 Francesca D’Auria1*
Francesca D’Auria1* Lucia Centurione2
Lucia Centurione2 Maria Antonietta Centurione3
Maria Antonietta Centurione3 Antonio Angelini2,4
Antonio Angelini2,4 Roberta Di Pietro2
Roberta Di Pietro2
- 1Department of Cardiac and Vascular Surgery, Campus Bio-Medico University of Rome, Rome, Italy
- 2Department of Medicine and Ageing Sciences, G. d’Annunzio University, Chieti, Italy
- 3Institute of Molecular Genetics, National Research Council-Pavia, Chieti, Italy
- 4Ageing Research Center, CeSI, G. d’Annunzio University Foundation, Chieti, Italy
Cyclic AMP response element binding (CREB) protein is a member of the CREB/activating transcription factor (ATF) family of transcription factors that play an important role in the cell response to different environmental stimuli leading to proliferation, differentiation, apoptosis, and survival. A number of studies highlight the involvement of CREB in the resistance to ionizing radiation (IR) therapy, demonstrating a relationship between IR-induced CREB family members’ activation and cell survival. Consistent with these observations, we have recently demonstrated that CREB and ATF-1 are expressed in leukemia cell lines and that low-dose radiation treatment can trigger CREB activation, leading to survival of erythro-leukemia cells (K562). On the other hand, a number of evidences highlight a proapoptotic role of CREB following IR treatment of cancer cells. Since the development of multiple mechanisms of resistance is one key problem of most malignancies, including those of hematological origin, it is highly desirable to identify biological markers of responsiveness/unresponsiveness useful to follow-up the individual response and to adjust anticancer treatments. Taking into account all these considerations, this mini-review will be focused on the involvement of CREB/ATF family members in response to IR therapy, to deepen our knowledge of this topic, and to pave the way to translation into a therapeutic context.
Introduction
Ionizing radiation (IR) induces loss of proliferative capacity as well as cell death by apoptosis and necrosis. Apoptosis can happen during interphase before division after the G2 block caused by IR (fast apoptosis) or after one or more divisions (late apoptosis). Some radiosensitive cells, such as lymphocytes, thymocytes, and intestinal crypt cells, undergo fast apoptosis without cell division after IR, while irradiated mouse leukemia cells undergo a G2 block and the apoptotic fraction begins to increase on the release from this G2 block (1–3). Furthermore, cells can respond by trying to counteract IR-induced damage to the membrane and to repair single- and double-stranded DNA breaks (respectively, SSB and DSB) by activating survival/signaling pathways. When the apoptosis ensues or the cell survives, genes that control complex pathways are activated (4). Amidst the proteins involved in cell responses to IR, a key role has been assigned to cyclic AMP response element binding (CREB) nuclear transcription factor (5, 6) and to members of the protein kinase C (PKC) family (7). CREB protein is a member of the CREB/ATF family of transcription factors that play a key role in the nuclear responses to a variety of external signals that lead to cell growth and proliferation, differentiation, apoptosis, and survival (8–10). Moreover, CREB has been also implicated in the adaptive response and, in particular, in the immune system balance (11–14). CREB protein is a 43-kDa basic leucine zipper (bzip) transcription factor composed of a C-terminal basic DNA binding domain, an adjacent leucine zipper domain, and a kinase-inducible transcriptional activation domain. The activation of CREB depends not only on the cell type but also on the stimulus, which has been administered (12, 15–18). CREB activation results from different posttranslational modifications, the main of which is the phosphorylation of serine-133 (Ser-133) that is mediated by several factors (19, 20). The signaling molecules responsible for CREB Ser-133 phosphorylation include Akt/protein kinase B (15, 21). Akt stimulates target gene expression via CREB in a phospho-Ser-133-dependent manner, promoting cell survival in response to growth factor stimulation (22). PKCs belong to a class of serine–threonine kinases, including at least 12 closely related iso-enzymes that have distinct, and in some cases opposing, roles in cell growth, differentiation, apoptosis, and survival (23). Among these 12 iso-enzymes, PKC delta is known for its activation in response to a variety of genotoxic stresses (24). PKC delta activity is controlled not only by binding diacylglycerol or phorbol ester but also by other molecular mechanisms, such as phosphorylation and proteolysis. In particular, an activation loop, a turn, and a hydrophobic motif site have been demonstrated at Thr505, Ser-643, and Ser-662, respectively, and these sites are phosphorylated in vivo (25). PKC can also undergo phosphorylation on tyrosine residues, such as Tyr-52, Tyr-155, Tyr-187, Tyr-311, Tyr-332, and Tyr-565, depending on the cell stimulus (26). Moreover, a catalytically active 40-kDa fragment of PKC delta arises from the proteolysis in cells exposed to IR, DNA damaging drugs, or anti-FAS antibody (27–31).
Based on these findings and on the complex and various molecular interactions involving CREB/ATF family members, this mini-review will be focused on the involvement of CREB/ATF family members in response to IR exposure to deepen our insight into this matter and contribute to the understanding of CREB-mediated radio-resistance/radio-sensitivity mechanisms. In fact, the knowledge of apoptotic and survival pathways recruited in tumor cells may be useful to establish targeted therapies by combining selective inhibitors or stimulators of fundamental signaling proteins with conventional chemotherapy, hormone therapy, and radiotherapy (RT) regimens.
The Cellular Response to IR
Cancer cells respond to IR in a heterogeneous manner depending on their intrinsic properties (DNA repair capability, proliferation status) or extrinsic environment (degree of hypoxia within the tumor population) (32). In other cases, the degree of radio-sensitivity depends on the cell heterogeneity within the tumor mass. Both in vitro and in vivo clonogenic assays were used in early studies to address various determinants of tumor radio-response (33). These investigations lead to the identification of cells capable to initiate the growth of new tumors after in vivo transplantation. These cells were named tumor clonogens, cancer-initiating cells, or cancer stem cells, and their frequency inside a tumor mass was related to radio-resistance (32).
