 Varda Shoshan-Barmatz
Varda Shoshan-Barmatz Soumasree De
Soumasree De Alon Meir
Alon Meir- Department of Life Sciences, National Institute for Biotechnology in the Negev, Ben-Gurion University of the Negev, Beer-Sheva, Israel
In the outer mitochondrial membrane, the voltage-dependent anion channel 1 (VDAC1) functions in cellular Ca2+ homeostasis by mediating the transport of Ca2+ in and out of mitochondria. VDAC1 is highly Ca2+-permeable and modulates Ca2+ access to the mitochondrial intermembrane space. Intramitochondrial Ca2+ controls energy metabolism by enhancing the rate of NADH production via modulating critical enzymes in the tricarboxylic acid cycle and fatty acid oxidation. Mitochondrial [Ca2+] is regarded as an important determinant of cell sensitivity to apoptotic stimuli and was proposed to act as a “priming signal,” sensitizing the organelle and promoting the release of pro-apoptotic proteins. However, the precise mechanism by which intracellular Ca2+ ([Ca2+]i) mediates apoptosis is not known. Here, we review the roles of VDAC1 in mitochondrial Ca2+ homeostasis and in apoptosis. Accumulated evidence shows that apoptosis-inducing agents act by increasing [Ca2+]i and that this, in turn, augments VDAC1 expression levels. Thus, a new concept of how increased [Ca2+]i activates apoptosis is postulated. Specifically, increased [Ca2+]i enhances VDAC1 expression levels, followed by VDAC1 oligomerization, cytochrome c release, and subsequently apoptosis. Evidence supporting this new model suggesting that upregulation of VDAC1 expression constitutes a major mechanism by which apoptotic stimuli induce apoptosis with VDAC1 oligomerization being a molecular focal point in apoptosis regulation is presented. A new proposed mechanism of pro-apoptotic drug action, namely Ca2+-dependent enhancement of VDAC1 expression, provides a platform for developing a new class of anticancer drugs modulating VDAC1 levels via the promoter and for overcoming the resistance of cancer cells to chemotherapy.
Overview
Intracellular Ca2+concentration ([Ca2+]i) regulates a number of cellular and intercellular events, such as the cell cycle, proliferation, gene transcription, and cell death pathways, as well as processes like muscle contractility and neuronal processing and transmission (1). The alteration of Ca2+ homeostasis is closely related with various cancer hallmarks, including proliferation, migration, angiogenesis, invasion abilities, and resistance to cell death (2).
Various systems and mechanisms have evolved to control and respond to minute changes in Ca2+ concentrations and localization (3). Moreover, many cellular compartments participate in the Ca2+ signaling network regulation. [Ca2+]i is controlled via its transport in and out of the cell or/and in and out of intracellular organelles. Within a given compartment, [Ca2+] can be buffered by binding to specific proteins and other molecules, as well as existing in its free form, albeit differentially across compartments (1). The major organelles that participate in controlling Ca2+ dynamics include the endoplasmic reticulum (ER) and mitochondria (4). Imbalance in the control of [Ca2+]i can lead to mitochondria Ca2+ overload and ultimately, to toxic effects. Tumor cells exhibit a well-developed capacity for modulating cytosolic Ca2+ levels by remodeling the cellular machinery that participates in processes that determine Ca2+ dynamics and homeostasis, as well as changes in sensitivity to the induction of cell death. This review is focused on the mitochondrial gatekeeper protein voltage-dependent anion channel 1 (VDAC1) and its role in Ca2+ transport and on Ca2+-mediated apoptosis involving regulation of VDAC1 expression levels.
Ca2+ and Mitochondria
Mitochondria not only play a key role in metabolism but also serve as a major hub for cellular Ca2+ homeostasis, regulating oxidative phosphorylation (OXPHOS) (4–7) and modulating cytosolic Ca2+ signals (8, 9), cell death (10), and secretion (11, 12). Enclosed by two different membranes, namely the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM), mitochondria thus present two aqueous compartments, the intermembrane space (IMS) and the matrix (M). To reach the matrix, Ca2+ must cross both the OMM and the IMM. Indeed, the mitochondrial matrix is one of the major cellular Ca2+ stores or buffers and is used to control [Ca2+]i and dynamics. Within the mitochondrial matrix, Ca2+ is precipitated as insoluble CaPO4, which exists in equilibrium with free Ca2+ (7, 13).
It is well established that mitochondria can rapidly sequester large and sudden increases in [Ca2+]i at the expense of the membrane potential across the IMM that is generated by the electron transport chain (6). Intramitochondrial Ca2+ controls energy metabolism by enhancing the rate of NADH production via modulating critical enzymes, such as those of the tricarboxylic acid (TCA) cycle and fatty acid oxidation (14, 15), linking glycolysis to the TCA cycle (16). Indeed, matrix Ca2+ is an essential cofactor for several rate-limiting TCA enzymes, namely pyruvate dehydrogenase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase.
Mitochondrial Ca2+ ([Ca2+]m) homeostasis is important not only for energy production but also for regulating [Ca2+]i and activating cell apoptotic pathways (10, 17). Several recent reviews have discussed the basic principles that govern [Ca2+]m homeostasis and maintenance of Ca2+ dynamics within organelles (18–22). The OMM and IMM pathways allowing Ca2+ entry into and exit from mitochondria are presented below.
Pathways Mediating Ca2+ Fluxes in the Mitochondria
Several mitochondria membrane proteins play central roles in Ca2+ signaling and/or Ca2+ influx and efflux in normal and disease conditions. Ca2+ transport across the IMM is mediated via several proteins, including the mitochondrial Ca2+ uniporter (MCU) (23, 24) and the Na+/Ca2+ exchanger, NCLX, the major Ca2+ efflux mediator (25, 26). In the OMM, VDAC1 was shown to control Ca2+ permeability (27–30).
VDAC1, the Ca2+ Channel in the OMM
Three different isoforms of VDAC have been identified, VDAC1, VDAC2, and VDAC3. VDAC1 has been best studied, whereas only limited information regarding the cellular functions of VDAC2 and VDAC3 is available (31). Thus, we focus here on VDAC1.
VDAC1, a Multifunctional Channel, Controls Cell Metabolism
VDAC1 at the OMM controls metabolic cross talk between mitochondria and rest of the cell by allowing the entry of metabolites (pyruvate, malate, succinate, nucleotides, and NADH) into the mitochondria and the exit of newly formed molecules, such as hemes (32, 33) (Figure 1). VDAC1 is also involved in cholesterol transport and mediates the fluxes of ions, including Ca2+ (34), serves as a ROS transporter, and contributes to regulating the redox states of mitochondria and the cytosol (32–34). Moreover, VDAC1 at the OMM interacts with proteins that mediate and regulate the integration of mitochondrial functions with other cellular activities. VDAC1 forms a complex with adenine nucleotide translocase (ANT) and creatine kinase (35). The interaction of VDAC1 with hexokinase (HK) allows for coupling between OXPHOS and glycolysis, an important factor in cancer cell energy homeostasis (the Warburg effect) (36). Thus, VDAC1 appears to be a convergence point for a variety of cell survival and death signals, mediated through its association with various ligands and proteins.
