 Eduardo Anguita
Eduardo Anguita Francisco J. Candel
Francisco J. Candel Alberto Chaparro
Alberto Chaparro Juan J. Roldán-Etcheverry
Juan J. Roldán-Etcheverry- 1Hematology Department, Hospital Clínico San Carlos, Instituto de Investigación Sanitaria San Carlos (IdISSC), Madrid, Spain
- 2Department of Medicine, Universidad Complutense de Madrid (UCM), Madrid, Spain
- 3Microbiology Department, Hospital Clínico San Carlos, Instituto de Investigación Sanitaria San Carlos (IdISSC), Madrid, Spain
Many human diseases arise through dysregulation of genes that control key cell fate pathways. Transcription factors (TFs) are major cell fate regulators frequently involved in cancer, particularly in leukemia. The GFI1B gene, coding a TF, was identified by sequence homology with the oncogene growth factor independence 1 (GFI1). Both GFI1 and GFI1B have six C-terminal C2H2 zinc fingers and an N-terminal SNAG (SNAIL/GFI1) transcriptional repression domain. Gfi1 is essential for neutrophil differentiation in mice. In humans, GFI1 mutations are associated with severe congenital neutropenia. Gfi1 is also required for B and T lymphopoiesis. However, knockout mice have demonstrated that Gfi1b is required for development of both erythroid and megakaryocytic lineages. Consistent with this, human mutations of GFI1B produce bleeding disorders with low platelet count and abnormal function. Loss of Gfi1b in adult mice increases the absolute numbers of hematopoietic stem cells (HSCs) that are less quiescent than wild-type HSCs. In keeping with this key role in cell fate, GFI1B is emerging as a gene involved in cancer, which also includes solid tumors. In fact, abnormal activation of GFI1B and GFI1 has been related to human medulloblastoma and is also likely to be relevant in blood malignancies. Several pieces of evidence supporting this statement will be detailed in this mini review.
Hematopoiesis, the process of formation of blood cellular components, provides an ideal model for cell differentiation of particular interest, as maturation block plays a crucial role in the pathogenesis of blood malignancies. It is regulated by transcription factors (TFs) that control gene expression, binding to specific DNA sequences and recruiting protein complexes that modify chromatin. Therefore, TFs are essential to establish a homeostatic balance, and they also play an essential role in disease.
Growth factor independence 1 (GFI1) and its homolog GFI1B are lineage-specific TFs required for hematopoiesis. GFI1 was discovered as a proviral integration site that confers IL-2-independency in T-cells infected by Moloney murine leukemia virus and was mapped to chromosome 1 of the human genome (1p22) (1, 2) (Figure 1A). The GFI1 paralog—GFI1B—was identified by its sequence homology with GFI1 and mapped to chromosome 9 of the human genome (9q34.13) (3, 4).
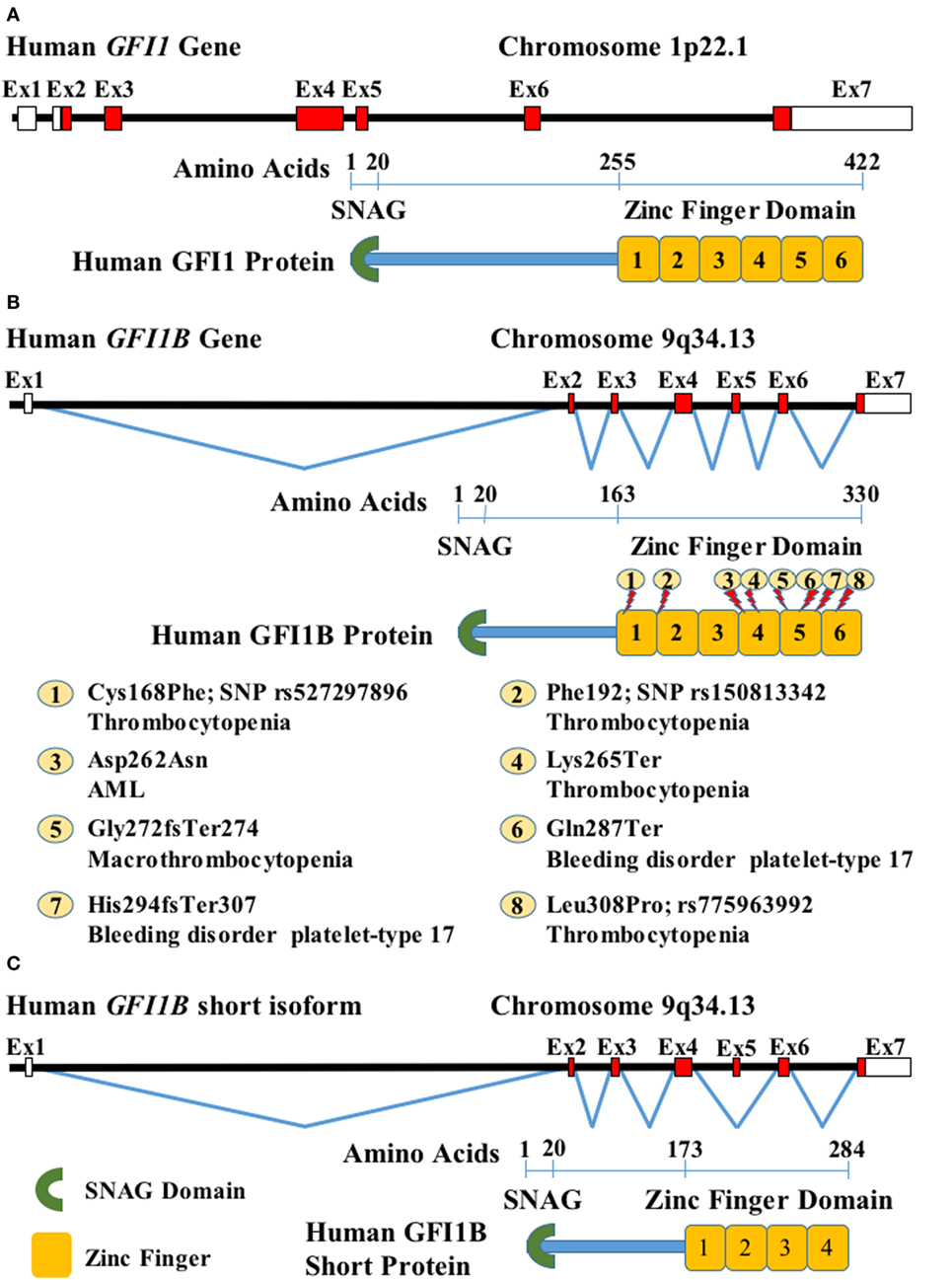
Figure 1. Schematic representation of human growth factor independence 1 (GFI1) and GFI1B genes and corresponding protein domains: (A) diagram of human GFI1 locus and protein. On top, the black horizontal line represents the DNA sequence, red boxes on the sequence are coding GFI1 exons (Ex), and white boxes indicate UTRs. Underneath, GFI1 protein domains are indicated (green crescent represents the SNAG domain, and orange boxes are zinc fingers), the numbers over the blue rule indicate the amino acid positions; (B) GFI1B locus and long protein isoforms are depicted as in panel (A). The zigzag lines joining the Ex represent the splicing. Known mutations are shown on the protein and briefly described [acute myeloid leukemia (AML)]. (C) Short GFI1B splice variant and protein represented as in panels (A,B).