Ionizing radiation-induced damage leads to the activation of complex signaling cascades, inducing with different mechanisms DNA repair, stress response genes, cell-cycle arrest, cell death, or cell survival (34). A number of key molecules involved in different signaling pathways are modulated by IR, as recently detailed in a comprehensive review (35). The activation of these molecules ultimately results in altered expression of series of target genes, whose promoters or enhancers may contain binding sites for one or more transcription factors, able, in turn, to influence the transcription of multiple genes. Among these factors, NF-κB is widely recognized for its antiapoptotic and pro-survival effects possibly leading to radio-resistance (36). NF-κB has been demonstrated to be a downstream target of DNA DSB-activated [ATM kinase- and DNA-dependent protein kinase (DNA-PK)-dependent] pathway since it is activated following ATM-mediated phosphorylation and ubiquitin-dependent degradation of IκBα. DNA-PK activates NF-κB in a different manner since it is able to hyper-phosphorylate IκBβ that binds to NF-κB in the nucleus, thus preventing the binding of IκBα to NF-κB (37). In spite of these well-characterized mechanisms, it has been demonstrated that different IR doses are able to recruit different signaling pathways in different systems to cause activation of NF-κB (38). Interestingly, different levels of constitutive NF-κB activity in colorectal cancer (CRC) cells show a strong correlation with different levels of radio-sensitivity and spontaneous apoptosis (39). NF-κB together with c-jun and c-fos are considered as immediate response-genes transcription factors since they are switched on within minutes after IR, possibly thwarting the general downregulation of transcription upon irradiation and allowing privileged transcription of specific genes. NF-κB and p300/CBP (CREB-binding protein) have been involved in the rescue effects of irradiated cells (40). Both of them are downstream targets of PI3K/Akt pathway, and both of them are able to trigger cell death or cell survival. In a just-published paper of our research group, we highlighted the involvement of CREB/ATF family members in the radiation response of two different lymphoma cell lines with a different level of radio-sensitivity (Daudi and Ramos) suggesting a proapoptotic role of CREB and, surprisingly and unexpectedly, a pro-necrotic role of NF-κB after 3-Gy IR dose (6). Other authors have recently demonstrated that CREB is phosphorylated in response to IR through p38 MAPK pathway in a time- and dose-dependent manner. They also showed that the inhibition of CREB transactivation by decoy oligonucleotides significantly increases the radio-sensitivity of multiple human cancer cell lines (AGS, MCF-7, HeLa, SaOs-2, DU-145, and HepG2) (41). Furthermore, cancer cells that ectopically expressed dominant-negative mutant CREB (KCREB) or were treated with p38 MAPK inhibitors displayed a higher sensitivity to IR than wild-type parental cells or control-treated cells. In light of this evidence, CREB appears to protect tumor cells from IR and, thus, the combination of CREB inhibition plus IR could be evaluated as a possible RT approach in some cellular systems. Interestingly, CREB-dependent radio-resistance was also shown in normal cells like primary human fibroblasts that are capable to weather high doses of IR and to avoid IR-induced apoptosis. To identify possible antiapoptotic pathways in human fibroblasts, Bluwstein et al. (42) carried out a kinetic pathway analysis based on reverse phase protein arrays combined with extensive western blot analysis. Through these methods, these authors showed the activation of DNA damage–response networks (e.g., phosphorylation of MKK3/6, p38, MK2, p53, Chk1, and Hsp27), pro-survival molecules (CREB, MEK-ERK, PKC), and antiapoptotic markers (Bcl-2 and Bad). In particular, they observed that the inhibition or downregulation of PKC in primary human fibroblasts caused IR-dependent downregulation of the identified pro-survival pathways, including CREB, and antiapoptotic pathways, like Bad and Bcl-2, leading to proliferation arrest and apoptosis (19, 21) and suggesting a regulatory function of these pathways in response to IR. The radiation damage to biological systems is caused by the type of radiation, the total dose of radiation, the dose rate, and the region of the body exposed. IR-induced cytotoxicity in mammalian cells involves various modes of cell death, including apoptosis (type 1 cell death), necrosis (oncosis), autophagy (type 2 cell death), accelerated senescence, and mitotic catastrophe (43–45). The different types of cell death occur both in vitro and in vivo in response to IR in cancer cells as well as in normal cells (46). The cellular choice of a specific mode depends on various factors, including the specific cell type involved, the dose of IR absorbed, and whether the cell is proliferating and/or transformed (35). Increased expression of p53 (in its phosphorylated or acetylated form) is certainly a critical step in mediating the cellular response to IR-induced DNA damage (34). In fact, p53 activation leads to de novo synthesis of proapoptotic molecules that mediate intrinsic (e.g., Bax, Puma) or extrinsic (e.g., Fas) apoptotic cell death. Moreover, p53-dependent induction of p21/waf1 or the upregulation of other cell-cycle inhibitory proteins (e.g., p16 INK4a) is able to interfere with cell-cycle machinery resulting in accelerated senescence. The precise mechanisms for the biological selection of a specific pathway of cell death following IR exposure have not been established unequivocally. Additionally, the mechanisms through which IR causes death of the cells directly affected by IR differ from those through which bystander cells undergo cell death. Mitotic catastrophe is the main form of cell death induced by IR that results from premature or inappropriate entry of the cells into mitosis. It has been recently proposed that it could result from a combination of deficient cell-cycle checkpoints (in particular the DNA structure checkpoints and the spindle assembly checkpoint) and cellular damage (47). Cell death occurring during the metaphase/anaphase transition (mitotic catastrophe) is characterized by the activation of caspase-2 and/or mitochondrial membrane permeabilization (MMP) with the release of cell death effectors, such as apoptosis-inducing factor and the caspase-9 and caspase-3 activator cytochrome c. It is still debated whether mitotic catastrophe can be considered a special case of apoptosis or a completely different phenomenon. In favor of the first hypothesis, findings obtained by Castedo et al. (45) in HeLa cell lines forming syncitia demonstrated that cells died during the metaphase of the cell cycle as a result of the activation of caspase-2 upstream of MMP and cytochrome c release, leading to caspase-3 activation and chromatin condensation. By contrast, Roninson et al. (48) argued that mitotic catastrophe would be fundamentally different from apoptosis since the prevention of apoptosis by overexpressing, for example, Bcl-2 in etoposide-treated HeLa cells (49) or by overexpressing MDR in irradiated tumor cells (50) can actually enhance the frequency of catastrophic mitoses.