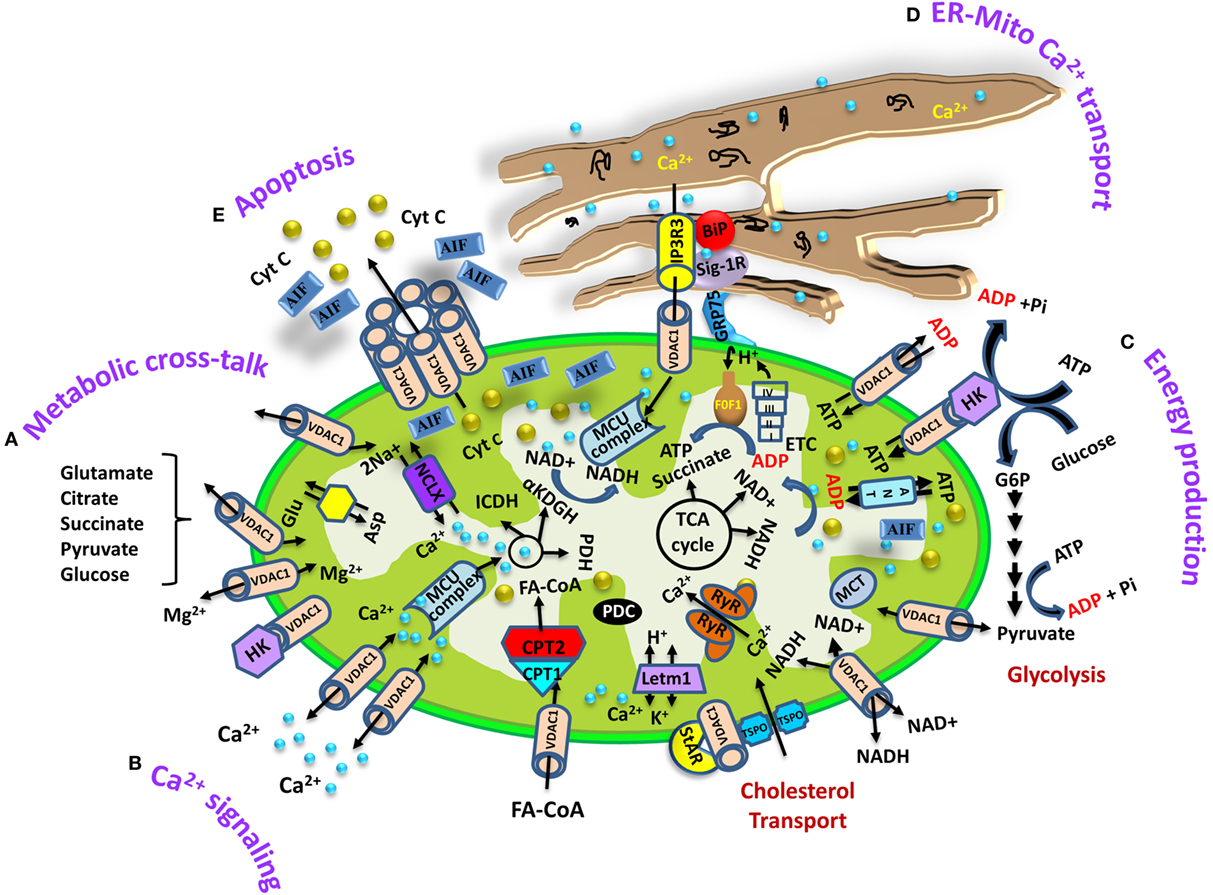
Figure 1. Schematic representation of voltage-dependent anion channel 1 (VDAC1) as a multifunctional protein involved in Ca2+ and metabolite transport, energy production, and the structural and functional association of mitochondria with the endoplasmic reticulum (ER). The various functions of VDAC1 in cell and mitochondria functions are presented. These include (A) Ca2+ signaling by transporting Ca2+; (B) control of metabolic cross talk between the mitochondria and the rest of the cell; (C) mediating cellular energy production by transporting ATP/ADP, NADH, and acyl-CoA from the cytosol to the intermembrane space and regulating glycolysis via the association with hexokinase (HK); (D) involvement in structural and functional association with the ER, mediating Ca2+ transport from the ER to mitochondria; (E) participation in apoptosis via its oligomerization to form a protein-conducting channel within a VDAC1 homo-oligomer, allowing Cyto c release and apoptotic cell death. Ca2+ influx and efflux transport systems in the outer mitochondrial membrane (OMM) and IMM are shown. In the OMM, VDAC1 is presented as a Ca2+ channel and also functions in the transport of Mg2+. In the IMM, Ca2+ uptake into the matrix is mediated by a Ca2+-selective transporter, the mitochondrial Ca2+ uniporter (MCU), regulated by a calcium-sensing accessory subunit (MCU1). Ryanodine receptor (RyR) in the IMM mediates Ca2+ influx. Ca2+ efflux is mediated by NCLX, an Na+/Ca2+ exchanger. High levels of matrix Ca2+ accumulation trigger the opening of the PTP, a fast Ca2+ release channel. The function of Ca2+ in regulation of energy production is mediated via tricarboxylic acid (TCA) cycle regulation. This includes activation of pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (αKGDH) by intramitochondrial Ca2+, leading to enhanced activity of the TCA cycle. The electron transport chain (ETC) and the ATP synthase (FoF1) are also presented. Molecular fluxes are indicated by arrows. VDAC1 mediates the transfer of fatty acid acyl-CoAs across the OMM to the IMS, where they are converted into acylcarnitine by CPT1a for further processing by β-oxidation. VDAC1 is involved in cholesterol transport by being constituent of a multi-protein complex, the transduceosome, containing Star/TSPO/VDAC1. The ER associated with the mitochondria is presented with key proteins indicated. These include the inositol 3 phosphate receptor type 3 (IP3R3), the sigma1 receptor (Sig1R) (a reticular chaperone), binding immunoglobulin protein (BiP), the ER heat shock protein (HSP70) chaperone, and glucose-regulated protein 75 (GRP75). IP3 activates the IP3R in the ER to release Ca2+ that is directly transferred to the mitochondrion via VDAC1.
VDAC1 As Ca2+ Transporter at OMM
Found in the OMM, VDAC1 regulates the transport of Ca2+ in and out of the mitochondria. VDAC1 is highly Ca2+-permeable and modulates the accessibility of Ca2+ to the IMS (27–30) (Figure 1). Bilayer-reconstituted VDAC1 under voltage-clamp conditions and in the presence of different CaCl2 concentration gradients showed well-defined voltage-dependent channel conductance as observed with either NaCl or KCl solution (27, 29). Bilayer-reconstituted VDAC1 showed higher permeability to Ca2+ in the low conductance state (29). The Ca2+ permeability of VDAC1 has also been established upon VDAC1 reconstitution into liposomes (27).
Various studies support the function of VDAC1 in the transport of Ca2+ and in cellular Ca2+ homeostasis. VDAC1 overexpression increases [Ca2+]m concentration in HeLa cells and skeletal myotubes (37), and silencing of VDAC1 expression by siRNA attenuates [Ca2+]m uptake and cell apoptosis induced by H2O2 or ceramide (38). It was also proposed that the magnitude of Ca2+ transfer into the mitochondrial matrix is regulated by protein–protein interactions between Bcl-xL and VDAC1 or VDAC3, with this interaction promoting matrix Ca2+ accumulation by increasing Ca2+ transfer across the OMM (39).
Silencing each of the VDAC isoforms in the presence of a pro-apoptotic stimulus revealed that each was differentially sensitive to H2O2, with VDAC1 silencing potentiating H2O2-induced apoptosis and impairing [Ca2+]m loading, while VDAC2 silencing had the opposite effects (38). In addition, several VDAC-interacting molecules like 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), 4-acetamido-4′-isothiocyanato-stilbene-2,2′-disulfonic acid, and dinitrostilbene-2,2′-disulfonic acid were shown to prevent apoptosis and also inhibit the rise in [Ca2+] levels associated with apoptosis induction (40). In another example, 5-aminolevulinic precluded Ca2+-mediated oxidative stress and apoptosis through VDAC1 inhibition (41).
VDAC1 Possesses Ca2+-Binding Sites
Several lines of evidence suggest that VDAC1 possesses divalent cation-binding site(s). [Ca2+], at micromolar concentrations, switched VDAC1 from a low to high conductance state (30). The trivalent ions La3+ and Tb3+, known to bind to Ca2+-binding proteins (42), reduced the channel conductance of bilayer-reconstituted VDAC (43). This and the direct demonstration of Tb3+ binding to purified VDAC1, as reflected in an enhanced green fluorescence, further suggest that VDAC1 possesses divalent cation-binding site(s) that its occupation by La3+ or Tb3+ lead to reduced channel conductance.
Similarly, molecules known to specifically interact with several Ca2+-binding proteins like ruthenium red (RuR) (43) and ruthenium amine binuclear complex (Ru360) (44), as well as a photo-reactive analog, azido ruthenium (AzRu) (45), induced VDAC1 channel closure in a time-dependent manner and stabilized the channel in a low conducting state. These compounds also inhibited apoptosis (43–45).
The putative VDAC1 metal binding site for RuR and AzRu was analyzed by mutation of specific VDAC1 residues (43). It has been demonstrated that E72 and E202 are essential for RuR-mediated reduction of bilayer-reconstituted VDAC1 conductance and for RuR-mediated protection against VDAC1-induced cell death (43, 46). This suggests that these two glutamate residues, located in two different β-strands, may form the VDAC1 Ca2+-binding site(s), or part thereof. However, their distant location and their being located in transmembrane sequences, suggest that these residues may stabilize VDAC1 in a conformation that is recognized by RuR and AzRu. Thus, these Ru-containing molecules may bind to a non-defined site in VDAC1 to induce conformation changes leading to reduced conductance and inhibited apoptosis.
The competition between Ca2+ and RuR (43), as well as the demonstration of VDAC gating regulation by physiological levels of Ca2+, whereby Ca2+ increases the conductance of the VDAC1 channel (30), supports the physiological function of the VDAC1 Ca2+-binding site(s).
VDAC1 Protein–Protein Interactions Regulate [Ca2+]m Transport
Interactions between VDAC1 and Bcl-2 family proteins, such as Bax/Bak, Bcl-2, and Bcl-xL, mediating the regulation of apoptosis, are well documented (33, 47–55). It has been shown that interaction of Bcl-xL with VDAC1 or VDAC3 promoted [Ca2+]m uptake (39). It was also reported that all three VDAC isoforms interact with regulator of microtubule dynamics protein 3 (56), a protein at the OMM involved in [Ca2+]i homeostasis regulation (57, 58). VDAC1 interacts with endothelial NO synthase (eNOS), with such interaction amplifying eNOS activity in a [Ca2+]i-mediated manner (59). VDAC also interacts with the L-type Ca2+ channel, and it was suggested that impaired communication between the L-type Ca2+ channel and mitochondrial VDAC contributes to cardiomyopathy (60). Thus, such interactions of VDAC with proteins associated with Ca2+ transport or activated by Ca2+ point to VDAC as functioning not only in [Ca2+]i homeostasis but also in many Ca2+-regulated cellular activities.
