostate cancer cell lines (22). Downregulation of these microRNAs by HDAC4, downstream of Nrf2, increased the expression of Krebs cycle and pentose phosphate pathway components. These findings support the hypothesis that the epigenetic effects of Nrf2 have redox and metabolic consequences.
D4 Receptors, Methylation, and Glioblastoma
Dolma et al. (12) recently reported that selective D4 receptor antagonists restrict the growth of glioblastoma stem cells in vitro and in vivo. Clonogenic activity of freshly isolated tumor cells was reduced 19- to 83-fold by these antagonists. Analysis of gene expression in cells exposed to a D4 receptor antagonist revealed significant changes in lipid/cholesterol biosynthetic, autophagic vacuole, and lysosomal pathways, leading massive accumulation of autophagic vacuoles and cholesterol owing to decreased autophagic flux and disrupted lysosomal function. These glioblastoma stem cell-specific effects were accompanied by decreased phosphorylation of multiple proteins, including platelet-derived growth factor receptor beta (PDGFRβ), which was previously shown to be transactivated by D4 receptor stimulation (34). Indeed, transactivation of PDGFRβ and its downstream signaling target ERK1/2 by the D4 receptor may contribute to increased Nrf2 activity, as described above, while D4 receptor antagonists have opposite effects. Since PDGFRβ levels are increased in proneural glioblastoma, more robust D4 receptor co-activation may be particularly important in this subtype. The inhibitory effects of D4 receptor antagonists were synergistic with those of temozolomide, and poorer survival rates were observed for tumors with greater D4 receptor expression.
Although the study by Dolma et al. (12) showed that the D4 receptor is an important new antitumor target for glioblastoma, the detailed molecular mechanism underlying its unique autophagy-based effectiveness remain unclear. Like other GPCRs, the D4 receptor exerts several G protein-mediated signaling activities, including inhibition of cyclic AMP formation. However, only the D4 receptor is involved in dopamine-dependent methylation of membrane phospholipids (76). This exclusive activity arises from a methionine residue (MET313) in transmembrane helix #6 of the D4 receptor, not present in other GPCRs, a locus that undergoes a large change in position in response to agonist occupation of the binding site (77). This dopamine-induced conformational change facilitates ATP-dependent S-adenosylation of the MET313, followed by transfer of its methyl group to adjacent phospholipids, especially phosphatidylethanolamine (78). After methyl transfer and removal of the adenosyl group, a replacement methyl group is provided to the resulting homocysteine form of the receptor by MS, in a cobalamin/methylfolate-dependent reaction, completing a cycle of dopamine-stimulated phospholipid methylation (PLM).
Notably, the cycle of D4 receptor-mediated PLM utilizes the same enzymes that support SAM-dependent methylation reactions (76), placing the D4 receptor in the position of regulating all methylation reactions in a dopamine-dependent manner, as illustrated in Figure 4. Indeed, D4 receptor-mediated stimulation of MS activity and increased global DNA methylation have been reported in human neuroblastoma cells (79), and these effects were blocked by inhibitors of the PI3K and ERK signaling pathways. It remains to be determined whether D4 receptors exert a similar influence in glioblastoma.
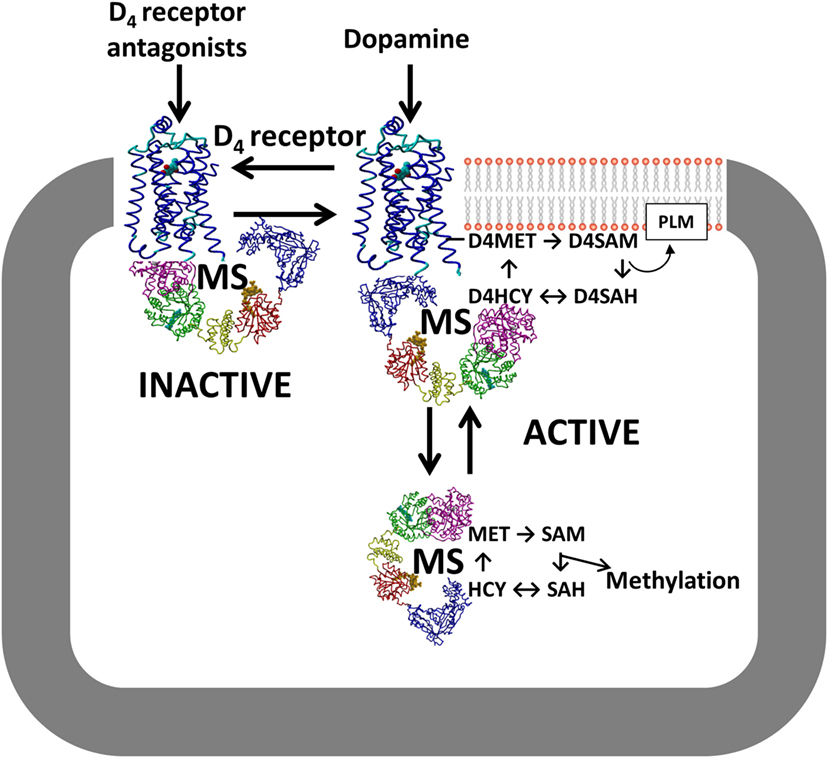
Figure 4. D4 receptor interaction with MS. Conformation-dependent exposure of the methionine residue in the D4 receptor allows it to catalyze phospholipid methylation (PLM), which is dependent upon folate-derived methyl groups supplied by MS. D4 receptor antagonists stabilize the inactive conformation and sequester MS, while D4 receptor agonists such as dopamine promote PLM and increase MS-dependent conversion of HCY to MET. Abbreviations: D4R, D4 receptor; HCY, homocysteine; MET, methionine; MS, methionine synthase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine.