IR and Hematological Malignancies
Myelodysplastic syndrome (MDS) is a group of blood disorders where hematopoietic stem cell undergoes an abnormal maturation and differentiation into one or more blood lineages. Disease progression is related to an increased genomic instability, and a high number of patients go on to develop acute myeloid leukemia (51–53). Primarily a disease of the elderly, it can also develop following chemotherapy. It has been reported that CBP heterozygous mice have an increased incidence of hematological malignancies, and it has been shown that CBP is one of the genes altered by chromosomal translocations found in patients suffering from therapy-related MDS (54). This evidence has led Zimmer et al. (55) to investigate whether hematopoietic tumor development in CBP+/− mice is preceded by a myelodysplastic phase and whether it could uncover molecular mechanisms that might contribute to its development. In particular, they observed that CBP+/− mice invariably develop myelodysplastic/myeloproliferative neoplasm within 9–12 months of age, are hypersensitive to IR, and show a marked decrease in PARP1 activity after IR. In addition, protein levels of XRCC1 and APEX1, key components of base-excision repair mechanism, appear reduced in CBP+/− cells not exposed to IR or upon targeted knockdown of CBP levels. This evidence provides validation of a novel myelodysplastic/myeloproliferative neoplasm mouse model and, more importantly, point at defective repair of DNA damage as a contributing factor to the pathogenesis of this currently incurable disease (55). In line with this evidence, the role of p38 MAPK in activating CREB metabolic pathway supported the events leading to erythroid differentiation (56). Furthermore, Cataldi et al. (57) demonstrated the effect of IR on PKC delta and Akt/CREB signaling pathways in Jurkat T human lymphoblastoid cells, known for being sensitive to IR (58, 59). More recently, our research group has shown the involvement of CREB family members in the IR response of Ramos B lymphoma cells, an Epstein–Barr virus-negative cell line derived from an American Burkitt’s lymphoma (6). In this case, CREB activation was unexpectedly linked to apoptotic cell death induced by IR whereas the early induction and activation of NF-κB1 in Ramos cells was associated with necrotic cell death and cell-cycle regulation (6). In another experimental model (represented by K562 erythro-leukemia cell lines), an IR dose of 1.5 Gy was considered as suboptimal, whereas a dose of 6 Gy was identified as the threshold dose above which cells lose clonogenicity, and the apoptosis rate increases (5). Interestingly, 6 Gy is the suboptimal dose delivered in RT protocols for the treatment of human hematological tumors, whereas it induces evident damage in many cancer cell lines (58). As previously demonstrated, K562 are radio-resistant human leukemia cells that express a high peroxiredoxins (Prx) stability (60). Prx are an extended family of small antioxidant proteins that conserve a thioredoxin-dependent catalytic function contributing to cell protection from reactive oxygen species, which are one of the deleterious intracellular effectors of IR damage. In fact, IR passing throughout living tissues generates reactive free radicals that can interact with critical macromolecules, such as DNA, proteins, or membrane lipids, inducing cell damage and, potentially, cell dysfunction and death (61).
IR and Gastrointestinal Cancer
The gastrointestinal epithelium (GI) is very sensitive to IR and chemotherapy (62), and is prone to IR-induced malignancy (63). CREB has a recognized role in neo-oncogenesis occurring in T-cell and myeloid leukemia, hepatocellular carcinoma, melanoma, clear cell sarcoma, lung adenocarcinoma, as well as CRC (64). Various external stimuli can induce CREB activation via various kinase pathways, including stress networks. These signaling pathways lead to CREB phosphorylation at Ser-133, causing interaction with co-activators, including CBP or p300 (65, 66). Ultimately, p300 and CBP are not only CREB co-factors but are also able to activate additional transcription factors, including Myb. Myb is a fundamental transcription factor for proliferation and differentiation of the intestinal epithelium (67). The transcriptional coregulator p300 might integrate the co-activation and transcriptional activities of both phospho-CREB and Myb, enabling finely tuned cellular responses in the GI. Sampurno et al. (68) carried out a study in which an inducible CREB knockout (KO) model was used to define the role of CREB in the GI tract of adult mice under both homeostasis and stress conditions. Extensive phospho-CREB activation was shown in Apc (Min/+) mouse adenomas. Furthermore, in CRC, phospho-CREB was found in most cells associated with elevated CREB-target genes, including MRP2 (multidrug resistance-associated protein 2) involved in the vesicular transport of a platinum-based anticancer drug known as Oxaliplatin, used in the chemotherapy treatment of CRC. These observations lead the authors to hypothesize a role for CREB in GI malignancy but not in GI homeostasis where Myb appears to be critical (69). The same authors showed that defects in cell proliferation are sometimes revealed or exacerbated by stressing the GI epithelium with IR, as it was observed in Myb-mutant mice (69), although Myb expression itself was unaffected in CREB KO GI mice not exposed to IR. In another study performed in rat colon transversum, Pejchal et al. (70) demonstrated that during the first 24 h after IR the activated forms of ATF-2 and CREB participate at the cellular response after whole-body gamma IR within the low-dose range of 0.25–1 Gy and up to 10 Gy. Based on these findings, these authors proposed the detection of phospho-ATF-2 (Thr-69/71) and phospho-CREB (Ser-133) as a prospective bio-dosimetric tool for irradiated enterocytes in vivo.