VDAC1 Function in Mitochondria—ER/Sarcoplasmic Reticulum (SR) Ca2+-Cross Talk
The participation of VDAC1 in supramolecular complexes and intracellular communication, including Ca2+ signal delivery between the ER and mitochondria, was postulated over a decade ago (28, 61, 62). The components involved in ER–mitochondria interaction include the IP3 receptor and grp75 on the ER as tethering components and VDAC1 on the OMM (63). VDAC1 (but not VDAC2 or VDAC3) was found to provide the route for Ca2+ entry into mitochondria upon apoptotic stimulus, representing a fundamental factor in mitochondria physiology (38). It was, moreover, proposed that the magnitude of Ca2+ transfer from the ER into the mitochondrial matrix is regulated by Bcl-xL (39, 51). ER–mitochondria cross talk regulates not only Ca2+ transfer but also different processes, such as mitochondrial fission, autophagy, and inflammation (64). Finally, Ca2+ dynamics are greatly enhanced where there is close apposition of the ER with mitochondria, as compared to the bulk cytosol. Such changes in Ca2+ signal profiles were modified by ROS, as monitored with genetically encoded redox indicators (65).
MCU and Auxiliary Subunits Form a Selective Ca2+ Transporter in the IMM
Ca2+ transport across the IMM and into the matrix is mediated via the MCU (23, 24), with the driving force being the steep mitochondrial membrane potential (8, 66, 67) (Figure 1). Such delivery is inhibited by RuR and its derivative, Ru360 (68).
The major channel-forming subunit of the MCU complex (CCDC109A) consists of two transmembrane and the N-terminal domains and forms a complex in the IMM with many gatekeeper membrane proteins (23, 24, 69–71). The calcium-sensing accessory subunits MICU1, MICU2, and MCUb are proposed to serve as negative regulators, while mitochondrial Ca2+ uniporter regulator 1 (MCUR1), essential MCU regulator, and SLC25A23 are essential for MCU activity (72–76). MCUR1 may, however, also play other roles, such as in cytochrome c oxidase assembly (77), as a cytosolic Ca2+-buffering agent (78), or in ROS generation (79).
The functional role of MCU under physiological conditions was extensively studied using several silencing techniques (80–85). Interestingly, MCU knockout mice did not exhibit obvious defects in mitochondrial number or morphology and any physiological function (82, 86, 87). MCU deletion was found to be lethal for C57BL/6 mice, whereas knockout mice with an outbred CD1 background were viable, albeit with reduced numbers (88). Basal organ functions were maintained, and impairment was only observed in the physiological adaptation of skeletal muscle to exercise (82). In cardiac-specific conditional MCU-deficient mice, the heart displayed increased resistance to ischemia–reperfusion injury (87, 89).
Na+/Ca2+ Exchanger Function in Ca2+ Efflux and Its Regulation
Mitochondrial Ca2+ is mainly determined by the balance between influx through the MCU and efflux via NCLX (90). To restore resting [Ca2+]m levels, Ca2+ efflux across IMM is mediated by the Na/Ca/Li exchanger (NCLX) or Na+/Ca2+ exchanger (25, 26, 91), and possibly by Letm1, under certain conditions, which functions as a Ca2+/H+ anti-porter in addition to being a H+/K+ anti-porter (see Other Proteins Proposed as Participating in or Mediating Ca2+ Efflux from Mitochondria) (92). NCLX mediates efflux of Ca2+ from the mitochondrial matrix to the IMS (20, 25, 93–95). In contrast to the plasma membrane Na+/Ca2+ exchanger, NCLX transports Li+ ions in addition to Na+ and Ca2+ (96). NCLX has also been proposed to regulate Ca2+-induced NAD(P)H production and matrix redox state modulation (97).
Mitochondrial Ca2+ regulates heart metabolism, where steady-state [Ca2+]m is determined by the dynamic balance between MCU-based Ca2+ influx and NCLX-based Ca2+ efflux (98). It has been proposed that a novel role of NCLX is to regulate the automaticity of cardiomyocytes via modulating SR Ca2+ handling (99). NCLX has been proposed to be involved in several pathological conditions. In ischemia, NCLX acts as a key regulator of [Ca2+]m accumulation (100), while in diabetic cardiac myocytes, NCLX is more susceptive to changes in the outside (cytosolic) Na+ concentration, as compared with controls (101). Phosphorylation of NCLX has been reported to reverse Ca2+ mitochondrial overload and promote survival of PINK1-deficient dopaminergic neurons (102).
Other Proteins Proposed as Participating in or Mediating Ca2+ Efflux from Mitochondria
The transient opening of the mitochondrial permeability transition pore (MPTP or PTP) represents another mechanism for Ca2+ release from mitochondria. However, its function is probably related to non-physiological Ca2+ overload that depolarizes mitochondria by an irreversible opening of the PTP, leading to apoptotic and necrotic cell death associated with disease pathogenesis (103, 104). Multiple proteins have been proposed to constitute the PTP and thus to play a role in PTP opening by Ca2+ or ROS challenge, such as VDAC1 in the OMM, ANT in the IMM, and cyclophilin D in the matrix (105, 106). However, silencing approaches have only confirmed cyclophilin D as being essential for Ca2+-sensitive PTP opening. One recent view considered parts of the FoF1 ATPase as components of the PTP, while other candidates has also emerged [for review, see Ref. (107)]. Recently, SPG7 at the IMM has been proposed as a key component of Ca2+- and ROS-induced PTP opening, forming a complex with VDAC1 at the OMM and cyclophilin D in the matrix (108).
A potential candidate for the [Ca2+]m/H+ anti-porter was suggested, the leucine zipper-EF-hand-containing transmembrane protein 1 (Letm1) (19, 92, 109–111). Letm1 has two Ca2+-binding EF hand domains and catalyzes the electronic exchange of Ca2+ for H+. Letm1 Ca2+ transport activity is pH-sensitive and is inhibited by RuR (111). Letm1 not only imports Ca2+ into the matrix through the IMM but can also extrude Ca2+ from the matrix when [Ca2+]m concentration is high (19, 109, 110).
Channels function in Ca2+ transport in membranes other than in the mitochondria as ryanodine receptors (RyRs) and the transient receptor potential 3 (TRPC3) channel were reported to function in Ca2+ homeostasis [for review, see Ref. (112, 113)]; RyRs, the main Ca2+-release channels in the SR/ER in excitable cells, were reported to be expressed at the IMM and mediate Ca2+ uptake in cardiomyocytes (114, 115). Recently, it was demonstrated that neuronal mitochondria express RyR at the IMM and accumulate Ca2+ in a manner that can be inhibited by dantrolene or ryanodine (116). Finally, canonical TRPC3 was shown to be located in the IMM and contributing to [Ca2+]m uptake and thus functions in regulating [Ca2+]m homeostasis (117).
VDAC1 at the Nexus of Mitochondria-Mediated Apoptosis
Mitochondria-mediated or intrinsic apoptotic pathway is activated via the release of mitochondrial pro-apoptotic proteins (e.g., Cyto c, AIF, Smac/DIABLO) from the IMS to the cytosol (32, 33, 52, 54, 118–126), leading to the activation of caspases. Some models for the release of apoptotic proteins suggest that release exclusively involves an increase in OMM permeability due to the formation of a channel large enough to allow the passage of apoptogenic proteins (32, 33, 124, 127–129), while others consider release to be due to disruption of OMM integrity (120, 130, 131). Recent studies demonstrated that upon apoptosis induction, VDAC1 is oligomerized to form a large pore, allowing the release of mitochondrial pro-apoptotic proteins (129, 132–139). VDAC1 oligomerization was found to be a general mechanism common to numerous apoptogens acting via different initiating cascades (135, 140, 141). Moreover, apoptosis inhibitors (135, 142) and recently identified VDAC1-interacting molecules [diphenylamine-2-carboxylate (DPC)] (40) and a molecule developed in our lab designated as VBIT-4 were found to prevent VDAC1 oligomerization and subsequent apoptosis (143). Furthermore, cyathin-R, a cyathane-type diterpenoid derived from a fungal secondary metabolite library from the medicinal fungus Cyathus africanus, was found to interact with purified VDAC1 and reduce its channel activity, as well as induce apoptosis via promoting VDAC1 oligomerization and the associated cytochrome c release in Bax/Bak-depleted cells but not when VDAC1 was depleted. Cyathin-R-induced apoptosis was inhibited by DPC (142).
VDAC1 also regulates apoptosis via the direct interaction with the anti-apoptotic protein HK (144–152), with apoptosis-regulating proteins, such as Bcl-2, Bcl-xL (33, 47, 48, 50, 144, 153–155), and with the pro-apoptotic proteins Bax and Bak (156).
Ca2+-Induced Apoptosis through VDAC1 Overexpression
Apoptosis induction affects cell Ca2+ homeostasis and energy production (157). The intrinsic apoptotic pathway, initiated in response to various stimuli, including high [Ca2+]i, oxygen radicals, activation of pro-apoptotic Bcl-2 family proteins, UV damage, and various anticancer drugs and cytotoxic agents, such as thapsigargin, staurosporine, As2O3, and selenite, disrupts cellular Ca2+ homeostasis and induces apoptosis (140). Indeed, the contribution of Ca2+ signals to cell death is well documented, and a few mechanisms that connect apoptotic stimuli, via a rise in [Ca2+]i, to cell death have been suggested (158–164).