Based on the unique capability of the D4 receptor to modulate methylation status, we suggest that the anti-glioblastoma tumor effects of D4 receptor antagonists described by Dolma et al. (12) may reflect inhibition of methylation reactions such as dopamine-stimulated PLM, which can influence the activity of receptors and transporters at the plasma membrane (80), and/or SAM-dependent methylation effects, which might include effects on autophagy. For example, increased membrane fluidity and/or membrane asymmetry promoted by D4 receptor-mediated PLM may contribute to PDGFRβ transactivation. PLM-related phospholipid adducts were also reported to play crucial regulatory roles in promoting mitophagy, a specialized form of autophagy (81). At high levels, methionine and SAM act as indicators of amino acid sufficiency and suppress autophagy (82). This hypothesis is supported by the finding that autophagy was attenuated by short hairpin RNA-mediated knockdown of MS reductase in SKOV3 and cisplatin-resistant SKOV3/DDP cells (83). Studies of dopamine-stimulated PLM may help our understanding of how the D4 receptor regulates autophagy in glioblastoma stem cells.
Potential Treatment Targets
Despite the development of temozolomide and bevacizumab, glioblastoma invariably has a poor prognosis, necessitating the development of more effective therapies. Several classes of drugs are being explored, including many that target the epigenetic landscape. Some epigenetic modifiers, such as BET proteins, are overexpressed in glioblastoma. Because BET proteins are indispensable for glioblastoma growth (10), BET inhibitors are an important area of research. JQ1 is a BET inhibitor that caused G1 cell cycle arrest and apoptosis in several glioblastoma cell lines, which were genetically modified to resist cell death (84). However, JQ1 has a short half-life, limiting its utility in glioblastoma treatment (85). Another BET inhibitor, OTX015, has entered dose optimization trials for glioblastoma therapy (http://ClinicalTrials.gov Identifier NCT02296476).
Histone deacetylases inhibitors have been also tested in glioblastoma (86), although their clinical use is generally limited to certain lymphomas. HDAC inhibitors reactivate the expression of genes, which either promote cell death or improve the efficacy of other drugs. For example, HDAC inhibitors can restore the expression of estrogen receptor α (ERα) in ERα negative breast cancer, potentially re-allowing the administration of tamoxifen to tumors that had become non-responsive to antiestrogen drugs (87). However, a concern of using HDAC inhibitors for treating glioblastoma is that they might re-activate expression of O6-alkylguanine DNA alkyltransferase (MGMT), a DNA repair methyltransferase associated with temozolomide resistance (88).
An interesting approach to circumvent the risk of re-activating MGMT in glioblastoma is to administer drugs capable of modulating the redox capacity, such as the flavonoid luteolin. Luteolin was reported to reduce Nrf2 mRNA and protein levels in tumors (89), thus affecting redox and epigenetic regulation. Owing to the antioxidant-boosting and antiapoptotic effects of Nrf2, the benefits of a Nrf2 inhibitor may outweigh any risks posed by altering the epigenetic signature it imparts to glioblastoma cells. Treatment of A549 cells with luteolin enhanced the cytotoxicity of doxorubicin, oxaliplatin, and bleomycin (89). Furthermore, a combination of luteolin and another flavonoid, silibinin, caused autophagy and apoptosis by inhibiting protein kinase Cα (PKCα) and inducible nitric oxide synthase (iNOS), respectively, in U87MG and T98G glioblastoma cells (90). Additionally, luteolin is considered to be neuroprotective and was ironically demonstrated to induce Nrf2 activity in a mouse model of traumatic brain injury (91). Therefore, this phytochemical may selectively target neoplastic tumor cells and protect surrounding brain tissue.