IR and Lung Cancer
Lung cancer is a malignant tumor arising from the respiratory epithelium that represents a leading cause of cancer death in both males and females worldwide. Lung cancer is divided into two morphologic groups: small cell lung cancer and non-small cell lung cancer (NSCLC). In spite of the number of therapeutic approaches (surgery, RT, and/or chemotherapy), the prognosis of lung cancer remains dismal, with a 5-year survival rate for NSCLC of around 15% (71). Thus, much research has been devoted to improving the efficiency of chemotherapy and RT based on a better understanding of the molecular mechanisms underlying cell proliferation and death (34, 72). In recent years, CBP and its homolog p300 have been demonstrated to play a role in respiratory epithelial tumorigenesis (73), and an increased expression in CREB protein has been associated with poor survival in NSCLC (74). However, controversial observations have been published regarding the role of CREB pathway in response to RT of lung cancer cells. In fact, Kim and Juhnn (72) demonstrated that cAMP signaling reduces sirtuin 6 (SIRT6) expression in human NSCLC by activating the PKA and CREB pathways. SIRT6 is able to remove acetyl groups from histones and to regulate genomic stability and cell viability, and when it is reduced, IR-induced apoptosis of NSCLC cells is increased. Moreover, Choi et al. (75) investigated the effect of the G-protein alpha signaling system on IR-induced apoptosis of p53-null H1299 lung adenocarcinoma cells, linking the increase in IR-induced apoptosis to the upregulation of Bak mediated by CREB and AP-1. By contrast, Cho et al. (76) reported that podophyllotoxin acetate (PA), a new anticancer drug candidate, inhibits IR-induced activation of CREB1/signal transducer and activator of transcription 3 (STAT3) pathway leading to the enhancement of apoptosis of wild-type p53 A459 NSCLC cancer cells, especially when used in combination with other chemotherapeutic compounds. In addition, they reported that IR increases migration/invasion and epithelial/mesenchymal transition (EMT) of A459 by increasing CREB1 and STAT3 activation and that PA blocks EGFR–p38/ERK–CREB-1/STAT3–EMT pathway to inhibit IR-induced invasion/migration. Taken together, these findings confirm that CREB/ATF family proteins behave as stress response signaling molecules but with pleiotropic effects depending on different experimental systems characteristics.
IR and Prostate Cancer
Radiotherapy is a first choice therapy when prostate cancer is a localized mass. Although some patients are good responders to the treatment, nearly 70% of the patients experience recurrent tumors. However, the molecular mechanisms underlying tumor recurrence remain largely unknown. Deng et al. (77) showed that fractionated IR induces differentiation of LNCaP prostate cancer cells into neuroendocrine (NE)-like cells. These cells are involved in prostate cancer progression, androgen-independent growth, and poor prognosis. Further analyses revealed that CREB and ATF-2 function as transcriptional activator and repressor, respectively, of NE-like differentiation and that IR induces NE-like differentiation by increasing the nuclear content of phospho-CREB and the cytoplasmic accumulation of ATF-2. Consistent with these notions, stable expression of a non-phosphorylatable CREB or a constitutively nuclear-localized ATF-2 in LNCaP cells inhibits the IR-induced NE-like cell differentiation. In a recent paper, Suarez et al. (78) showed the increased activation of CREB protein during the course of fractionated IR-induced NE differentiation (NED). To determine whether targeting NED could be explored as an IR sensitization approach, these authors employed two CREB targeting strategies: (i) CREB knockdown and (ii) overexpression of ACREB, a dominant-negative mutant of CREB. Their results showed that ACREB expression increased fractioned IR-induced cell death and sensitized prostate cancer cells to IR. Consistent with this, knockdown of CREB also inhibited fractioned IR-induced NED. Interestingly, the authors observed that CREB targeting primarily increases IR-induced pre-mitotic apoptosis. Taken together, these results suggest that targeting CREB could be considered as an IR sensitization approach for prostate cancer treatment.
Conclusion
Understanding the specific cellular mechanisms and signal transduction networks responsible for radio-resistance or radio-sensitivity is important in improving human cancer RT since it may help in establishing more efficient targeted therapies by combining selective inhibitors or stimulators of key signaling proteins with conventional RT. In this regard, different potential therapeutic targets have been identified in modulating IR sensitivity of various malignancies among which poly(ADP-ribose) polymerase (PARP), DNA-PK, DNA ligase IV, Mre11-Rad50-Nbs1 (MRN complex), and checkpoint kinase (79). Moreover, although this is outside the focus of this paper, a number of agents not directly involved in DNA damage repair or DNA damage response have been demonstrated to increase radio-sensitivity by inhibiting homology directed repair, many of these, including inhibitors of histone deacetylase (CHR-3996, CHF-2845, 4SC-202, etc.) and HSP90 (IPI-504, AUY922, STA-9090, etc.), are under evaluation in ongoing clinical trials (79). Accumulating evidence suggests that CREB/ATF family proteins act as signaling molecules activated in response to stress (Table 1). In various cancers, CREB protects tumor cells from IR in others it promotes apoptosis. Different approaches (CREB mutants, CRE decoy oligonucleotides) and a number of small molecules (Ro 31-8220, NSC 12155, NSC 45576, naphthol AS-E phosphate) have been used to inhibit CREB function and transcriptional activity, as recently reported in a comprehensive review (80). Nevertheless, according to http://ClinicalTrials.gov website no clinical trials combining CREB inhibitors with RT are currently ongoing. The combination of CREB inhibition and IR can be certainly considered as a promising RT approach but a careful evaluation of appropriate conditions is needed before going from bench to bedside. Indeed, we think that further preclinical work is still necessary to identify and better characterize the molecular networks involved by this pleiotropic actor in exerting cell death or cell survival in response to IR treatment of cancer cells.