Recently, it was demonstrated that a panel of apoptotic inducers, such as UV irradiation, H2O2, etoposide, cisplatin, or selenite, elevated [Ca2+]i and upregulated VDAC1 expression levels in a Ca2+-dependent manner (Table 1), resulting in VDAC1 oligomerization, Cyto c release, and subsequent apoptosis (140, 141) (Figure 2). Furthermore, direct elevation of [Ca2+]i by the Ca2+-mobilizing agents A23187, ionomycin, or thapsigargin led to VDAC1 overexpression, VDAC1 oligomerization, and apoptosis, while decreasing [Ca2+]i using the cell-permeable Ca2+-chelating reagent BAPTA-AM inhibited these events (141).
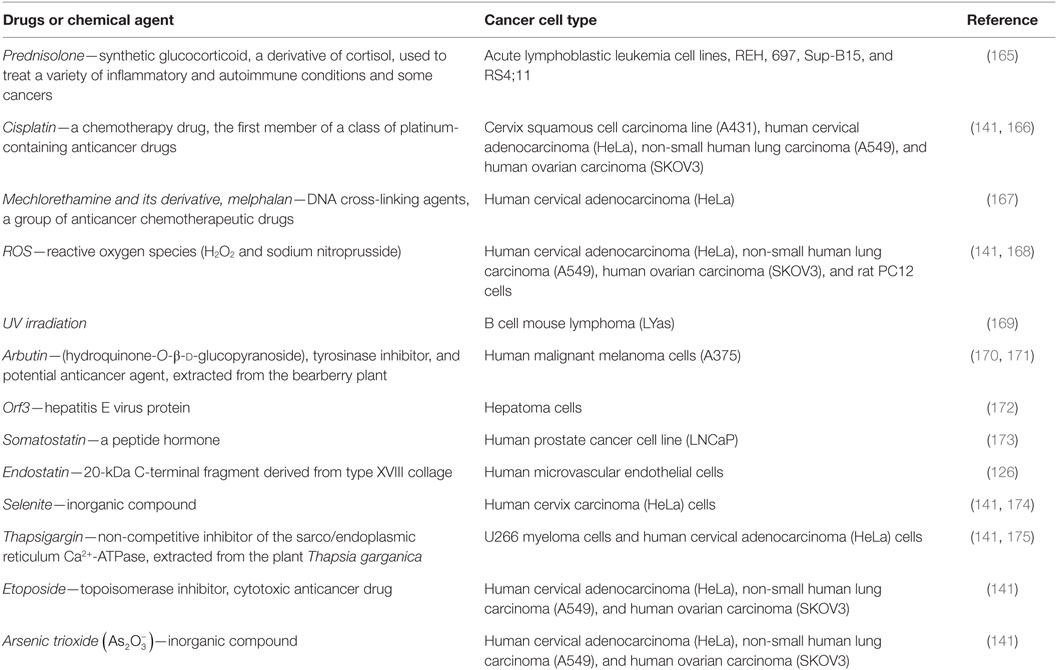
Table 1. Anticancer, pro-apoptotic drugs, and chemical agents that increase voltage-dependent anion channel 1 expression level in cancer cells.
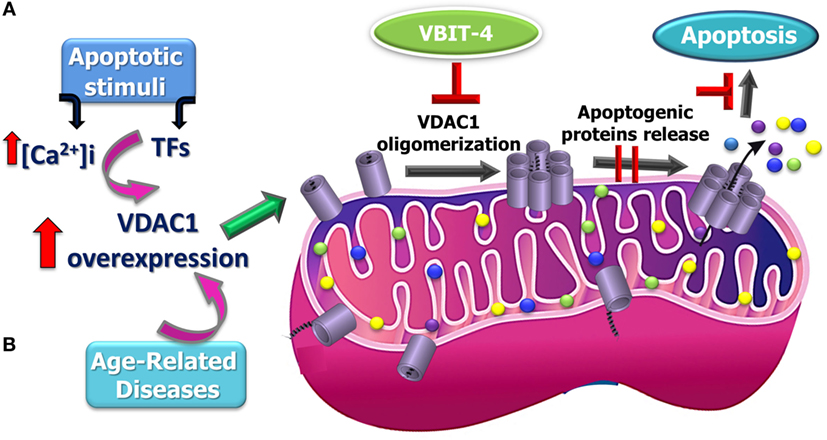
Figure 2. Proposed model for apoptosis stimuli-induced increase in voltage-dependent anion channel 1 (VDAC1) expression levels leading to VDAC1 oligomerization, Cyto c release, and apoptosis and possible inhibition steps. A schematic model describing the novel pathway proposed for apoptosis induction involving elevation of [Ca2+]i leading to VDAC1 overexpression. (A) This facilitates VDAC1 oligomerization to form a large channel mediating cytochrome c release from the mitochondrion into the cytosol, resulting in apoptosis activation. It is proposed that the overexpression of VDAC1 in diseases such as Alzheimer’s disease, cardiovascular diseases, and diabetes is associated mitochondrial dysfunction, including apoptosis induction (B).
It has also been shown that the sensitivity of the CD45-positive (CD45+) U266 myeloma cell line to various apoptotic stimuli is well correlated with the elevated levels of VDAC1 expression that follow Ca2+ signals in response to apoptosis stimulation (169, 175). This suggests that apoptosis-inducing agents act by increasing [Ca2+]i and that this, in turn, leads to an upregulation of VDAC1 expression, which is connected to apoptosis induction (Table 1). The proposed sequence of events leading to VDAC1-mediated apoptosis can be schematically depicted as:

Support for this model comes with the findings that several VDAC-interacting molecules prevent its oligomerization, the elevation in [Ca2+]i associated with apoptosis induction, Cyto c release, and apoptosis (40, 41, 141, 151, 165, 166, 170, 171, 173, 176–178). DIDS was shown to prevent the apoptosis stimuli-inducing increase in [Ca2+]i levels (40) and Ca2+-mediated oxidative stress and apoptosis, as induced by 5-aminolevulinic (41). The small molecules AKOS-022 and VBIT-4 that bind to VDAC1 prevent its oligomerization, the elevation [Ca2+]i associated with apoptosis induction, Cyto c release, and apoptosis (166). Furthermore, mitochondria-mediated apoptosis was correlated with VDAC1 expression levels (141, 165–175). Thus, although different apoptosis inducers elicit cell death via different mechanisms, all induce VDAC1 overexpression in a Ca2+-dependent manner, raising the possibility that elevating [Ca2+]i represents a common mechanism for various apoptosis stimuli, subsequently leading to an elevation in VDAC1 expression. We, therefore, suggest that the upregulation of VDAC1 expression constitutes a major mechanism by which apoptosis inducers lead to apoptosis (Figure 2).
Although many studies in various experimental systems have demonstrated increased VDAC1 expression levels following apoptosis stimulation (Table 1), only a few have linked VDAC1 overexpression to the rise in [Ca2+]i following apoptosis induction. Indeed, the expression level of VDAC1 has been shown to be a crucial factor in the process of mitochondria-mediated apoptosis (141, 165–175). Moreover, exogenous VDAC1 expression leads to apoptosis in the absence of any apoptotic stimulus (32, 34, 137, 141, 144, 151, 165, 179). There are several potential Ca2+-dependent steps that could contribute to the process of gene expression and a few, such as mRNA transcription, elongation, splicing, stability, and translation, have been suggested as being regulated by Ca2+ (180, 181).
This new mode of action for apoptosis stimulus involving increased expression of VDAC1 leading to dynamic VDAC1 oligomerization, release of Cyto c, and apoptosis provides a platform for developing a new class of anticancer drugs modulating VDAC1 expression via its promoter.
VDAC1 and Ca2+ in Cancer and Other Diseases
Various cancer hallmarks, such as proliferation, migration, angiogenesis, invasion abilities, and resistance to cell death, are associated with alterations of Ca2+ homeostasis (2). As a transporter of metabolites and Ca2+, VDAC1 contributes to the metabolic phenotype of cancer cells, possibly as reflected in its overexpression in many cancer types (182, 183). Moreover, its downregulation resulted in reduced metabolite exchanges between mitochondria and cytosol and inhibited cell and tumor growth (122, 176, 182, 184, 185).
Tumor cells exhibit a well-developed capacity for modulating [Ca2+]i levels by remodeling the cellular machinery that participates in processes that determine Ca2+ dynamics and homeostasis, as well as changes in sensitivity to cell death induction (186). It was recently demonstrated that the basal [Ca2+]m uptake via the ER–mitochondria junction is essential for tumorigenic cell viability, and that inhibition of this pathway in cancer cells might be used as a therapeutic approach (187). Moreover, some cancer cells are addicted to such constitutive Ca2+ transfer to sustain their mitochondrial metabolism, particularly nucleoside production (188). Thus, the increase in VDAC1 levels in cancer (182, 183) also contributes to this enhanced transport of Ca2+.
In diabetic mouse coronary vascular endothelial cells (MCECs), VDAC levels were increased, as were [Ca2+]m, mitochondrial O2 production, and PTP opening activity (189). Downregulation of VDAC1 in diabetic MCECs decreased [Ca2+]m and subsequently normalized the levels of PTP activity and mitochondrial ROS production (190). VDAC1 has proposed to mediate the protective effects of hesperidin, a bioactive flavonoid compound, against amyloid β-induced mitochondrial dysfunction, mitochondrial PTP opening, [Ca2+]i increase, and ROS production (191). It has also shown that blocking of VDAC1-mediated [Ca2+]m release in Schwann cells prevented demyelinating neuropathies (192). Thus, VDAC function in Ca2+ homeostasis is connected to several diseases.