Because cystine transporter activity is upregulated in glioblastoma (48), selective transporter inhibitors might be useful treatment options. Indeed, several studies have reported beneficial effects of sulfasalazine, an inhibitor of systemic Xc−-mediated cystine uptake (92, 93). However, in a small clinical trial, sulfasalazine was without benefit in recurrent glioblastoma (94). In addition, sulfasalazine did not improve the outcomes of combined temozolomide and radiotherapy in patients with newly diagnosed glioblastoma (95). This therapeutic failure may be due to Nrf2-mediated upregulation of Xct induced by temozolomide (96), offsetting the inhibitory effects of sulfasalazine (92). Erastin, another Xct inhibitor, was reported to be superior to sulfasalazine for induction of cell death when administered in combination with temozolomide to C6 glioma cells, owing to its ability to decrease the expression of the Nrf2-induced transsulfuration enzyme cystathionine-γ-lyase (92), thereby depleting GSH formation via two mechanisms.
Inhibition of GSH synthesis can be compensated for by Nrf2-induced upregulation of the NADPH-dependent thioredoxin pathway (20). However, combined treatment with inhibitors of GSH synthesis (e.g., buthionine sulfoximine) and thioredoxin reductase (e.g., auranofin) decreased tumor survival in A172 glioblastoma cells (20). Although a combination of GSH and thioredoxin pathway inhibitors was efficacious in vitro, it may be unsuitable for patients considering the potential safety and tolerability of this combination. However, it may be possible to limit its toxicity by utilizing targeted drug delivery techniques.
Conclusion
We have discussed the roles of redox and epigenetic regulation in glioblastoma. The findings described here suggest that glioblastoma is sustained by an intricate network of Nrf2, receptor tyrosine kinases, cobalamins and cobalamin-dependent enzymes, along with the D4 receptor and other factors, such as cystine/cysteine transporters and TET enzymes (Figure 5). Mutations in receptor tyrosine kinases or their downstream targets can result in hyperactivity of the PI3K and ERK pathways, leading to constitutively higher Nrf2 activity. Higher Nrf2 activity induces overexpression of genes involved in antioxidant production, such as cystine/cysteine transporters and promotes 2-HG formation in IDH1 mutants. Nrf2 can also upregulate its own activity by promoting p62 transcription. The excessively reduced redox state of glioblastoma cells enhances the uptake and processing of cobalamins and promotes the activity of redox-sensitive proteins (i.e., HDAC2). In methionine-independent cells, the cobalamin-dependent enzyme, MS, enhances the formation of SAM, which influences methyltransferase activity. The other cobalamin-dependent enzyme in humans, MCM, promotes the formation of oncometabolites, affecting a wide range of proteins, including TET1-3, KDM2-7, prolyl hydroxylases, and Keap1. D4 receptors may contribute to this network by regulating MS activity via stimulation of the ERK/PI3K pathways, either directly or indirectly by transactivation of PDGFRβ. These interactions result in an amalgamation of epigenetic changes, which contribute to the induction of the cancer phenotype and drive tumor growth. Indeed, the model described for glioblastoma can be applied to all cancers, and we hope that the novel concepts presented here will spur new research in this context.
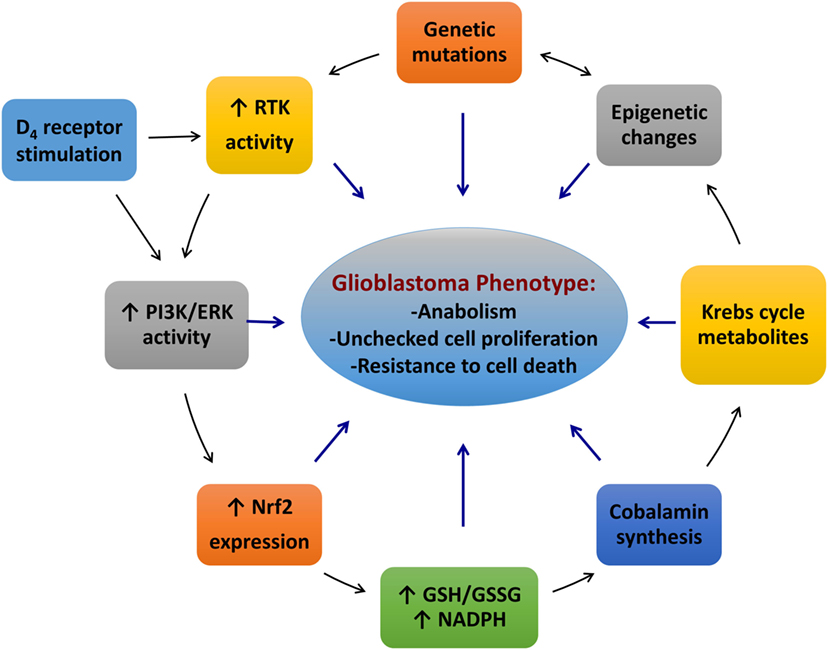
Figure 5. An interactive network of the redox and methylation factors contributing to the glioblastoma phenotype. Abbreviations: ERK, extracellular signal-regulated kinase; GSH, glutathione; GSSG, glutathione disulfide; Nrf2, nuclear factor (erythroid-derived 2)-like 2; PI3K, phosphoinositide 3-kinase.