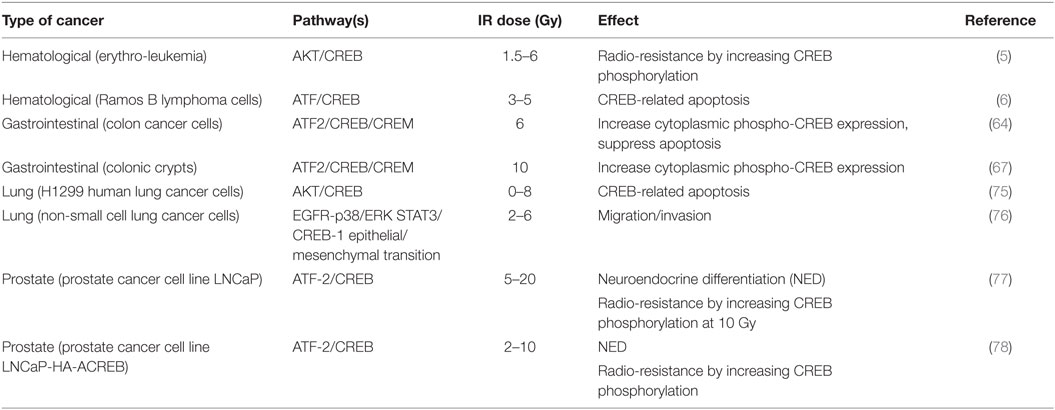
Table 1. Involvement of cyclic AMP response element binding (CREB) pathway in different types of cancer cells after exposure to X-ray ionizing radiation (IR) treatment.
Author Contributions
FDA: study and analysis of scientific literature and preparation of the manuscript and Table 1. LC: critical reading of the manuscript. MC: preparation of the reference list and Table 1. AA: analysis of the manuscript. RDP: supervision, correction, and improvement in the preparation of the manuscript and Table 1.
Conflict of Interest Statement
This review has been done in the absence of any commercial or financial relationships that can be considered as a potential conflict of interest.
Acknowledgments
This manuscript was payed with MIUR (Italian Ministry of University and Research) funds granted to RDP in 2016.
References
1. Radford IR, Murphy TK, Radley JM, Ellis SL. Radiation response of mouse lymphoid and myeloid cell lines. Part II. Apoptotic death is shown by all lines examined. Int J Radiat Biol (1994) 65:217–27. doi: 10.1080/09553009414550251
2. Tauchi H, Sawada S. Analysis of mitotic cell death caused by radiation in mouse leukaemia L5178Y cells: apoptosis is the ultimate form of cell death following mitotic failure. Int J Radiat Biol (1994) 65:449–55. doi:10.1080/09553009414550521
3. Merritt A, Allen TD, Potten CS, Hickman JA. Apoptosis in small intestinal epithelia from p-53-null mice: evidence for a delayed p53-independent G2/M associated cell death after γ-irradiation. Oncogene (1997) 14:2759–66. doi:10.1038/sj.onc.1201126
4. Miura Y. Oxidative stress, radiation-adaptive responses, and aging. J Radiat Res (2004) 45:357–72. doi:10.1269/jrr.45.357
5. Cataldi A, di Giacomo V, Rapino M, Genovesi D, Rana RA. Cyclic nucleotide response element binding protein (CREB) activation promotes survival signal in human K562 erythroleukemia cells exposed to ionising radiation/etoposide combined treatment. J Radiat Res (2006) 47:113–20. doi:10.1269/jrr.47.113
6. Di Nisio C, Sancilio S, di Giacomo V, Rapino M, Sancillo L, Genovesi D, et al. Involvement of cyclic-nucleotide response element-binding family members in the radiation response of Ramos B lymphoma cells. Int J Oncol (2016) 48(1):28–36. doi:10.3892/ijo.2015.3238
7. Cataldi A, Miscia S, Centurione L, Rapino M, Bosco D, Grifone G, et al. Role of nuclear PKC delta in mediating caspase-3-upregulation in Jurkat T leukemic cells exposed to ionizing radiation. J Cell Biochem (2002) 86:553–60. doi:10.1002/jcb.10251
8. Di Pietro R, di Giacomo V, Caravatta L, Sancilio S, Rana RA, Cataldi A. Cyclic nucleotide response element binding (CREB) protein activation is involved in K562 erythroleukemia cells differentiation. J Cell Biochem (2007) 100:1070–9. doi:10.1002/jcb.21106
9. Caravatta L, Sancilio S, di Giacomo V, Rana RA, Cataldi A, Di Pietro R. PI3-K/Akt-dependent activation of cAMP-response element-binding (CREB) protein in Jurkat T leukemia cells treated with TRAIL. J Cell Physiol (2008) 214:192–200. doi:10.1002/jcp.21186
10. D’Auria F, Di Pietro R. Role of CREB protein family members in human haematological malignancies. In: Gali-Muhtasib H, editor. Cancer Treatment/Book 2. Rijeka: InTech (2013). p. 201–26.