Author Contributions
VS-B wrote the review; SD and AM helped in writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This research was supported by a grant from the Israel Science Foundation 307/13 and by Sima and Philip Needleman research funds.
References
1. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol (2003) 4:517–29. doi: 10.1038/nrm1155
2. Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer (2008) 8:361–75. doi:10.1038/nrc2374
3. Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol (2000) 1:11–21. doi:10.1038/35036191
4. Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol (2012) 13:566–78. doi:10.1038/nrm3412
5. Cox DA, Matlib MA. A role for the mitochondrial Na(+)-Ca2+ exchanger in the regulation of oxidative phosphorylation in isolated heart mitochondria. J Biol Chem (1993) 268:938–47.
6. Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry (2012) 51:2959–73. doi:10.1021/bi2018909
7. Nicholls DG. Mitochondria and calcium signaling. Cell Calcium (2005) 38:311–7. doi:10.1016/j.ceca.2005.06.011
8. Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium (2000) 28:285–96. doi:10.1054/ceca.2000.0168
9. Xia H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med (2004) 10:816–20. doi:10.1038/nm1076
10. Giacomello M, Drago I, Pizzo P, Pozzan T. Mitochondrial Ca2+ as a key regulator of cell life and death. Cell Death Differ (2007) 14:1267–74. doi:10.1038/sj.cdd.4402147
11. Maechler P, Kennedy ED, Pozzan T, Wollheim CB. Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic beta-cells. EMBO J (1997) 16:3833–41. doi:10.1093/emboj/16.13.3833
12. Lee B, Miles PD, Vargas L, Luan P, Glasco S, Kushnareva Y, et al. Inhibition of mitochondrial Na+-Ca2+ exchanger increases mitochondrial metabolism and potentiates glucose-stimulated insulin secretion in rat pancreatic islets. Diabetes (2003) 52:965–73. doi:10.2337/diabetes.52.4.965
13. Prins D, Michalak M. Organellar calcium buffers. Cold Spring Harb Perspect Biol (2011) 3:1–16. doi:10.1101/cshperspect.a004069
14. Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta (2009) 1787:1309–16. doi:10.1016/j.bbabio.2009.01.005
15. Nichols BJ, Denton RM. Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol Cell Biochem (1995) 149:203–12. doi:10.1007/BF01076578
16. Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell (2010) 142:270–83. doi:10.1016/j.cell.2010.06.007
17. Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol (2000) 529:37–47. doi:10.1111/j.1469-7793.2000.00037.x
18. De Stefani D, Rizzuto R, Pozzan T. Enjoy the trip: calcium in mitochondria back and forth. Annu Rev Biochem (2016) 85:161–92. doi:10.1146/annurev-biochem-060614-034216
19. Santo-Domingo J, Demaurex N. Calcium uptake mechanisms of mitochondria. Biochim Biophys Acta (2010) 1797:907–12. doi:10.1016/j.bbabio.2010.01.005
20. Sekler I. Standing of giants shoulders the story of the mitochondrial Na+Ca2+ exchanger. Biochem Biophys Res Commun (2015) 460:50–2. doi:10.1016/j.bbrc.2015.02.170
21. Takeuchi A, Kim B, Matsuoka S. The destiny of Ca(2+) released by mitochondria. J Physiol Sci (2015) 65:11–24. doi:10.1007/s12576-014-0326-7
22. Nita LI, Hershfinkel M, Sekler I. Life after the birth of the mitochondrial Na+/Ca2+ exchanger, NCLX. Sci China Life Sci (2015) 58:59–65. doi:10.1007/s11427-014-4789-9
23. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature (2011) 476:341–5. doi:10.1038/nature10234
24. De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature (2011) 476:336–40. doi:10.1038/nature10230
25. Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A (2010) 107:436–41. doi:10.1073/pnas.0908099107
26. Boyman L, Williams GS, Khananshvili D, Sekler I, Lederer WJ. NCLX: the mitochondrial sodium calcium exchanger. J Mol Cell Cardiol (2013) 59:205–13. doi:10.1016/j.yjmcc.2013.03.012
27. Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J (2001) 358:147–55. doi:10.1042/bj3580147
28. Rapizzi E, Pinton P, Szabadkai G, Wieckowski MR, Vandecasteele G, Baird G, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol (2002) 159:613–24. doi:10.1083/jcb.200205091
29. Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta (2007) 1768:2510–5. doi:10.1016/j.bbamem.2007.06.002
30. Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J Biol Chem (2006) 281:17347–58. doi:10.1074/jbc.M600906200
31. De Pinto V, Guarino F, Guarnera A, Messina A, Reina S, Tomasello FM, et al. Characterization of human VDAC isoforms: a peculiar function for VDAC3? Biochim Biophys Acta (2010) 1797:1268–75. doi:10.1016/j.bbabio.2010.01.031
32. Shoshan-Barmatz V, Ben-Hail D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion (2012) 12:24–34. doi:10.1016/j.mito.2011.04.001
33. Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med (2010) 31:227–85. doi:10.1016/j.mam.2010.03.002
34. Shoshan-Barmatz V, Mizrachi D. VDAC1: from structure to cancer therapy. Front Oncol (2012) 2:164. doi:10.3389/fonc.2012.00164
35. Dolder M, Wendt S, Wallimann T. Mitochondrial creatine kinase in contact sites: interaction with porin and adenine nucleotide translocase, role in permeability transition and sensitivity to oxidative damage. Biol Signals Recept (2001) 10:93–111. doi:10.1159/000046878
36. Wu W, Zhao S. Metabolic changes in cancer: beyond the Warburg effect. Acta Biochim Biophys Sin (Shanghai) (2013) 45:18–26. doi:10.1093/abbs/gms104
37. Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol (2001) 155:1003–15. doi:10.1083/jcb.200105057
38. De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, et al. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ (2012) 19:267–73. doi:10.1038/cdd.2011.92
39. Huang H, Hu X, Eno CO, Zhao G, Li C, White C. An interaction between Bcl-xL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. J Biol Chem (2013) 288:19870–81. doi:10.1074/jbc.M112.448290
40. Ben-Hail D, Shoshan-Barmatz V. VDAC1-interacting anion transport inhibitors inhibit VDAC1 oligomerization and apoptosis. Biochim Biophys Acta (2016) 1863:1612–23. doi:10.1016/j.bbamcr.2016.04.002
41. Chen H, Gao W, Yang Y, Guo S, Wang H, Wang W, et al. Inhibition of VDAC1 prevents Ca(2)(+)-mediated oxidative stress and apoptosis induced by 5-aminolevulinic acid mediated sonodynamic therapy in THP-1 macrophages. Apoptosis (2014) 19:1712–26. doi:10.1007/s10495-014-1045-5
42. Charuk JH, Pirraglia CA, Reithmeier RA. Interaction of ruthenium red with Ca2(+)-binding proteins. Anal Biochem (1990) 188:123–31. doi:10.1016/0003-2697(90)90539-L
43. Israelson A, Abu-Hamad S, Zaid H, Nahon E, Shoshan-Barmatz V. Localization of the voltage-dependent anion channel-1 Ca2+-binding sites. Cell Calcium (2007) 41:235–44. doi:10.1016/j.ceca.2006.06.005
44. Gincel D, Vardi N, Shoshan-Barmatz V. Retinal voltage-dependent anion channel: characterization and cellular localization. Invest Ophthalmol Vis Sci (2002) 43:2097–104.
45. Israelson A, Arzoine L, Abu-hamad S, Khodorkovsky V, Shoshan-Barmatz V. A photoactivable probe for calcium binding proteins. Chem Biol (2005) 12:1169–78. doi:10.1016/j.chembiol.2005.08.006
46. Israelson A, Zaid H, Abu-Hamad S, Nahon E, Shoshan-Barmatz V. Mapping the ruthenium red-binding site of the voltage-dependent anion channel-1. Cell Calcium (2008) 43:196–204. doi:10.1016/j.ceca.2007.05.006
47. Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. J Biol Chem (2012) 287:23152–61. doi:10.1074/jbc.M112.345918
48. Arbel N, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem (2010) 285:6053–62. doi:10.1074/jbc.M109.082990
49. Kholmukhamedov EL, Czerny C, Lovelace G, Beeson KC, Baker T, Johnson CB, et al. [The role of the voltage-dependent anion channels in the outer membrane of mitochondria in the regulation of cellular metabolism]. Biofizika (2010) 55:822–33.