GFI1 and GFI1B Structure and Function
Growth factor independence 1 and GFI1B share two domains with over 95% identity. The conserved N-terminal SNAG domain contains 20 amino acids, which recruit proteins that modify histones (4–6). This domain has a nuclear localization motif and plays an important role in transcriptional repression through the binding of cofactors lysine-specific histone demethylase 1A (KDM1A, also known as LSD1) and RCOR1/2 (COREST) (6–8). However, the SNAG domain is not required for GFI1 interaction with corepressors euchromatic histone lysine methyltransferase 2 (EHMT2, G9A) and histone deacetylase HDAC 1 (9), suggesting that different portions of these proteins cooperate in gene repression, possibly with different peculiarities in each TF.
The C-terminal domain is formed by a highly conserved region with six C2H2 zinc fingers. Fingers 1, 2, and 6 are required for protein interaction, whereas fingers 3–5 are necessary to bind DNA at an AATC containing motif [TA AATCAC(T/A)GC(A/T)] (10, 11).
Between both domains, there is a less-characterized region that completely differs in both proteins, the function of which is still unknown.
This region is responsible for the size difference in both proteins: GFI1 has 422 amino acids (55 kDa), while GFI1B consists of 330 amino acids (37 kDa, CCDS6957) (Figure 1B). There is also a short 284 amino acid GFI1B isoform (CCDS48049) (Figure 1C) that lacks the first two zinc-finger domains as a result of an alternative splicing, skipping exon 5 (ENST00000372123.4).
Although GFI1 and GFI1B are similar in structure and share functional mechanisms, they show distinct cell expression patterns. Both GFI1 and GFI1B have an important role in the endothelial cell to hematopoiesis transition, the process by which endothelial cells become blood cells during the third wave of blood development. This generates the first hematopoietic stem cells (HSCs) in the intraembryonic aorta–gonad–mesonephros region, silencing the endothelial program. Interestingly, the expression pattern of both genes is different: Gfi1 is specifically expressed within the dorsal aorta in endothelial cells and cells within emerging intra-aortic hematopoietic clusters, whereas Gfi1b expression is more associated with the fully formed intra-aortic hematopoietic clusters (12). This suggests that although both proteins can apparently compensate for the loss of one gene by the other, they play unique differential roles in vivo.
Knockout (KO) mice have shown that Gfi1 is essential for neutrophil differentiation (13, 14); consistently, in humans, severe congenital neutropenia is associated with GFI1 mutations (15). Gfi1 is also required for B and T lymphopoiesis (Figure 2). Besides being expressed in the hematopoietic system, Gfi1 is also expressed in precursors of sensory neurons, the retina, specific lung cells, and in the central nervous system (16).