11. Wilson BE, Mochon E, Boxer L. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol (1996) 16:5546–56. doi:10.1128/MCB.16.10.5546
12. De Cesare D, Fimia GM, Sassone Corsi P. Signaling routes to CREM and CREB: plasticity in transcriptional activation. Trends Biochem Sci (1999) 24:281–5. doi:10.1016/S0968-0004(99)01414-0
13. Saeki K, You A, Suzuki E, Yazaki Y, Takaku F. Aberrant expression of c-AMP-response-element-binding protein (CREB) induces apoptosis. Biochem J (1999) 343:249–55. doi:10.1042/bj3430249
14. Andreatta CP, Nahreini P, Hanson AJ, Prasad KN. Regulated expression of VP16CREB in neuroblastoma cells: analysis of differentiation and apoptosis. J Neurosci Res (2004) 78:570–9. doi:10.1002/jnr.20304
15. Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem (1998) 273:32377–9. doi:10.1074/jbc.273.49.32377
16. Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J Neurochem (1999) 73:1836–42.
17. Dodson GE, Tibbetts RS. DNA replication stress induced phosphorylation of cyclic AMP response element-binding protein mediated by ATM. J Biol Chem (2006) 281:1692–7. doi:10.1074/jbc.M509577200
18. Di Giulio C, Rapino M, Zingariello M, Antonucci A, Cataldi A. PKC alpha-mediated CREB activation is oxygen- and age-dependent in rat myocardial tissue. Histochem Cell Biol (2007) 127:327–33. doi:10.1007/s00418-006-0245-7
19. Muthusamy N, Leiden JM. A protein kinase C-, Ras-, and RSK2 dependent signal transduction pathway activates the cAMP-responsive element binding protein transcription factor following T cell receptor engagement. J Biol Chem (1998) 273:22841–7. doi:10.1074/jbc.273.35.22841
20. Shi Y, Venkataraman SL, Dodson GE, Mabb AM, LeBlanc S, Tibbetts RS. Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc Natl Acad Sci U S A (2004) 101:5898–903. doi:10.1073/pnas.0307718101
21. Pugazenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE, et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem (2000) 275:10761–6. doi:10.1074/jbc.275.15.10761
22. Uchiyama T, Engelman RM, Maulik N, Das DK. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation (2004) 109:3042–9. doi:10.1161/01.CIR.0000130647.29030.90
23. Musashi M, Ota S, Shiroshita N. The role of protein kinase C isoforms in cell proliferation and apoptosis. Int J Hematol (2000) 72:12–9.
24. Brodie C, Blumberg PM. Regulation of cell apoptosis by PKC delta. Apoptosis (2003) 8:19–27. doi:10.1023/A:1021640817208
25. Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, et al. Phosphorylation sites of protein kinase C in H2O2-treated cells and its activation by tyrosine kinase in vitro. Proc Natl Acad Sci U S A (2001) 98:6587–92. doi:10.1073/pnas.111158798
26. Li W, Mischak H, Yu JC, Wang LM, Mushinski JF, Heidaran MA, et al. Tyrosine phosphorylation of protein kinase C-δ in response to its activation. J Biol Chem (1994) 269:2349–52.
27. Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, et al. Proteolytic activation of protein kinase C-δ by an ICE-like protease in apoptotic cells. EMBO J (1995) 14:6148–56.
28. Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, et al. Proteolytic activation of protein kinase C delta by an ICE-CED 3-like protease induces characteristics of apoptosis. J Exp Med (1996) 184:2399–404. doi:10.1084/jem.184.6.2399
29. Mizuno K, Noda K, Araki T, Imaoka T, Kobayashi Y, Akita Y, et al. The proteolytic cleavage of protein kinase C isotypes, which generates kinase and regulatory fragments, correlates with Fas-mediated and 12-O-tetradecanoyl-phorbol-13-acetate-induced apoptosis. Eur J Biochem (1997) 250:7–18. doi:10.1111/j.1432-1033.1997.00007.x
30. Takahashi M, Mukai H, Toshimori M, Miyamoto M, Ono Y. Proteolytic activation of PKN by caspase-3 or related protease during apoptosis. Proc Natl Acad Sci U S A (1998) 95:11566–71. doi:10.1073/pnas.95.20.11566
31. Lewis AE, Susarla R, Wong BC, Langman MJ, Eggo MC. Protein kinase C delta is not activated by caspase-3 and its inhibition is sufficient to induce apoptosis in the colon cancer line, COLO 205. Cell Signal (2005) 17:253–62. doi:10.1016/j.cellsig.2004.07.005
32. Hittelman WN, Liao Y, Wang L, Milas L. Are cancer stem cells radioresistant? Future Oncol (2010) 6(10):1563–76. doi:10.2217/fon.10.121
33. Milas L, Hittelman WN. Cancer stem cells and tumor response to therapy: current problems and future prospects. Semin Radiat Oncol (2009) 19(2):96–105. doi:10.1016/j.semradonc.2008.11.004
34. Impicciatore G, Sancilio S, Miscia S, Di Pietro R. Nutlins and ionizing radiation in cancer therapy. Curr Pharm Des (2010) 16(12):1427–42. doi:10.2174/138161210791033932
35. Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci (2016) 17:102–34. doi:10.3390/ijms17010102
36. Bai M, Ma X, Li X, Wang X, Mei Q, Li X, et al. The accomplices of NF-κB lead to radioresistance. Curr Protein Pept Sci (2015) 16(4):279–94. doi:10.2174/138920371604150429152328
37. Tran K, Merika M, Thanos D. Distinct functional properties of IkappaB alpha and IkappaB beta. Mol Cell Biol (1997) 17(9):5386–99. doi:10.1128/MCB.17.9.5386
38. Criswell T, Leskov K, Miyamoto S, Luo G, Boothman DA. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene (2003) 22(37):5813–27. doi:10.1038/sj.onc.1206680
39. Voboril R, Weberova-Voborilova J. Constitutive NF-kappaB activity in colorectal cancer cells: impact on radiation-induced NF-kappaB activity, radiosensitivity, and apoptosis. Neoplasma (2006) 53(6):518–23.