50. Malia TJ, Wagner G. NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry (2007) 46:514–25. doi:10.1021/bi061577h
51. Monaco G, Decrock E, Arbel N, van Vliet AR, La Rovere RM, De Smedt H, et al. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. J Biol Chem (2015) 290:9150–61. doi:10.1074/jbc.M114.622514
52. Shimizu S, Konishi A, Kodama T, Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc Natl Acad Sci U S A (2000) 97:3100–5. doi:10.1073/pnas.97.7.3100
53. Sugiyama T, Shimizu S, Matsuoka Y, Yoneda Y, Tsujimoto Y. Activation of mitochondrial voltage-dependent anion channel by a pro-apoptotic BH3-only protein Bim. Oncogene (2002) 21:4944–56. doi:10.1038/sj.onc.1205621
54. Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene (2008) 27:4221–32. doi:10.1038/onc.2008.63
55. Tsujimoto Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. J Cell Physiol (2003) 195:158–67. doi:10.1002/jcp.10254
56. Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell (2015) 163:712–23. doi:10.1016/j.cell.2015.09.053
57. Lv BF, Yu CF, Chen YY, Lu Y, Guo JH, Song QS, et al. Protein tyrosine phosphatase interacting protein 51 (PTPIP51) is a novel mitochondria protein with an N-terminal mitochondrial targeting sequence and induces apoptosis. Apoptosis (2006) 11:1489–501. doi:10.1007/s10495-006-8882-9
58. De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau K-F, Ackerley S, et al. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet (2012) 21:1299–311. doi:10.1093/hmg/ddr559
59. Sun J, Liao JK. Functional interaction of endothelial nitric oxide synthase with a voltage-dependent anion channel. Proc Natl Acad Sci U S A (2002) 99:13108–13. doi:10.1073/pnas.202260999
60. Viola HM, Adams AM, Davies SM, Fletcher S, Filipovska A, Hool LC. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc Natl Acad Sci U S A (2014) 111:E2905–14. doi:10.1073/pnas.1402544111
61. Shoshan-Barmatz V, Israelson A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: characterization, modulation and possible function. J Membr Biol (2005) 204:57–66. doi:10.1007/s00232-005-0749-4
62. Shoshan-Barmatz V, Zalk R, Gincel D, Vardi N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim Biophys Acta (2004) 1657:105–14. doi:10.1016/j.bbabio.2004.02.009
63. Csordás G, Renken C, Várnai P, Walter L, Weaver D, Buttle KF, et al. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol (2006) 174:915–21. doi:10.1083/jcb.200604016
64. Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta (2014) 1837:461–9. doi:10.1016/j.bbabio.2013.10.015
65. Booth DM, Joseph SK, Hajnoczky G. Subcellular ROS imaging methods: relevance for the study of calcium signaling. Cell Calcium (2016) 60:65–73. doi:10.1016/j.ceca.2016.05.001
66. Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev (1999) 79:1127–55.
67. Foskett JK, Philipson B. The mitochondrial Ca2 + uniporter complex. J Mol Cell Cardiol (2015) 78:3–8. doi:10.1016/j.yjmcc.2014.11.015
68. Vasington FD, Gazzotti P, Tiozzo R, Carafoli E. The effect of ruthenium red on Ca 2+ transport and respiration in rat liver mitochondria. Biochim Biophys Acta (1972) 256:43–54. doi:10.1016/0005-2728(72)90161-2
69. Kamer KJ, Sancak Y, Mootha VK. The uniporter: from newly identified parts to function. Biochem Biophys Res Commun (2014) 449:370–2. doi:10.1016/j.bbrc.2014.04.143
70. Marchi S, Pinton P. The mitochondrial calcium uniporter complex: molecular components, structure and physiopathological implications. J Physiol (2014) 592:829–39. doi:10.1113/jphysiol.2013.268235
71. Lee Y, Min CK, Kim TG, Song HK, Lim Y, Kim D, et al. Structure and function of the N-terminal domain of the human mitochondrial calcium uniporter. EMBO Rep (2015) 16:1318–33. doi:10.15252/embr.201540436
72. Csordás G, Golenár T, Seifert EL, Kamer KJ, Sancak Y, Perocchi F, et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca(2+) uniporter. Cell Metab (2013) 17:976–87. doi:10.1016/j.cmet.2013.04.020
73. Hoffman NE, Chandramoorthy HC, Shanmughapriya S, Zhang XQ, Vallem S, Doonan PJ, et al. SLC25A23 augments mitochondrial Ca2+ uptake, interacts with MCU, and induces oxidative stress-mediated cell death. Mol Biol Cell (2014) 25:936–47. doi:10.1091/mbc.E13-08-0502
74. Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell (2012) 151:630–44. doi:10.1016/j.cell.2012.10.011
75. Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One (2013) 8:e55785. doi:10.1371/journal.pone.0055785
76. Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science (2013) 342:1379–82. doi:10.1126/science.1242993
77. Paupe V, Prudent J, Dassa EP, Rendon OZ, Shoubridge EA. CCDC90A (MCUR1) is a cytochrome c oxidase assembly factor and not a regulator of the mitochondrial calcium uniporter. Cell Metab (2015) 21:109–16. doi:10.1016/j.cmet.2014.12.004
78. Pozzan T, Magalhaes P, Rizzuto R. The comeback of mitochondria to calcium signalling. Cell Calcium (2000) 28:279–83. doi:10.1054/ceca.2000.0166
79. Xu S, Chisholm AD. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev Cell (2014) 31:48–60. doi:10.1016/j.devcel.2014.08.002
80. Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, et al. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J Biol Chem (2012) 287:34445–54. doi:10.1074/jbc.M112.392084
81. Drago I, De Stefani D, Rizzuto R, Pozzan T. Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes. Proc Natl Acad Sci U S A (2012) 109:12986–91. doi:10.1073/pnas.1210718109
82. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol (2013) 15:1464–72. doi:10.1038/ncb2868
83. Qiu J, Tan Y-W, Hagenston AM, Martel M-A, Kneisel N, Skehel PA, et al. Mitochondrial calcium uniporter MCU controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat Commun (2013) 4:2034. doi:10.1038/ncomms3034
84. Quan X, Nguyen TT, Choi S-K, Xu S, Das R, Cha S-K, et al. Essential role of mitochondrial Ca2+ uniporter in the generation of mitochondrial pH gradient and metabolism-secretion coupling in insulin-releasing cells. J Biol Chem (2015) 290:4086–96. doi:10.1074/jbc.M114.632547
85. Rasmussen TP, Wu Y, Joiner ML, Koval OM, Wilson NR, Luczak ED, et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc Natl Acad Sci U S A (2015) 112:9129–34. doi:10.1073/pnas.1504705112
86. Holmström KM, Pan X, Liu JC, Menazza S, Liu J, Nguyen TT, et al. Assessment of cardiac function in mice lacking the mitochondrial calcium uniporter. J Mol Cell Cardiol (2015) 85:178–82. doi:10.1016/j.yjmcc.2015.05.022
87. Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep (2015) 12:23–34. doi:10.1016/j.celrep.2015.06.017
88. Murphy E, Pan X, Nguyen T, Liu J, Holmstrom KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem Biophys Res Commun (2014) 449:384–5. doi:10.1016/j.bbrc.2014.04.144
89. Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, et al. The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep (2015) 12:15–22. doi:10.1016/j.celrep.2015.06.002
90. Murphy E, Eisner DA. Regulation of intracellular and mitochondrial Na in health and disease. Circ Res (2009) 104:292–303. doi:10.1161/CIRCRESAHA.108.189050
91. Kim B, Takeuchi A, Hikida M, Matsuoka S. Roles of the mitochondrial Na(+)-Ca(2+) exchanger, NCLX, in B lymphocyte chemotaxis. Sci Rep (2016) 6:28378. doi:10.1038/srep28378
92. Jiang D, Zhao L, Clish CB, Clapham DE. Letm1, the mitochondrial Ca2+/H+ antiporter, is essential for normal glucose metabolism and alters brain function in Wolf-Hirschhorn syndrome. Proc Natl Acad Sci U S A (2013) 110:E2249–54. doi:10.1073/pnas.1308558110
93. Palty R, Hershfinkel M, Sekler I. Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. J Biol Chem (2012) 287:31650–7. doi:10.1074/jbc.R112.355867
94. Arco AD, Satrústegui J. New mitochondrial carriers: an overview. Cell Mol Life Sci (2005) 62:2204–27. doi:10.1007/s00018-005-5197-x
95. McCormack JG, Denton RM. Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev Neurosci (1993) 15:165–73. doi:10.1159/000111332
96. Palty R, Ohana E, Hershfinkel M, Volokita M, Elgazar V, Beharier O, et al. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J Biol Chem (2004) 279:25234–40. doi:10.1074/jbc.M401229200
97. De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. J Biol Chem (2014) 289:20377–85. doi:10.1074/jbc.M113.540898
98. Williams GS, Boyman L, Lederer WJ. Mitochondrial calcium and the regulation of metabolism in the heart. J Mol Cell Cardiol (2015) 78:35–45. doi:10.1016/j.yjmcc.2014.10.019
99. Takeuchi A, Kim B, Matsuoka S. The mitochondrial Na+-Ca2+ exchanger, NCLX, regulates automaticity of HL-1 cardiomyocytes. Sci Rep (2013) 3:2766. doi:10.1038/srep02766
100. Murphy E, Cross H, Steenbergen C. Sodium regulation during ischemia versus reperfusion and its role in injury. Circ Res (1999) 84:1469–70. doi:10.1161/01.RES.84.12.1469
101. Babsky A, Doliba N, Doliba N, Savchenko A, Wehrli S, Osbakken M. Na+ effects on mitochondrial respiration and oxidative phosphorylation in diabetic hearts. Exp Biol Med (2001) 226:543–51.