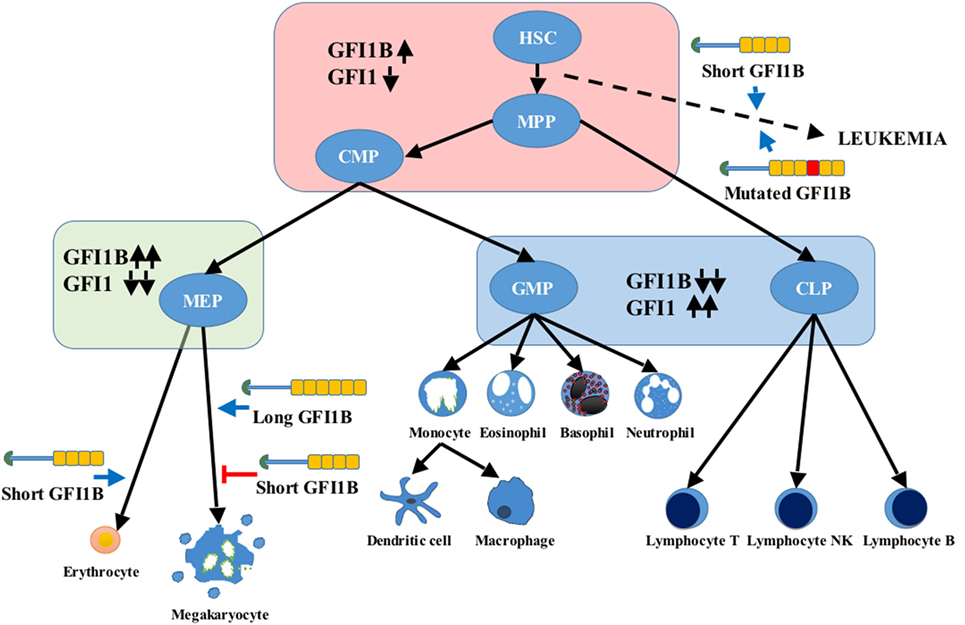
Figure 2. Differential expression of growth factor independence 1 (GFI1) and GFI1B during hematopoiesis. Upward pointing black arrows indicate high expression, and downward pointing black arrows represent low expression. The long and short GFI1B isoforms are represented as in Figure 1. The blue arrows indicate a positive impact on cell differentiation, and the red line reflects an inhibitory effect. Several evidences point to the role of GFI1B mutations and increase expression of the short variant in leukemogenesis (dashed arrow). HSC, hematopoietic stem cell; MPP, multipotent progenitor; CMP, common myeloid progenitor; MEP, megakaryocyte and erythroid progenitor; GMP, granulocyte and monocyte progenitor; CLP, common lymphoid progenitor.
GFI1B is expressed in HSCs, common myeloid and megakaryocyte/erythroid progenitors, and erythroid and megakaryocytic lineages. Moderate levels of expression are also found in immature B-cells, a subset of early T-cell precursors (17, 18) and peripheral blood granulocytes and monocytes. GFI1B is very low or absent in lymphoid-primed multipotent, common lymphoid, early thymocyte, and granulocyte–monocyte progenitors (19) (Figure 2).
GFI1B role in erythropoiesis is crucial for expansion and differentiation of erythroid progenitors. Gfi1b deficiency in mice results in embryonic lethality by day E15 (20). Gfi1b null embryos display a delay maturation of erythrocytes and die because of the lack of enucleated erythrocytes. Gfi1b KO mice also fail to develop megakaryocytes but have arrested erythroid and megakaryocytic precursors in the fetal liver. Loss of Gfi1b in adult mice increases the absolute numbers of HSCs that are less quiescent than wild-type ones (21), ablates erythroid development at an early progenitor stage, and blocks terminal megakaryocytic differentiation in the polyploid promegakaryocytes that fail to produce platelets (22). Lineage-specific KOs have shown that the role of Gfi1b in megakaryocyte polyploidization and motility can be achieved by inhibiting p21 activated kinases, and the effect on proplatelet formation, by controlling α-tubulin expression, which is highly decreased in the KO cells (23).
The short GFI1B variant seems important for erythroid development and to show a stronger repressor activity than the long one (24). Instead, hyperexpression and knockdown experiments in human primary cells have shown that the long GFI1B is required for megakaryopoiesis, but not the short form (25), which may have an inhibitory effect on platelet production (26) (Figure 2).
It has also been observed that the absence of Gfi1 and Gfi1b expression produces a severe block in B-cell development. On the contrary, in vitro overexpression of Gfi1b inhibits myeloid differentiation of a cultured myelomonocytic cell line (4).
However, GFI1 and GFI1B are not just different in terms of cell specificity, as demonstrated by sequence interchange (27). Consistent with this, Gfi1 hyperexpression can rescue erythroid and early megakaryocytic differentiation from adult mouse Gfi1b KO, but terminal megakaryocyte maturation defect cannot be compensated by Gfi1 or Gfi1b hybrid containing the Gfi1 N-terminal portion (22). These differences are more patent in the inner ear than in hematopoietic cells (27).
GFI1B Networks
At the DNA level, GFI1B represses the following: CDKN1A (4); proto-oncogenes myc, myb (28); GFI1 (29) and Meis1 (7); tumor suppressor genes Socs1 and Socs2 (30); the antiapoptotic gene BCL2L1 (BCXL) (31); TGFβR3 (32); GFI1B itself (29); T lymphopoiesis regulator GATA3 (33); hematopoietic master regulators SPI1 (PU.1) (34); and GATA2 (19), among other genes such as MGEA5, CSDE1, GABPB2, and ITSN1 (24). Recent insights into the role of Gfi1b in megakaryopoiesis have revealed new targets repressed by this TF. These include the regulator of G-protein signaling 18, which is differentially regulated in erythroid and megakaryocytic cells, with low expression in the former and high expression in the latter (35). It also represses Fermt3 (Kindlin3) required for integrin-mediated platelet adhesion and Talin1, involved in integrin–cytoskeleton connections (36). This is consistent with observations in the megakaryocyte-specific KO model, indicating the importance of Gfi1b control of cytoskeletal and integrin-binding proteins (23).
Besides its repressive function, GFI1B may directly or indirectly activate gene expression; for example, MEF2C upregulation in T-cells by GFI1B binding to this gene promoter and MLLT3, whose expression correlates with GFI1B hyperexpression or functional block (34).
At the protein level, GFI1B interacts with hematopoietic TFs such as SCL, E2A, and GATA1 and with corepressors RUNX1T1 (ETO), CBFA2T3 (ETO2) (37–39), SUV39H1 (40), KDM1A, RCOR1/2, HDACs, and EHMT2.