40. Lam RKK, Han W, Yu KN. Unirradiated cells rescue cells exposed to ionizing radiation: activation of NF-kB pathway in irradiated cells. Mutat Res (2015) 782:23–33. doi:10.1016/j.mrfmmm.2015.10.004
41. Park SI, Park SJ, Lee J, Kim HE, Park SJ, Sohn JW, et al. Inhibition of cyclic AMP response element-directed transcription by decoy oligonucleotides enhances tumor-specific radiosensitivity. Biochem Biophys Res Commun (2016) 469(3):363–9. doi:10.1016/j.bbrc.2015.11.122
42. Bluwstein A, Kumar N, Léger K, Traenkle J, van Oostrum J, Rehrauer H, et al. PKC signaling prevents irradiation-induced apoptosis of primary human fibroblasts. Cell Death Dis (2013) 4(2):e498. doi:10.1038/cddis.2013.15
43. Leist M, Jäättelä M. Four deaths and a funeral: from caspases to alternative mechanisms. Nat Rev Mol Cell Biol (2001) 2(8):589–98. doi:10.1038/35085008
44. Lockshin RA, Zakeri Z. Caspase-independent cell deaths. Curr Opin Cell Biol (2002) 14(6):727–33. doi:10.1016/S0955-0674(02)00383-6
45. Castedo M, Perfettini J-L, Roumier T, Valent A, Raslova H, Yakushijin K, et al. Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene (2004) 23:4362–70. doi:10.1038/sj.onc.1207572
46. Panganiban RA, Snow AL, Day RM. Mechanisms of radiation toxicity in transformed and non-transformed cells. Int J Mol Sci (2013) 14(8):15931–58. doi:10.3390/ijms140815931
47. Castedo M, Perfettini J-L, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene (2004) 23:2825–37. doi:10.1038/sj.onc.1207528
48. Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat (2001) 4(5):303–13. doi:10.1054/drup.2001.0213
49. Lock RB, Stribinskiene L. Dual modes of death induced by etoposide in human epithelial tumor cells allow Bcl-2 to inhibit apoptosis without affecting clonogenic survival. Cancer Res (1996) 56(17):4006–12.
50. Ruth AC, Roninson IB. Effects of the multidrug transporter P-glycoprotein on cellular responses to ionizing radiation. Cancer Res (2000) 60(10):2576–8.
51. Corey SJ, Minden MD, Barber DL, Kantarjian H, Wang JC, Schimmer AD. Myelodysplastic syndromes: the complexity of stem-cell diseases. Nat Rev Cancer (2007) 7:118–29. doi:10.1038/nrc2047
52. Godley LA, Larson RA. Therapy-related myeloid leukemia. Semin Oncol (2008) 35:418–29. doi:10.1053/j.seminoncol.2008.04.012
53. Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood (2002) 100:2292–302. doi:10.1182/blood-2002-04-1199
54. Bedford DC, Kasper LH, Fukuyama T, Brindle PK. Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases. Epigenetics (2010) 5:9–15. doi:10.4161/epi.5.1.10449
55. Zimmer SN, Lemieux ME, Karia BP, Day C, Zhou T, Zhou Q. Mice heterozygous for CREB binding protein are hypersensitive to γ-radiation and invariably develop myelodysplastic/myeloproliferative neoplasm. Exp Hematol (2012) 40(4):295–306.e5. doi:10.1016/j.exphem.2011.12.004
56. di Giacomo V, Sancilio S, Caravatta L, Rana RA, Pietro R. Di, Cataldi A. Regulation of CREB activation by p38 mitogen activated protein kinase during human primary erythroblast differentiation. Int J Immunopathol Pharmacol (2009) 22(3):679–88. doi:10.1177/039463200902200313
57. Cataldi A, di Giacomo V, Rapino M, Zara S, Rana RA. Ionizing radiation induces apoptotic signal through protein kinase C-δ (delta) and survival signal through Akt and cyclic-nucleotide response element-binding protein (CREB) in Jurkat T cells. Biol Bull (2009) 217(2):202–12. doi:10.1086/BBLv217n2p202
58. Dahlberg WK, Azzam EI, Yu Y, Little JB. Response of human tumor cells of varying radiosensitivity and radiocurability to fractionated irradiation. Cancer Res (1999) 59:5365–9.
59. Di Pietro R, Centurione L, Sabatini N, Bosco D, Sancilio S, Garaci F, et al. Caspase-3 is dually regulated by apoptogenic factors mitochondrial release and by SAPK/JNK metabolic pathway in leukemic cells exposed to etoposide-ionizing radiation combined treatment. Int J Immunopathol Pharmacol (2004) 17:181–90.