102. Kostic M, Ludtmann MH, Bading H, Hershfinkel M, Steer E, Chu CT, et al. PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep (2015) 13:376–86. doi:10.1016/j.celrep.2015.08.079
103. Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis (2007) 12:835–40. doi:10.1007/s10495-006-0525-7
104. Rasola A, Bernardi P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium (2011) 50:222–33. doi:10.1016/j.ceca.2011.04.007
105. Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest (2010) 120:3680–7. doi:10.1172/JCI43171
106. Galluzzi L, Kroemer G. Mitochondrial apoptosis without VDAC. Nat Cell Biol (2007) 9:487–9. doi:10.1038/ncb0507-487
107. Biasutto L, Azzolini M, Szabò I, Zoratti M. The mitochondrial permeability transition pore in AD 2016: an update. Biochim Biophys Acta (2016) 1863:2515–30. doi:10.1016/j.bbamcr.2016.02.012
108. Shanmughapriya S, Rajan S, Hoffman NE, Higgins AM, Tomar D, Nemani N, et al. SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol Cell (2015) 60:47–62. doi:10.1016/j.molcel.2015.08.009
109. Doonan PJ, Chandramoorthy HC, Hoffman NE, Zhang X, Cardenas C, Shanmughapriya S, et al. LETM1-dependent mitochondrial Ca2+ flux modulates cellular bioenergetics and proliferation. FASEB J (2014) 28:4936–49. doi:10.1096/fj.14-256453
110. Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science (2009) 326:144–7. doi:10.1126/science.1175145
111. Mailloux RJ, Harper M-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic Biol Med (2011) 51:1106–15. doi:10.1016/j.freeradbiomed.2011.06.022
112. Hoppe UC. Mitochondrial calcium channels. FEBS Lett (2010) 584:1975–81. doi:10.1016/j.febslet.2010.04.017
113. Wagner S, De Bortoli S, Schwarzlander M, Szabo I. Regulation of mitochondrial calcium in plants versus animals. J Exp Bot (2016) 67:3809–29. doi:10.1093/jxb/erw100
114. Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem (2001) 276:21482–8. doi:10.1074/jbc.M101486200
115. Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim Biophys Acta (2005) 1717:1–10. doi:10.1016/j.bbamem.2005.09.016
116. Jakob R, Beutner G, Sharma VK, Duan Y, Gross RA, Hurst S, et al. Molecular and functional identification of a mitochondrial ryanodine receptor in neurons. Neurosci Lett (2014) 575:7–12. doi:10.1016/j.neulet.2014.05.026
117. Feng S, Li H, Tai Y, Huang J, Su Y, Abramowitz J, et al. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc Natl Acad Sci U S A (2013) 110:11011–6. doi:10.1073/pnas.1309531110
118. De Pinto V, Messina A, Accardi R, Aiello R, Guarino F, Tomasello MF, et al. New functions of an old protein: the eukaryotic porin or voltage dependent anion selective channel (VDAC). Ital J Biochem (2003) 52:17–24.
119. Granville DJ, Gottlieb RA. The mitochondrial voltage-dependent anion channel (VDAC) as a therapeutic target for initiating cell death. Curr Med Chem (2003) 10:1527–33. doi:10.2174/0929867033457214
120. Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie (2002) 84:153–66. doi:10.1016/S0300-9084(02)01375-5
121. Shoshan-Barmatz V, Gincel D. The voltage-dependent anion channel: characterization, modulation, and role in mitochondrial function in cell life and death. Cell Biochem Biophys (2003) 39:279–92. doi:10.1385/CBB:39:3:279
122. Shoshan-Barmatz V, Golan M. Mitochondrial VDAC1: function in cell life and death and a target for cancer therapy. Curr Med Chem (2012) 19:714–35. doi:10.2174/092986712798992110
123. Shoshan-Barmatz V, Israelson A, Brdiczka D, Sheu SS. The voltage-dependent anion channel (VDAC): function in intracellular signalling, cell life and cell death. Curr Pharm Des (2006) 12:2249–70. doi:10.2174/138161206777585111
124. Tsujimoto Y, Shimizu S. The voltage-dependent anion channel: an essential player in apoptosis. Biochimie (2002) 84:187–93. doi:10.1016/S0300-9084(02)01370-6
125. Vyssokikh MY, Brdiczka D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim Pol (2003) 50:389–404.
126. Yuan S, Fu Y, Wang X, Shi H, Huang Y, Song X, et al. Voltage-dependent anion channel 1 is involved in endostatin-induced endothelial cell apoptosis. FASEB J (2008) 22:2809–20. doi:10.1096/fj.08-107417
127. Doran E, Halestrap AP. Cytochrome c release from isolated rat liver mitochondria can occur independently of outer-membrane rupture: possible role of contact sites. Biochem J (2000) 348 Pt 2:343–50. doi:10.1042/bj3480343
128. Martinou JC, Desagher S, Antonsson B. Cytochrome c release from mitochondria: all or nothing. Nat Cell Biol (2000) 2:E41–3. doi:10.1038/35004069
129. Zalk R, Israelson A, Garty ES, Azoulay-Zohar H, Shoshan-Barmatz V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem J (2005) 386:73–83. doi:10.1042/BJ20041356
130. Bernardi P. The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death. Biochim Biophys Acta (1996) 1275:5–9. doi:10.1016/0005-2728(96)00041-2
131. Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J (1999) 341(Pt 2):233–49. doi:10.1042/bj3410233
132. Betaneli V, Petrov EP, Schwille P. The role of lipids in VDAC oligomerization. Biophys J (2012) 102:523–31. doi:10.1016/j.bpj.2011.12.049
133. Goncalves RP, Buzhynskyy N, Prima V, Sturgis JN, Scheuring S. Supramolecular assembly of VDAC in native mitochondrial outer membranes. J Mol Biol (2007) 369:413–8. doi:10.1016/j.jmb.2007.03.063
134. Hoogenboom BW, Suda K, Engel A, Fotiadis D. The supramolecular assemblies of voltage-dependent anion channels in the native membrane. J Mol Biol (2007) 370:246–55. doi:10.1016/j.jmb.2007.04.073
135. Keinan N, Tyomkin D, Shoshan-Barmatz V. Oligomerization of the mitochondrial protein voltage-dependent anion channel is coupled to the induction of apoptosis. Mol Cell Biol (2010) 30:5698–709. doi:10.1128/MCB.00165-10
136. Shoshan-Barmatz V, Arbel N, Arzoine L. VDAC, the voltage-dependent anion channel: function, regulation & mitochondrial signaling in cell life and death. Cell Sci (2008) 4:74–118.
137. Shoshan-Barmatz V, Keinan N, Zaid H. Uncovering the role of VDAC in the regulation of cell life and death. J Bioenerg Biomembr (2008) 40:183–91. doi:10.1007/s10863-008-9147-9
138. Shoshan-Barmatz V, Mizrachi D, Keinan N. Oligomerization of the mitochondrial protein VDAC1: from structure to function and cancer therapy. Prog Mol Biol Transl Sci (2013) 117:303–34. doi:10.1016/B978-0-12-386931-9.00011-8
139. Ujwal R, Cascio D, Chaptal V, Ping P, Abramson J. Crystal packing analysis of murine VDAC1 crystals in a lipidic environment reveals novel insights on oligomerization and orientation. Channels (Austin) (2009) 3:167–70. doi:10.4161/chan.3.3.9196
140. Keinan N, Pahima H, Ben-Hail D, Shoshan-Barmatz V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim Biophys Acta (2013) 1833:1745–54. doi:10.1016/j.bbamcr.2013.03.017
141. Weisthal S, Keinan N, Ben-Hail D, Arif T, Shoshan-Barmatz V. Ca(2+)-mediated regulation of VDAC1 expression levels is associated with cell death induction. Biochim Biophys Acta (2014) 1843:2270–81. doi:10.1016/j.bbamcr.2014.03.021
142. Huang L, Han J, Ben-Hail D, He L, Li B, Chen Z, et al. A new fungal diterpene induces VDAC1-dependent apoptosis in Bax/Bak-deficient cells. J Biol Chem (2015) 290:23563–78. doi:10.1074/jbc.M115.648774
143. Ben-Hail D, Begas-Shvartz R, Shalev M, Shteinfer-Kuzmine A, Gruzman A, Reina S, et al. Novel compounds targeting the mitochondrial protein VDAC1 inhibit apoptosis and protect against mitochondria dysfunction. J Biol Chem (2016) 291:24986–5003. doi:10.1074/jbc.M116.744284
144. Abu-Hamad S, Arbel N, Calo D, Arzoine L, Israelson A, Keinan N, et al. The VDAC1 N-terminus is essential both for apoptosis and the protective effect of anti-apoptotic proteins. J Cell Sci (2009) 122:1906–16. doi:10.1242/jcs.040188
145. Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J Biol Chem (2009) 284:3946–55. doi:10.1074/jbc.M803614200
146. Azoulay-Zohar H, Israelson A, Abu-Hamad S, Shoshan-Barmatz V. In self-defence: hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem J (2004) 377:347–55. doi:10.1042/bj20031465
147. Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene (2006) 25:4777–86. doi:10.1038/sj.onc.1209603
148. Pastorino JG, Hoek JB, Shulga N. Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res (2005) 65:10545–54. doi:10.1158/0008-5472.CAN-05-1925
149. Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim Biophys Acta (2002) 1555:14–20. doi:10.1016/S0005-2728(02)00248-7
150. Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem (2002) 277:7610–8. doi:10.1074/jbc.M109950200
151. Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ (2005) 12:751–60. doi:10.1038/sj.cdd.4401599
152. Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem (2008) 283:13482–90. doi:10.1074/jbc.M708216200
153. Shimizu S, Ide T, Yanagida T, Tsujimoto Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J Biol Chem (2000) 275:12321–5.
154. Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature (1999) 399:483–7. doi:10.1038/20959
155. Shi Y, Chen J, Weng C, Chen R, Zheng Y, Chen Q, et al. Identification of the protein-protein contact site and interaction mode of human VDAC1 with Bcl-2 family proteins. Biochem Biophys Res Commun (2003) 305:989–96. doi:10.1016/S0006-291X(03)00871-4
156. Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta (2011) 1813:521–31. doi:10.1016/j.bbamcr.2010.12.019
158. Gerasimenko JV, Gerasimenko OV, Palejwala A, Tepikin AV, Petersen OH, Watson AJM. Menadione-induced apoptosis: roles of cytosolic Ca2+ elevations and the mitochondrial permeability transition pore. J Cell Sci (2002) 115:485–97.
159. Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol (2003) 5:1051–61. doi:10.1038/ncb1063
160. Miyamoto S, Howes AL, Adams JW, Dorn GW, Brown JH. Ca2+ dysregulation induces mitochondrial depolarization and apoptosis: role of Na+/Ca2+ exchanger and AKT. J Biol Chem (2005) 280:38505–12. doi:10.1074/jbc.M505223200
161. Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol (2008) 70:73–91. doi:10.1146/annurev.physiol.70.021507.105852
162. Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc Natl Acad Sci U S A (2015) 112:1779–84. doi:10.1073/pnas.1410723112
163. Borahay MA, Kilic GS, Yallampalli C, Snyder RR, Hankins GDV, Al-Hendy A, et al. Simvastatin potently induces calcium-dependent apoptosis of human leiomyoma cells. J Biol Chem (2014) 289:35075–86. doi:10.1074/jbc.M114.583575
164. Hedgepeth SC, Garcia MI, Wagner LE, Rodriguez AM, Chintapalli SV, Snyder RR, et al. The BRCA1 tumor suppressor binds to inositol 1,4,5-trisphosphate receptors to stimulate apoptotic calcium release. J Biol Chem (2015) 290:7304–13. doi:10.1074/jbc.M114.611186
165. Jiang N, Kham SK, Koh GS, Suang Lim JY, Ariffin H, Chew FT, et al. Identification of prognostic protein biomarkers in childhood acute lymphoblastic leukemia (ALL). J Proteomics (2011) 74:843–57. doi:10.1016/j.jprot.2011.02.034
166. Castagna A, Antonioli P, Astner H, Hamdan M, Righetti SC, Perego P, et al. A proteomic approach to cisplatin resistance in the cervix squamous cell carcinoma cell line A431. Proteomics (2004) 4:3246–67. doi:10.1002/pmic.200400835
167. Sharaf el dein O, Gallerne C, Brenner C, Lemaire C. Increased expression of VDAC1 sensitizes carcinoma cells to apoptosis induced by DNA cross-linking agents. Biochem Pharmacol (2012) 83:1172–82. doi:10.1016/j.bcp.2012.01.017
168. Jung JY, Han CR, Jeong YJ, Kim HJ, Lim HS, Lee KH, et al. Epigallocatechin gallate inhibits nitric oxide-induced apoptosis in rat PC12 cells. Neurosci Lett (2007) 411:222–7. doi:10.1016/j.neulet.2006.09.089
169. Voehringer DW, Hirschberg DL, Xiao J, Lu Q, Roederer M, Lock CB, et al. Gene microarray identification of redox and mitochondrial elements that control resistance or sensitivity to apoptosis. Proc Natl Acad Sci U S A (2000) 97:2680–5. doi:10.1073/pnas.97.6.2680
170. Cheng SL, Liu RH, Sheu JN, Chen ST, Sinchaikul S, Tsay GJ. Toxicogenomics of A375 human malignant melanoma cells treated with arbutin. J Biomed Sci (2007) 14:87–105. doi:10.1007/s11373-006-9130-6
171. Nawarak J, Huang-Liu R, Kao SH, Liao HH, Sinchaikul S, Chen ST, et al. Proteomics analysis of A375 human malignant melanoma cells in response to arbutin treatment. Biochim Biophys Acta (2009) 1794:159–67. doi:10.1016/j.bbapap.2008.09.023
172. Moin SM, Panteva M, Jameel S. The hepatitis E virus Orf3 protein protects cells from mitochondrial depolarization and death. J Biol Chem (2007) 282:21124–33. doi:10.1074/jbc.M701696200
173. Liu Z, Bengtsson S, Krogh M, Marquez M, Nilsson S, James P, et al. Somatostatin effects on the proteome of the LNCaP cell-line. Int J Oncol (2007) 30:1173–9.
174. Tomasello F, Messina A, Lartigue L, Schembri L, Medina C, Reina S, et al. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res (2009) 19:1363–76. doi:10.1038/cr.2009.98
175. Liu S, Ishikawa H, Tsuyama N, Li FJ, Abroun S, Otsuyama KI, et al. Increased susceptibility to apoptosis in CD45(+) myeloma cells accompanied by the increased expression of VDAC1. Oncogene (2006) 25:419–29. doi:10.1038/sj.onc.1208982
176. Abu-Hamad S, Sivan S, Shoshan-Barmatz V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc Natl Acad Sci U S A (2006) 103:5787–92. doi:10.1073/pnas.0600103103
177. Ghosh T, Pandey N, Maitra A, Brahmachari SK, Pillai B. A role for voltage-dependent anion channel VDAC1 in polyglutamine-mediated neuronal cell death. PLoS One (2007) 2:e1170. doi:10.1371/journal.pone.0001170
178. Godbole A, Varghese J, Sarin A, Mathew MK. VDAC is a conserved element of death pathways in plant and animal systems. Biochim Biophys Acta (2003) 1642:87–96. doi:10.1016/S0167-4889(03)00102-2
179. Shoshan-Barmatz V, Keinan N, Abu-Hamad S, Tyomkin D, Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim Biophys Acta (2010) 1797:1281–91. doi:10.1016/j.bbabio.2010.03.003
180. Mellstrom B, Savignac M, Gomez-Villafuertes R, Naranjo JR. Ca2+-operated transcriptional networks: molecular mechanisms and in vivo models. Physiol Rev (2008) 88:421–49. doi:10.1152/physrev.00041.2005
181. Naranjo JR, Mellström B. Ca2+-dependent transcriptional control of Ca2+ homeostasis. J Biol Chem (2012) 287:31674–80. doi:10.1074/jbc.R112.384982
182. Arif T, Vasilkovsky L, Refaely Y, Konson A, Shoshan-Barmatz V. Silencing VDAC1 expression by siRNA inhibits cancer cell proliferation and tumor growth in vivo. Mol Ther Nucleic Acids (2014) 3:e159. doi:10.1038/mtna.2014.9
183. Shoshan-Barmatz V, Ben-Hail D, Admoni L, Krelin Y, Tripathi SS. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim Biophys Acta (2015) 1848:2547–75. doi:10.1016/j.bbamem.2014.10.040
184. Koren I, Raviv Z, Shoshan-Barmatz V. Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol Ther (2010) 9:1046–52. doi:10.4161/cbt.9.12.11879
185. Arif T, Kerlin Y, Nakdimon I, Benharroch D, Paul A, Dadon-Klein D, et al. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol (2017). doi:10.1093/neuonc/now297
186. Marchi S, Pinton P. Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol (2016) 29:1–6. doi:10.1016/j.coph.2016.03.002
187. Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep (2016) 14:2313–24. doi:10.1016/j.celrep.2016.02.030
188. Rimessi A, Patergnani S, Bonora M, Wieckowski MR, Pinton P. Mitochondrial Ca(2+) remodeling is a prime factor in oncogenic behavior. Front Oncol (2015) 5:143. doi:10.3389/fonc.2015.00143
189. Sasaki K, Donthamsetty R, Heldak M, Cho YE, Scott BT, Makino A. VDAC: old protein with new roles in diabetes. Am J Physiol Cell Physiol (2012) 303:C1055–60. doi:10.1152/ajpcell.00087.2012
190. Truong AH, Murugesan S, Youssef KD, Makino A. Mitochondrial ion channels in metabolic disease. In: Levitan I, Dopico MA, editors. Vascular Ion Channels in Physiology and Disease. Switzerland: Springer International Publishing (2016). p. 397–419.
191. Wang DM, Li SQ, Zhu XY, Wang Y, Wu WL, Zhang XJ. Protective effects of hesperidin against amyloid-beta (Abeta) induced neurotoxicity through the voltage dependent anion channel 1 (VDAC1)-mediated mitochondrial apoptotic pathway in PC12 cells. Neurochem Res (2013) 38:1034–44. doi:10.1007/s11064-013-1013-4
Keywords: apoptosis, Ca2+ transporters, mitochondria, oligomerization, voltage-dependent anion channel
Citation: Shoshan-Barmatz V, De S and Meir A (2017) The Mitochondrial Voltage-Dependent Anion Channel 1, Ca2+ Transport, Apoptosis, and Their Regulation. Front. Oncol. 7:60. doi: 10.3389/fonc.2017.00060
Received: 20 December 2016; Accepted: 17 March 2017;
Published: 10 April 2017
Edited by:
Paolo Pinton, University of Ferrara, ItalyReviewed by:
Ildikò Szabò, University of Padua, ItalyCatherine Brenner Jan, Institut national de la santé et de la recherche médicale (INSERM), France
Copyright: © 2017 Shoshan-Barmatz, De and Meir. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Varda Shoshan-Barmatz, dmFyZGFzYkBiZ3UuYWMuaWw=