These findings suggest that GFI1B plays a major role in hematopoiesis. Its importance is also reflected by the tight control of its expression. We have identified several evolutionary conserved non-coding elements (CNEs) containing multiple erythroid/megakaryocytic-specific TF-binding sequences, three downstream and one in the first intron (37). Chromatin immunoprecipitation studies in human and mouse demonstrated that GATA1, TAL1 (SCL), NFE2, LDB1, LMO2, SPI1 (PU.1), MYB (37), TCF3 (E2A) (33, 37), and GATA2 (19, 41) are bound to the promoter and/or these distant regulatory elements in vivo in human and mouse. This is particularly significant at CNEs +1 and +3, corresponding to mouse DNase1 hypersensitivity sites +13 and +17 (coordinates in relation to the ATG start codon in Kb) (37). These downstream elements have been validated as hematopoietic enhancers in transgenic mouse assays (19). Interestingly, these sites also bind repressors and corepressors GFI1B, CBFA2T3 (ETO2), RCOR1 (COREST), and KDM1A and could behave as negative regulatory elements depending on the GFI1B levels (37).
Platelet Deficiencies and Bleeding Disorders
Consistent with the Gfi1b KO effect on megakaryopoiesis (20, 22, 23), different mutations in GFI1B are involved in platelet-related bleeding disorders (42).
A mutation described by Stevenson et al. (43), which consists of a single-nucleotide insertion in GFI1B exon 7 (c.880_881insC) that produces a frameshift and protein premature termination (His294fsTer307, g.135866324dupC in GRCh37/hg19 assembly) (Figure 1B: 7), disrupts the integrity of the fifth zinc finger and eliminates the coding sequence for the sixth zinc-finger domain. The mutated protein cannot bind to DNA and loses its repressor activity on target genes. This mutation was described in patients with an inherited dominant bleeding disorder with moderate macrothrombocytopenia and anisopoikilocytosis. Platelets of affected patients had substantial reductions in the α-granule components, P-selectin, and Fg, and somewhat less glycoproteins GPIba and GPIIIa.
Next, Monteferrario et al. (44) detected a nonsense hereditary mutation (c.859C>T) in the amino acid 287 (Gln287Ter, g.135866303C>T in GRCh37/hg19) (Figure 1B: 6) in a family with similar clinical features. This mutation also produces a stop codon and a truncated protein that lacks 44 amino acids at the carboxyl terminus. Normal levels of mRNA are expressed in the mutant allele, but the truncated protein is inactive as a repressor.
The condition produced by both mutations was considered a type of gray platelet syndrome, as platelets look gray under optical microscopy owing to their lack of alpha granules. However, the variable and, in general, less severe α-granule deficiency and the red cell phenotype differ from the classic gray platelet syndrome (45). This has led to classify these conditions as bleeding disorder platelet-type 17 (OMIM #187900).
Recently, another GFI1B mutation Gly272fsTer274 (c.814+ 1G>C, g.135865294G>C, GRCh37/hg19) (Figure 1B: 5), generating a truncated protein, has been associated with congenital macrothrombocytopenia linked to α-granule deficiency. This mutation affects 58 amino acids of the C-terminal, which results in complete deletion of zinc finger 5. Platelets in these patients present an increased level of CD34 expression and decreased levels of thrombospondin-1 (46).
GFI1B mutations have also been found in two patients from unrelated families with a combined alpha–delta storage pool deficiency, with reduction of α and dense (δ) granules. Both cases had thrombocytopenia. One of them had also anemia and a granulocytic left shift that corrected itself spontaneously in a few months. One patient also had urogenital and heart abnormalities and developed seizures. The other patient had persistent ductus arteriosus. A whole-exon sequencing of the first patient demonstrated a de novo heterozygous GFI1B nonsense mutation—Lys265Ter (c.793A>T, g. 135865273A>T in GRCh37/hg19) (Figure 1B: 4). Targeted GFI1B sequencing in the second case revealed a homozygous mutation at this gene—Leu308Pro (c.923T>C, rs775963992, g.135866367T>C in GRCh37/hg19) (Figure 1B: 8). These mutations too were located at zinc-finger domains 4 and 6, respectively. It is still unclear if the non-hematological congenital abnormalities observed in these patients were related to the GFI1B mutations (47).
A GFI1B sequence study of 529 patients with atypical platelet phenotypes also allowed for the identification of seven cases with non-synonymous single-nucleotide polymorphism affecting this locus, which was absent in 11,216 unaffected individuals. Four of them were located at zinc fingers 1 and 2, highlighting the importance of these domains. One of these variants was a homozygous Cys168Phe (c.503G>T, rs527297896, g.135863848G>T in GRCh37/hg19) (Figure 1B: 1) that was associated with abnormal function and reduced platelets in an individual of Asian Indian ancestry; however, the variant was not found in 321 Indian Asian genomes (25).
Whole-exome sequence in 15,459 unselected individuals revealed a synonymous GFI1B variant—Phe192 (c.576C>T, rs150813342, g.135864513C>T in GRCh37/hg19, MAF = 0.009) (Figure 1B: 2)—that was associated with low platelet count. Heterozygous carriers had an average platelet reduction of 25–30 × 109/L. This change promotes the short splicing form, affecting megakaryocyte differentiation and platelets, but not red cell production (26).
All these mutations demonstrate the fundamental role of GFI1B in the biogenesis of human platelets.
GFI1B and Malignancy
Mutations that block differentiation and those that promote cell survival or proliferation have been considered necessary for developing acute leukemia (48). Therefore, the major role of GFI1B in hematopoiesis makes it a good candidate to be involved in blood cancers (49). Besides its role in cell differentiation, GFI1B has been reported to possess proapoptotic activity when expressed in human CD34+ cells (50); disruption of this function may also contribute to leukemogenesis.