60. Di Pietro R, Fang H, Fields K, Miller S, Flora M, Petricoin EC, et al. Peroxiredoxin genes are not induced in myeloid leukemia cells exposed to ionizing radiation. Int J Immunopathol Pharmacol (2006) 19(3):517–24. doi:10.1177/039463200601900307
61. Hosseinimehr SJ. Trends in the development of radioprotective agents. Drug Discov Today (2007) 12:794–805. doi:10.1016/j.drudis.2007.07.017
62. Novak JM, Collins JT, Donowitz M, Farman J, Sheahan DG, Spiro HM. Effects of radiation on the human gastrointestinal tract. J Clin Gastroenterol (1979) 1:9–39. doi:10.1097/00004836-197903000-00003
63. Chang ML, Hou JK. Cancer risk related to gastrointestinal diagnostic radiation exposure. Curr Gastroenterol Rep (2011) 13:449–57. doi:10.1007/s11894-011-0214-8
64. Nishihara H, Hwang M, Kizaka-Kondoh S, Eckmann L, Insel PA. Cyclic AMP promotes cAMP-responsive element-binding protein-dependent induction of cellular inhibitor of apoptosis protein-2 and suppresses apoptosis of colon cancer cells through ERK1/2 and p38 MAPK. J Biol Chem (2004) 279:26176–83. doi:10.1074/jbc.M313346200
65. Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature (1993) 365:855–9. doi:10.1038/365855a0
66. Lundblad JR, Kwok RP, Laurance ME, Harter ML, Goodman RH. Adenoviral E1 associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature (1995) 374:85–8. doi:10.1038/374085a0
67. Malaterre J, Carpinelli M, Ernst M, Alexander W, Cooke M, Sutton S, et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci U S A (2007) 104:3829–34. doi:10.1073/pnas.0610055104
68. Sampurno S, Bijenhof A, Cheasley D, Xu H, Robine S, Hilton D, et al. The Myb-p300-CREB axis modulates intestine homeostasis, radiosensitivity and tumorigenesis. Cell Death Dis (2013) 4(4):e605. doi:10.1038/cddis.2013.119
69. Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer (2008) 8:523–34. doi:10.1038/nrc2439
70. Pejchal J, Osterreicher J, Vilasová Z, Tichý A, Vàvrovà J. Expression of activated ATF-2, CREB and c-Myc in rat colon transversum after whole-body gamma-irradiation and its contribution to pathogenesis and biodosimetry. Int J Radiat Biol (2008) 84(4):315–24. doi:10.1080/09553000801953367
71. Govindan R, Page N, Morgensztern D, Read W, Tierney R, Vlahiotis A, et al. Changing epidemiology of small-cell lung cancer in the United States over the last 30 years: analysis of the surveillance, epidemiologic, and end results database. J Clin Oncol (2006) 24:4539–44. doi:10.1200/JCO.2005.04.4859
72. Kim EJ, Juhnn YS. Cyclic AMP signaling reduces sirtuin 6 expression in non-small cell lung cancer cells by promoting ubiquitin-proteasomal degradation via inhibition of the Raf-MEK-ERK (Raf/mitogen-activated extracellular signal-regulated kinase/extracellular signal-regulated kinase) pathway. J Biol Chem (2015) 290(15):9604–13. doi:10.1074/jbc.M114.633198
73. Karamouzis MV, Konstantinopoulos PA, Papavassiliou AG. Roles of CREB-binding protein (CBP)/p300 in respiratory epithelium tumorigenesis. Cell Res (2007) 17:324–32. doi:10.1038/cr.2007.10
74. Chao BH, Traynor AM, Yang DT, Zhang C, Pier T, Kim KM, et al. Automated quantitative analyses (AQUA) of cyclic-AMP-response-element-binding protein (CREB), phosphorylated CREB (pCREB), and ataxia-telangiectasia-mutated protein kinase (ATM) protein expression in patients (pts) undergoing thoracic radiation for locally advanced non-small cell lung cancer (NSCLC). J Clin Oncol (2012) 30(Suppl):abstr e13537.
75. Choi YJ, Kim SY, Oh JM, Juhnn YS. Stimulatory heterotrimeric G protein augments gamma ray-induced apoptosis by up-regulation of Bak expression via CREB and AP-1 in H1299 human lung cancer cells. Exp Mol Med (2009) 41(8):592–600. doi:10.3858/emm.2009.41.8.065
76. Cho JH, Hong WG, Jung YJ, Lee J, Lee E, Hwang SG. G-Ionizing radiation-induced activation of the EGFR-p38/ERK-STAT3/CREB-1-EMT pathway promotes the migration/invasion of non-small cell lung cancer cells and is inhibited by podophyllotoxin acetate. Tumour Biol (2015) 37(6):7315–25. doi:10.1007/s13277-015-4548-y
77. Deng X, Elzey BD, Poulson JM, Morrison WB, Ko SC, Hahn NM. Ionizing radiation induces prostate cancer neuroendocrine differentiation through interplay of CREB and ATF2: implications for disease progression. Am J Cancer Res (2011) 1(7):834–44. doi:10.1158/0008-5472.CAN-08-2229
78. Suarez CD, Deng X, Hu CD. Targeting CREB inhibits radiation-induced neuroendocrine differentiation and increases radiation-induced cell death in prostate cancer cells. Am J Cancer Res (2014) 4(6):850–61.
79. Gavande SN, Vander-Vere-Carozza PS, Hisnhaw HD, Jalal SI, Sears CR, Pawelczak KS, et al. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol Ther (2016) 160:65–83. doi:10.1016/j.pharmthera.2016.02.003
Keywords: cyclic AMP response element binding/activating transcription factor, radiotherapy, ionizing radiation, radio-resistance, radio-sensitivity, DNA repair, nuclear transcription factors, cancer
Citation: D’Auria F, Centurione L, Centurione MA, Angelini A and Di Pietro R (2017) Regulation of Cancer Cell Responsiveness to Ionizing Radiation Treatment by Cyclic AMP Response Element Binding Nuclear Transcription Factor. Front. Oncol. 7:76. doi: 10.3389/fonc.2017.00076
Received: 06 February 2017; Accepted: 07 April 2017;
Published: 05 May 2017
Edited by:
John Varlotto, University of Massachusetts Medical Center, USAReviewed by:
M. Christine Hollander, National Institutes of Health, USAMichael Wayne Epperly, University of Pittsburgh, USA
Copyright: © 2017 D’Auria, Centurione, Centurione, Angelini and Di Pietro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca D’Auria, Zi5kYXVyaWFAaG90bWFpbC5jb20=