Although existing treatment options for glioblastoma have not achieved clinically meaningful remission rates, epigenetic- and redox-modifying agents may augment the repertoire of available life-saving drugs. HDAC inhibitors have demonstrated utility in treating certain forms of cancer. However, these drugs may reactivate a DNA repair mechanism in glioblastoma. BET inhibitors may be immune to this unwanted effect of HDAC inhibitors. Luteolin and related drugs may sensitize glioblastoma cells to the effects of radiotherapy or chemotherapy and mitigate damage to surrounding neurons and non-cancerous glia. The ability of D4 receptor antagonists to induce growth arrest and apoptosis of stem cells is an encouraging development and suggests that a better understanding of the relationships between cancer metabolism, redox status, and epigenetics may lead to significantly higher remission rates. Mutations in redox and methylation pathway genes may complement canonical mutations for existing glioblastoma subtypes and could be incorporated into a panel for pharmacogenomic screening for the optimization of drug treatment. We envision an expansion in the near future of drugs targeting antioxidant production and related biochemical and molecular pathways, offering hope of improved treatment outcomes for glioblastoma.
Author Contributions
MS, MT, and RD reviewed relevant literature, drafted and revised the manuscript, designed the figures, and read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer (2007) 110:13–24. doi:10.1002/cncr.22741
2. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (2016) 131:803–20. doi:10.1007/s00401-016-1545-1
3. Kotliarova S, Fine HA. SnapShot: glioblastoma multiforme. Cancer Cell (2012) 21:710–710.e1. doi:10.1016/j.ccr.2012.04.031
4. Clarke J, Penas C, Pastori C, Komotar RJ, Bregy A, Shah AH, et al. Epigenetic pathways and glioblastoma treatment. Epigenetics (2013) 8:785–95. doi:10.4161/epi.25440
5. Stupp R, Mason WP, Van Den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med (2005) 352:987–96. doi:10.1056/NEJMoa043330
6. Lau D, Magill ST, Aghi MK. Molecularly targeted therapies for recurrent glioblastoma: current and future targets. Neurosurg Focus (2014) 37:E15. doi:10.3171/2014.9.FOCUS14519
7. Mummaneni P, Shord SS. Epigenetics and oncology. Pharmacother J Hum Pharmacol Drug Ther (2014) 34:495–505. doi:10.1002/phar.1408
8. Pastori C, Kapranov P, Penas C, Peschansky V, Volmar C-H, Sarkaria JN, et al. The Bromodomain protein BRD4 controls HOTAIR, a long noncoding RNA essential for glioblastoma proliferation. Proc Natl Acad Sci U S A (2015) 112:8326–31. doi:10.1073/pnas.1424220112
9. Li G, Warden C, Zou Z, Neman J, Krueger JS, Jain A, et al. Altered expression of polycomb group genes in glioblastoma multiforme. PLoS One (2013) 8:e80970. doi:10.1371/journal.pone.0080970
10. Pastori C, Daniel M, Penas C, Volmar C-H, Johnstone AL, Brothers SP, et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics (2014) 9:611–20. doi:10.4161/epi.27906
11. Morin A, Letouzé E, Gimenez-Roqueplo A-P, Favier J. Oncometabolites-driven tumorigenesis: from genetics to targeted therapy. Int J Cancer (2014) 135:2237–48. doi:10.1002/ijc.29080
12. Dolma S, Selvadurai HJ, Lan X, Lee L, Kushida M, Voisin V, et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell (2016) 29:859–73. doi:10.1016/j.ccell.2016.05.002
13. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev (2013) 27:2179–91. doi:10.1101/gad.225680.113
14. Baldelli S, Aquilano K, Ciriolo MR. Punctum on two different transcription factors regulated by PGC-1α: nuclear factor erythroid-derived 2-like 2 and nuclear respiratory factor 2. Biochim Biophys Acta (2013) 1830:4137–46. doi:10.1016/j.bbagen.2013.04.006
15. Holmstrom KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open (2013) 2:761–70. doi:10.1242/bio.20134853
16. Shelton P, Jaiswal AK. The transcription factor NF-E2-related Factor 2 (Nrf2): a protooncogene? FASEB J (2013) 27:414–23. doi:10.1096/fj.12-217257
17. Son Y-O, Pratheeshkumar P, Roy RV, Hitron JA, Wang L, Zhang Z, et al. Nrf2/p62 signaling in apoptosis resistance and its role in cadmium-induced carcinogenesis. J Biol Chem (2014) 289:28660–75. doi:10.1074/jbc.M114.595496