In keeping with this, GFI1B expression has been found in high levels in some primary CD34+ human acute myeloid leukemias (AMLs) and leukemic cell lines. GFI1B silencing in these cell lines decreased proliferation and increased apoptosis (51). In chronic myeloid leukemia (CML), other myeloproliferative neoplasms (MPNs), AML, and B-lymphoblastic leukemias, GFI1B expression has also been observed to increase. Remarkably, the short GFI1B isoform is highly expressed in the leukemic cells. However, both isoforms were higher in CML after treating with tyrosine kinase inhibitors (52). Simultaneous silencing of BCR-ABL1 and GFI1B in CML cells showed a cooperative antiproliferative and proapoptotic effect in the K562 CML cell line (53). In this context, the short form may be acting as a repressor over the long species. The caveat of these experiments is the low number of patients and controls analyzed.
JAK2 V617F mutation is frequent in MPNs but has also been found in the general population (0.14–0.2%). Consistent with the importance of the GFI1B downstream sequence in its regulation and the role of this gene in blood cancer, a genome-wide association study identified a C>G variation in this region (rs621940, g.135870130C>G in GRCh37/hg19), which is associated with MPN patients and normal carriers of JAK2 V617F, but not with normal unmutated individuals (p = 1.9 × 10−7) (54). Similarly, we reported GFI1B promoter mutations in human leukemias. However, no clear link has yet been established between these mutations and hematopoietic neoplasms (55).
Gfi1b repression of oncogene Meis1 also suggests that GFI1B is involved in leukemia when its repressor function is abolished (7).
In light of these evidences, we described a dominant-negative GFI1B mutation, Asp262Asn (c.784G>A, g. 135865264G>A in GRCh37/hg19) (Figure 1B: 3), associated with transition to AML from antecedent myelodysplastic syndrome (MDS). This mutation promotes the survival of normal and MDS human bone marrow CD34+ cells and skews lineage output of these normal adult primary cells and human cord blood common myeloid progenitors toward myeloid cells. This mutant works mainly through master hematopoietic regulator SPI1 (PU.1) (34). In agreement with this, SPI1 is upregulated in JAK2 V617F-positive MPNs (56).
Similar to GFI1 (57, 58), GFI1B has been linked to lymphomagenesis. TF TCF3 (E2A) is involved in T-cell human leukemias, and Tcf3 KO develops T-cell lymphoma. Ectopic expression of Tcf3 in this context induces growth arrest and apoptosis, together with direct Gfi1b upregulation. Gfi1b-increased expression in Tcf3−/− cells produces similar consequences. Therefore, consistent with the importance of GFI1B block in myeloid leukemias, TCF3 inhibition in T-cell malignancies may work through GFI1B downregulation (33). Another piece of evidence of the implication of GFI1B reduction in lymphoma comes from its relation with B-cell lymphoma 6 (BCL6), a gene frequently expressed in T- and B-cell lymphomas. BCL6 chromosomal rearrangements and/or mutations are associated with human lymphomas, up to 73% in diffuse large B-cell lymphoma (59). Gfi1b has been identified as a retrovirus integration site in diffuse large B-cell lymphomas of mice containing the human BCL6 transgene, but this was not the case in retroviral injected non-transgenic control lymphomas. Again, in this context, Gfi1b expression was decreased in the first lymphomas compared with the latest. Additionally, GFI1B was decreased in human BCL6-positive T- and B-cell lymphomas, analyzed by immunohistochemistry (60).
Unlike blood malignancies, GFI1B and GFI1 activation has been associated with solid tumors, in particular medulloblastoma. In most cases, GFI1B/GFI1 mutually exclusive abnormal expression was produced by structural variants, showing that abnormal expression of GFI1B is definitely linked with human cancer (61–63). Further investigation may establish a wider role in blood or solid malignancy.
Other genes have been related to malignancy both when upregulated and functionally inactivated, including key regulators of hematopoiesis CEBPA (64) and SPI1 (PU.1) (56, 65, 66). This may be the case with GFI1B too. However, more data will be needed to get a full insight into the mechanisms involved in GFI1B’s role in cancer, particularly in the blood setting and the importance of its two isoforms.
Author Contributions
All authors listed have made substantial, direct, and intellectual contribution to the work and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was partially supported by the “Hay Esperanza” foundation.
References
1. Gilks CB, Bear SE, Grimes HL, Tsichlis PN. Progression of interleukin-2 (IL-2)-dependent rat T cell lymphoma lines to IL-2-independent growth following activation of a gene (Gfi-1) encoding a novel zinc finger protein. Mol Cell Biol (1993) 13(3):1759–68. doi: 10.1128/MCB.13.3.1759
2. Roberts T, Cowell JK. Cloning of the human Gfi-1 gene and its mapping to chromosome region 1p22. Oncogene (1997) 14(8):1003–5. doi:10.1038/sj.onc.1200910
3. Rödel B, Wagner T, Zörnig M, Niessing J, Möröy T. The human homologue (GFI1B) of the chicken GFI gene maps to chromosome 9q34. 13-A locus frequently altered in hematopoietic diseases. Genomics (1998) 54(3):580–2. doi:10.1006/geno.1998.5601
4. Tong B, Grimes HL, Yang TY, Bear SE, Qin Z, Du K, et al. The Gfi-1B proto-oncoprotein represses p21WAF1 and inhibits myeloid cell differentiation. Mol Cell Biol (1998) 18(5):2462–73. doi:10.1128/MCB.18.5.2462
5. Grimes HL, Chan TO, Zweidler-McKay PA, Tong B, Tsichlis PN. The Gfi-1 proto-oncoprotein contains a novel transcriptional repressor domain, SNAG, and inhibits G1 arrest induced by interleukin-2 withdrawal. Mol Cell Biol (1996) 16(11):6263–72. doi:10.1128/MCB.16.11.6263
6. Saleque S, Kim J, Rooke HM, Orkin SH. Epigenetic regulation of hematopoietic differentiation by Gfi-1 and Gfi-1b is mediated by the cofactors CoREST and LSD1. Mol Cell (2007) 27(4):562–72. doi:10.1016/j.molcel.2007.06.039
7. Chowdhury AH, Ramroop JR, Upadhyay G, Sengupta A, Andrzejczyk A, Saleque S. Differential transcriptional regulation of meis1 by Gfi1b and its co-factors LSD1 and CoREST. PLoS One (2013) 8(1):e53666. doi:10.1371/journal.pone.0053666
8. Upadhyay G, Chowdhury AH, Vaidyanathan B, Kim D, Saleque S. Antagonistic actions of Rcor proteins regulate LSD1 activity and cellular differentiation. Proc Natl Acad Sci U S A (2014) 111(22):8071–6. doi:10.1073/pnas.1404292111
9. Duan Z, Zarebski A, Montoya-Durango D, Grimes HL, Horwitz M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol (2005) 25(23):10338–51. doi:10.1128/MCB.25.23.10338-10351.2005
10. Lee S, Doddapaneni K, Hogue A, McGhee L, Meyers S, Wu Z. Solution structure of Gfi-1 zinc domain bound to consensus DNA. J Mol Biol (2010) 397(4):1055–66. doi:10.1016/j.jmb.2010.02.006
11. Zweidler-McKay PA, Grimes HL, Flubacher MM, Tsichlis PN. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol Cell Biol (1996) 16(8):4024–34. doi:10.1128/MCB.16.8.4024
12. Thambyrajah R, Patel R, Mazan M, Lie-a-Ling M, Lilly A, Eliades A, et al. New insights into the regulation by RUNX1 and GFI1 (s) proteins of the endothelial to hematopoietic transition generating primordial hematopoietic cells. Cell Cycle (2016) 15(16):2108–14. doi:10.1080/15384101.2016.1203491
13. Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, et al. Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet (2002) 30(3):295–300. doi:10.1038/ng831
14. Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S, et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity (2003) 18(1):109–20. doi:10.1016/S1074-7613(02)00501-0
15. Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet (2003) 34(3):308–12. doi:10.1038/ng1170
16. Wallis D, Hamblen M, Zhou Y, Venken KJ, Schumacher A, Grimes HL, et al. The zinc finger transcription factor Gfi1, implicated in lymphomagenesis, is required for inner ear hair cell differentiation and survival. Development (2003) 130(1):221–32. doi:10.1242/dev.00190
17. Tabrizifard S, Olaru A, Plotkin J, Fallahi-Sichani M, Livak F, Petrie HT. Analysis of transcription factor expression during discrete stages of postnatal thymocyte differentiation. J Immunol (2004) 173(2):1094–102. doi:10.4049/jimmunol.173.2.1094
18. Vassen L, Okayama T, Möröy T. Gfi1b: green fluorescent protein knock-in mice reveal a dynamic expression pattern of Gfi1b during hematopoiesis that is largely complementary to Gfi1. Blood (2007) 109(6):2356–64. doi:10.1182/blood-2006-06-030031
19. Moignard V, Macaulay IC, Swiers G, Buettner F, Schütte J, Calero-Nieto FJ, et al. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nat Cell Biol (2013) 15(4):363–72. doi:10.1038/ncb2709
20. Saleque S, Cameron S, Orkin SH. The zinc-finger proto-oncogene Gfi-1b is essential for development of the erythroid and megakaryocytic lineages. Genes Dev (2002) 16(3):301–6. doi:10.1101/gad.959102
21. Khandanpour C, Sharif-Askari E, Vassen L, Gaudreau MC, Zhu J, Paul WE, et al. Evidence that growth factor independence 1b regulates dormancy and peripheral blood mobilization of hematopoietic stem cells. Blood (2010) 116(24):5149–61. doi:10.1182/blood-2010-04-280305
22. Foudi A, Kramer DJ, Qin J, Ye D, Behlich AS, Mordecai S, et al. Distinct, strict requirements for Gfi-1b in adult bone marrow red cell and platelet generation. J Exp Med (2014) 211(5):909–27. doi:10.1084/jem.20131065