18. Ichimura Y, Waguri S, Sou Y, Kageyama S, Hasegawa J, Ishimura R, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell (2013) 51:618–31. doi:10.1016/j.molcel.2013.08.003
19. Zhou S, Ye W, Shao Q, Zhang M, Liang J. Nrf2 is a potential therapeutic target in radioresistance in human cancer. Crit Rev Oncol Hematol (2013) 88:706–15. doi:10.1016/j.critrevonc.2013.09.001
20. Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell (2015) 27:211–22. doi:10.1016/j.ccell.2014.11.019
21. Ito K, Hanazawa T, Tomita K, Barnes P, Adcock I. Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun (2004) 315:240–5. doi:10.1016/j.bbrc.2004.01.046
22. Singh A, Happel C, Manna SK, Acquaah-Mensah G, Carrerero J, Kumar S, et al. Transcription factor NRF2 regulates miR-1 and miR-206 to drive tumorigenesis. J Clin Invest (2013) 123:2921–34. doi:10.1172/JCI66353
23. Hurd TR, Prime TA, Harbour ME, Lilley KS, Murphy MP. Detection of reactive oxygen species-sensitive thiol proteins by redox difference gel electrophoresis: implications for mitochondrial redox signaling. J Biol Chem (2007) 282:22040–51. doi:10.1074/jbc.M703591200
24. Lo S-C, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res (2008) 314:1789–803. doi:10.1016/j.yexcr.2008.02.014
25. Lo S-C, Hannink M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem (2006) 281:37893–903. doi:10.1074/jbc.M606539200
26. Ji X, Wang H, Zhu J, Tang Y, Zhou Y, Zhu L, et al. Correlation of Nrf2 and HIF-1α in glioblastoma and their relationships to clinicopathologic features and survival. Neurol Res (2013) 35:1044–50. doi:10.1179/1743132813Y.0000000251
27. Kanamori M, Higa T, Sonoda Y, Murakami S, Dodo M, Kitamura H, et al. Activation of the NRF2 pathway and its impact on the prognosis of anaplastic glioma patients. Neuro Oncol (2015) 17:555–65. doi:10.1093/neuonc/nou282
28. Kim J, Gherasim C, Banerjee R. Decyanation of vitamin B12 by a trafficking chaperone. Proc Natl Acad Sci U S A (2008) 105:14551–4. doi:10.1073/pnas.0805989105
29. Gherasim C, Lofgren M, Banerjee R. Navigating the B12 road: assimilation, delivery, and disorders of cobalamin. J Biol Chem (2013) 288:13186–93. doi:10.1074/jbc.R113.458810
30. Jiang W, Sequeira JM, Nakayama Y, Lai S-C, Quadros EV. Characterization of the promoter region of TCblR/CD320 gene, the receptor for cellular uptake of transcobalamin-bound cobalamin. Gene (2010) 466:49–55. doi:10.1016/j.gene.2010.07.004
31. Flodh H, Ullberg S. Accumulation of labelled vitamin B12 in some transplanted tumours. Int J Cancer (1968) 3:694–9. doi:10.1002/ijc.2910030518
32. Sysel AM, Valli VE, Bauer JA. Immunohistochemical quantification of the cobalamin transport protein, cell surface receptor and Ki-67 in naturally occurring canine and feline malignant tumors and in adjacent normal tissues. Oncotarget (2015) 6:2331–48. doi:10.18632/oncotarget.3206
33. Kim J, Hannibal L, Gherasim C, Jacobsen DW, Banerjee RA. Human vitamin B12 trafficking protein uses glutathione transferase activity for processing alkylcobalamins. J Biol Chem (2009) 284:33418–24. doi:10.1074/jbc.M109.057877
34. Oak JN, Lavine N, Van Tol HHM. Dopamine D4 and D2L receptor stimulation of the mitogen-activated protein kinase pathway is dependent on trans-activation of the platelet-derived growth factor receptor. Mol Pharmacol (2001) 60:92–103. doi:10.1124/mol.60.1.92
35. Zhang Y, Hodgson N, Trivedi M, Deth R. Neuregulin 1 promotes glutathione-dependent neuronal cobalamin metabolism by stimulating cysteine uptake. Oxid Med Cell Longev (2016) 2016:1–13. doi:10.1155/2016/3849087
36. Escartin C, Joon Won S, Malgorn C, Auregan G, Berman AE, Chen P-C, et al. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J Neurosci (2011) 31:7392–401. doi:10.1523/JNEUROSCI.6577-10.2011
37. Cong Z-X, Wang H-D, Wang J-W, Zhou Y, Pan H, Zhang D-D, et al. ERK and PI3K signaling cascades induce Nrf2 activation and regulate cell viability partly through Nrf2 in human glioblastoma cells. Oncol Rep (2013) 30:715–22. doi:10.3892/or.2013.2485