23. Beauchemin H, Shooshtarizadeh P, Vadnais C, Vassen L, Pastore YD, Möröy T. Gfi1b controls integrin signaling-dependent cytoskeleton dynamics and organization in megakaryocytes. Haematologica (2017) 102(3):484–97. doi:10.3324/haematol.2016.150375
24. Laurent B, Randrianarison-Huetz V, Frisan E, Andrieu-Soler C, Soler E, Fontenay M, et al. A short Gfi-1B isoform controls erythroid differentiation by recruiting the LSD1–CoREST complex through the dimethylation of its SNAG domain. J Cell Sci (2012) 125(4):993–1002. doi:10.1242/jcs.095877
25. Chen L, Kostadima M, Martens JH, Canu G, Garcia SP, Turro E, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science (2014) 345(6204):1251033. doi:10.1126/science.1251033
26. Polfus LM, Khajuria RK, Schick UM, Pankratz N, Pazoki R, Brody JA, et al. Whole-exome sequencing identifies loci associated with blood cell traits and reveals a role for alternative GFI1B splice variants in human hematopoiesis. Am J Hum Genet (2016) 99(2):481–8. doi:10.1016/j.ajhg.2016.06.016
27. Fiolka K, Hertzano R, Vassen L, Zeng H, Hermesh O, Avraham KB, et al. Gfi1 and Gfi1b act equivalently in haematopoiesis, but have distinct, non-overlapping functions in inner ear development. EMBO Rep (2006) 7(3):326–33. doi:10.1038/sj.embor.7400618
28. Rodriguez P, Bonte E, Krijgsveld J, Kolodziej KE, Guyot B, Heck A, et al. GATA-1 forms distinct activating and repressive complexes in erythroid cells. EMBO J (2005) 24(13):2354–66. doi:10.1038/sj.emboj.7600702
29. Vassen L, Fiolka K, Mahlmann S, Möröy T. Direct transcriptional repression of the genes encoding the zinc-finger proteins Gfi1b and Gfi1 by Gfi1b. Nucleic Acids Res (2005) 33(3):987–98. doi:10.1093/nar/gki243
30. Jegalian AG, Wu H. Regulation of Socs gene expression by the proto-oncoprotein GFI-1B two routes for stat5 target gene induction by erythropoietin. J Biol Chem (2002) 277(3):2345–52. doi:10.1074/jbc.M105575200
31. Kuo YY, Chang ZF. GATA-1 and Gfi-1B interplay to regulate Bcl-xL transcription. Mol Cell Biol (2007) 27(12):4261–72. doi:10.1128/MCB.02212-06
32. Randrianarison-Huetz V, Laurent B, Bardet V, Blobe GC, Huetz F, Duménil D. Gfi-1B controls human erythroid and megakaryocytic differentiation by regulating TGF-β signaling at the bipotent erythro-megakaryocytic progenitor stage. Blood (2010) 115(14):2784–95. doi:10.1182/blood-2009-09-241752
33. Xu W, Kee BL. Growth factor independent 1B (Gfi1b) is an E2A target gene that modulates Gata3 in T-cell lymphomas. Blood (2007) 109(10):4406–14. doi:10.1182/blood-2006-08-043331
34. Anguita E, Gupta R, Olariu V, Valk PJ, Peterson C, Delwel R, et al. A somatic mutation of GFI1B identified in leukemia alters cell fate via a SPI1 (PU. 1) centered genetic regulatory network. Dev Biol (2016) 411(2):277–86. doi:10.1016/j.ydbio.2016.02.002
35. Sengupta A, Upadhyay G, Sen S, Saleque S. Reciprocal regulation of alternative lineages by Rgs18 and its transcriptional repressor Gfi1b. J Cell Sci (2016) 129(1):145–54. doi:10.1242/jcs.177519
36. Singh D, Upadhyay G, Sengupta A, Biplob MA, Chakyayil S, George T, et al. Cooperative stimulation of megakaryocytic differentiation by Gfi1b gene targets Kindlin3 and Talin1. PLoS One (2016) 11(10):e0164506. doi:10.1371/journal.pone.0164506
37. Anguita E, Villegas A, Iborra F, Hernández A. GFI1B controls its own expression binding to multiple sites. Haematologica (2010) 95(1):36–46. doi:10.3324/haematol.2009.012351
38. Hamlett I, Draper J, Strouboulis J, Iborra F, Porcher C, Vyas P. Characterization of megakaryocyte GATA1-interacting proteins: the corepressor ETO2 and GATA1 interact to regulate terminal megakaryocyte maturation. Blood (2008) 112(7):2738–49. doi:10.1182/blood-2008-03-146605
39. Schuh AH, Tipping AJ, Clark AJ, Hamlett I, Guyot B, Iborra FJ, et al. ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol Cell Biol (2005) 25(23):10235–50. doi:10.1128/MCB.25.23.10235-10250.2005
40. Vassen L, Fiolka K, Möröy T. Gfi1b alters histone methylation at target gene promoters and sites of γ-satellite containing heterochromatin. EMBO J (2006) 25(11):2409–19. doi:10.1038/sj.emboj.7601124
41. May G, Soneji S, Tipping AJ, Teles J, McGowan SJ, Wu M, et al. Dynamic analysis of gene expression and genome-wide transcription factor binding during lineage specification of multipotent progenitors. Cell Stem Cell (2013) 13(6):754–68. doi:10.1016/j.stem.2013.09.003
42. Songdej N, Rao AK. Inherited platelet dysfunction and hematopoietic transcription factor mutations. Platelets (2017) 28(1):20–6. doi:10.1080/09537104.2016.1203400
43. Stevenson WS, Morel-Kopp MC, Chen Q, Liang HP, Bromhead CJ, Wright S, et al. GFI1B mutation causes a bleeding disorder with abnormal platelet function. J Thromb Haemost (2013) 11(11):2039–47. doi:10.1111/jth.12368
44. Monteferrario D, Bolar NA, Marneth AE, Hebeda KM, Bergevoet SM, Veenstra H, et al. A dominant-negative GFI1B mutation in the gray platelets syndrome. N Engl J Med (2014) 370(3):245–53. doi:10.1056/NEJMoa1308130
45. Stevenson WS, Morel-Kopp MC, Ward CM. Platelets are not all gray in GFI1B disease. Clin Genet (2015) 87(3):299–299. doi:10.1111/cge.12424
46. Kitamura K, Okuno Y, Yoshida K, Sanada M, Shiraishi Y, Muramatsu H, et al. Functional characterization of a novel GFI1B mutation causing congenital macrothrombocytopenia. J Thromb Haemost (2016) 14(7):1462–9. doi:10.1111/jth.13350
47. Ferreira CR, Chen D, Abraham SM, Adams DR, Simon KL, Malicdan MC, et al. Combined alpha-delta platelet storage pool deficiency is associated with mutations in GFI1B. Mol Genet Metab (2017) 120(3):288–94. doi:10.1016/j.ymgme.2016.12.006
48. Gilliland DG. Molecular genetics of human leukemias: new insights into therapy. Semin Hematol (2002) 39(4):6–11. doi:10.1053/shem.2002.36921
49. Sanders MA, Valk PJ. The evolving molecular genetic landscape in acute myeloid leukaemia. Curr Opin Hematol (2013) 20(2):79–85. doi:10.1097/MOH.0b013e32835d821c
50. Osawa M, Yamaguchi T, Nakamura Y, Kaneko S, Onodera M, Sawada KI, et al. Erythroid expansion mediated by the Gfi-1B zinc finger protein: role in normal hematopoiesis. Blood (2002) 100(8):2769–77. doi:10.1182/blood-2002-01-0182
51. Elmaagacli AH, Koldehoff M, Zakrzewski JL, Steckel NK, Ottinger H, Beelen DW. Growth factor-independent 1B gene (GFI1B) is overexpressed in erythropoietic and megakaryocytic malignancies and increases their proliferation rate. Br J Haematol (2007) 136(2):212–9. doi:10.1111/j.1365-2141.2006.06407.x
52. Vassen L, Khandanpour C, Ebeling P, van der Reijden BA, Jansen JH, Mahlmann S, et al. Growth factor independent 1b (Gfi1b) and a new splice variant of Gfi1b are highly expressed in patients with acute and chronic leukemia. Int J Hematol (2009) 89(4):422–30. doi:10.1007/s12185-009-0286-5
53. Koldehoff M, Zakrzewski JL, Beelen DW, Elmaagacli AH. Additive antileukemia effects by GFI1B-and BCR–ABL-specific siRNA in advanced phase chronic myeloid leukemic cells. Cancer Gene Ther (2013) 20(7):421–7. doi:10.1038/cgt.2013.31
54. Hinds DA, Barnholt KE, Mesa RA, Kiefer AK, Do CB, Eriksson N, et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood (2016) 128(8):1121–8. doi:10.1182/blood-2015-06-652941
55. Hernández A, Villegas A, Anguita E. Human promoter mutations unveil Oct-1 and GATA-1 opposite action on Gfi1b regulation. Ann Hematol (2010) 89(8):759–65. doi:10.1007/s00277-009-0900-x
56. Irino T, Uemura M, Yamane H, Umemura S, Utsumi T, Kakazu N, et al. JAK2 V617F-dependent upregulation of PU. 1 expression in the peripheral blood of myeloproliferative neoplasm patients. PLoS One (2011) 6(7):e22148. doi:10.1371/journal.pone.0022148
57. Scheijen B, Jonkers J, Acton D, Berns A. Characterization of pal-1, a common proviral insertion site in murine leukemia virus-induced lymphomas of c-myc and Pim-1 transgenic mice. J Virol (1997) 71(1):9–16.
58. Zörnig M, Schmidt T, Karsunky H, Grzeschiczek A, Möröy T. Zinc finger protein GFI-1 cooperates with myc and pim-1 in T-cell lymphomagenesis by reducing the requirements for IL-2. Oncogene (1996) 12(8):1789–801.
59. Migliazza A, Martinotti S, Chen W, Fusco C, Ye BH, Knowles DM, et al. Frequent somatic hypermutation of the 5′ noncoding region of the BCL6 gene in B-cell lymphoma. Proc Natl Acad Sci U S A (1995) 92(26):12520–4. doi:10.1073/pnas.92.26.12520
60. Baron BW, Anastasi J, Bies J, Reddy PL, Joseph L, Thirman MJ, et al. GFI1B, EVI5, MYB—additional genes that cooperate with the human BCL6 gene to promote the development of lymphomas. Blood Cells Mol Dis (2014) 52(1):68–75. doi:10.1016/j.bcmd.2013.07.003
61. Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol (2011) 29(11):1408–14. doi:10.1200/JCO.2009.27.4324
62. Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer (2012) 12(12):818–34. doi:10.1038/nrc3410
63. Northcott PA, Lee C, Zichner T, Stütz AM, Erkek S, Kawauchi D, et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature (2014) 511(7510):428–34. doi:10.1038/nature13379
64. Paz-Priel I, Friedman A. C/EBPα dysregulation in AML and ALL. Crit Rev Oncog (2011) 16(1–2):93–102. doi:10.1615/CritRevOncog.v16.i1-2.90
65. Moreau-Gachelin F, Wendling F, Molina T, Denis N, Titeux M, Grimber G, et al. Spi-1/PU. 1 transgenic mice develop multistep erythroleukemias. Mol Cell Biol (1996) 16(5):2453–63. doi:10.1128/MCB.16.5.2453
Keywords: GFI1B, hematopoiesis, cancer, leukemia, platelets disease
Citation: Anguita E, Candel FJ, Chaparro A and Roldán-Etcheverry JJ (2017) Transcription Factor GFI1B in Health and Disease. Front. Oncol. 7:54. doi: 10.3389/fonc.2017.00054
Received: 29 November 2016; Accepted: 13 March 2017;
Published: 28 March 2017
Edited by:
Christian Thiede, Technische Universität Dresden, GermanyReviewed by:
Tarik Moroy, Institut de recherches cliniques de Montreal (IRCM), CanadaCyrus Khandanpour, Essen University Hospital, Germany
Copyright: © 2017 Anguita, Candel, Chaparro and Roldán-Etcheverry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo Anguita, ZWR1YXJkby5hbmd1aXRhQHNhbHVkLm1hZHJpZC5vcmc=