38. Yu H-N, Park W-K, Nam K-H, Song D-Y, Kim H-S, Baik T-K, et al. Neuregulin 1 controls glutamate uptake by upregulating excitatory amino acid carrier 1 (EAAC1). J Biol Chem (2015) 290:20233–44. doi:10.1074/jbc.M114.591867
39. Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol (2012) 2:200. doi:10.3389/fonc.2012.00200
40. Agyeman AS, Chaerkady R, Shaw PG, Davidson NE, Visvanathan K, Pandey A, et al. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat (2012) 132:175–87. doi:10.1007/s10549-011-1536-9
41. Arnér ESJ. Focus on mammalian thioredoxin reductases – important selenoproteins with versatile functions. Biochim Biophys Acta (2009) 1790:495–526. doi:10.1016/j.bbagen.2009.01.014
42. Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell (2012) 21:297–308. doi:10.1016/j.ccr.2012.02.014
43. Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem (2002) 277:44765–71. doi:10.1074/jbc.M208704200
44. Shi J, He Y, Hewett SJ, Hewett JA. Interleukin 1β regulation of the system xc-substrate-specific subunit, xCT, in primary mouse astrocytes involves the RNA-binding protein HuR. J Biol Chem (2016) 291:1643–51. doi:10.1074/jbc.M115.697821
45. No JH, Kim Y-B, Song YS. Targeting Nrf2 signaling to combat chemoresistance. J Cancer Prev (2014) 19:111–7. doi:10.15430/JCP.2014.19.2.111
46. Hayes JD, Ashford MLJ. Nrf2 orchestrates fuel partitioning for cell proliferation. Cell Metab (2012) 16:139–41. doi:10.1016/j.cmet.2012.07.009
47. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer (2011) 11:85–95. doi:10.1038/nrc2981
48. Tsuchihashi K, Okazaki S, Ohmura M, Ishikawa M, Sampetrean O, Onishi N, et al. The EGF receptor promotes the malignant potential of glioma by regulating amino acid transport system xc(–). Cancer Res (2016) 76:2954–63. doi:10.1158/0008-5472.CAN-15-2121
49. Cellarier E, Durando X, Vasson M, Farges M, Demiden A, Maurizis J, et al. Methionine dependency and cancer treatment. Cancer Treat Rev (2003) 29:489–99. doi:10.1016/S0305-7372(03)00118-X
50. Loewy AD, Niles KM, Anastasio N, Watkins D, Lavoie J, Lerner-Ellis JP, et al. Epigenetic modification of the gene for the vitamin B12 chaperone MMACHC can result in increased tumorigenicity and methionine dependence. Mol Genet Metab (2009) 96:261–7. doi:10.1016/j.ymgme.2008.12.011
51. Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab (2015) 22:861–73. doi:10.1016/j.cmet.2015.08.024
52. Lu SC, Mato JM. S-adenosylmethionine in liver health, injury, and cancer. Physiol Rev (2012) 92:1515–42. doi:10.1152/physrev.00047.2011
53. Jiang R, Choi W, Hu L, Gerner EW, Hamilton SR, Zhang W. Activation of polyamine catabolism by N1, N11-diethylnorspermine alters the cellular localization of mTOR and downregulates mTOR protein level in glioblastoma cells. Cancer Biol Ther (2007) 6:1644–8. doi:10.4161/cbt.6.10.4800
54. Yang Y, Bedford MT. Protein arginine methlytransferases and cancer. Nat Rev Cancer (2013) 13:37–50. doi:10.1038/nrc3409
55. Liu X, Li H, Liu L, Lu Y, Gao Y, Geng P, et al. Methylation of arginine by PRMT1 regulates Nrf2 transcriptional activity during the antioxidative response. Biochim Biophys Acta (2016) 1863:2093–103. doi:10.1016/j.bbamcr.2016.05.009
56. Guo Y, Yu S, Zhang C, Kong A-NT. Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic Biol Med (2015) 88:337–49. doi:10.1016/j.freeradbiomed.2015.06.013
57. Nowicki S, Gottlieb E. Oncometabolites: tailoring our genes. FEBS J (2015) 282:2796–805. doi:10.1111/febs.13295
58. Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov (2013) 3:648–57. doi:10.1158/2159-8290.CD-13-0092
59. Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell (2013) 23:739–52. doi:10.1016/j.ccr.2013.04.018
60. Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet (2014) 23:2440–6. doi:10.1093/hmg/ddt639
61. Raabe EH, Eberhart CG. Methylome alterations “Mark” new therapeutic opportunities in glioblastoma. Cancer Cell (2012) 22:417–8. doi:10.1016/j.ccr.2012.10.001
62. Ohno M, Narita Y, Miyakita Y, Matsushita Y, Arita H, Yonezawa M, et al. Glioblastomas with IDH1/2 mutations have a short clinical history and have a favorable clinical outcome. Jpn J Clin Oncol (2016) 46:31–9. doi:10.1093/jjco/hyv170
63. Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung Y-L, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell (2005) 8:143–53. doi:10.1016/j.ccr.2005.06.017
64. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell (2005) 7:77–85. doi:10.1016/j.ccr.2004.11.022
65. Ooi A, Furge KA. Fumarate hydratase inactivation in renal tumors: HIF1 alpha, NRF2, and “cryptic targets” of transcription factors. Chin J Cancer (2012) 31:413–20. doi:10.5732/cjc.012.10102
66. Zhou Y, Zhou Y, Shingu T, Feng L, Chen Z, Ogasawara M, et al. Metabolic alterations in highly tumorigenic glioblastoma cells: preference for hypoxia and high dependency on glycolysis. J Biol Chem (2011) 286:32843–53. doi:10.1074/jbc.M111.260935
67. Nagy A, Eder K, Selak MA, Kalman B. Mitochondrial energy metabolism and apoptosis regulation in glioblastoma. Brain Res (2015) 1595:127–42. doi:10.1016/j.brainres.2014.10.062
68. Salminen A, Haapasalo A, Kauppinen A, Kaarniranta K, Soininen H, Hiltunen M. Impaired mitochondrial energy metabolism in Alzheimer’s disease: impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Prog Neurobiol (2015) 131:1–20. doi:10.1016/j.pneurobio.2015.05.001
69. Kozono D, Li J, Nitta M, Sampetrean O, Gonda D, Kushwaha DS, et al. Dynamic epigenetic regulation of glioblastoma tumorigenicity through LSD1 modulation of MYC expression. Proc Natl Acad Sci U S A (2015) 112:E4055–64. doi:10.1073/pnas.1501967112
70. Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature (2010) 466:1129–33. doi:10.1038/nature09303
71. Scourzic L, Mouly E, Bernard OA. TET proteins and the control of cytosine demethylation in cancer. Genome Med (2015) 7:9. doi:10.1186/s13073-015-0134-6
72. Haffner MC, Chaux A, Meeker AK, Esopi DM, Gerber J, Pellakuru LG, et al. Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget (2011) 2:627–37. doi:10.18632/oncotarget.316
73. Bardella C, Al-Dalahmah O, Krell D, Brazauskas P, Al-Qahtani K, Tomkova M, et al. Expression of Idh1R132H in the murine subventricular zone stem cell niche recapitulates features of early gliomagenesis. Cancer Cell (2016) 30:578–94. doi:10.1016/j.ccell.2016.08.017
74. Ago T, Liu T, Zhai P, Chen W, Li H, Molkentin JD, et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell (2008) 133:978–93. doi:10.1016/j.cell.2008.04.041
75. Lucio-Eterovic AK, Cortez MA, Valera ET, Motta FJ, Queiroz RG, Machado HR, et al. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer (2008) 8:243. doi:10.1186/1471-2407-8-243
76. Sharma A, Kramer ML, Wick PF, Liu D, Chari S, Shim S, et al. D 4 dopamine receptor-mediated phospholipid methylation and its implications for mental illnesses such as schizophrenia. Mol Psychiatry (1999) 4:235–46. doi:10.1038/sj.mp.4000522
77. Manglik A, Kim TH, Masureel M, Altenbach C, Yang Z, Hilger D, et al. Structural insights into the dynamic process of β2-adrenergic receptor signaling. Cell (2015) 161:1101–11. doi:10.1016/j.cell.2015.04.043
78. Zhao R, Chen Y, Tan W, Waly M, Sharma A, Stover P, et al. Relationship between dopamine-stimulated phospholipid methylation and the single-carbon folate pathway. J Neurochem (2001) 78:788–96. doi:10.1046/j.1471-4159.2001.00471.x
79. Waly M, Olteanu H, Banerjee R, Choi S-W, Mason JB, Parker BS, et al. Activation of methionine synthase by insulin-like growth factor-1 and dopamine: a target for neurodevelopmental toxins and thimerosal. Mol Psychiatry (2004) 9:358–70. doi:10.1038/sj.mp.4001476
80. Kuznetsova AY, Deth RC. A model for modulation of neuronal synchronization by D4 dopamine receptor-mediated phospholipid methylation. J Comput Neurosci (2008) 24:314–29. doi:10.1007/s10827-007-0057-3
81. Sakakibara K, Eiyama A, Suzuki SW, Sakoh-Nakatogawa M, Okumura N, Tani M, et al. Phospholipid methylation controls Atg32-mediated mitophagy and Atg8 recycling. EMBO J (2015) 34:2703–19. doi:10.15252/embj.201591440
82. Laxman S, Sutter BM, Tu BP. Methionine is a signal of amino acid sufficiency that inhibits autophagy through the methylation of PP2A. Autophagy (2014) 10:386–7. doi:10.4161/auto.27485
83. Chen J, Wang Q, Yin F-Q, Zhang W, Yan L-H, Li L. MTRR silencing inhibits growth and cisplatin resistance of ovarian carcinoma via inducing apoptosis and reducing autophagy. Am J Transl Res (2015) 7:1510–27.
84. Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res (2013) 19:1748–59. doi:10.1158/1078-0432.CCR-12-3066
85. Wadhwa E, Nicolaides T. Bromodomain inhibitor review: bromodomain and extra-terminal family protein inhibitors as a potential new therapy in central nervous system tumors. Cureus (2016) 8:e620. doi:10.7759/cureus.620
86. Bezecny P. Histone deacetylase inhibitors in glioblastoma: pre-clinical and clinical experience. Med Oncol (2014) 31:985. doi:10.1007/s12032-014-0985-5
87. Plotkin A, Volmar C-H, Wahlestedt C, Ayad N, El-Ashry D. Transcriptional repression of ER through hMAPK dependent histone deacetylation by class I HDACs. Breast Cancer Res Treat (2014) 147:249–63. doi:10.1007/s10549-014-3093-5
88. Kitange GJ, Mladek AC, Carlson BL, Schroeder MA, Pokorny JL, Cen L, et al. Inhibition of histone deacetylation potentiates the evolution of acquired temozolomide resistance linked to MGMT upregulation in glioblastoma xenografts. Clin Cancer Res (2012) 18:4070–9. doi:10.1158/1078-0432.CCR-12-0560
89. Tang X, Wang H, Fan L, Wu X, Xin A, Ren H, et al. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med (2011) 50:1599–609. doi:10.1016/j.freeradbiomed.2011.03.008
90. Chakrabarti M, Ray SK. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis (2016) 21:312–28. doi:10.1007/s10495-015-1198-x
91. Xu J, Wang H, Ding K, Zhang L, Wang C, Li T, et al. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol Med (2014) 71:186–95. doi:10.1016/j.freeradbiomed.2014.03.009
92. Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol Rep (2015) 33:1465–74. doi:10.3892/or.2015.3712
93. Sleire L, Skeie BS, Netland IA, Forde HE, Dodoo E, Selheim F, et al. Drug repurposing: sulfasalazine sensitizes gliomas to gamma knife radiosurgery by blocking cystine uptake through system Xc-, leading to glutathione depletion. Oncogene (2015) 34:5951–9. doi:10.1038/onc.2015.60
94. Robe PA, Martin DH, Nguyen-Khac MT, Artesi M, Deprez M, Albert A, et al. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of progressing malignant gliomas in adults. BMC Cancer (2009) 9:372. doi:10.1186/1471-2407-9-372
95. Takeuchi S, Wada K, Nagatani K, Otani N, Osada H, Nawashiro H. Sulfasalazine and temozolomide with radiation therapy for newly diagnosed glioblastoma. Neurol India (2014) 62:42–7. doi:10.4103/0028-3886.128280
Keywords: cobalamin, D4 dopamine receptor, glioblastoma, Nrf2, DNA methylation, glutathione, methionine synthase, transsulfuration
Citation: Schrier MS, Trivedi MS and Deth RC (2017) Redox-Related Epigenetic Mechanisms in Glioblastoma: Nuclear Factor (Erythroid-Derived 2)-Like 2, Cobalamin, and Dopamine Receptor Subtype 4. Front. Oncol. 7:46. doi: 10.3389/fonc.2017.00046
Received: 19 January 2017; Accepted: 06 March 2017;
Published: 30 March 2017
Edited by:
Nader Sanai, Barrow Neurological Institute, USAReviewed by:
Carsten Friedrich, University of Rostock, GermanyKamalakannan Palanichamy, Ohio State University at Columbus, USA
Copyright: © 2017 Schrier, Trivedi and Deth. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard Carlton Deth, cmRldGhAbm92YS5lZHU=